 Qi Qi1†Mengxin Zhao1†
Qi Qi1†Mengxin Zhao1† Shiping Wang2,3Xingyu Ma1Yuxuan Wang4,5Ying Gao1
Shiping Wang2,3Xingyu Ma1Yuxuan Wang4,5Ying Gao1 Qiaoyan Lin6
Qiaoyan Lin6 Xiangzhen Li7
Xiangzhen Li7 Baohua Gu8Guoxue Li9
Baohua Gu8Guoxue Li9 Jizhong Zhou1,10,11,12
Jizhong Zhou1,10,11,12 Yunfeng Yang1*
Yunfeng Yang1*- 1State Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing, China
- 2Key Laboratory of Alpine Ecology and Biodiversity, Institute of Tibetan Plateau Research, Chinese Academy of Sciences, Beijing, China
- 3CAS Center for Excellence in Tibetan Plateau Earth Science, Beijing, China
- 4Department of Earth System Science, Tsinghua University, Beijing, China
- 5Department of Earth and Atmospheric Sciences, University of Houston, Houston, TX, United States
- 6Key Laboratory of Adaptation and Evolution of Plateau Biota, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining, China
- 7Key Laboratory of Environmental and Applied Microbiology, Environmental Microbiology Key Laboratory of Sichuan Province, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, China
- 8Environmental Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN, United States
- 9College of Resources and Environmental Science, China Agricultural University, Beijing, China
- 10Institute for Environmental Genomics and Department of Microbiology and Plant Biology, University of Oklahoma, Norman, OK, United States
- 11Earth Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, United States
- 12Collaborative Innovation Center for Regional Environmental Quality, School of Environment, Tsinghua University, Beijing, China
As the highest place of the world, the Tibetan plateau is a fragile ecosystem. Given the importance of microbial communities in driving soil nutrient cycling, it is of interest to document the microbial biogeographic pattern here. We adopted a microarray-based tool named GeoChip 4.0 to investigate grassland microbial functional genes along an elevation gradient from 3200 to 3800 m above sea level open to free grazing by local herdsmen and wild animals. Interestingly, microbial functional diversities increase with elevation, so does the relative abundances of genes associated with carbon degradation, nitrogen cycling, methane production, cold shock and oxygen limitation. The range of Shannon diversities (10.27–10.58) showed considerably smaller variation than what was previously observed at ungrazed sites nearby (9.95–10.65), suggesting the important role of livestock grazing on microbial diversities. Closer examination showed that the dissimilarity of microbial community at our study sites increased with elevations, revealing an elevation-decay relationship of microbial functional genes. Both microbial functional diversity and the number of unique genes increased with elevations. Furthermore, we detected a tight linkage of greenhouse gas (CO2) and relative abundances of carbon cycling genes. Our biogeographic study provides insights on microbial functional diversity and soil biogeochemical cycling in Tibetan pastures.
Introduction
It is estimated that two–thirds of the Tibetan plateau, the highest and largest plateau on earth characterized by cold climate, low oxygen level, strong UV irradiation and poor primary productivity, are comprised of alpine grasslands (Qiu, 2008). In the past decade, Tibet has witnessed more than three-fold increases in average Gross Domestic Product per capita and a 10% increase in human population, resulting in pronounced influence on natural environments (National Bureau of Statistics of China, 2015-2006). In addition, global warming has led to the substantial glacier and permafrost thaw, which impose risks of drought and flooding (Qiu, 2008). Since soil is considered to be the most complex ecosystem on earth and plays a vital role in mediating biogeochemical cycling (Falkowski et al., 2008; Bardgett and van der Putten, 2014), it is of great interest to examine the biogeography of soil microbial communities in such a fragile ecosystem (Wu et al., 2016a).
To date, biogeographic variations of microbial communities have predominantly been studied at the taxonomic level (Bryant et al., 2008; Singh et al., 2012). Phylogenetic diversities along elevation gradients showed a dip pattern (Singh et al., 2014) or a bulge pattern (Singh et al., 2012, 2016) at mid elevations, a monotonic decrease pattern (Bryant et al., 2008), or no pattern (Lauber et al., 2009; Fierer et al., 2011). The major controlling factors in driving the phylogenetic diversity can be attributed to temperature (Nottingham et al., 2016), soil pH (Zhang et al., 2015), C/N (Siles and Margesin, 2016), carbon content, climate variability (Delgado-Baquerizo et al., 2016) and plant diversity (Zhang et al., 2015). Nonetheless, there is a renewed interest in understanding the biogeography of microbial functional traits, which sheds light on microbial community functions (Green et al., 2008). GeoChip, a microarray-based tool, is excellent in detecting microbial functional genes. It contains DNA probes targeting a variety of functional genes, including those associated with elemental cycling, stress response, metal resistance and complex carbon degradation (He et al., 2007). It has been successfully used to profile functional genes of microbial communities in different habitats (Van Nostrand et al., 2009; Chu et al., 2014; Ding et al., 2015), which provides important insights into microbe-mediated processes. It was shown that measurements of relative abundances of functional genes were predictive of in situ CO2 and N2O emissions (Morales et al., 2010) and nitrification potential (Zhao et al., 2014) to certain extent.
Recently, we have used GeoChip to examine functional genes of soil microbial communities in an enclosed Tibetan alpine grassland to prevent anthropogenic disturbance, which revealed strikingly tight linkages between microbial functional gene structure and several environmental variables (Yang et al., 2014). Livestock grazing is the most prevalent economic activity in Tibet (Zhou et al., 2006), resulting in significant ecological effects. For example, it reduces vegetation biomass, switches dominant plant species to short and prostrate forb species (Lkhagva et al., 2013), expands the range of soil temperature fluctuation, increases soil mean temperature, enhances forage quality (Odriozola et al., 2014), and decreases microbial biomass carbon and nitrogen (Fu et al., 2012). Furthermore, owing to strong ruminant CH4 emission, total greenhouse gas budget of high grazing densities (-1034 g CO2eq m-2 yr-1) was nearly four times to that of low grazing densities (-260 g CO2eq m-2 yr-1) (Skiba et al., 2013). Thus, it is important to examine microbial biogeography of functional genes in the grassland subjected to livestock grazing, which is more common in Tibet.
In this study, we collected soil samples along an altitudinal gradient of 3200, 3400, 3600, and 3800 m asl in a Tibetan grassland subjected to open free livestock grazing. We adopted GeoChip 4.0 to examine microbial functional genes, aiming to address the following hypotheses: (i) because vegetation removal, manure deposition, and trampling by grazing impose multi-dimensional disturbance to soil, microbial biogeographic pattern in pasture would substantially differ from what is observed in the enclosed grassland; (ii) the relative abundance of specific microbial functional genes can be used to explain CO2 and N2O fluxes. The relationship between gene abundance and process rates has been a topic of dispute. It was shown recently that the activity of C degradation enzymes (ß-D-cellulosidase, ß-Xylosidase, α-Glucosidase and N-acetyl-ß-Glucosaminidase) in grain-producing soils was significant correlated to the relative abundance of microbial functional gene abundance determined by GeoChip 4.0 (R2> 0.90, P < 0.001) (Trivedi et al., 2016), and amoA gene relative abundances had significant positive correlations with soil nitrification potentials (R > 0.48, P < 0.05) in agricultural soils (Liu et al., 2015). However, a meta-analysis showed that the linkages between gene abundance and processes are often missed in many studies, which can be ascribed to habitat type or differences among genes (Rocca et al., 2015). Therefore, it is interesting to examine whether there is such linkage in this study.
Materials and Methods
Site Description and Soil Sampling
This study was carried out at the Haibei Alpine Meadow Ecosystem Research Station located in the northeast of the Tibetan Plateau. The region has a typical plateau continental climate characterized by cold and long winter but warm and short summer, with annual mean air temperature of -1.7°C and annual mean precipitation of 570 mm (Zhao et al., 2006). The vegetation growing season is from May to September. The major soil type is Mat Cryic Cambisol based on Chinese Soil Taxonomic Classification System (Arthur et al., 2008).
Soil samples were collected at elevations of 3200, 3400, 3600, and 3800 m asl. Geographical distances between adjacent elevations were 1.3 km (3600–3800 m), 4.2 km (3400–3600 m), and 6.2 km (3200–3400 m). To reduce soil heterogeneity, five soil cores at the 0–10 cm and 10–20 cm depths were randomly taken from a location with a size of 1 × 1 m and thoroughly mixed to make a composite sample. Three such locations at each elevation, which were several to dozens of meters away from each other, were chosen to create biological triplicates. All soil samples were collected in August 2009 and immediately transported on ice to the laboratory. After being sieved with 2 mm mesh to remove visible vegetation roots, residues and stones, parts of samples were stored at -80°C for DNA extraction and 4°C for environmental variable measurements, respectively.
DNA Extraction, Purification, and Quantitation
After mixing soil cores from 0 to 10 cm and 10 to 20 cm depths, we extracted soil DNA with FastDNA spin kits (MP Biomedical, Carlsbad, CA, United States) following manufacturer’s instructions and then purified the extracted DNA in a mixture of 2.0 volume of 100% ice-cold ethanol and 0.1 volume of 3 M NaOAc (pH 5.2). After overnight incubation at -20°C, DNA solutions were centrifuged at 13000 g for 30 min and then washed with 1 ml 70% ethanol. Then DNA solutions were air dried for 30 min and dissolved in nuclease-free water. We used NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, United States) to determine DNA quality and a PicoGreen method using a FLUOstar Optima (BMG Labtech, Jena, Germany) to determine DNA concentrations (Ahn et al., 1996).
