 Yongxin Lin1,2
Yongxin Lin1,2 Weixin Ding
Weixin Ding- 1State Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science, Chinese Academy of Sciences, Nanjing, China
- 2University of the Chinese Academy of Sciences, Beijing, China
The effects of elevated atmospheric CO2 concentration on soil microbial communities have been previously recorded. However, limited information is available regarding the response of methanogenic communities to elevated CO2 in freshwater marshes. Using high-throughput sequencing and real-time quantitative PCR, we compared the abundance and community structure of methanogens in different compartments (bulk soil, rhizosphere soil, and roots) of Calamagrostis angustifolia and Carex lasiocarpa growing marshes under ambient (380 ppm) and elevated CO2 (700 ppm) atmospheres. C. lasiocarpa rhizosphere was a hotspot for potential methane production, based on the 10-fold higher abundance of the mcrA genes per dry weight. The two marshes and their compartments were occupied by different methanogenic communities. In the C. lasiocarpa marsh, archaeal family Methanobacteriaceae, Rice Cluster II, and Methanosaetaceae co-dominated in the bulk soil, while Methanobacteriaceae was the exclusively dominant methanogen in the rhizosphere soil and roots. Families Methanosarcinaceae and Methanocellaceae dominated in the bulk soil of C. angustifolia marsh. Conversely, Methanosarcinaceae and Methanocellaceae together with Methanobacteriaceae dominated in the rhizosphere soil and roots, respectively, in the C. angustifolia marsh. Elevated atmospheric CO2 increased plant photosynthesis and belowground biomass of C. lasiocarpa and C. angustifolia marshes. However, it did not significantly change the abundance (based on mcrA qPCR), diversity, or community structure (based on high-throughput sequencing) of methanogens in any of the compartments, irrespective of plant type. Our findings suggest that the population and species of the dominant methanogens had weak responses to elevated atmospheric CO2. However, minor changes in specific methanogenic taxa occurred under elevated atmospheric CO2. Despite minor changes, methanogenic communities in different compartments of two contrasting freshwater marshes were rather stable under elevated atmospheric CO2.
Introduction
Atmospheric CO2 concentration has increased dramatically over the past 250 years, and is expected to continue to rise in the future (Denman et al., 2007). Elevated atmospheric CO2 concentrations are predicted to affect plant growth and alter soil properties. For example, atmospheric CO2 enrichment has been found to increase plant photosynthesis (Sasaki et al., 2005) and biomass (Hungate et al., 1997). These changes subsequently regulate the quality and quantity of the labile organic compounds that are secreted (Drigo et al., 2008) and influence downstream consumers. Wetland ecosystems play a pivotal role in global biogeochemical cycles and are a major source of atmospheric methane (CH4), having contributed 32% of the global annual CH4 emissions in the 2000s (International Panel on Climate Change [IPCC], 2013). Elevated CO2 can increase the carbon supply to wetland sediment (Freeman et al., 2004a), which in turn may increase CH4 emissions from wetlands (Vann and Patrick Megonigal, 2003; van Groenigen et al., 2011). Thus, understanding the belowground process response to elevated CO2 in a wetland ecosystem may help to accurately evaluate ecosystem feedbacks to global climate change.
Freshwater marshes are a substantial source of atmospheric CH4, with high global CH4 emission rates (Ding and Cai, 2007; Flury et al., 2010). For example, although freshwater marshes only account for 27% of the total natural wetland area in China, they contribute up to 66% of the annual natural wetland CH4 emissions (Ding et al., 2004). Calamagrostis angustifolia and Carex lasiocarpa are two dominant plants in a freshwater marsh, but with different standing water depths on the Sanjiang plain, China. The methanogenic substrates in the C. lasiocarpa marsh have been identified to mainly derive from plant litter, and from root exudates in the C. angustifolia marsh (Lin et al., 2015). A previous study has shown that dominant methanogenic communities in the bulk soil of C. lasiocarpa and C. angustifolia marshes are clearly different (Liu et al., 2012a). It is not clear if and how methanogenic communities in the rhizosphere and roots in the C. lasiocarpa and C. angustifolia marshes respond to elevated CO2.
Methanogenic archaea belong to the phylum Euryarchaeota (Garcia et al., 2000; Liu and Whitman, 2008), and there are generally three types in a wetland ecosystem. Members of order Methanomicrobiales, Methanobacteriales, and Methanocellales (Rice Cluster I) have been identified to exclusively use H2/CO2 as a substrate for CH4 production, and are known as hydrogenotrophic methanogens (Conrad, 2007). Methanosaetaceae within the order Methanosarcinales use acetate as an energy substrate, and are named acetoclastic methanogens (Liu and Whitman, 2008). In addition, Methanosarcinaceae within the order of Methanosarcinales use H2/CO2, acetate, and methyl compounds, and are ascribed to metabolically versatile methanogens (Thauer et al., 2008). Different habitats within wetland ecosystems may provide different niches for different types of methanogens. For example, Methanocellales were found to dominate in the rice rhizosphere and roots (Lu and Conrad, 2005; Lu et al., 2005; Conrad et al., 2008) while Methanoregulaceae (Fen Cluster) were dominant in boreal fen (Galand et al., 2002; Juottonen et al., 2005; Cadillo-Quiroz et al., 2006). Previous studies have shown that elevated atmospheric CO2 altered the soil microbial community (Deng et al., 2012; Drigo et al., 2013; He et al., 2014; Lee et al., 2015). It has been suggested that these changes were indirectly induced by changes in root growth, followed by changes in the quality and quantity of exudates under elevated CO2 (Drigo et al., 2008). Studying a temperate marsh microcosm, Lee et al. (2012) found that elevated CO2 concentrations increased the relative abundance of hydrogenotrophic methanogens. In contrast, other studies did not identify any apparent effect on the methanogen community (Angel et al., 2012; Liu et al., 2016). Drigo et al. (2008) found that elevated atmospheric CO2 did not affect the microbial community in the bulk soil, but did near the roots. Therefore, the effects of elevated CO2 concentration on methanogen communities are still debated. It is therefore necessary to evaluate whether and how the methanogenic community structure in the bulk soil, rhizosphere and roots will respond to elevated CO2 in wetland ecosystems.
In the present study, the methanogenic community structure and its response to elevated atmospheric CO2 was measured in the bulk soil, rhizosphere soil and roots of C. lasiocarpa and C. angustifolia marshes using high-throughput sequencing. We hypothesized that (1) there is a substantial difference in the methanogenic community structure among the different compartments, including the bulk soil, rhizosphere and roots; and (2) elevated atmospheric CO2 concentrations will inevitably induce higher methanogenic abundance by increasing substrate supply, including root exudates, due to an increase in plant photosynthesis, and also alter the methanogenic community composition in wetland ecosystems.
Materials and Methods
Experimental Site
The study site is located in the Sanjiang Mire Wetland Experimental Station, Tongjiang City, Heilongjiang Province (47°35′N, 133°31′E). The area of freshwater marsh in the test region is about 105 km2. The elevation is 56 m above sea level, and the mean annual precipitation is approximately 600 mm. The mean annual temperature was 1.9°C, with a range of -18.8°C in January and 20.8°C in July. The dominant vegetation in the freshwater marsh varies from Calamagrostis angustifolia to Carex meyeriana, and to Carex lasiocarpa as the standing water depth increases (Ding et al., 2002). Standing water depth ranged from 0 to 20 cm in the Carex lasiocarpa marsh, from 0 to 15 cm in the Carex meyeriana marsh, and from 0 to 5 cm in the Calamagrostis angustifolia marsh. We selected a permanently inundated C. lasiocarpa marsh and a summer inundated C. angustifolia marsh for the elevated CO2 experiment.
Experimental Setup
Sixteen intact soil columns with 25-cm diameter and 30-cm depth were collected using a stainless steel sampler from each plant growing wetland on 10 May 2015. These columns were immediately transferred into specially designed PVC macrocosms (40-cm height and 25-cm inner diameter) and transported to the experimental site. The standing water depth in the C. lasiocarpa and C. angustifolia marshes was maintained at 8 and 2 cm (average standing water depth in summer), respectively, during the experimental period from May to October.
All macrocosms were divided into two groups based on measured methane emission rates and transferred into two open top chambers (OTC) in the field. The methane emission rates were nearly identical in the two groups of the same plant type marsh. The OTC chambers were constructed from a PVC frame and clear, infrared-transmissive Aclar film walls (Shanghai, China) to allow natural light exposure (>95%). The tops and bottoms of the chambers were left open to allow air circulation. One chamber received an elevated CO2 treatment (eCO2), while the other received ambient air (aCO2). Continuous injection of pure CO2 into the OTC was carried out to maintain the elevated levels of CO2 in the OTC at 700 ± 20 ppm. The air sample was drawn from the middle of the chamber by an automated system and the CO2 concentration was monitored using an infrared gas analyzer (Li-7000; Licor, Lincoln, NE, United States). A rain-diversion system was constructed over the experiment site, and automatically opened on sunny days and closed on rainy days. The chamber air temperature was automatically adjusted to ±1°C of the real air temperature by a temperature control system. At the end of the growing season in October, the aboveground biomass was determined by carefully clipping plants in each macrocosm in one group at the soil surface. The plants were then washed with distilled water and oven-dried for 24 h at 70°C to constant mass. To estimate underground plant biomass, the live roots and rhizome material were manually picked out from the soil columns and then oven-dried as mentioned above.
