
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 29 May 2017
Sec. Evolutionary and Genomic Microbiology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.00852
 James L. Crainey1
James L. Crainey1 Jacob Hurst2
Jacob Hurst2 Poppy H. L. Lamberton3
Poppy H. L. Lamberton3 Robert A. Cheke4,5
Robert A. Cheke4,5 Claire E. Griffin6Michael D. Wilson7Cláudia P. Mendes de Araújo1
Claire E. Griffin6Michael D. Wilson7Cláudia P. Mendes de Araújo1 María-Gloria Basáñez5*†Rory J. Post8,9†
María-Gloria Basáñez5*†Rory J. Post8,9†Research interest in Wolbachia is growing as new discoveries and technical advancements reveal the public health importance of both naturally occurring and artificial infections. Improved understanding of the Wolbachia bacteriophages (WOs) WOcauB2 and WOcauB3 [belonging to a sub-group of four WOs encoding serine recombinases group 1 (sr1WOs)], has enhanced the prospect of novel tools for the genetic manipulation of Wolbachia. The basic biology of sr1WOs, including host range and mode of genomic integration is, however, still poorly understood. Very few sr1WOs have been described, with two such elements putatively resulting from integrations at the same Wolbachia genome loci, about 2 kb downstream from the FtsZ cell-division gene. Here, we characterize the DNA sequence flanking the FtsZ gene of wDam, a genetically distinct line of Wolbachia isolated from the West African onchocerciasis vector Simulium squamosum E. Using Roche 454 shot-gun and Sanger sequencing, we have resolved >32 kb of WO prophage sequence into three contigs representing three distinct prophage elements. Spanning ≥36 distinct WO open reading frame gene sequences, these prophage elements correspond roughly to three different WO modules: a serine recombinase and replication module (sr1RRM), a head and base-plate module and a tail module. The sr1RRM module contains replication genes and a Holliday junction recombinase and is unique to the sr1 group WOs. In the extreme terminal of the tail module there is a SpvB protein homolog—believed to have insecticidal properties and proposed to have a role in how Wolbachia parasitize their insect hosts. We propose that these wDam prophage modules all derive from a single WO genome, which we have named here sr1WOdamA1. The best-match database sequence for all of our sr1WOdamA1-predicted gene sequences was annotated as of Wolbachia or Wolbachia phage sourced from an arthropod. Clear evidence of exchange between sr1WOdamA1 and other Wolbachia WO phage sequences was also detected. These findings provide insights into how Wolbachia could affect a medically important vector of onchocerciasis, with potential implications for future control methods, as well as supporting the hypothesis that Wolbachia phages do not follow the standard model of phage evolution.
It is estimated that Wolbachia naturally infect about 40% of arthropods, including many important disease vectors (Bourtzis et al., 2014; Zug and Hammerstein, 2015). As these infections have an impact on several epidemiologically-relevant aspects of disease vector biology, such as longevity, insecticide resistance, and refractoriness to infection, it has been argued that Wolbachia are likely to influence disease epidemiology (Echaubard et al., 2010; Slatko et al., 2014; Hoffmann et al., 2015). Much of the present public health interest in arthropod-infecting Wolbachia focuses on how artificial infections can be manipulated as tools for effective disease control (Bourtzis et al., 2014; Hoffmann et al., 2015; Jeffries and Walker, 2015).
Wolbachia bacteriophages (WOs) have received far less attention than their bacterial hosts, with some research focusing on how they could influence disease ecology and epidemiology (Tanaka et al., 2009; Metcalf and Bordenstein, 2012; LePage and Bordenstein, 2013; Wang et al., 2013) and, most commonly, how they might be utilized for disease control (Metcalf and Bordenstein, 2012; LePage and Bordenstein, 2013; Slatko et al., 2014). Several authors have advocated the possibility of developing artificial WO vectors for the genetic modification of Wolbachia. Despite the potential of WO-based tools and the growing interest in the use of Wolbachia for vector-borne disease control, there are presently no genetic manipulation tools available for the genetic engineering of Wolbachia (LePage and Bordenstein, 2013; Bourtzis et al., 2014; Slatko et al., 2014; Hoffmann et al., 2015; Jeffries and Walker, 2015). There is, thus, a growing need for a better understanding of the basic biology, diversity and distribution of naturally occurring WOs to assess the feasibility and potential utility of WO-based Wolbachia manipulation tools (Tanaka et al., 2009; LePage and Bordenstein, 2013; Wang et al., 2013). Similarly, there is also a pressing need to improve our understanding about how naturally occurring WOs influence vector-borne disease epidemiology and what risks (if any) they pose to the safety of using artificial Wolbachia infections for disease control (Bourtzis et al., 2014; Hoffmann et al., 2015; Jeffries and Walker, 2015; Caragata et al., 2016).