GeoChip 4.0 Experiments
DNA was labeled with Dye Cy-5 by a random priming method as previously described (Yang et al., 2017) and the labeled DNA was purified with QIA quick purification kits (Qiagen, Valencia, CA, United States). SpeedVac (ThermoSavant, Milford, MA, United States) was used to dry DNA at 45°C for 45 min. We hybridized DNA with GeoChip 4.0 on a MAUI hybridization station (BioMicro, Salt Lake City, UT, United States) at 42°C for 16 h, as described previously (Yue et al., 2015). The slides were scanned using a NimbleGen MS200 scanner (Roche, Madison, WI, United States) at 633 nm using 100% laser power and 75% photomultiplier tube gain. Signal intensities were quantified by scanned images.
The Elevation-Decay Relationship (EDR)
Sorensen similarity was calculated for generating the EDR z-value. An equation of log(SS) = constant-2zlog(D) was used to calculate power-law exponent z, in which D was the Euclidean distance between two elevations and SS was the pairwise similarity in microbial compositions by using Sorensen’s index. Elevation differences for samples at the same elevation were zero, which were changed into 0.01 to enable logarithm transformation (Legendre and Legendre, 1998). Since our pairwise comparison data were not independent with each other, we determined whether the slope was significant different from zero by bootstrapping (999 times) (Efron and Tibshirani, 1994). To this end, the original dataset was randomly paired (with replacement) and the slope of EDR was calculated by the new dataset. Using one sample t-test, we determined whether the slopes of the regressions based on bootstrapping differed from the observed slope followed by BH correction for P-value (Zhou et al., 2008).
Data Analysis
We processed raw data as follows: (i) removing spots with a signal to noise ratio of less than 2.0; (ii) removing spots detected only once in three replicates; (iii) normalizing intensity of each spot by dividing total signal intensity of a sample and multiplying by a constant; and (iv) taking natural logarithm (Wu et al., 2016b). Subsequently, the detrended correspondence analysis (DCA), a multivariate statistical method (Ayoub-Hannaa et al., 2013), was used to compare overall functional gene structures across different samples. The hierarchical clustering analysis was used to cluster different microbial functional gene structures among soil samples. The dissimilarity test of adonis, one of the permutational multivariate analysis using distance matrices (Ricotta and Burrascano, 2009), was used to test differences of functional gene structures among different elevations. Alpha diversities were presented by Shannon and Simpson indexes. The canonical correspondence analysis (CCA) was used to identify main environmental drivers in shaping microbial communities. Variance inflation factors (VIF) of less than 20 were used as a threshold to remove redundant variables before performing CCA. The variation partitioning analysis (VPA) was used to partition environmental variables selected by CCA into three groups. Simple Mantel tests were performed to determine the relationships between microbial communities and environmental variables. Correlations between relative abundances of functional genes and environmental variables were determined by Pearson correlation and then adjusted by false discovery rate (FDR) method. These analyses were carried out using functions in the vegan package (v. 2.3-1) in R-studio v. 3.2.2. Association network analysis was performed with a subset of GeoChip data since the whole dataset was beyond the computational capacity. A random matrix theory (RMT)-based algorithm was used to construct networks using the coefficients of Spearman correlations to define similarity matrix (Deng et al., 2012).
Measurements of Soil and Vegetation Variables
We recorded in situ soil temperatures using type-K thermocouples (Campbell Scientific, Logan, UT, United States) with a CR1000 datalogger at the depth of 10 cm, which represents 0–20 cm. We measured in situ soil moisture at the depth of 10 cm using a time domain reflectometry (Model Diviner-2000, Sentek Pty Ltd., Stepney, SA, Australia). Soil pH data were missing for our sample. Therefore, we used soils nearby our plots collected in 2012 as substitutes since soil pH in Tibet was a generally stable variable, which had decreased only by 0.5 units since 1980 (Zhou et al., 2009). We measured soil pH with a combination glass electrode in soil/water solution of volume ratio 1:5. The concentrations of ammonia and nitrate were detected by a FIAstar 5000 Analyzer (Yang et al., 2013). To generate finer measurements of nitrogen, carbon and phosphorus, we examined total nitrogen (TN), total organic carbon (TOC) and total phosphorus at both depths of 0–10 cm and 10–20 cm. We determined TN by a Vario EL III Elemental Analyzer (Elementar, Hanau, Germany), TOC by a TOC-5000A analyzer (Shimadzu Corp., Kyoto, Japan) and total phosphorus by a UV-visible spectrophotometer (Agilent 8453, Agilent Co., Santa Clara, CA, United States). Using a Agilent 4890D gas chromatograph (Agilent Co., Santa Clara, CA, United States) equipped with Flame Ionization Detector (FID) and Electron Capture Detector (ECD) (Wang and Wang, 2003), we quantified greenhouse gasses of CO2, CH4 and N2O except the 3600 m site, where rat holes were too rampant to perform accurate measurements of greenhouse gasses (Yang et al., 2014). During gas sampling, static stainless steel chambers (40 cm × 40 cm × 40 cm) were manually closed for half an hour. Then gas samples were taken every 10 min by plastic syringes and sampled within 24 h (Hu et al., 2010). We determined vegetation variables of species number, aboveground vegetation biomass and vegetation diversity (Shannon index).
Results
Environmental Variables
Soil temperature (T) and pH decreased along the altitudinal gradient (Table 1), while soil moisture, nitrate (NO3-) and TN measured at the 10–20 cm depth (TN10-20cm) increased with elevations. TOC measured at the 10–20 cm depth (TOC10-20cm), TN measured at the 0–10 cm depth (TN0-10 cm) and vegetation diversity were higher at 3400 and 3600 m than those at 3200 and 3800 m. Notably, there were correlations (P < 0.050) among TOC, TN, and total phosphorous (TP) (Supplementary Table S1).
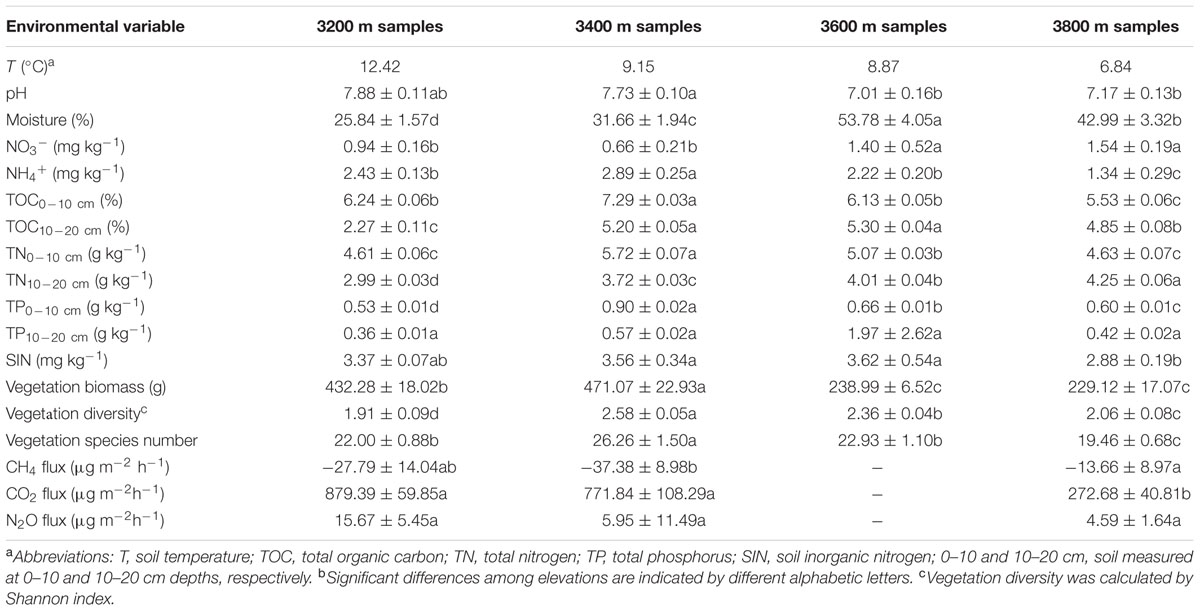
TABLE 1. Summary of environmental variables.
The Biogeographic Pattern of Microbial Functional Genes
A total of 52864 genes were detected by GeoChip, ranging from 31528 to 43816 for each site (Supplementary Table S2). The results of the dissimilarity test of adonis showed that samples were grouped by elevations (Supplementary Table S3). Microbial functional diversities, represented as Shannon indices, ranged in 10.27–10.58 and increased (P < 0.050) with elevations (Supplementary Table S2). However, the evenness indices of microbial functional genes were similar across elevations (Supplementary Table S2).