Measurement of Net Photosynthetic Rate and Soil Pore Water DOC Concentration
The net photosynthetic rate (Pn) was measured on flag leaves from 09:00 to 11:00 am by a LI-6400 portable photosynthesis system (LiCor Inc., Lincoln, NE, United States). For each treatment, Pn of nine leaves was determined at each measurement and the measurement repeated twice. A Rhizon MOM pore water sampler (Rhizosphere Research Products, Wageningen, the Netherlands), which consisted of a 5 cm porous part and 20 cm tubing PVC hose, was used to collect soil pore water at 10 cm depth of marsh profile. Prior to collecting samples, approximately 5 mL of pore water were extracted and discarded and then 50 mL of pore water was collected. Dissolved organic carbon (DOC) in soil pore water was analyzed on a Shimadzu C analyzer (TOC Vcph, Shimadzu, Kyoto, Japan).
Plant and Soil Sampling and DNA Extraction
The plants and soil in the four macrocosms of each treatment in another group were destructively sampled on 8 August 2015 when the plants showed the highest activity. The plants and soil were manually separated into three compartments; bulk soil (non-rooted soil), rhizosphere soil (soil on root surface), and root. The samples from the three compartments were collected according to Bao et al. (2014). Briefly, the near-bottom and non-rooted soil was collected and designated as bulk soil. Then, the roots with surface soils were washed before and after sonication, and all soils were pooled and centrifuged for 10 min at 10,000 × g at 4°C to generate the rhizosphere soil samples. After the rhizosphere soil was removed, the roots were transferred to new 50-mL Falcon tubes containing sterilized water and centrifuged for 10 min at 10,000 × g at 4°C; the pellet in this centrifugation tube was designated as the root component. All samples were stored at -80°C for no more than 2 weeks prior to molecular analysis. DNA was extracted from all samples using the Fast DNA SPIN for soil kit (BIO 178 101, Qbiogene, Carlsbad, CA, United States) according to the manufacturer’s instructions. For the root samples, the frozen tissues were ground to powder in liquid nitrogen before DNA extraction.
Real-Time Quantitative PCR
To quantify the functional marker genes (mcrA) of methanogens, we performed q-PCR using a CFX96 Optical Real-Time Detection System (Bio-Rad Laboratories Inc., Hercules, CA, United States). Each reaction in the 25 μl contained the specific primer set for the methanogen: MLf (GGTGGTGTMGGATTCACACARTAYGCWACAGC) – MLr (TTCATTGCRTAGTTWGGRTAGTT) (Juottonen et al., 2006). Each reaction mixture (25 μl) consisted of 12.5 μl 1 × SYBR Premix Ex Taq (Takara, Tokyo, Japan), 0.25 μl of each primer, and 1 μl of DNA template containing approximately 1–10 ng of DNA. A negative control was always run with sterilized distilled water as the template instead of a DNA sample. The amplification was initiated by denaturation at 95°C for 2 min, followed by 35 cycles of denaturation at 95°C for 10 s, annealing at 55°C for 30 s, extension at 72°C for 30 s, and the plate was read at 80°C. The standard curves were created using 10-fold dilution series of plasmid DNA containing the mcrA gene from environmental samples for methanogen. A serial dilution of the DNA template was also used to assess whether the PCR was inhibited during the amplification. The amplification always resulted in a single peak, and amplification efficiencies of 106.7–108.7% were obtained with R2 values of 0.992–0.999.
High-Throughput Sequencing and Data Analysis
For methanogens, the primer set 1106F and 1378R was used to amplify approximately 280 bp of methanogenic archaeal 16S rRNA gene fragments (Feng et al., 2013). The forward primer contained a sample-identifying barcode unique to each sample. PCR amplifications were performed in 50 μl reaction mixtures containing approximately 40 ng of template DNA, 25 μl Phusion High-Fidelity PCR Master Mix (New England Biolabs), 20.5 μl H2O, 0.5 μl 1% bovine serum albumin (BSA), and 0.2 μM of each primer. Thirty cycles (95°C for 45 s, 56°C for 45 s, and 72°C for 60 s) were performed with a final extension at 72°C for 7 min. PCR products were mixed in equidensity ratios. Then, the mixed PCR products were purified with the Qiagen Gel Extraction Kit (Qiagen, Germany). Sequencing libraries were generated using the TruSeq DNA PCR-Free Sample Preparation Kit (Illumina, United States) following the manufacturer’s recommendations. The library quality was assessed on the Qubit 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 systems. Finally, high-throughput sequencing of the methanogenic archaeal 16S rRNA genes was carried out on the Illumina HiSeq2500 platform at the Novogene (Beijing, China), and 250 bp paired-end reads were generated.
The high-throughput sequencing data were processed using the Quantitative Insights Into Microbial Ecology (QIIME) 1.7.0-dev pipeline (Caporaso et al., 2010) using default parameters, unless otherwise indicated. In brief, low quality sequence reads were trimmed and the barcode was examined to assign the multiplexed reads to samples. Next, the sequences were compared with the reference database (Edgar et al., 2011) using the UCHIME algorithm to detect chimeric sequences, and the chimeric sequences were removed. Sequences with ≥97% similarity were assigned to the same OTUs using the Uparse software (Uparse v7.0.1001) (Edgar, 2013), and the most abundant sequence from each OTU was selected as a representative sequence for that OTU. Taxonomy was assigned to methanogenic archaeal OTUs against the GreenGene Database (DeSantis et al., 2006) using a RDP classifier (Wang et al., 2007). To remove the heterogeneity of the number of sequences per sample, the sequences were rarefied prior to the calculation of the alpha diversity statistics. Alpha diversity was assessed by calculating Chao1 (Chao and Bunge, 2002), Observed species, Phylogenetic Diversity (PD), and Shannon metrics in QIIME. All sequences have been deposited in the DNA Data Bank of Japan under the accession number DRA005365.
Statistical Analysis
The SPSS 18.0 software package for Windows (SPSS Inc., Chicago, IL, United States) was used to perform statistical analyses. All data were expressed on the basis of the oven-dried soil mass. Before analysis of variance (ANOVA), the data were checked for normality and homogeneity of variance (Levene’s test), and were subsequently ln-transformed to meet the assumptions of the ANOVA analyses, if necessary. Statistically significant differences among treatments and compartments were determined by one-way ANOVA and least significant difference (LSD) calculations at a 5% confidence level. A Non-Metric Multidimensional Scaling (NMDS) plot was constructed from the analyzed samples based on the pairwise shared OTUs to determine the change in the community structure. Three different non-parametric analysis methods, including non-parametric multivariate analysis of variance using distance matrices (adonis), analysis of similarities (ANOSIM), and a multi-response permutation procedure (MRPP), were used to examine whether there were significant differences in the community structures between the treatments. The Bray-Curtis distance was used for the adonis, ANOSIM, and MRPP analyses. NMDS, adonis, ANOSIM, and MRPP were processed with the “vegan” package in the R software version 3.0.2.
Results
Plant Growth and Physiological Characteristics
The net photosynthetic rate, DOC concentration, and aboveground and belowground biomass in the C. lasiocarpa and C. angustifolia marshes under ambient and elevated CO2 concentration were shown in Figure 1. The net photosynthetic rates in the C. lasiocarpa and C. angustifolia marshes were increased under elevated CO2 treatment compared with ambient CO2 treatment, irrespective of plant types. Elevated CO2 concentration increased belowground biomass in the C. lasiocarpa and C. angustifolia marshes. It also increased DOC concentration in soil pore water of C. angustifolia marsh.