In previous studies, we identified a genetically isolated strain of Wolbachia from the West African onchocerciasis vector Simulium squamosum E (a member of the S. damnosum sensu lato [s.l.] species complex [Diptera: Simuliidae]) and identified bacterial artificial chromosomes (BACs) containing its FtsZ cell-division gene (Crainey et al., 2010a,b). As shown in Table 1, the FtsZ gene is part of a conserved block (spanning ~3 kb) immediately adjacent to where two closely-related prophages (WOcauB2 and a WOri phage relic) have been identified in two genetically-distinct Wolbachia genomes: wCau and wRi genomes (Tanaka et al., 2009; Kent et al., 2011a; Ellegaard et al., 2013). This six-gene block begins in both cases with superoxide dismutase and terminates with the magnesium chelatase-related protein, which occurs immediately adjacent to the prophages' serine recombinase gene. If these WOs belong to a group of site-specific bacteriophages, large cloned fragments of the wDam genome, containing FtsZ gene sequences (Crainey et al., 2010a) could be expected also to contain Wolbachia prophage sequences. Similarly, if, as proposed, certain WOs have a role in male-killing (and male-killing is affecting the S. damnosum s.l. complex), any WOs might also be expected to harbor SpvB genes (Crainey et al., 2010a; Kent et al., 2011a; Metcalf and Bordenstein, 2012; LePage and Bordenstein, 2013). In this study, we have characterized the genomic DNA of wDam flanking its FtsZ-gene and have recovered three WO phage sequence elements, including one that encodes a SpvB-like gene, that we propose all derive from a single WO prophage genome that we have named sr1WOdamA1.
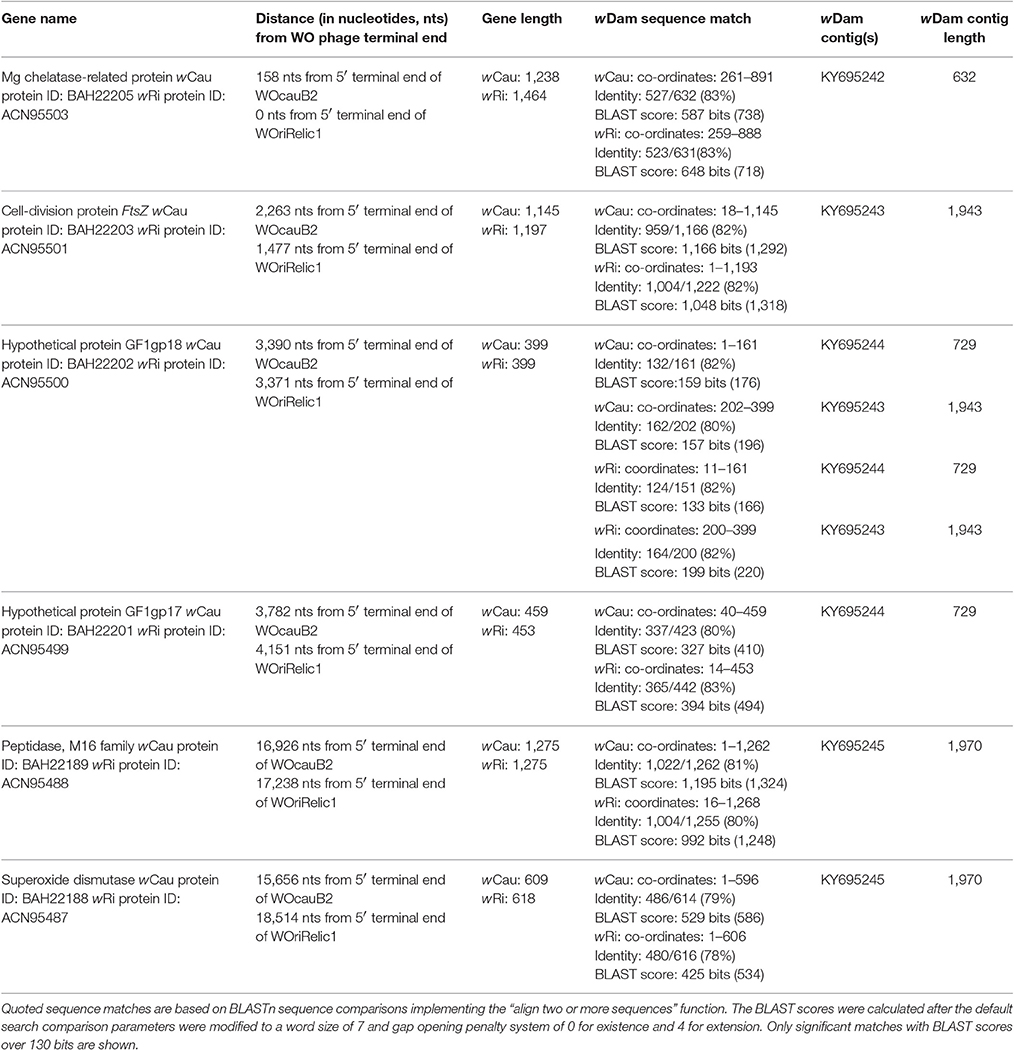
Table 1. wDam shot-gun sequence contigs from matching known and predicted serine recombinase WO upstream integration sites.
Large Wolbachia-DNA-containing BAC clone mini cultures from seven FtsZ-positive BACs (identified previously) were grown shaking over-night in BAC library growth media (Crainey et al., 2010b). Thick mini-culture preparations from each BAC colony were pooled and their BAC DNA was isolated in single preparations using a QIAGEN large-construct kit and protocol (https://www.qiagen.com/kr/resources/resourcedetail?id=8f67b644-6d21-4ef3-b33e-a60f32623785&lang=en). A 10-mg sample of purified BAC DNA was shot-gun sequenced commercially using a Roche 454 FLX system sequencer at the Cambridge University Biochemistry Department. Sequence reads were quality-checked using