The Elevation-Decay Relationship (EDR)
A significant EDR (r = -0.80, P < 0.001) was observed for the whole functional community (Figure 1) with a z-value of 0.0093 (Supplementary Table S4). We found that almost all z-values of carbon or nitrogen cycling processes were lower than 0.01, with the highest z-value (0.0127) of anammox and the lowest z-value (0.0055) of nitrogen fixation.
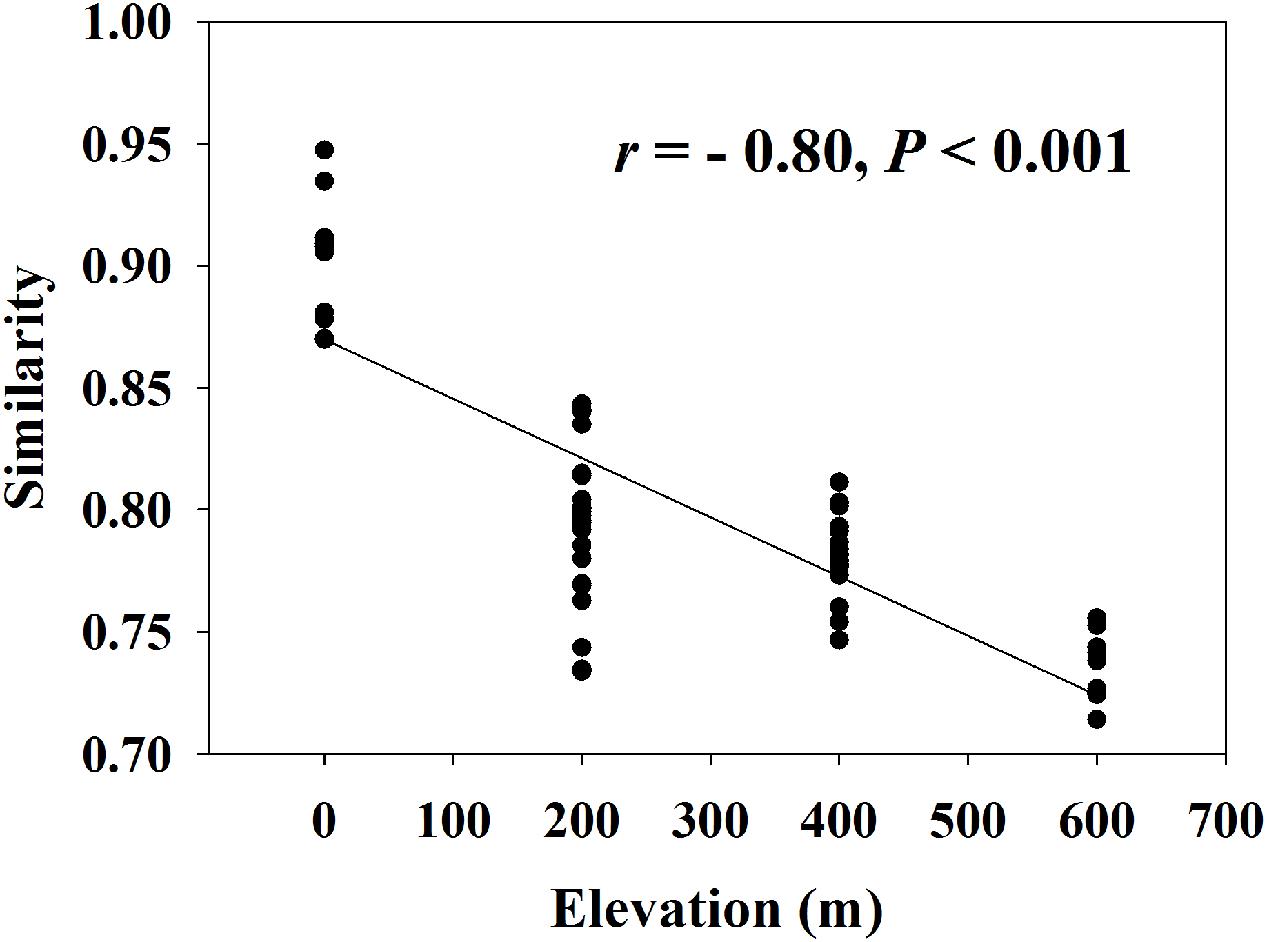
FIGURE 1. Elevation-decay relationship for whole microbial community (z-value = 0.0093, t = –209.6, n = 999, P < 0.001). Similarity represents Sorensen’s similarity index.
Ubiquitous genes, defined as genes detected in all of four elevations, accounted for 50.41% of all genes detected by GeoChip (Supplementary Figure S1). In contrast, unique genes, defined as genes detected only in one elevation, accounted for only 20.98% of all detected genes (Supplementary Figure S1). The number of unique genes increased with elevations, which was in accordance with the increasing ratio of the unique gene number to the detected gene number at each elevation (Supplementary Table S5). The percentage of shared genes between different elevations was the highest between 3600 and 3800 m asl (65.47%), which was the shortest in distance (Supplementary Table S6). In contrast, it was the lowest between 3200 and 3800 m asl (54.57%), which was the highest in distance. A negative correlation between distance and the number of shared genes (r = -0.71, P < 0.001) (Supplementary Figure S2) was detected.
The Linkage of Overall Microbial Functional Gene Structure and Environmental Variables
We performed simple Mantel tests to examine major factors linking to overall microbial functional gene structure. The results showed that elevation was the most important factor in affecting functional genes structure (Table 2). To discriminate whether elevational influence is attributable to environmental variables, we performed CCA to determine the linkage between microbial functional gene structure and environmental variables (Supplementary Figure S3). Seven environmental variables (T, pH, NO3-, TP10-20cm, aboveground vegetation biomass, vegetation species number and vegetation diversity), were identified to have a significant influence on functional microbial structure, which was also verified by Mantel tests (P < 0.05) (Table 2). The VPA showed that these seven variables explained a total of 82.64% variations of functional gene structure (Supplementary Figure S4).
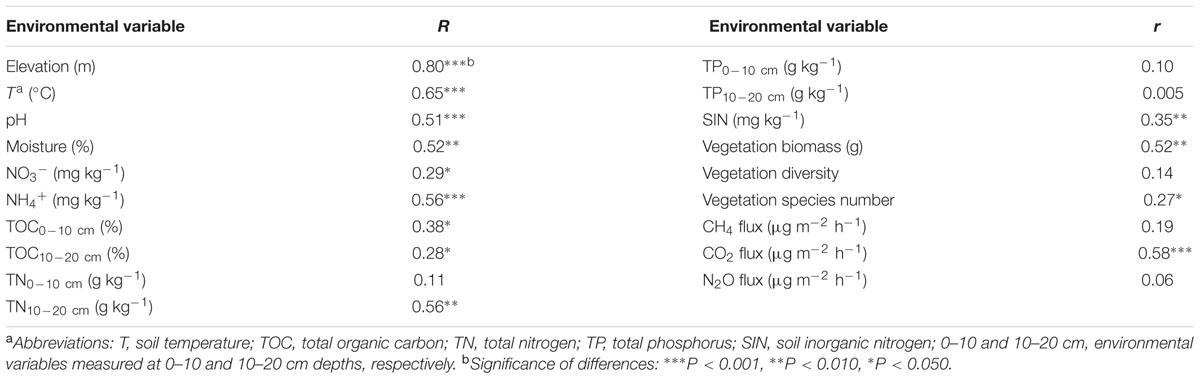
TABLE 2. Simple Mantel tests between functional genes and environmental variables.
Carbon Cycling Genes
Almost all of carbon cycling genes (Supplementary Figure S5) increased in relative abundance with elevations. However, there are certain exceptions. The relative abundances of pulA genes (encoding extracellular polyurethane/esterase) (Supplementary Figure S5A), and rubisco genes (encoding ribulose-1, 5-bisphosphate carboxylase/oxygenase) were lowest at the 3800 m site (Supplementary Figure S5B).
We found that soil moisture has a strong positive correlation with mcrA genes (encoding methyl coenzyme M reductase subunit A, r = 0.75, P = 0.010) and pmoA genes (encoding particulate methane monooxygenase A, r = 0.84, P = 0.001) (Figures 2A,B). In addition, the important role of microbes on CO2 flux was verified by Mantel tests (r= 0.58, P < 0.001) (Table 2). In-depth investigation showed that CO2 flux negatively correlated with aclB genes (encoding ATP citrate lyase involved in reductive TCA cycle, r = -0.83, P = 0.009), CODH genes (encoding carbon monoxide dehydrogenase involved in biosynthesis of acetyl-CoA, r = -0.89, P = 0.001) and pcc genes (involved in hydroxypropionate pathways, r = -0.77, P = 0.022) (Figures 2C–E).