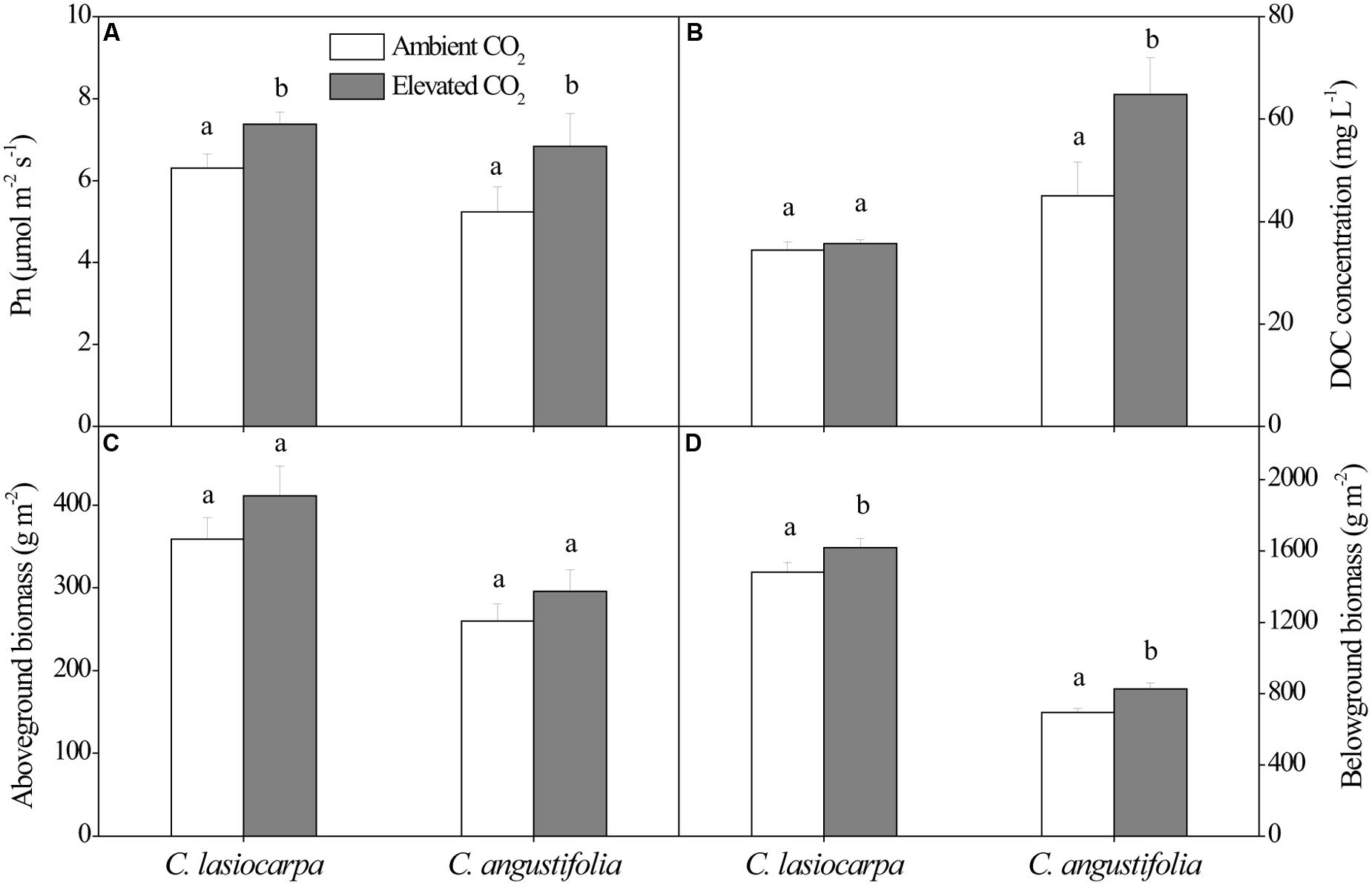
FIGURE 1. Effect of elevated CO2 concentration on net photosynthetic rate (A), dissolved organic C (DOC) concentration in soil pore water (B), aboveground biomass (C) and belowground biomass (D) in the C. lasiocarpa and C. angustifolia marshes. Different letters denote significant differences between ambient and elevated CO2 treatments at P < 0.05. Vertical bars denote the standard error of the mean (n = 4).
Abundance of Methanogenic Archaea
The copy numbers of the methanogenic archaeal mcrA gene varied from 1.59 ± 0.98 × 106 to 2.74 ± 0.56 × 1010 copies per g d.w.s. There appeared to be a greater abundance of the methanogenic archaeal mcrA gene in the rhizosphere soil than in the bulk soil and roots, especially in the C. lasiocarpa marsh (Figure 2). The mcrA gene abundance in the rhizosphere soil of C. lasiocarpa marsh was at least one order of magnitude greater than that in the bulk soil and roots. The mcrA gene abundance in the C. lasiocarpa marsh was significantly higher than that in the C. angustifolia marsh in all three compartments. However, mcrA gene abundance did not differ between ambient and elevated CO2 treatments in all the compartments, irrespective of plant types.
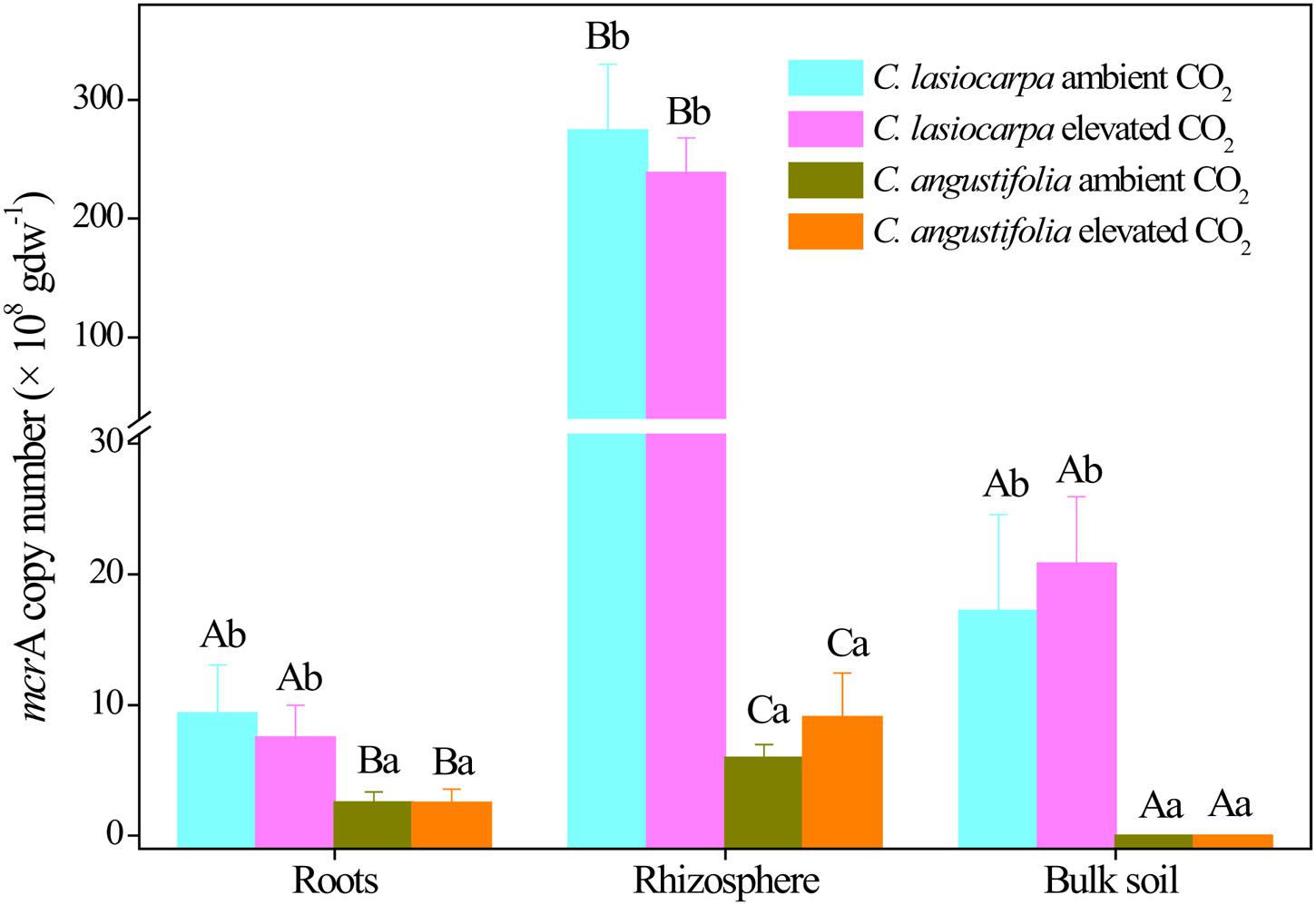
FIGURE 2. Change in the abundances of methanogenic mcrA genes in the C. lasiocarpa and C. angustifolia marshes from ambient to elevated CO2 concentrations based on qPCR. Different letters denote significant differences between compartments in the same treatment (capital letters) and treatments in the same compartment (lowercase letters) at P < 0.05. Vertical bars denote the standard error of the mean (n = 4).
Diversity, Richness and Taxonomic Distribution of Methanogenic Archaea
The Chao1, PD, observed species, and Shannon indices derived from high-throughput sequencing provided estimates of the microbial diversity and richness among different samples. Chao1 and observed species consistently showed that the maximum richness and diversity of methanogeic archaea in the bulk soil were much higher than those in the rhizosphere soil and roots (Supplementary Figure S1). However, there was no significant difference in methanogenic archaeal richness and diversity between rhizosphere soil and roots (Table 1). The elevated CO2 concentration reduced the PD and observed species of methanogenic archaea in the bulk soil in the C. angustifolia marsh, but did not affect the corresponding values in the C. lasiocarpa marsh. The richness and diversity of methanogenic archaea appeared similar between the elevated and ambient CO2 treatments in the rhizosphere soil and roots in both plant marshes.