Phred Software (http://www.phrap.org/phredphrapconsed.html) and assembled into 8,238 contigs using Phrap (Ewing and Green, 1998; Ewing et al., 1998). Shot-gun sequence contigs were screened for the presence of WOcauB2 and WOcauB2-flanking sequences using BLAST (Basic Local Alignment Search Tool) homology searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and the NCBI sequence deposits AB478515 and AB478516 as well as a library of previously proposed Wolbachia phage sequences (Supplementary File 1).
Contigs showing significant matches were classified as being of bacteriophage origin if two of their three best matches in the NCBI's non-redundant sequence data bank were annotated as a Wolbachia phage sequence. Contigs identified as containing possible phage sequences were aligned to the WOcauB2 reference genome to identify putative gap sequences. Primers were designed to amplify predicted phage genome “gap” DNA sequences. All “gap-closing” PCRs that produced PCR products of the expected size had their PCR fragments Sanger sequenced in the forward and reverse directions (http://www.lifesciences.sourcebioscience.com/genomic-services/sanger-sequencing-service/). A full list of the primers used for this step is provided in Supplementary File 2. The primer design and PCR conditions used to amplify these “gap regions” followed an approach described previously (Post et al., 2009). “Gap-closing” Sanger-sequence reads were aligned to those generated from 454 sequence runs and used to extend the original contigs into a total of three large non-contiguous sequences, spanning what is proposed here to be a complete WO genome sequence (i.e., from its first gene sequence to its last).
A BLAST search using the S. squamosum E FtsZ sequence (FN563974) confirmed that the Roche 454 sequence reads were from the targeted S. squamosum E Wolbachia described in Crainey et al. (2010a). To confirm that the WO prophage sequences occur adjacent to the wDam FtsZ gene, all seven FtsZ-positive BAC colonies used in the shotgun sequence run were individually PCR-screened for the presence of WO genes using four primer sets (Supplementary File 2). Two of the primer sets targeted the serine recombinase gene (i.e., the phage's WOcauB2 gp1 paralog), which was predicted to occur at the 5′ end of the bacteriophage (as in WOcauB2 and WOcauB3) and the other two primer sets targeted the gene sequences from the tail end of the phage (corresponding to WOcauB2 gp32 and gp33 paralogous sequences).