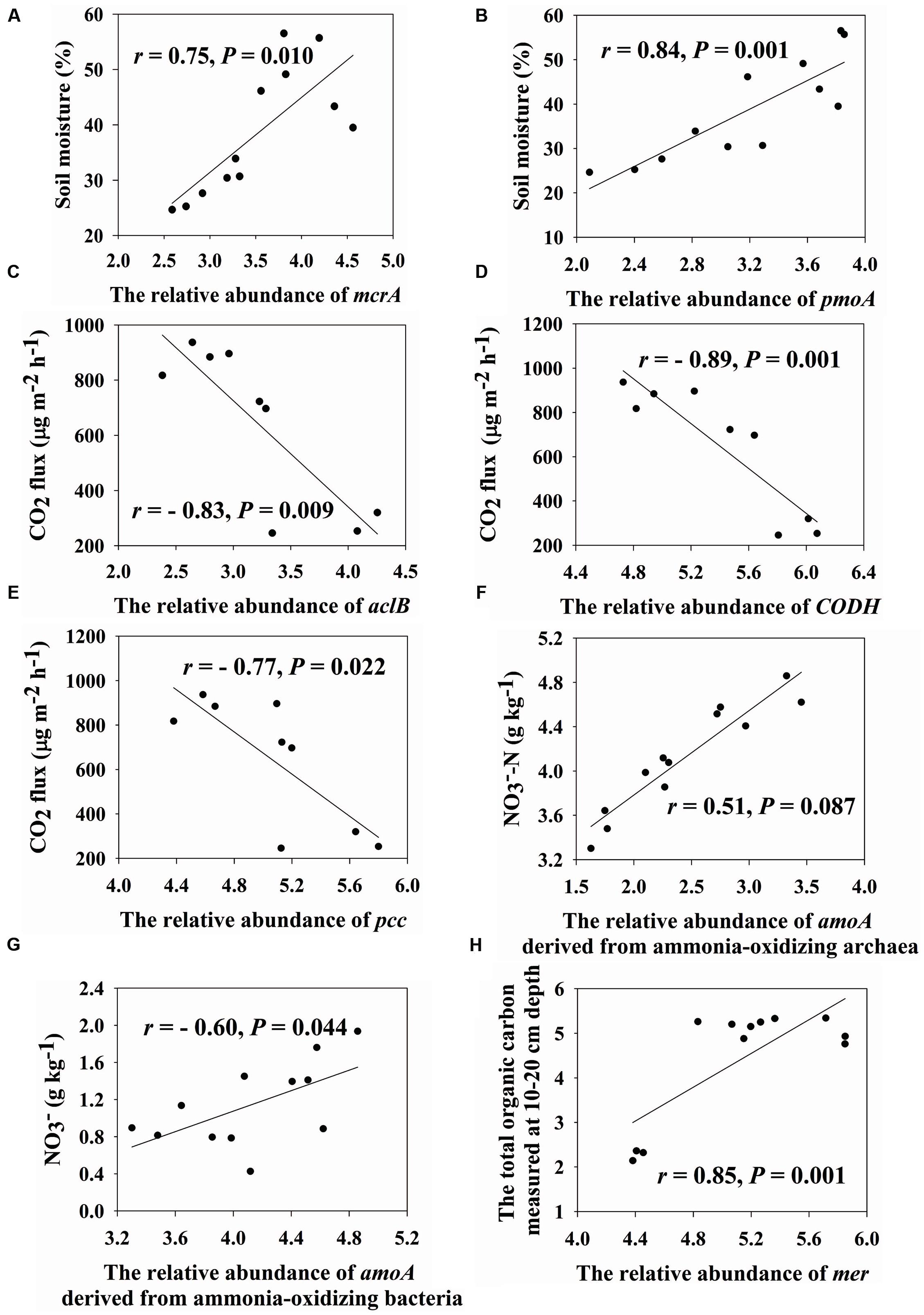
FIGURE 2. Correlations between soil moisture and the relative abundance of mcrA (A) and pmoA (B), CO2 flux and the relative abundance of aclB (C), CODH (D) and pcc (E), nitrate and the relative abundance of amoA drived from ammonia-oxidizing archaea (F) and bacteria (G) and the total organic carbon measured at 10–20 cm depth and the relative abundance of mer (H). Pearson correlation and TDIST tests were used to calculate r and P values, respectively.
To identify major carbon fixation bacteria responsible for CO2 flux, we performed a molecular ecological network analysis with carbon fixation genes and environmental variables. The resulting network showed typical topological features of scale-free (power law = 0.92), small world (average path distance = 3.49), modular (modularity = 0.64) and hierarchical (average clustering coefficient = 0.21) (Supplementary Table S7). Notably, soil CO2 flux showed correlations with rubisco genes derived from Roseovarius, Synechococcus, and Nakamurella multipartita, and pcc genes derived from Mycobacterium and Comamonas testosteroni (Figure 3A).
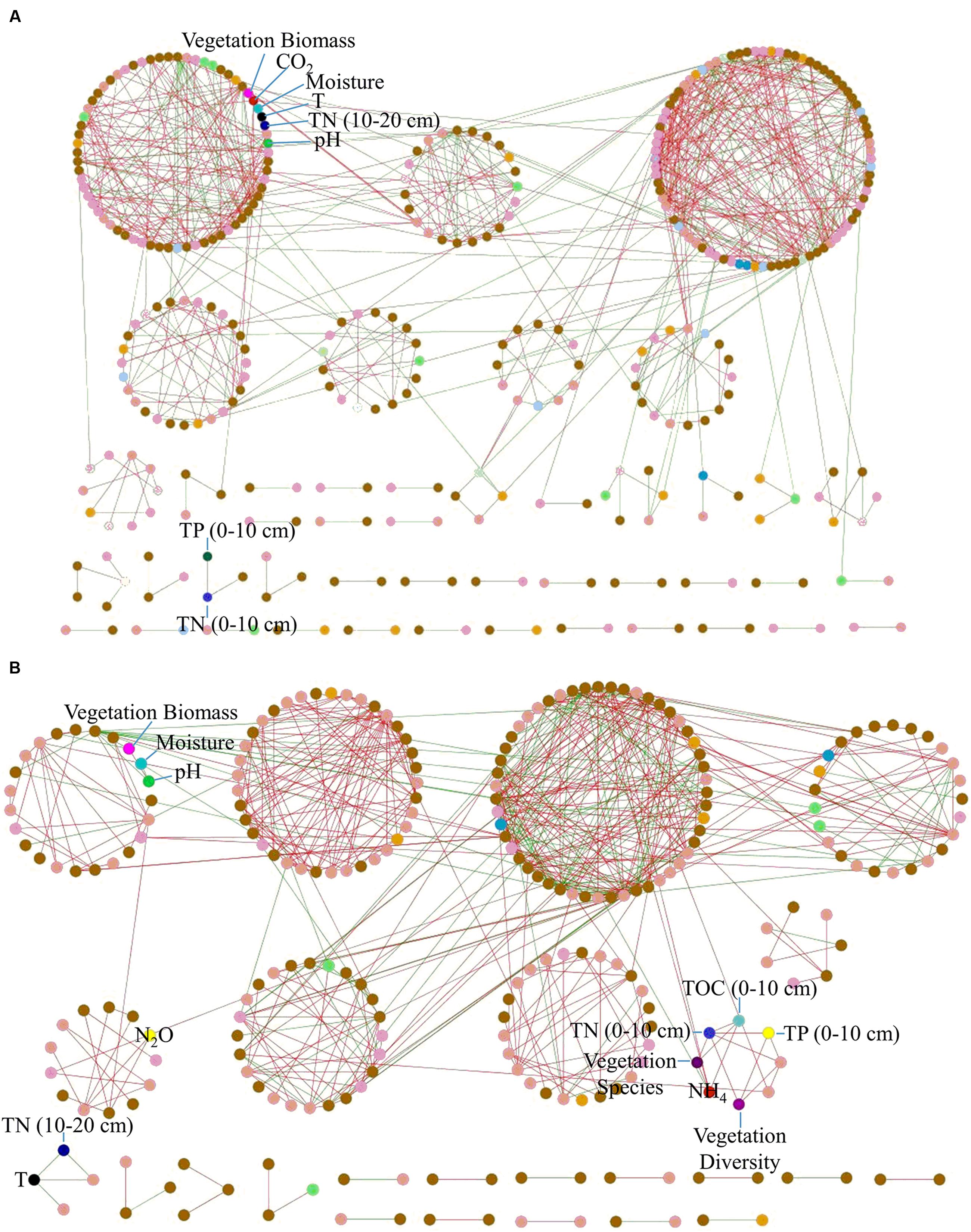
FIGURE 3. Association network analyses of (A) carbon fixation genes and (B) amoA genes.
Nitrogen Cycling Genes
Almost all of nitrogen cycling genes increased in relative abundances with elevations, except that nosZ genes (encoding nitrous oxide reductase) were similar across four elevations (Supplementary Figure S6). NO3- positively correlated with amoA (encoding active site polypeptide of the ammonia monooxygenase)-AOA (ammonia-oxidizing archaea; r = 0.51, P = 0.087) but negatively correlated with amoA-AOB (ammonia-oxidizing bacteria; r = -0.60, P = 0.044) (Figures 2F,G). In addition, NO3- positively correlated with a number of nitrogen cycling genes, such as amoA genes (r = 0.57, P = 0.051), nirK/S genes (encoding a nitrite reductase, r > 0.58, P < 0.048), napA genes (encoding periplasmic nitrate reductase, r = 0.68, P = 0.024) and gdh genes (encoding glutamate dehydrogenase, r = 0.60, P = 0.046), and ammonia (NH4+) negatively correlated with nirK/S genes (r < -0.58, P < 0.046) (Supplementary Table S8). The ratio of nirK to nirS gene abundances positively correlated with NH4+ (r = 0.59, P = 0.046) and negatively correlated with moisture (r = -0.46, P = 0.123) (Supplementary Table S8). N2O flux was positively correlated with nosZ gene derived from Roseobacter denitrificans OCh 114 (r = 0.71, P = 0.034) and another two probes derived from uncultured bacteria (r > 0.69, P < 0.036) (Supplementary Table S9), and negatively correlated with norB gene derived from Moraxella catarrhalis (r = -0.88, P = 0.002) (Supplementary Table S9).