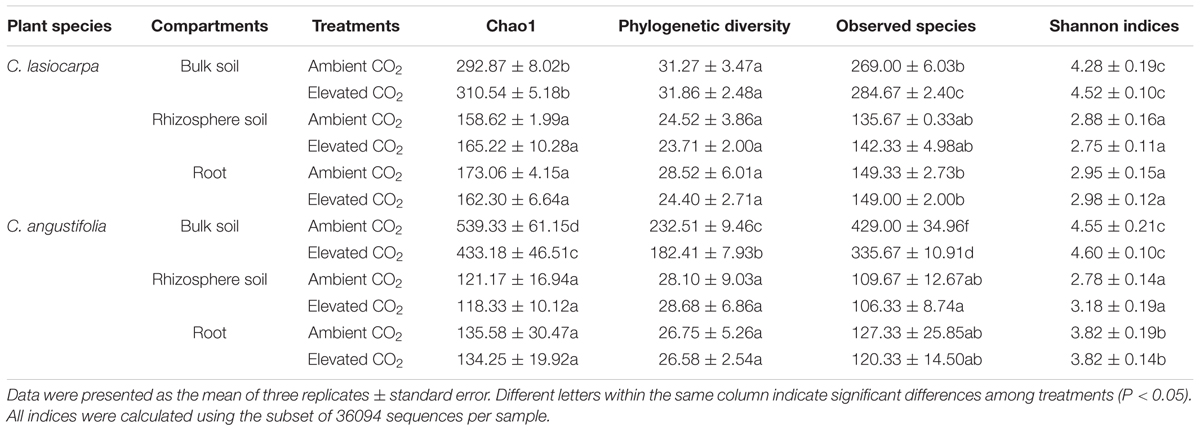
TABLE 1. Comparison of the estimated OTU richness and diversity indices of the 16S rRNA gene libraries from the high-throughput sequencing analysis.
High-throughput sequencing revealed that the methanogenic archaeal community was dominated by several families (Figure 3). In the bulk soil, the methanogenic archaeal community was dominated by Methanobacteriaceae (21.6%), Rice Cluster II (29.9%), and Methanosaetaceae (22.7%) in the C. lasiocarpa marsh; and by Methanosarcinaceae (7.3%) and Methanocellaceae (10.9%) in the C. angustifolia marsh. In the rhizosphere soil, the dominant methanogenic archaeal families were Methanobacteriaceae (60.5%) and Methanosarcinaceae (18.8%) in the C. lasiocarpa marsh; and Methanosarcinaceae (69.6%) and Methanoregulaceae (9.4%) in the C. angustifolia marsh. In the roots, the methanogenic archaeal community was dominated by Methanobacteriaceae (61.7%) and Methanoregulaceae (18.4%) in the C. lasiocarpa marsh; and by Methanobacteriaceae (35.1%), Methanosarcinaceae (18.8%), and Methanocellaceae (34.2%) in the C. angustifolia marsh. A significant difference was found in the methanogenic archaeal community between the bulk soil and roots in the same plant marsh (Table 2). Significant differences also occurred in the methanogenic archaeal community between the C. lasiocarpa and C. angustifolia marshes. However, there was only a small, and insignificant, variation in the methanogenic archaeal community between the elevated and ambient CO2 treatments in the two tested marshes.
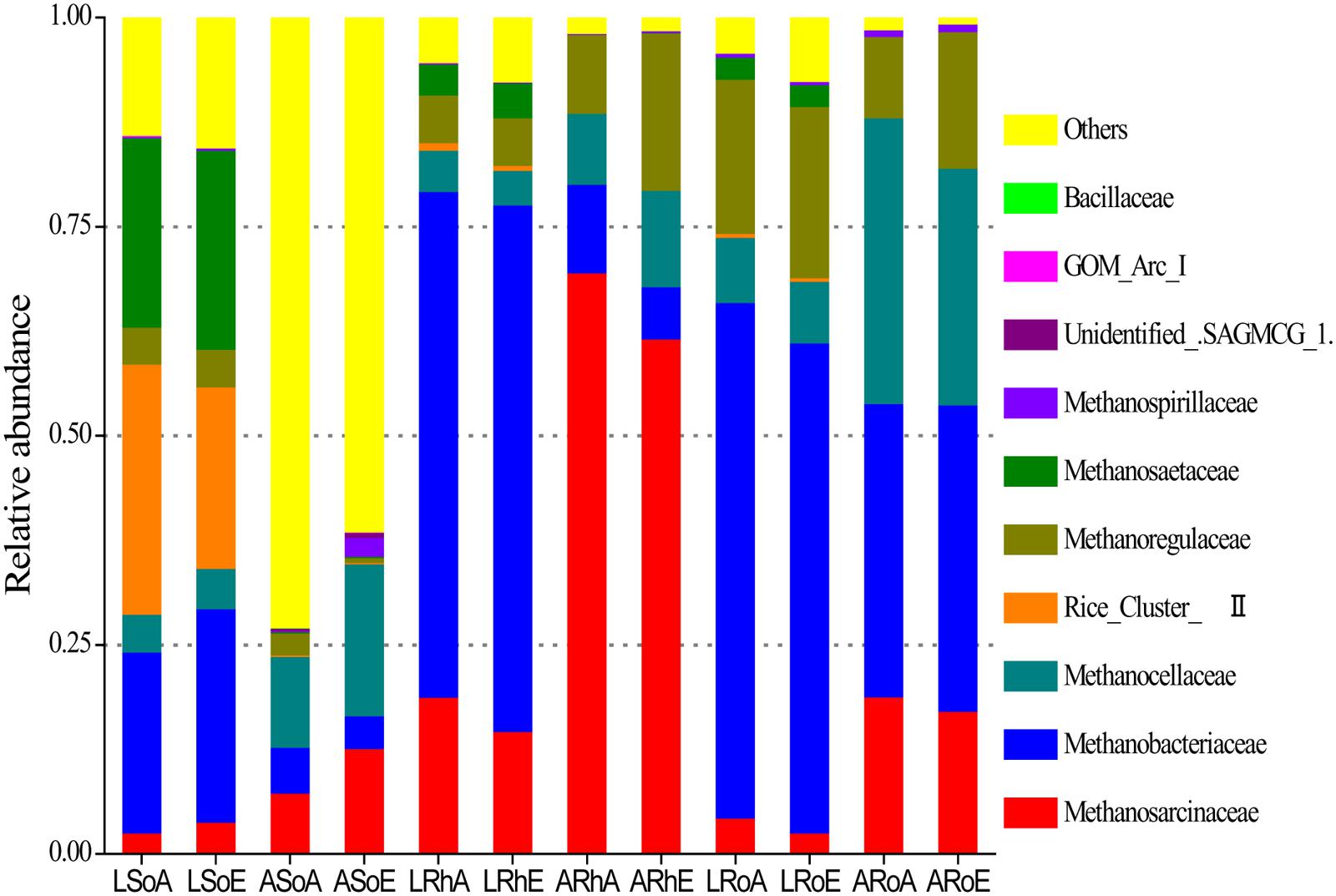
FIGURE 3. A 100% stack column chart of the dominant methanogenic archaeal families in each sample from high-throughput sequencing. The value of each family percentage is the mean of four replicates. Group names have been selected according to the treatments: L and A denote the C. lasiocarpa and C. angustifolia marshes, respectively; So, Rh, and Ro indicate the soil, rhizosphere soil, and root compartments, respectively; and A and E represent ambient and elevated CO2 concentrations, respectively.
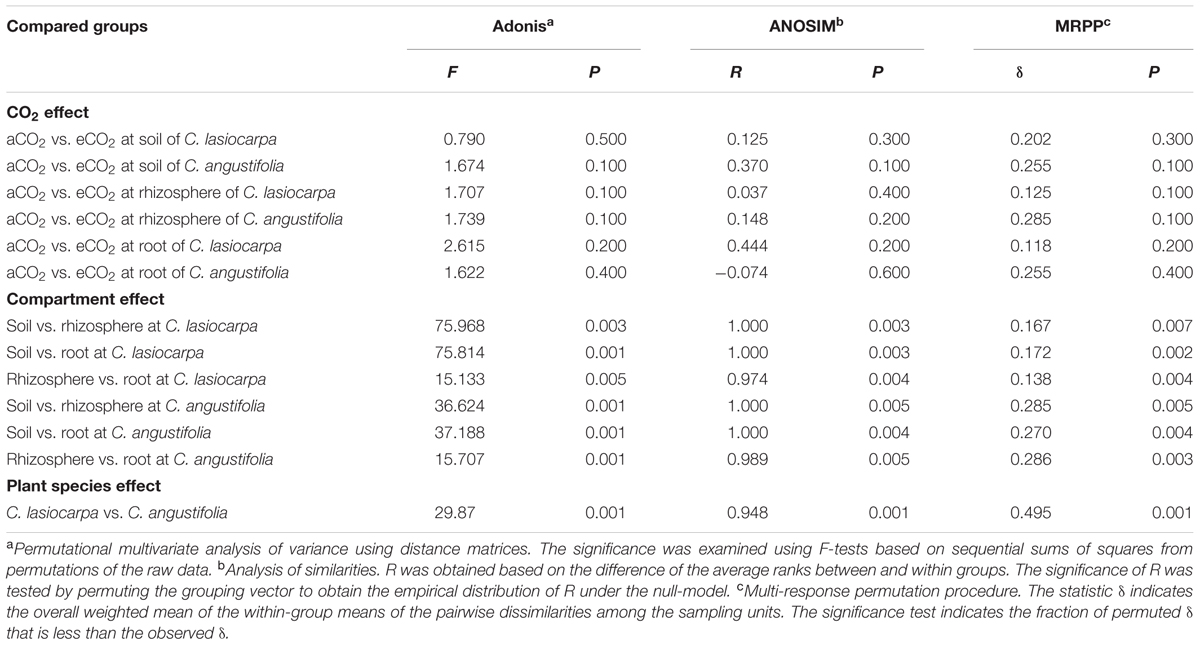
TABLE 2. Significance tests of the effects of the CO2, soil or plant compartments and plant species on the overall methanogenic community structure with three statistical approaches.