The phylogenetic classification of the WO prophage sequences was performed using the minor capsid (sometimes referred to as the WO orf7 gene) and recombinase genes corresponding to WOcauB2 gp17 and gp1 paralogs, respectively. Clustal X (Thompson et al., 1997) was used to align the serine recombinase amino acid sequence of our WO gp1 paralog to the serine recombinases amino acid sequences of WO phage used in the recombinase analysis of Kent et al. (2011a). Clustal X was also used to align the nucleotide sequence of the minor capsid gene of our WO gp17 paralog to the minor capsid genes of the same WO phage, as well as the genes of three other Wolbachia prophages that lack integrase/recombinase genes.
The resulting alignments were used to construct maximum likelihood trees using the software from the PHYLIP package (Felsenstein, 2002). The robustness of the constructed trees' topologies was tested with 1000 pseudoreplicates (http://evolution.genetics.washington.edu/phylip.html). Final alignment files used in the tree construction are provided in Supplementary Files 3, 4.
In total, 22 contigs were identified as containing putative bacteriophage sequences and showing homology with 33 WOcauB2 genes. In each case, only one allele for each phage gene was identified, which led to the hypothesis that the sequences recovered from the shot-gun sequence analysis had originated from just one phage genome sequence that might be resolvable by gap-closing PCR amplification and Sanger sequencing. Following gap-closing PCRs, the 22 original phage contigs were extended and assembled into three large contigs totalling 32.439 kb of unique sequence, which we propose here represents the near-complete genome of sr1WOdamA1 and which has been deposited at the NCBI with accession numbers KY695239–KY695241.
wDam WO prophage contig number 1 (NCBI accession number KY695239) is 11.689 kb in length and is predicted to contain a block of 12 genes that show high levels of sequence identity with the first 12 predicated genes in WOcauB2 [WOcauB2 gp1–gp12 (Table 2)]. The WOcauB2 gp1 paralog (sr1WOdamA1 gp1p) occurs at the extreme 5' terminal end of this contig and corresponds to the sr1WOdamA1 recombinase gene, whose phylogenetic analysis robustly groups with the four previously described WO group serine recombinases (Figure 1). The next 11 gene sequences occur in the same order and orientation as in WOcauB2, representing the conserved group 1 serine recombinase and replication module (sr1RRM) which is unique to and highly conserved among, the sr1WO group bacteriophages (Figure 2 and below). The extreme 3′ terminal end of contig 1 shows very high levels of sequence identity with the WOcauB2 predicated gene protein 13 (WOcauB2 gp13). The first 545 base pairs of WOcauB2 gp13 thus correspond with the last 469 nucleotides of contig 1 (Table 2). As the first 354 nucleotides of contig 2 correspond to the last 369 nucleotides of the same gene (WOcauB2 gp13), we assumed that the two contigs would be easily joined by PCR (Table 2). Despite repeated efforts (using eight different primer sets), we were unable to bridge what we predicted would correspond to a 584 base-pair (bp) gap of WOcauB2 gp13 paralog gene sequence, which we expected to occur between contigs 1 and 2 (Table 2). It is, thus, most probable that the sr1WOdamA1 genome is not orientated as the WOcauB2 genome (Figure 2 shows its similarity to the other serine recombinase WOs).
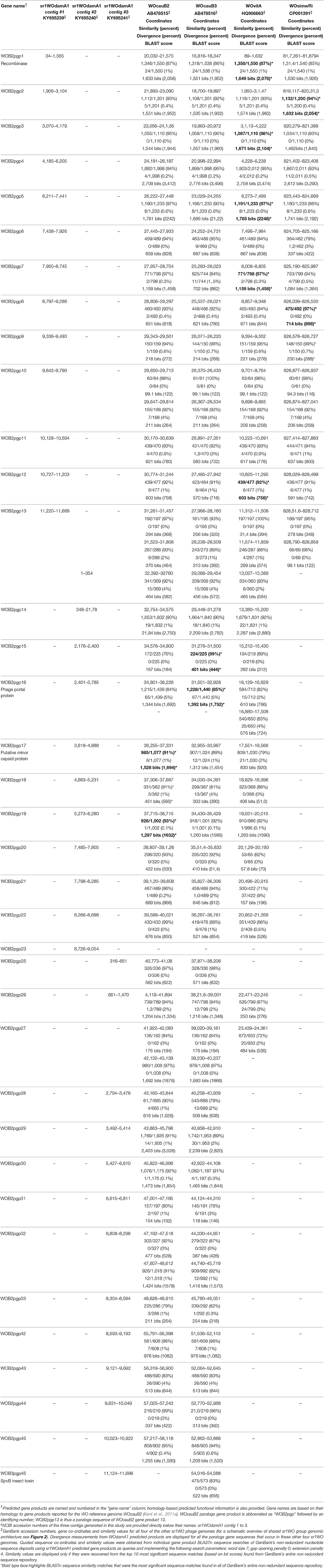
Table 2. Conservation of gene content, synteny and sequence similarity in the sr1WO group Wolbachia phage, and gene-content inventory for the three sr1WOdamA1 Wolbachia phage contigs generated and characterized in this study.