The association network with amoA genes and environmental variables showed topological features of scale-free, small world, modular and hierarchical (Supplementary Table S7). Notably, there was a positive correlation between N2O and amoA gene derived from Pseudomonas (Figure 3B), which was abundant in Tibet (Zhang et al., 2007; Tong et al., 2008). In addition, soil pH was negatively correlated with amoA gene derived from Aurantimonas.
Stress Response Genes
Induced under cold environment, cold shock proteins help maintain cell phenotypes and viabilities through preventing damage of ice crystal (Willimsky et al., 1992). In our study, increased cold shock genes of cspA and cspB (encoding cold shock proteins) along the altitudinal gradient suggested stronger capability of microorganisms in acclimation of cold environments (Supplementary Figure S7A). In addition, we found that soil temperature correlated with cspA genes (r = -0.74, P = 0.010) and cspB genes (r = -0.90, P < 0.001) (Supplementary Table S8).
Relative abundances of genes associated with oxygen limitation, radiation stress and mercury generally increased with elevations (Supplementary Figures S7B,C,E), such as arcA genes (encoding the cytosolic response regulator) and arcB genes (encoding the membrane-bound senor kinase) derived from Shewanella, Vibrio, Nitrosospira, and Oxalobacter. In contrast, genes associated with glucose limitation were similar in relative abundances (Supplementary Figure S7D). We found that TOC10-20 cm was positively correlated with mer genes encoding a mercury-resist operon (r = 0.85, P = 0.001) (Figure 2H). Soil pH was negatively correlated with mer (r = -0.78, P = 0.001) and merG (r = -0.64, P = 0.021) (Supplementary Table S8).
Discussion
The Effect of Grazing on Microbial Biogeographic Pattern
It has been shown that soil microbial biomass, taxonomic compositions, and bacterial diversities differed along the altitudinal gradient (Faoro et al., 2010; Fu et al., 2012). Our studies provided evidence that functional gene structures of soil microbial communities also differed along the altitudinal gradient (Supplementary Table S3). We observed increased soil moisture, TN10-20 cm and NO3- at higher elevations (Table 1), which could provide more electron acceptors and available substrates to stimulate elemental cycling (Xu et al., 2014). In addition, it has been shown that water-extractable organic carbon, microbial biomass carbon, and carbon storage increased with elevations (Pabst et al., 2013; Tsui et al., 2013). Therefore, the increased pattern of microbial diversity could mainly be attributed to the more resources at higher elevations.
The diversity patterns of microbial communities in this study differed from those in enclosed grasslands without livestock grazing using the same data analysis method (Yang et al., 2014). We found microbial functional diversity increased with elevations (Supplementary Table S2). However, microbial functional diversity showed a humpback shape in enclosed grasslands, suggesting that land uses imposed a significant impact on soil microbial communities. Environmental stresses may not contribute to differences in microbial biogeographic patterns, as genes associated with cold shock also increased with elevations in the enclosed grassland. Rather, we speculate that differences in soil nutrient inputs could lead to differences in microbial communities. Animal excreta from livestock provide available nitrogen to vegetation and microbial communities and consequently stimulate nitrogen cycling (Kohler et al., 2005; Ambus et al., 2007). Aboveground vegetation grazed by livestock could reduce litter input to soil, which ameliorates nitrogen limitation for microbial communities because vegetation C/N ratios are higher. These two reasons could collectively stimulate substrate availability to soil microbial communities in pastures.
Livestock grazing could result in more homogenous soil environments because of soil compaction induced by trampling of livestock (Zhao et al., 2007). Similarly, we found that variations of environmental variables in grazing sites, such as NH4+ (1.34–2.89 mg kg-1), soil inorganic nitrogen (SIN; 2.88–3.62 mg kg-1) and vegetation species numbers (19.46–26.26), were smaller than variations of NH4+ (1.5–3.7 mg kg-1), SIN (2.88–4.63 mg kg-1) and vegetation species numbers (13.67–25.00) in enclosed grasslands (Table 1). At the functional gene level, the range of Shannon diversities (10.27–10.58) was less than half of previous observations at the enclosed grassland (9.95–10.65) (Yang et al., 2014), suggesting the important role of livestock grazing on microbial diversities. However, we could not exclude the possibility that some functional genes in soils might not be represented on GeoChip, which would lead to underestimation of microbial functional diversities (Zhou et al., 2015a). Notably, GeoChip is also limited in discovering novel genes in the environment and subjected to errors derived from cross hybridization.
Field simulation studies showed that soils exposed to warmer climates decreased functional diversities (Yue et al., 2015). In addition, warming decreased microbial diversities under normal precipitation conditions (Sheik et al., 2011). It is likely that dominant species would be more advantageous while rare species would extinct, resulting in decreased diversities (Fussmann et al., 2014). Alternatively but not exclusively, warming might increase nutrient availability (Rinnan et al., 2008). Since our results show that microbial functional diversities are higher in colder environments, we predict that warming in Tibetan pastures will decrease microbial functional diversities and shift soil carbon and nitrogen cycling. However, given strong metabolic flexibility and adaptability of soil microbial communities (Bardgett and van der Putten, 2014), it remains to be an open question whether warming will exert permanent effects on functional potentials of microbial communities and other parts of the biosphere (Lau and Lennon, 2011).
Linkages between Vegetation and Microbial Communities
Unique vegetation species number increased with elevations between 1000 and 4000 m asl in the Tibetan Plateau (Vetaas and Grytnes, 2002). Similarly, we found that numbers of unique genes, which are genes detected only at one elevation, increased with elevations (Supplementary Table S5). The co-occurrence diversification of vegetation and microbes at higher elevations can be attributed to stronger environmental selective forces (Hanson et al., 2012), which was supported by revealing seven environmental variables explaining a total of 82.64% variations of functional gene structure (Supplementary Figure S4) and strong correlation between environmental variables and functional genes (Table 2). In contrast, evolutionary or ecological drift might play a minor role.
The tight linkages between vegetation and microbial communities, as indicated by CCA (Supplementary Figure S3) and Mantel tests (Table 2), can result from complex interactions between them. Vegetation root exudates and litter input could offer diverse organic resources to influence soil microbial communities (Wardle et al., 2004; Chu et al., 2014), while microbial communities provide feedbacks to vegetation through altering nutrient availability (Lau and Lennon, 2011). In this study, aboveground vegetation biomass correlated with microbial community structures (r = 0.52, P < 0.010) (Table 2), which has been observed elsewhere (Yang et al., 2013; Yue et al., 2015). In contrast, vegetation diversity was not correlated with microbial community structures (r = 0.14, P > 0.050) (Table 2), which could be explained by recent observations that vegetation diversity could predict beta but not alpha diversity of microbial communities in grasslands (Prober et al., 2015). More aboveground vegetation biomass confers more litter and root exudate to soil, which consequently changes C/N ratio of substrates for microbial utilization and alters microbial communities.
C Cycling Genes
Negative correlations between CO2 flux and carbon fixation genes (Figures 2C–E) suggested that changes in CO2 flux might be attributed to anaerobic acetyl-CoA pathway, reverse TCA cycle and hydroxypropionate pathways. In contrast, Calvin-Benson-Bassham cycle, as indicated by microbial rubisco genes encoding an enzyme coupling oxygenolysis of ribulose-1, 5-bisphophate and CO2 reduction, were similar along the altitudinal gradient. However, this observation does not refute the possibility that rubisco genes play a substantial role in soil CO2 flux. Recently, microbial rubisco gene abundance and enzyme activities were shown to be high in Tibetan grasslands (Guo et al., 2015). In addition, we have identified correlations between CO2 flux and rubisco genes derived from Roseovarius, Synechococcus, and N. multipartita by the association network analysis. Therefore, individual functional species possessing rubisco genes might contribute to variations of CO2 flux, despite the lack of overall changes in total abundance of rubisco genes.
Higher soil moisture strengthens substrate utilization of microbial communities (Chen et al., 2007). Furthermore, TOC, readily mineralizable carbon, water-soluble organic carbon and reducing sugar carbon increase when soil water-holding capability varies in 20–60% (Hassan et al., 2015), owing to increase in microbial accessibility to soluble nutrients, especially autotrophic carbon, ammonia and nitrate (Bell et al., 2008; Van Horn et al., 2014). Accordingly, we observed an increase of carbon degradation genes (Supplementary Figure S5A).