Shift in the Composition of the Methanogenic Archaeal Community
To better visualize the shifts in the methanogenic archaeal community composition of the two plant marshes in response to elevated CO2, non-metric multidimensional scaling (NMDS) was performed (Figure 4). Elevated CO2 did not alter the methanogenic archaeal community composition in the bulk soil, rhizosphere soil or roots in either plant dominated marshes. This finding was further confirmed by three non-parametric multivariate analyses (adonis, ANOSIM, and MRPP). In contrast, the methanogenic communities were significantly different between the different compartments, regardless of CO2 levels (Table 2). For example, a potential shift was observed in the methanogenic archaeal community between the bulk and rhizosphere soils, irrespective of plant type. Furthermore, the methanogenic archaeal communities in the C. lasiocarpa marsh were grouped together and significantly separated from those in the C. angustifolia marsh (Figure 4).
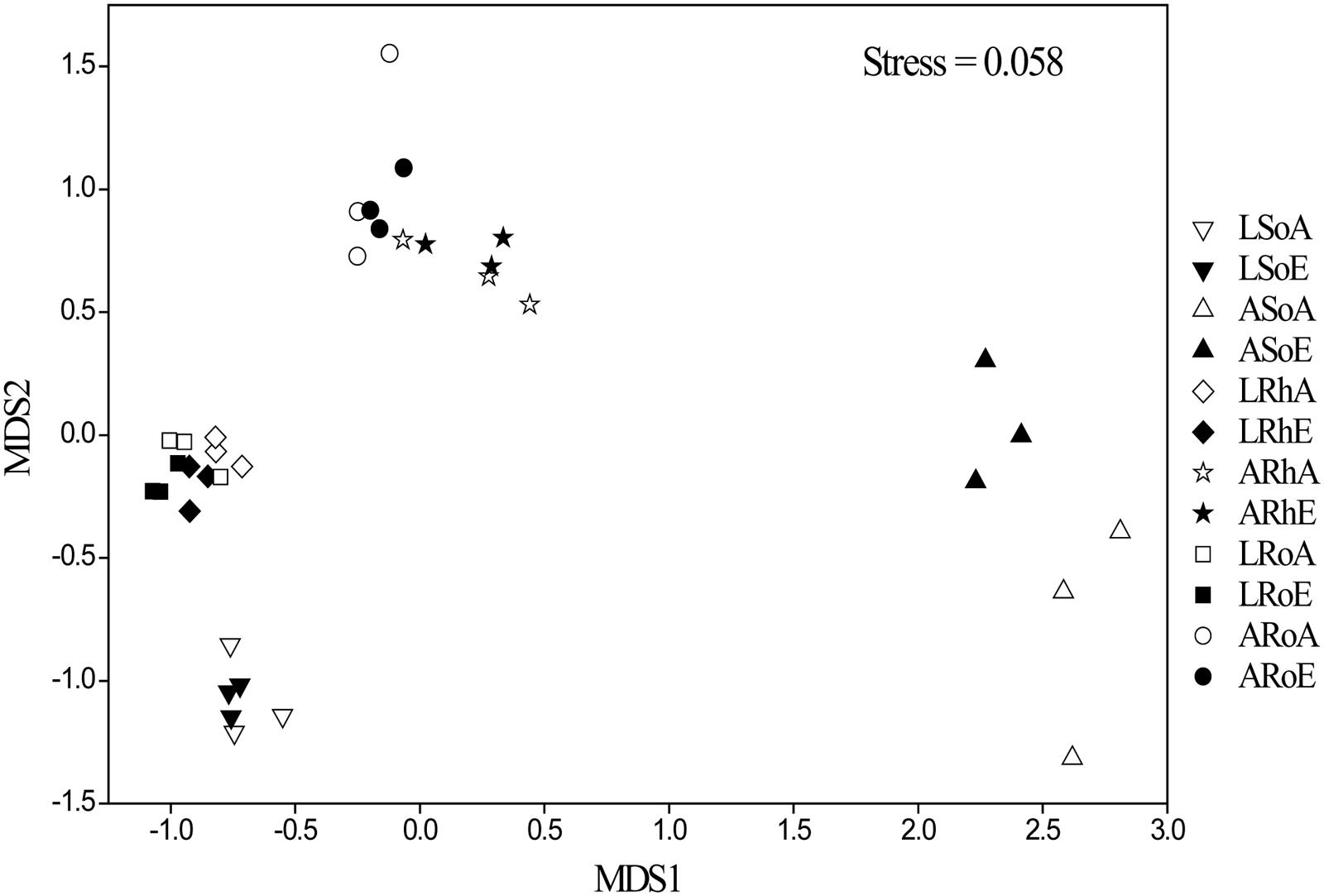
FIGURE 4. Community composition structure of methanogenic archaea in different compartments from high-throughput sequencing, indicated by a non-metric multi-dimensional scaling plot among the different samples. Group names have been selected according to the treatments: L and A denote the C. lasiocarpa and C. angustifolia marshes, respectively; So, Rh and Ro indicate the soil, rhizosphere soil, and root compartments, respectively; and A and E represent ambient and elevated CO2 concentrations, respectively.
Methanogenic Archaeal Community Response to CO2 Enrichment
The response of the methanogenic archaea to elevated CO2 was calculated based on their sequence numbers obtained by high-throughput sequencing to identify the respective responses of dominant methanogenic archaeal communities to elevated CO2. Elevated CO2 decreased the relative abundance of Methanosarcinaceae in the root compartment in the C. lasiocarpa marsh and Methanoregulaceae in the bulk soil in the C. angustifolia marsh (Figure 5). In addition, some trends were seen in the relative abundance of Methanosarcinaceae, Methanobacteriaceae, and Methanosaetaceae (Figure 5), but due to high variation and low sample numbers they were not statistically significant.
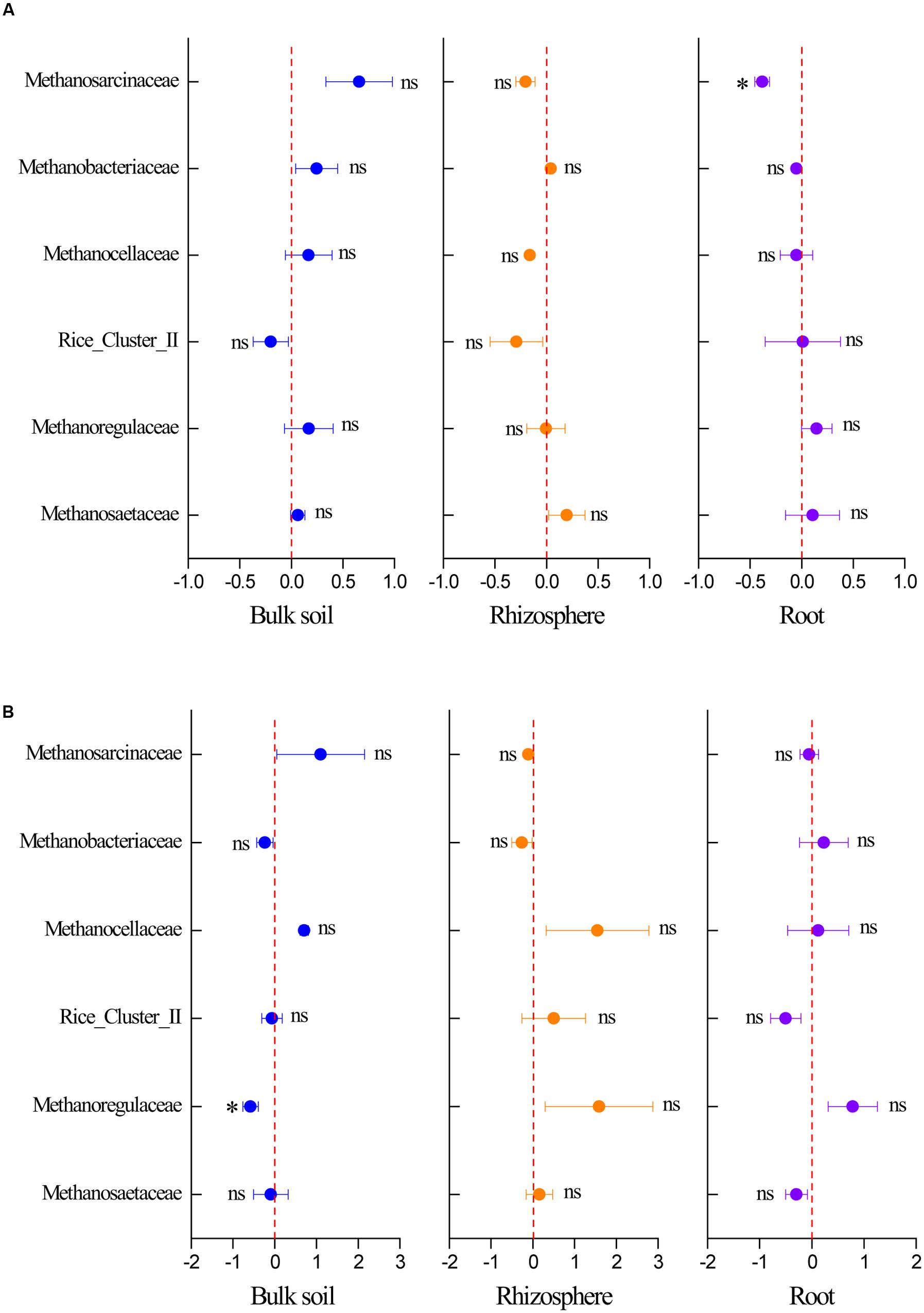
FIGURE 5. Effect of elevated CO2 on the relative abundance of methanogenic taxa at the family level in the C. lasiocarpa (A) and C. angustifolia (B) marshes based on high-throughput sequencing. The elevated CO2 effect was measured using the net difference in the relative abundance of methanogenic taxa between the elevated and ambient CO2 treatments and then divided by the relative abundance of the phylotype under ambient CO2 treatment. The error bars represent the standard error of the mean (n = 3). The asterisk indicates significant difference at P < 0.05; ns indicates no significant difference (P > 0.05).