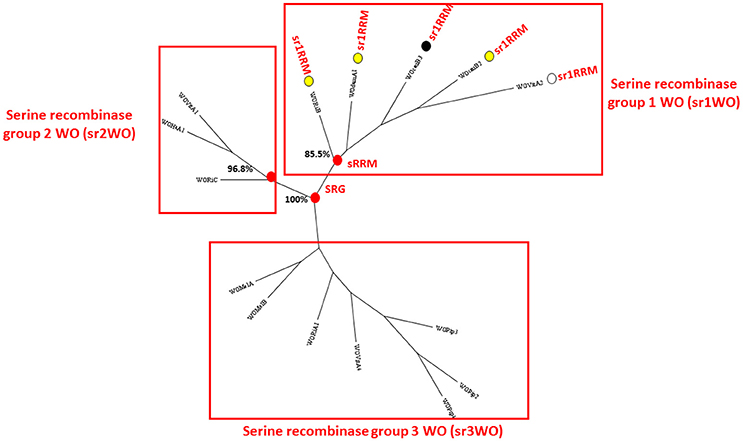
Figure 1. Maximum likelihood consensus tree constructed from an alignment of Wolbachia phage recombinase amino acid sequences. Three bootstrap-supported sequence clusters (labeled sr1WO-sr3WO) recovered in the analysis of Kent et al. (2011a) and Wang et al. (2013) were recovered in this analysis and are indicated. All the WOs known to have the structural group 1 serine recombinase replication module (sr1RRM) can be seen to occur in the sr1WO group. Sr1WO group recombinases are marked with a circle. When these circles are colored in yellow, the phage is known to occur adjacent to the FtsZ cell-division gene, white coloring indicates that the phage's genomic location is unknown and black is used to indicate that the phage does not appear to be located close to the FtsZ gene. The recombinase amino acid sequences are provided in Supplementary File 3.
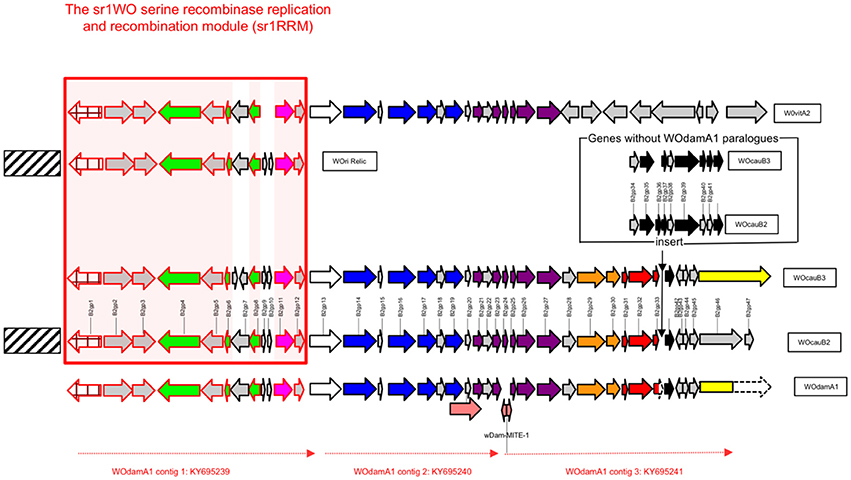
Figure 2. Schematic representation of the predicted structural organization of sr1WOdamA1 in relation to all four other members of the sr1WO group bacteriophage group (sr1WO). Arrows are used to indicate the direction in which predicted gene sequences are encoded and shading is used to indicate functional homology. The gene sequences of the WOcauB2 reference genome (used in this paper and in Kent et al., 2011a) are annotated with numbers and the gene product (gp) abbreviation. A conserved block of genes spanning from WOcauB2 gp1 to WOcauB2 gp12 with functions involved in recombination and replication functions, and referred to in the main text as “sr1RMM,” is highlighted with a red box. See also Figure 1. Nine perfectly conserved genes found within this box are indicated with pink highlighting. Predicted gene protein functional groups have been colored following the classification of Kent et al. (2011a). Blue is used to show “head” proteins; purple is used for “base-plate” proteins; orange is used for virulence proteins, and black is used for “tail” proteins. Red coloring is used to highlight a block of three predicted gene proteins, which appears only to occur in sr1WOdamA1, WOcauB2, and WOcauB3, suggesting a special relationship between these phages. The inset with WOcauB2 and WOcauB3 genes that have no WOdamA1 paralogs represents that there is no non-coding DNA separating the 3′-truncated gp33 paralog and the gp42 paralog in sr1WOdamA1 contig 3. The two proposed transposable elements referred to in the main text are indicated with pink arrows; the wDam-MITE-1 element is also labeled with its name.