Soil moisture also has a strong effect on net soil CH4 uptake rate, which is the balance between CH4 production and oxidation (Shrestha et al., 2012). As CH4 production and oxidation are two tightly intertwined processes (Inagaki et al., 2004), it is no surprise to note an increase in relative abundances of both CH4 production and oxidation genes with elevations and soil moisture (Supplementary Figure S5C). Similarly, more CH4 and available substrates induced by soil moisture can stimulate microbial methanotrophs as well as CH4 oxidation gene abundance (Shrestha et al., 2012). Conversely, CH4 oxidation stimulates the growth of methanogenic bacteria by supplying energy (Hoehler et al., 1994).
N Cycling Genes
Many studies used nirS and (or) nirK genes as functional markers to profile diversities or structures of denitrifiers because nitrite reductase is the rate-limiting step of denitrification (Braker et al., 1998; Liu et al., 2003). The ratio of nirK to nirS gene abundance negatively and positively correlated with soil moisture (r = -0.46, P = 0.123) and NH4+ (r = 0.59, P = 0.046), respectively (Supplementary Table S8), which was consistent with a recent finding that water logging decreased the nirK/nirS ratio (Yoshida et al., 2009). In addition, both NO3- and NH4+ are associated with community compositions of nirS-type denitrifiers (Zheng et al., 2015).
The correlation between amoA and NO3- (r = 0.57, P = 0.051) has been similarly documented (Ding et al., 2015). Intriguingly, amoA-AOB negatively correlated with NO3- (r = -0.60, P = 0.044) and amoA-AOA positively correlated with NO3- (r = 0.51, P = 0.087) (Figures 2F,G), which could be attributed to different roles of ammonia monooxygenase, physiological pathways and electron transport mechanisms in AOA and AOB (Hatzenpichler, 2012). Consistently, it was shown in groundwater that nitrate positively correlated with copy numbers of amoA-AOA and negatively correlated with those of amoA-AOB (Qin et al., 2014). The positive correlations between NO3- and hzo and napA genes provide further evidence to the finding that NO3- can regulate the anammox and dissimilarity nitrate reduction processes (Silver et al., 2001; Trimmer et al., 2005).
Heavy Metal
Livestock manure was one of heavy metal sources to soil, including mercury (Nicholson et al., 2003; Wang et al., 2016). At higher elevations, decreased turnover rate of soil organic carbon could accelerate mercury accumulation and cold temperature could decrease mercury evasion, causing the increasing abundance of mer genes (Zhou et al., 2015b). Consistently, mer gene positively correlated with TOC10-20 cm (r = 0.85, P = 0.001) (Figure 2H), which might be attributed to mercury affinity to organic matter (Zhou et al., 2015b). In addition, the negative correlations between pH and mer (r = -0.78, P = 0.001) and merG (r = -0.64, P = 0.021) genes indicated that soil acidification constrained Hg volatilization (Supplementary Table S8).
Conclusion
This study represents an in-depth investigation of soil microbial functional gene profiles in an alpine pasture, based on GeoChip. Compared to enclosed grassland, stronger elevation-decay relationships of microbial functional genes were detected, which could be ascribed to strong environment selection in the alpine environment. By focusing on microbial functional genes, our study provides valuable insights for understanding microbe-mediated mechanisms of soil biogeochemical cycling.
Data Accessibility
GeoChip 4.0 data is available online1 with the accession number GSE52425.
Author Contributions
This study was conceived and led by SW, JZ, and YY. YY, SW, QL, and XL contributed to GeoChip experiments and environmental measurements. QQ, MZ, and YG analyzed data. QQ and MZ led the efforts to synthesize the data and write manuscript. YY, YW, BG, GL, and XM revised this manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer WK declared a shared affiliation, though no other collaboration, with the author SW and additionally different institutes to the authors QL, XL to the handling Editor, who ensured that the process nevertheless met the standards of a fair and objective review.
The reviewer SW declared a shared affiliation, though no other collaboration and different institutes, with several of the authors SW, QL, XL to the handling Editor, who ensured that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We would like to thank the staff in Haibei Research Station for helping with soil sampling. This research was supported by grants to YY from the open funds of state key laboratory of urban and region ecology of China (No. SKLURE2015-2-3), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB15010102), National Key Basic Research Program of China (2013CB956601), National Science Foundation of China (41471202), and to JZ from the National Science Foundation of China (41430856).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00976/full#supplementary-material
Footnotes
References
Ahn, S. J., Costa, J., and Emanuel, J. R. (1996). PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res. 24, 2623–2625. doi: 10.1093/nar/24.13.2623
Ambus, P., Petersen, S. O., and Soussana, J. F. (2007). Short-term carbon and nitrogen cycling in urine patches assessed by combined carbon-13 and nitrogen-15 labelling. Agric. Ecosyst. Environ. 121, 84–92. doi: 10.1016/j.agee.2006.12.007
Arthur, A. D., Pech, R. P., Davey, C., Yanming, Z., and Hui, L. (2008). Livestock grazing, plateau pikas and the conservation of avian biodiversity on the Tibetan plateau. Biol. Conserv. 141, 1972–1981. doi: 10.1016/j.biocon.2008.05.010
Ayoub-Hannaa, W., Huntley, J. W., and Fürsich, F. T. (2013).Significance of detrended correspondence analysis (DCA) in palaeoecology and biostratigraphy: a case study from the upper cretaceous of Egypt. J. Afr. Earth Sci. 80, 48–59. doi: 10.1016/j.jafrearsci.2012.11.012
Bardgett, R. D., and van der Putten, W. H. (2014). Belowground biodiversity and ecosystem functioning. Nature 515, 505–511. doi: 10.1038/nature13855
Bell, C., McIntyre, N., Cox, S., Tissue, D., and Zak, J. (2008). Soil microbial responses to temporal variations of moisture and temperature in a Chihuahuan Desert Grassland. Microb. Ecol. 56, 153–167. doi: 10.1007/s00248-007-9333-z
Braker, G., Fesefeldt, A., and Witzel, K. P. (1998). Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl. Environ. Microbiol. 64, 3769–3775.
Bryant, J. A., Lamanna, C., Morlon, H., Kerkhoff, A. J., Enquist, B. J., and Green, J. L. (2008). Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc. Natl. Acad. Sci. U.S.A. 105, 11505–11511. doi: 10.1073/pnas.0801920105
Chen, M., Zhu, Y., Su, Y., Chen, B., Fu, B., and Marschner, P. (2007). Effects of soil moisture and plant interactions on the soil microbial community structure. Eur. J. Soil Biol. 43, 31–38. doi: 10.1016/j.ejsobi.2006.05.001
Chu, H., Wang, S., Yue, H., Lin, Q., Hu, Y., Li, X., et al. (2014). Contrasting soil microbial community functional structures in two major landscapes of the Tibetan alpine meadow. Microbiologyopen 3, 585–594. doi: 10.1002/mbo3.190
Delgado-Baquerizo, M., Maestre, F. T., Reich, P. B., Jeffries, T. C., Gaitan, J. J., Encinar, D., et al. (2016). Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Commun. 7:10541. doi: 10.1038/ncomms10541
Deng, Y., Jiang, Y., Yang, Y., He, Z., Luo, F., and Zhou, J. (2012). Molecular ecological network analyses. BMC Bioinformatics 13:113. doi: 10.1186/1471-2105-13-113
Ding, J., Zhang, Y., Deng, Y., Cong, J., Lu, H., Sun, X., et al. (2015). Integrated metagenomics and network analysis of soil microbial community of the forest timberline. Sci. Rep. 5:7994. doi: 10.1038/srep07994
Efron, B., and Tibshirani, R. J. (1994). An Introduction to the Bootstrap. Boca Raton, FL: CRC press.