Discussion
Methanogenic Archaea in Different Niches
The composition of the methanogenic archaea community was clearly different between the soil and roots of the two plant marshes, as reported by Ke et al. (2013, 2014). In the two plant roots, Methanobacteriaceae were dominant although their relative abundance was lower in C. angustifolia. Methanoregulaceae were more abundant than Methanocellaceae in C. lasiocarpa roots and the opposite was observed in C. angustifolia roots, suggesting a plant-specific enrichment. This result was consistent with a previous finding in another two contrasting vascular plants that Methanoregulaceae were more abundant than Methanocellaceae on Dulichium arundinaceum and the opposite was observed on Sarracenia purpurea, in an acidic bog (Cadillo-Quiroz et al., 2010). In the C. lasiocarpa marsh, Methanobacteriaceae, Rice Cluster II, and Methanosaetaceae were co-dominant in the bulk soil. However, Methanobacteriaceae were the exclusively dominant methanogenic archaea (>60%) in the rhizosphere soil and roots. The Methanobacteriaceae family has been widely studied, and all species are known to use H2/CO2 as a substrate (Bonin and Boone, 2006). Previous studies have demonstrated that the specific communities of methanogens that had colonized rice roots preferred hydrogenotrophic over acetoclastic methanogens (Chin et al., 2004; Wu et al., 2009; Liu G.C. et al., 2012). Methanocellaceae were found to dominate particularly in the rice roots, as they have a multiple set of genes encoding for antioxidant enzymes (Erkel et al., 2006) and can produce CH4 even under aerobic conditions (Angel et al., 2011). In the present study, however, Methanobacteriaceae rather than Methanocellaceae dominated in the roots of C. lasiocarpa, which has a strong gas transport capacity delivering a lot of oxygen downward to the rhizosphere (Ding et al., 2005). Some members of the Methanobacteriaceae were found to survive after an exposure to oxygen (Kiener and Leisinger, 1983). Using a DNA-based stable isotope probing (DNA-SIP) technique, our previous study (Lin et al., 2015) identified Methanobacteriaceae as a metabolically active methanogen in the C. lasiocarpa marsh at the same site. These findings suggest that Methanobacteriaceae may play a key role in the C. lasiocarpa marsh. However, further study is required to evaluate the physiological characteristics and molecular mechanisms of how Methanobacteriaceae act in the rhizosphere and roots of the C. lasiocarpa marshes, which have high CH4 emissions (Song et al., 2009).
In the C. angustifolia marsh, Methanosarcinaceae and Methanocellaceae were dominant in the bulk soil, while Methanosarcinaceae and Methanocellaceae dominated in the rhizosphere soil and roots, respectively. Methanosarcinaceae and Methanocellaceae were also found to be the main community members on rice roots and associated soil (Krüger et al., 2005), and to be metabolically active methanogens in the rice roots measured by DNA-SIP (Lu et al., 2005). In addition, Methanocellaceae have the ability to produce CH4 under aerobic conditions (Angel et al., 2011) as they contain antioxidant enzymes encoded genes. It is therefore not surprising that Methanocellaceae dominated in the C. angustifolia roots. Methanosarcinaceae have the most versatile substrate spectrum, including H2/CO2, acetate, and methyl compounds (Thauer et al., 2008). They flourish in the rhizosphere supported by their ability to use various byproducts from root exudates and decaying plant debris (Shrestha et al., 2011). Furthermore, Methanosarcinaceae have the capacity to tolerate low oxygen levels and possess catalase genes that actively transcribe for oxygen detoxification (Angel et al., 2011). Consequently, C. angustifolia rhizosphere soil is a suitable habitat for Methanosarcinaceae.
Effect of Elevated CO2 on Methanogenic Archaea
In this study, the difference in the mcrA gene abundance in the three compartments of both marshes was statistically not significant under elevated CO2 when compared to ambient CO2. In a previous paddy soil study, Liu et al. (2016) similarly did not observe an effect of elevated CO2 on the abundance of the methanogenic archaeal community in the bulk soil. In contrast, some studies have reported that elevated CO2 significantly increased the abundance of methanogenic archaea in rice cultivated soil (Inubushi et al., 2003; Okubo et al., 2015) because of the increase in the carbon supply as root exudates with elevated CO2 (Bhattacharyya et al., 2013). In the present study, elevated CO2 concentration significantly increased the plant photosynthesis and root biomass in both plant marshes (Figure 1) and these changes would subsequently increase C substrate for methanogens. However, the increased C substrate under elevated CO2 did not result in a significant increase in the abundance of methanogenic archaea (Figure 2). The reasons for this include: first, the increased belowground C substrate input may have not been large enough to enhance growth of the methanogenic population, or such C-input increases may have only promoted methanogenic metabolic rates rather than increased population size. The effects of elevated CO2 on CH4 emission rates in the two marshes, increasing CH4 emissions by 27.60% in C. angustifolia marsh and suppressing them by 18.67% in the C. lasiocarpa marsh (to be discussed in a future publication) were unrelated to responses of the methanogenic population (Lin et al., unpublished data). Secondly, the potential stimulating effect of increased substrate supply under elevated CO2 on the abundance of methanogenic archaea may have been counteracted by the inhibiting effect of increased delivery of O2 to the roots and rhizosphere (Freeman et al., 2004b). Elevated CO2 may enlarge aerenchyma conduits and increase root porosity and subsequently increase O2 concentration in the rhizosphere (Schrope et al., 1999; Cheng et al., 2000). Furthermore, the significantly increased root biomass and photosynthesis (Figure 1) due to elevated CO2 may also have more effectively aerated the soil and inhibited methanogen growth.
To date, studies have shown different responses of the soil microbial communities to elevated CO2. Some studies have found that elevated CO2 induced shifts in the microbial community composition (Lagomarsino et al., 2008; Lee et al., 2012, 2015), while others did not find any significant influence (Grüter et al., 2006; Angel et al., 2012). Gschwendtner et al. (2015) pointed out that the intensity of the effect of elevated CO2 on soil microbial communities was dependent on the distance from the roots. Hayden et al. (2012) and Okubo et al. (2015) suggested that elevated CO2 induced changes in the community structures of microorganisms mainly in the rhizosphere and roots. In the present study; however, we did not find a significant effect of elevated CO2 on the methanogenic community composition in the bulk soil, rhizosphere soil, or roots (Figure 4). The resilience of the methanogenic community to elevated CO2 may be due to the general robustness of the community against changes in the surrounding environment (Liu et al., 2012b, 2016), or the changes in the organic substrate input and environmental factors were potentially not large enough to change the methanogen community composition (Angel et al., 2012). Likewise, in a 10-year atmospheric CO2 enrichment experiment, Angel et al. (2012) did not observe changes to the methanogenic communities in a waterlogged grassland under elevated CO2. Therefore, it is unlikely that the lack of apparent effect of elevated CO2 on the methanogenic community in this study was due to the short duration of the field experimental period. Thus, we suggest that the methanogenic community was relatively stable in the two contrasting marshes even if the atmospheric CO2 concentration doubled. Although we did not find significant changes in the methanogenic community in response to elevated CO2 concentrations in either of the marshes or their compartments, minor trends were observed.