Falkowski, P. G., Fenchel, T., and Delong, E. F. (2008). The microbial engines that drive Earth’s biogeochemical cycles. Science 320, 1034–1039. doi: 10.1126/science.1153213
Faoro, H., Alves, A. C., Souza, E. M., Rigo, L. U., Cruz, L. M., Al-Janabi, S. M., et al. (2010). Influence of soil characteristics on the diversity of bacteria in the southern brazilian atlantic forest. Appl. Environ. Microbiol. 76, 4744–4749. doi: 10.1128/AEM.03025-09
Fierer, N., McCain, C. M., Meir, P., Zimmermann, M., Rapp, J. M., Silman, M. R., et al. (2011). Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92, 797–804. doi: 10.1890/10-1170.1
Fu, G., Shen, Z., Zhang, X., Zhou, Y., and Zhang, Y. (2012). Response of microbial biomass to grazing in an alpine meadow along an elevation gradient on the Tibetan plateau. Eur. J. Soil Biol. 52, 27–29. doi: 10.1155/2014/265142
Fussmann, K. E., Schwarzmueller, F., Brose, U., Jousset, A., and Rall, B. C. (2014). Ecological stability in response to warming. Nat. Clim. Change 4, 206–210. doi: 10.1038/nclimate2134
Green, J. L., Bohannan, B. J. M., and Whitaker, R. J. (2008). Microbial biogeography: rrom taxonomy to traits. Science 320, 1039–1043. doi: 10.1126/science.1153475
Guo, G., Kong, W., Liu, J., Zhao, J., Du, H., Zhang, X., et al. (2015). Diversity and distribution of autotrophic microbial community along environmental gradients in grassland soils on the Tibetan plateau. Appl. Microbiol. Biotechnol. 99, 8765–8776. doi: 10.1007/s00253-015-6723-x
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., and Martiny, J. B. H. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506. doi: 10.1038/nrmicro2795
Hassan, W., Bano, R., Khatak, B. U., Hussain, I., Yousaf, M., and David, J. (2015). Temperature sensitivity and soil organic carbon pools decomposition under different moisture regimes: effect on total microbial and enzymatic activity. Clean-Soil Air Water 43, 391–398. doi: 10.1002/clen.201300727
Hatzenpichler, R. (2012). Diversity, physiology, and niche differentiation of ammonia-oxidizing archaea. Appl. Environ. Microbiol. 78, 7501–7510. doi: 10.1128/AEM.01960-12
He, Z., Gentry, T. J., Schadt, C. W., Wu, L., Liebich, J., Chong, S. C., et al. (2007). GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J. 1, 67–77. doi: 10.1038/ismej.2007.2
Hoehler, T. M., Alperin, M. J., Albert, D. B., and Martens, C. S. (1994). Field and laboratory studies of methane oxidation in an anoxic marine sediment - evidence for a methanogen-sulfate reducer consortium. Glob. Biogeochem. Cycles 8, 451–463. doi: 10.1029/94GB01800
Hu, Y., Chang, X., Lin, X., Wang, Y., Wang, S., Duan, J., et al. (2010). Effects of warming and grazing on N2O fluxes in an alpine meadow ecosystem on the Tibetan plateau. Soil Biol. Biochem. 42, 944–952. doi: 10.1016/j.soilbio.2010.02.011
Inagaki, F., Tsunogai, U., Suzuki, M., Kosaka, A., Machiyama, H., Takai, K., et al. (2004). Characterization of C-1-metabolizing prokaryotic communities in methane seep habitats at the kuroshima knoll, southern Ryukyu arc, by analyzing pmoA, mmoX, mxaF, mcrA, and 16S rRNA genes. Appl. Environ. Microbiol. 70, 7445–7455. doi: 10.1128/AEM.70.12.7445-7455.2004
Kohler, F., Hamelin, J., Gillet, F., Gobat, J. M., and Buttler, A. (2005). Soil microbial community changes in wooded mountain pastures due to simulated effects of cattle grazing. Plant Soil 278, 327–340. doi: 10.1007/s11104-005-8809-1
Lau, J. A., and Lennon, J. T. (2011). Evolutionary ecology of plant-microbe interactions: soil microbial structure alters selection on plant traits. New Phytol. 192, 215–224. doi: 10.1111/j.1469-8137.2011.03790.x
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/AEM.00335-09
Liu, S., Wang, F., Xue, K., Sun, B., Zhang, Y., He, Z., et al. (2015). The interactive effects of soil transplant into colder regions and cropping on soil microbiology and biogeochemistry. Environ. Microbiol. 17, 566–576. doi: 10.1111/1462-2920.12398
Liu, X. D., Tiquia, S. M., Holguin, G., Wu, L. Y., Nold, S. C., Devol, A. H., et al. (2003). Molecular diversity of denitrifying genes in continental margin sediments within the oxygen-deficient zone off the Pacific coast of Mexico. Appl. Environ. Microbiol. 69, 3549–3560. doi: 10.1128/AEM.69.6.3549-3560.2003
Lkhagva, A., Boldgiv, B., Goulden, C. E., Yadamsuren, O., and Lauenroth, W. K. (2013). Effects of grazing on plant community structure and aboveground net primary production of semiarid boreal steppe of northern Mongolia. Grass. Sci. 59, 135–145. doi: 10.1111/grs.12022
Morales, S. E., Cosart, T., and Holben, W. E. (2010). Bacterial gene abundances as indicators of greenhouse gas emission in soils. ISME J. 4, 799–808. doi: 10.1038/ismej.2010.8
National Bureau of Statistics of China (2015-2006). China Statistical Yearbook. Shenzhen: China Statistics Press.
Nicholson, F. A., Smith, S. R., Alloway, B. J., Carlton-Smith, C., and Chambers, B. J. (2003). An inventory of heavy metals inputs to agricultural soils in England and Wales. Sci. Total Environ. 311, 205–219. doi: 10.1016/S0048-9697(03)00139-6
Nottingham, A., Fierer, N., Turner, B., Whitaker, J., Ostle, N., McNamara, N., et al. (2016). Temperature drives plant and soil microbial diversity patterns across an elevation gradient from the Andes to the Amazon. bioRxiv doi: 10.1101/079996
Odriozola, I., García-Baquero, G., Laskurain, N. A., and Aldezabal, A. (2014). Livestock grazing modifies the effect of environmental factors on soil temperature and water content in a temperate grassland. Geoderma 23, 347–354. doi: 10.1016/j.geoderma.2014.08.002
Pabst, H., Kuehnel, A., and Kuzyakov, Y. (2013). Effect of land-use and elevation on microbial biomass and water extractable carbon in soils of Mt. Kilimanjaro ecosystems. Appl. Soil Ecol. 67, 10–19. doi: 10.1016/j.apsoil.2013.02.006
Prober, S. M., Leff, J. W., Bates, S. T., Borer, E. T., Firn, J., Harpole, W. S., et al. (2015). Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide. Ecol. Lett. 18, 85–95. doi: 10.1111/ele.12381
Qin, H., Quan, Z., Yuan, H., Liu, X., Zhu, Y., Chen, C., et al. (2014). Response of ammonium-oxidizing (amoA) and nitrate-reducing (narG) gene abundances in groundwater to land use change. Water Air Soil Pollut. 225, 1–8. doi: 10.1007/s11270-014-1908-y
Ricotta, C., and Burrascano, S. (2009). Testing for differences in beta diversity with asymmetric dissimilarities. Ecol. Indic. 9, 719–724. doi: 10.1016/j.ecolind.2008.09.003
Rinnan, R., Michelsen, A., and Jonasson, S. (2008). Effects of litter addition and warming on soil carbon, nutrient pools and microbial communities in a subarctic heath ecosystem. Appl. Soil Ecol. 39, 271–281. doi: 10.1016/j.apsoil.2007.12.014
Rocca, J. D., Hall, E. K., Lennon, J. T., Evans, S. E., Waldrop, M. P., Cotner, J. B., et al. (2015). Relationships between protein-encoding gene abundance and corresponding process are commonly assumed yet rarely observed. ISME J. 9, 1693–1699. doi: 10.1038/ismej.2014.252
Sheik, C. S., Beasley, W. H., Elshahed, M. S., Zhou, X., Luo, Y., and Krumholz, L. R. (2011). Effect of warming and drought on grassland microbial communities. ISME J. 5, 1692–1700. doi: 10.1038/ismej.2011.32
Shrestha, P. M., Kammann, C., Lenhart, K., Dam, B., and Liesack, W. (2012). Linking activity, composition and seasonal dynamics of atmospheric methane oxidizers in a meadow soil. ISME J. 6, 1115–1126. doi: 10.1038/ismej.2011.179
Siles, J. A., and Margesin, R. (2016). Abundance and diversity of bacterial, archaeal, and fungal communities along an altitudinal gradient in alpine forest soils: what are the driving factors? Microb. Ecol. 72, 207–220. doi: 10.1007/s00248-016-0748-2
Silver, W. L., Herman, D. J., and Firestone, M. K. (2001). Dissimilatory nitrate reduction to ammonium in upland tropical forest soils. Ecology 82, 2410–2416. doi: 10.1890/0012-9658(2001)082[2410:DNRTAI]2.0.CO;2
Singh, D., Lee-Cruz, L., Kim, W.-S., Kerfahi, D., Chun, J.-H., and Adams, J. M. (2014). Strong elevational trends in soil bacterial community composition on Mt. Halla, South Korea. Soil Biol. Biochem. 68, 140–149. doi: 10.1016/j.soilbio.2013.09.027
Singh, D., Takahashi, K., and Adams, J. M. (2012). Elevational patterns in archaeal diversity on Mt. Fuji. PLoS ONE 7:e44494. doi: 10.1371/journal.pone.0044494
Singh, D., Takahashi, K., Park, J., and Adams, J. M. (2016). Similarities and contrasts in the archaeal community of two japanese mountains: Mt. Norikura Compared to Mt. Fuji. Microb. Ecol. 71, 428–441. doi: 10.1007/s00248-015-0681-9
Skiba, U., Jones, S. K., Drewer, J., Helfter, C., Anderson, M., Dinsmore, K., et al. (2013). Comparison of soil greenhouse gas fluxes from extensive and intensive grazing in a temperate maritime climate. Biogeosciences 10, 1231–1241. doi: 10.5194/bg-10-1231-2013
Tong, X., Chen, F., Yu, J., Hua, S., Asan, C., Luosang, J., et al. (2008). Phylogenetic identification and microbial diversity in snow of the summit (8201 m) of Cho Oyu Mountain, Tibet. Chin. Sci. Bull. 53, 3317–3323. doi: 10.1007/s11434-008-0424-0
Trimmer, M., Nicholls, J. C., Morley, N., Davies, C. A., and Aldridge, J. (2005). Biphasic behavior of anammox regulated by nitrite and nitrate in an estuarine sediment. Appl. Environ. Microbiol. 71, 1923–1930. doi: 10.1128/AEM.71.4.1923-1930.2005
Trivedi, P., Delgado-Baquerizo, M., Trivedi, C., Hu, H., Anderson, I. C., Jeffries, T. C., et al. (2016). Microbial regulation of the soil carbon cycle: evidence from gene-enzyme relationships. ISME J. 10, 2593–2604. doi: 10.1038/ismej.2016.65
Tsui, C., Tsai, C., and Chen, Z. (2013). Soil organic carbon stocks in relation to elevation gradients in volcanic ash soils of Taiwan. Geoderma 209, 119–127. doi: 10.1016/j.geoderma.2013.06.013
Van Horn, D. J., Okie, J. G., Buelow, H. N., Gooseff, M. N., Barrett, J. E., and Takacs-Vesbach, C. D. (2014). Soil microbial responses to increased moisture and organic resources along a salinity gradient in a polar desert. Appl. Environ. Microbiol. 80, 3034–3043. doi: 10.1128/AEM.03414-13
Van Nostrand, J. D., Wu, W., Wu, L., Deng, Y., Carley, J., Carroll, S., et al. (2009). GeoChip-based analysis of functional microbial communities during the reoxidation of a bioreduced uranium-contaminated aquifer. Environ. Microbiol. 11, 2611–2626. doi: 10.1111/j.1462-2920.2009.01986.x
Vetaas, O. R., and Grytnes, J. A. (2002). Distribution of vascular plant species richness and endemic richness along the Himalayan elevation gradient in Nepal. Glob. Ecol. Biogeogr. 11, 291–301. doi: 10.1046/j.1466-822X.2002.00297.x
Wang, Q., Zhang, J., Xin, X., Zhao, B., Ma, D., and Zhang, H. (2016). The accumulation and transfer of arsenic and mercury in the soil under a long-term fertilization treatment. J. Soils Sediments 16, 427–437. doi: 10.1007/s11368-015-1227-y
Wang, Y., and Wang, Y. (2003). Quick measurement of CH4, CO2 and N2O emissions from a short-plant ecosystem. Adv. Atmos. Sci. 20, 842–844. doi: 10.1007/BF02915410
Wardle, D. A., Bardgett, R. D., Klironomos, J. N., Setala, H., van der Putten, W. H., and Wall, D. H. (2004). Ecological linkages between aboveground and belowground biota. Science 304, 1629–1633. doi: 10.1126/science.1094875
Willimsky, G., Bang, H., Fischer, G., and Marahiel, M. A. (1992). Characterization of scpB, a Bacillus-subtilis inducible cold shock gene affecting cell viability at low-temperatures. J. Bacteriol. 174, 6326–6335. doi: 10.1128/jb.174.20.6326-6335.1992
Wu, L., Ma, X., and Yang, Y. (2016a). Livestock grazing and greenhouse gas emission in tibet. Geoinfor. Geostat. Overv. 4, 430–440.
Wu, L., Yang, Y., Chen, S., Zhao, M., Zhu, Z., Yang, S., et al. (2016b). Long-term successional dynamics of microbial association networks in anaerobic digestion processes. Water Res. 104, 1–10. doi: 10.1016/j.watres.2016.07.072
Xu, M., Zhang, Q., Xia, C., Zhong, Y., Sun, G., Guo, J., et al. (2014). Elevated nitrate enriches microbial functional genes for potential bioremediation of complexly contaminated sediments. ISME J. 8, 1932–1944. doi: 10.1038/ismej.2014.42
Yang, S., Zhang, Y., Cong, J., Wang, M., Zhao, M., Lu, H., et al. (2017). Variations of soil microbial community structures beneath broadleaved forest trees in temperate and subtropical climate zones. Front. Microbiol. 8:200. doi: 10.3389/fmicb.2017.00200
Yang, Y., Gao, Y., Wang, S., Xu, D., Yu, H., Wu, L., et al. (2014). The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J. 8, 430–440. doi: 10.1038/ismej.2013.146
Yang, Y., Wu, L., Lin, Q., Yuan, M., Xu, D., Yu, H., et al. (2013). Responses of the functional structure of soil microbial community to livestock grazing in the Tibetan alpine grassland. Glob. Change Biol. 19, 637–648. doi: 10.1111/gcb.12065
Yoshida, M., Ishii, S., Otsuka, S., and Senoo, K. (2009). Temporal shifts in diversity and quantity of nirS and nirK in a rice paddy field soil. Soil Biol. Biochem. 41, 2044–2051. doi: 10.1016/j.soilbio.2009.07.012
Yue, H., Wang, M., Wang, S., Gilbert, J. A., Sun, X., Wu, L., et al. (2015). The microbe-mediated mechanisms affecting topsoil carbon stock in Tibetan grasslands. ISME J. 9, 2012–2020. doi: 10.1038/ismej.2015.19
Zhang, G., Niu, F., Ma, X., Liu, W., Dong, M., Feng, H., et al. (2007). Phylogenetic diversity of bacteria isolates from the Qinghai-Tibet plateau permafrost region. Can. J. Microbiol. 53, 1000–1010. doi: 10.1139/W07-031
Zhang, Y., Cong, J., Lu, H., Li, G., Xue, Y., Deng, Y., et al. (2015). Soil bacterial diversity patterns and drivers along an elevational gradient on Shennongjia Mountain, China. Microb. Biotechnol. 8, 739–746. doi: 10.1111/1751-7915.12288
Zhao, L., Li, Y., Xu, S., Zhou, H., Gu, S., Yu, G., et al. (2006). Diurnal, seasonal and annual variation in net ecosystem CO2 exchange of an alpine shrubland on Qinghai-Tibetan plateau. Glob. Change Biol. 12, 1940–1953. doi: 10.1111/j.1365-2486.2006.01197.x
Zhao, M., Xue, K., Wang, F., Liu, S., Bai, S., Sun, B., et al. (2014). Microbial mediation of biogeochemical cycles revealed by simulation of global changes with soil transplant and cropping. ISME J. 8, 2045–2055. doi: 10.1038/ismej.2014.46
Zhao, Y., Peth, S., Krümmelbein, J., Horn, R., Wang, Z., Steffens, M., et al. (2007). Spatial variability of soil properties affected by grazing intensity in Inner Mongolia grassland. Ecol. Modell. 205, 241–254. doi: 10.1016/j.ecolmodel.2007.02.019
Zheng, Y., Hou, L., Liu, M., Gao, J., Yin, G., Li, X., et al. (2015). Diversity, abundance, and distribution of nirS-harboring denitrifiers in intertidal sediments of the yangtze estuary. Microb. Ecol. 70, 30–40. doi: 10.1007/s00248-015-0567-x
Zhou, H., Tang, Y., Zhao, X., and Zhou, L. (2006). Long-term grazing alters species composition and biomass of a shrub meadow on the Qinghai-Tibet Plateau. Pak. J. Bot. 38, 1055–1069. doi: 10.1111/j.1744-7909.2008.00676.x
Zhou, J., He, Z., Yang, Y., Deng, Y., Tringe, S. G., and Alvarez-Cohen, L. (2015a). High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. MBio 6:e2288-14. doi: 10.1128/mBio.02288-14
Zhou, J., Kang, S., Schadt, C. W., and Garten, C. T. (2008). Spatial scaling of functional gene diversity across various microbial taxa. Proc. Natl. Acad. Sci. U.S.A. 105, 7768–7773. doi: 10.1073/pnas.0709016105
Zhou, J., Wang, Z., Zhang, X., and Chen, J. (2015b). Distribution and elevated soil pools of mercury in an acidic subtropical forest of southwestern China. Environ. Pollut. 202, 187–195. doi: 10.1016/j.envpol.2015.03.021
Zhou, Z., Du, S., and Liu, G. (2009). “Acidification of surface soil in croplands in the semiarid middle tibet plateau, china,” in Proceeding ESIAT ’09 Proceedings of the 2009 International Conference on Environmental Science and Information Application Technology, Vol. 2, (Washington, DC: IEEE Computer Society), 209–212. doi: 10.1109/ESIAT.2009.72
Keywords: microbial biogeography, soil microbial community, microbial functional potential, alpine grassland, altitudinal gradient
Citation: Qi Q, Zhao M, Wang S, Ma X, Wang Y, Gao Y, Lin Q, Li X, Gu B, Li G, Zhou J and Yang Y (2017) The Biogeographic Pattern of Microbial Functional Genes along an Altitudinal Gradient of the Tibetan Pasture. Front. Microbiol. 8:976. doi: 10.3389/fmicb.2017.00976
Received: 11 January 2017; Accepted: 15 May 2017;
Published: 13 June 2017.
Edited by:
Etienne Yergeau, University of Quebec, CanadaReviewed by:
Shang Wang, Research Center for Eco-Environmental Science, Chinese Academy of Sciences (CAS), ChinaWeidong Kong, Institute of Tibetan Plateau Research, Chinese Academy of Sciences (CAS), China
Terrence H. Bell, Pennsylvania State University, United States
Copyright © 2017 Qi, Zhao, Wang, Ma, Wang, Gao, Lin, Li, Gu, Li, Zhou and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunfeng Yang, eWFuZ3lmQHRzaW5naHVhLmVkdS5jbg==
†These authors have contributed equally to this work.