Overall, our data demonstrate that there is a clear difference in abundance and community structure of methanogens among the different compartments of two contrasting plant dominated marshes. Abundance and community structure of methanogens also differed between C. lasiocarpa and C. angustifolia marsh. C. lasiocarpa rhizosphere was a hotspot for potential methane production, based on the 10-fold higher abundance of the mcrA genes per dry weight. However, in contrast to our hypothesis, although minor difference occurred in specific methanogenic taxa in the different compartments and plant dominated marshes, the abundance and community structure of methanogens were rather stable when atmospheric CO2 concentrations was increased.
Author Contributions
Conceived and designed the experiments: DL and WD. Performed the experiments: YL, JY, and GY. Analyzed the data: YL and DL. Wrote the paper: YL and WD. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to gratefully acknowledge the Sanjiang Mire Wetland Experimental Station, Chinese Academy of Sciences for providing the study site and assistance in field experiment. This study was financially supported by the Chinese Academy of Sciences (XDB15020100) and the National Natural Science Foundation of China (31561143011, 41171190).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00932/full#supplementary-material
References
Angel, R., Kammann, C., Claus, P., and Conrad, R. (2012). Effect of long-term free-air CO2 enrichment on the diversity and activity of soil methanogens in a periodically waterlogged grassland. Soil Biol. Biochem. 51, 96–103. doi: 10.1016/j.soilbio.2012.04.010
Angel, R., Matthies, D., and Conrad, R. (2011). Activation of methanogenesis in arid biological soil crusts despite the presence of oxygen. PLoS ONE 6:e20453. doi: 10.1371/journal.pone.0020453
Bao, Z., Watanabe, A., Sasaki, K., Okubo, T., Tokida, T., Liu, D., et al. (2014). A rice gene for microbial symbiosis, Oryza sativa CCaMK, reduces CH4 flux in a paddy field with low nitrogen input. Appl. Environ. Microbiol. 80, 1995–2003. doi: 10.1128/AEM.03646-13
Bhattacharyya, P., Roy, K. S., Neogi, S., Manna, M. C., Adhya, T. K., Rao, K. S., et al. (2013). Influence of elevated carbon dioxide and temperature on belowground carbon allocation and enzyme activities in tropical flooded soil planted with rice. Environ. Monit. Assess. 185, 8659–8671. doi: 10.1007/s10661-013-3202-7
Bonin, A., and Boone, D. (2006). “The order methanobacteriales,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer).
Cadillo-Quiroz, H., Bräuer, S., Yashiro, E., Sun, C., Yavitt, J., and Zinder, S. (2006). Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central New York State. USA. Environ. Microbiol. 8, 1428–1440. doi: 10.1111/j.1462-2920.2006.01036.x
Cadillo-Quiroz, H., Yavitt, J. B., Zinder, S. H., and Thies, J. E. (2010). Diversity and community structure of Archaea inhabiting the rhizoplane of two contrasting plants from an acidic bog. Microb. Ecol. 59, 757–767. doi: 10.1007/s00248-009-9628-3
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chao, A., and Bunge, J. (2002). Estimating the number of species in a stochastic abundance model. Biometrics 58, 531–539. doi: 10.1111/j.0006-341X.2002.00531.x
Cheng, W., Chander, K., and Inubushi, K. (2000). “Effects of elevated CO2 and temperature on methane production and emission from submerged soil microcosm,” in Methane Emissions from Major Rice Ecosystems in Asia, eds R. Wassmann, R. S. Lantin, and H.-U. Neue (Dordrecht: Springer), 339–347. doi: 10.1007/978-94-010-0898-3_29
Chin, K. J., Lueders, T., Friedrich, M. W., Klose, M., and Conrad, R. (2004). Archaeal community structure and pathway of methane formation on rice roots. Microb. Ecol. 47, 59–67. doi: 10.1007/s00248-003-2014-7
Conrad, R. (2007). “Microbial ecology of methanogens and methanotrophs,” in Advances in Agronomy, ed. L. S. Donald (Cambridge, MA: Academic Press).
Conrad, R., Klose, M., Noll, M., Kemnitz, D., and Bodelier, P. L. E. (2008). Soil type links microbial colonization of rice roots to methane emission. Glob. Change Biol. 14, 657–669. doi: 10.1111/j.1365-2486.2007.01516.x
Deng, Y., He, Z., Xu, M., Qin, Y., Van Nostrand, J. D., Wu, L., et al. (2012). Elevated carbon dioxide alters the structure of soil microbial communities. Appl. Environ. Microbiol. 78, 2991–2995. doi: 10.1128/AEM.06924-11
Denman, K. L., Brasseur, G., Chidthaisong, A., et al. (2007). “Couplings between changes in the climate system and biogeochemistry,” in Proceedings of the Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, eds S. Solomon, D. Qin, M. Manning, Z. Chen, M. Marquis, K. B. Averyt, et al. (Cambridge: Cambridge University Press).
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Ding, W., and Cai, Z. (2007). Methane emission from natural wetlands in China: summary of years 1995–2004 studies. Pedosphere 17, 475–486. doi: 10.1016/S1002-0160(07)60057-5
Ding, W., Cai, Z., and Tsuruta, H. (2005). Plant species effects on methane emissions from freshwater marshes. Atmos. Environ. 39, 3199–3207. doi: 10.1016/j.atmosenv.2005.02.022
Ding, W., Cai, Z., Tsuruta, H., and Li, X. (2002). Effect of standing water depth on methane emissions from freshwater marshes in northeast China. Atmos. Environ. 36, 5149–5157. doi: 10.1016/S1352-2310(02)00647-7
Ding, W., Cai, Z., and Wang, D. (2004). Preliminary budget of methane emissions from natural wetlands in China. Atmos. Environ. 38, 751–759. doi: 10.1016/j.atmosenv.2003.10.016
Drigo, B., Kowalchuk, G. A., Knapp, B. A., Pijl, A. S., Boschker, H. T. S., and Van Veen, J. A. (2013). Impacts of 3 years of elevated atmospheric CO2 on rhizosphere carbon flow and microbial community dynamics. Glob. Change Biol. 19, 621–636. doi: 10.1111/gcb.12045
Drigo, B., Kowalchuk, G. A., and Van Veen, J. A. (2008). Climate change goes underground: effects of elevated atmospheric CO2 on microbial community structure and activities in the rhizosphere. Biol. Fertil. Soils 44, 667–679. doi: 10.1007/s00374-008-0277-3
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Erkel, C., Kube, M., Reinhardt, R., and Liesack, W. (2006). Genome of Rice Cluster I archaea—the key methane producers in the rice rhizosphere. Science 313, 370–372. doi: 10.1126/science.1127062
Feng, Y., Lin, X., Yu, Y., Zhang, H., Chu, H., and Zhu, J. (2013). Elevated ground-level O3 negatively influences paddy methanogenic archaeal community. Sci. Rep. 3:3193. doi: 10.1038/srep03193
Flury, S., Mcginnis, D. F., and Gessner, M. O. (2010). Methane emissions from a freshwater marsh in response to experimentally simulated global warming and nitrogen enrichment. J. Geophys. Res. Biogeosci. 115:G01007. doi: 10.1029/2009jg001079
Freeman, C., Fenner, N., Ostle, N. J., Kang, H., Dowrick, D. J., Reynolds, B., et al. (2004a). Export of dissolved organic carbon from peatlands under elevated carbon dioxide levels. Nature 430, 195–198.
Freeman, C., Kim, S. Y., Lee, S. H., and Kang, H. (2004b). Effects of elevated atmospheric CO2 concentrations on soil microorganisms. J. Microbiol. 42, 267–277.
Galand, P. E., Saarnio, S., Fritze, H., and Yrjälä, K. (2002). Depth related diversity of methanogen Archaea in Finnish oligotrophic fen. FEMS Microbiol. Ecol. 42, 441–449. doi: 10.1111/j.1574-6941.2002.tb01033.x
Garcia, J. L., Patel, B. K. C., and Ollivier, B. (2000). Taxonomic, phylogenetic, and ecological diversity of methanogenic archaea. Anaerobe 6, 205–226. doi: 10.1006/anae.2000.0345
Grüter, D., Schmid, B., and Brandl, H. (2006). Influence of plant diversity and elevated atmospheric carbon dioxide levels on belowground bacterial diversity. BMC Microbiol. 6:68. doi: 10.1186/1471-2180-6-68
Gschwendtner, S., Leberecht, M., Engel, M., Kublik, S., Dannenmann, M., Polle, A., et al. (2015). Effects of elevated atmospheric CO2 on microbial community structure at the plant-soil interface of young beech trees (Fagus sylvatica L.) grown at two sites with contrasting climatic conditions. Microb. Ecol. 69, 867–878. doi: 10.1007/s00248-014-0527-x
Hayden, H. L., Mele, P. M., Bougoure, D. S., Allan, C. Y., Norng, S., Piceno, Y. M., et al. (2012). Changes in the microbial community structure of bacteria, archaea and fungi in response to elevated CO2 and warming in an Australian native grassland soil. Environ. Microbiol. 14, 3081–3096. doi: 10.1111/j.1462-2920.2012.02855.x
He, Z., Xiong, J., Kent, A. D., Deng, Y., Xue, K., Wang, G., et al. (2014). Distinct responses of soil microbial communities to elevated CO2 and O3 in a soybean agro-ecosystem. ISME J. 8, 714–726. doi: 10.1038/ismej.2013.177
Hungate, B. A., Holland, E. A., Jackson, R. B., Chapin, F. S., Mooney, H. A., and Field, C. B. (1997). The fate of carbon in grasslands under carbon dioxide enrichment. Nature 388, 576–579. doi: 10.1038/41550
International Panel on Climate Change [IPCC] (2013). “Climate change 2013: the physical science basis,” in Proceedings of the Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (Cambridge: Cambridge University Press), 465–570.
Inubushi, K., Cheng, W., Aonuma, S., Hoque, M. M., Kobayashi, K., Miura, S., et al. (2003). Effects of free-air CO2 enrichment (FACE) on CH4 emission from a rice paddy field. Glob. Change Biol. 9, 1458–1464. doi: 10.1046/j.1365-2486.2003.00665.x
Juottonen, H., Galand, P. E., Tuittila, E.-S., Laine, J., Fritze, H., and Yrjälä, K. (2005). Methanogen communities and bacteria along an ecohydrological gradient in a northern raised bog complex. Environ. Microbiol. 7, 1547–1557. doi: 10.1111/j.1462-2920.2005.00838.x
Juottonen, H., Galand, P. E., and Yrjälä, K. (2006). Detection of methanogenic Archaea in peat: comparison of PCR primers targeting the mcrA gene. Res. Microbiol. 157, 914–921. doi: 10.1016/j.resmic.2006.08.006
Ke, X., Angel, R., Lu, Y., and Conrad, R. (2013). Niche differentiation of ammonia oxidizers and nitrite oxidizers in rice paddy soil. Environ. Microbiol. 15, 2275–2292. doi: 10.1111/1462-2920.12098
Ke, X., Lu, Y., and Conrad, R. (2014). Different behaviour of methanogenic archaea and Thaumarchaeota in rice field microcosms. FEMS Microbiol. Ecol. 87, 18–29. doi: 10.1111/1574-6941.12188
Kiener, A., and Leisinger, T. (1983). Oxygen sensitivity of methanogenic bacteria. Syst. Appl. Microbiol. 4, 305–312. doi: 10.1016/S0723-2020(83)80017-4
Krüger, M., Frenzel, P., Kemnitz, D., and Conrad, R. (2005). Activity, structure and dynamics of the methanogenic archaeal community in a flooded Italian rice field. FEMS Microbiol. Ecol. 51, 323–331. doi: 10.1016/j.femsec.2004.09.004
Lagomarsino, A., Moscatelli, M. C., Hoosbeek, M. R., De Angelis, P., and Grego, S. (2008). Assessment of soil nitrogen and phosphorous availability under elevated CO2 and N-fertilization in a short rotation poplar plantation. Plant Soil 308, 131–147. doi: 10.1007/s11104-008-9614-4
Lee, S. H., Kim, S. Y., Ding, W., and Kang, H. (2015). Impact of elevated CO2 and N addition on bacteria, fungi, and archaea in a marsh ecosystem with various types of plants. Appl. Microbiol. Biotechnol. 99, 5295–5305. doi: 10.1007/s00253-015-6385-8
Lee, S. H., Kim, S. Y., and Kang, H. (2012). Effects of elevated CO2 on communities of denitrifying bacteria and methanogens in a temperate marsh microcosm. Microb. Ecol. 64, 485–498. doi: 10.1007/s00248-012-0036-8
Lin, Y., Liu, D., Ding, W., Kang, H., Freeman, C., Yuan, J., et al. (2015). Substrate sources regulate spatial variation of metabolically active methanogens from two contrasting freshwater wetlands. Appl. Microbiol. Biotechnol. 99, 10779–10791. doi: 10.1007/s00253-015-6912-7
Liu, D., Ding, W., Jia, Z., and Cai, Z. (2012a). The impact of dissolved organic carbon on the spatial variability of methanogenic archaea communities in natural wetland ecosystems across China. Appl. Microbiol. Biotechnol. 96, 253–263. doi: 10.1007/s00253-011-3842-x
Liu, D., Suekuni, C., Akita, K., Ito, T., Saito, M., Watanabe, T., et al. (2012b). Effect of winter-flooding on methanogenic archaeal community structure in paddy field under organic farming. Soil Sci. Plant Nutr. 58, 553–561. doi: 10.1080/00380768.2012.726598
Liu, G. C., Tokida, T., Matsunami, T., Nakamura, H., Okada, M., Sameshima, R., et al. (2012). Microbial community composition controls the effects of climate change on methane emission from rice paddies. Environ. Microbiol. Rep. 6, 648–654. doi: 10.1111/j.1758-2229.2012.00391.x
Liu, D., Tago, K., Hayatsu, M., Tokida, T., Sakai, H., Nakamura, H., et al. (2016). Effect of elevated CO2 concentration, elevated temperature and no nitrogen fertilization on methanogenic archaeal and methane-oxidizing bacterial community structures in paddy soil. Microbes Environ. 31, 349–356. doi: 10.1264/jsme2.ME16066
Liu, Y., and Whitman, W. B. (2008). Metabolic, phylogenetic, and ecological diversity of the methanogenic Archaea. Ann. N. Y. Acad. Sci. 1125, 171–189. doi: 10.1196/annals.1419.019
Lu, Y., Lueders, T., Friedrich, M. W., and Conrad, R. (2005). Detecting active methanogenic populations on rice roots using stable isotope probing. Environ. Microbiol. 7, 326–336. doi: 10.1111/j.1462-2920.2005.00697.x
Lu, Y. H., and Conrad, R. (2005). In situ stable isotope probing of methanogenic archaea in the rice rhizosphere. Science 309, 1088–1090. doi: 10.1126/science.1113435
Okubo, T., Liu, D., Tsurumaru, H., Ikeda, S., Asakawa, S., Tokida, T., et al. (2015). Elevated atmospheric CO2 levels affect community structure of rice root-associated bacteria. Front. Microbiol. 6:136. doi: 10.3389/fmicb.2015.00136
Sasaki, H., Hara, T., Ito, S., Miura, S., Hoque, M. M., Lieffering, M., et al. (2005). Seasonal changes in canopy photosynthesis and respiration, and partitioning of photosynthate, in rice (Oryza sativa L.) grown under free-Air CO2 enrichment. Plant Cell Physiol. 46, 1704–1712. doi: 10.1093/pcp/pci186
Schrope, M. K., Chanton, J. P., Allen, L. H., and Baker, J. T. (1999). Effect of CO2 enrichment and elevated temperature on methane emissions from rice, Oryza sativa. Glob. Change Biol. 5, 587–599. doi: 10.1111/j.1365-2486.1999.00252.x
Shrestha, M., Shrestha, P. M., and Conrad, R. (2011). Bacterial and archaeal communities involved in the in situ degradation of 13C-labelled straw in the rice rhizosphere. Environ. Microbiol. Rep. 3, 587–596. doi: 10.1111/j.1758-2229.2011.00267.x
Song, C., Xu, X., Tian, H., and Wang, Y. (2009). Ecosystem-atmosphere exchange of CH4 and N2O and ecosystem respiration in wetlands in the Sanjiang Plain, Northeastern China. Glob. Change Biol. 15, 692–705. doi: 10.1111/j.1365-2486.2008.01821.x
Thauer, R. K., Kaster, A. K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591. doi: 10.1038/nrmicro1931
van Groenigen, K. J., Osenberg, C. W., and Hungate, B. A. (2011). Increased soil emissions of potent greenhouse gases under increased atmospheric CO2. Nature 475, 214–216. doi: 10.1038/nature10176
Vann, C. D., and Patrick Megonigal, J. (2003). Elevated CO2 and water depth regulation of methane emissions: comparison of woody and non-woody wetland plant species. Biogeochemistry 63, 117–134. doi: 10.1023/A:1023397032331
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Keywords: bulk soil, elevated CO2, freshwater marsh, methanogen, rhizosphere, roots
Citation: Lin Y, Liu D, Yuan J, Ye G and Ding W (2017) Methanogenic Community Was Stable in Two Contrasting Freshwater Marshes Exposed to Elevated Atmospheric CO2. Front. Microbiol. 8:932. doi: 10.3389/fmicb.2017.00932
Received: 22 December 2016; Accepted: 08 May 2017;
Published: 24 May 2017.
Edited by:
Marja Tiirola, University of Jyväskylä, FinlandReviewed by:
Katharina Palmer, University of Oulu, FinlandBruce A. Hungate, Northern Arizona University, United States
Copyright © 2017 Lin, Liu, Yuan, Ye and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weixin Ding, d3hkaW5nQGlzc2FzLmFjLmNu