wDam WO prophage contig 2 (NCBI accession number KY695240) is 9.054 kb and spans from the 3′ end of our WOcauB2 gp13 paralog to the middle of our WOcauB2 gp23 paralog (Figure 2). It contains a block of 10 WOcauB2 gene sequences which, as shown in Figure 2, code for genes corresponding to what Kent et al. (2011a) defined as WO head and base-plate modules. It also includes gene sequence coding for the minor capsid (orf7) protein, which is a B2gp17 paralog and has been used to construct the phylogenetic tree shown in Figure 3. The 10 whole gene sequences that occur in contig 2 appear in the same order and orientation as their paralogs in the WOcauB2 genome. The synteny between the WOcauB2 and sr1WOdamA1 genomes is only interrupted by the existence of a transposable element-like sequence occurring between the sr1WOdamA1 WOcauB2 gene protein paralogs B2gp19 and B2gp20.
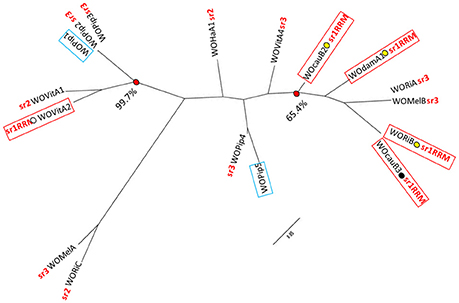
Figure 3. Representative maximum likelihood tree constructed from a 1031 nucleotide position alignment of 17 WO minor capsid protein gene sequence. The tree (historically used for WO classification) includes sequences from all the WOs used in Figure 1, as well as two additional WO sequences (from WOPip1 and WOPip5), the genomes of which lack integrase/recombinase genes (Kent et al., 2011a). The gene sequences of WOPip1 and WOPip5 are highlighted with blue boxing. Minor capsid gene sequences originating from WOs with the group 1 serine replication recombinase module (sr1RRM) referred to in the main text are indicated, as are their integration sites (labeled as in Figure 1). The serine recombinase-based phylogenetic classification of these WOs is also highlighted, using branch tip suffixes written in red (sr1–sr3). The bootstrap support for two monophyletic groups containing WO sequence representatives with varying integrase/recombinase gene sequences is also shown. The minor capsid protein sequences alignment is provided in Supplementary File 4.
wDam WO prophage contig 3 (NCBI accession number KY695241) is 11.696 kb in length and contains 14 predicted gene sequences, spanning from a WOcauB2 gene protein 25 paralog at one end through to a WOcauB3 gp45 gene protein paralog at the other end (Table 2 and Figure 2). As can be seen in Figure 2, this contig contains the 3′ prime end of the phage base-plate gene modules as well as its virulence and tail regions (Kent et al., 2011a). The first predicted nine gene sequences in this contig correspond to paralogs of WOcauB2 gene protein sequences spanning from 25 to 33 (Table 2 and Figure 2). This wDam WO prophage sequence element then appears to have a deletion. Thus, after this nine-gene block, there is a 3′-truncated gene protein sequence (a paralog of the WOcauB2 gene protein 33), which is immediately followed by a block of five gene sequences corresponding to paralogs of WOcauB2 gene proteins B2gp42 to B2gp45 (Table 2 and Figure 2). At the extreme 3′ end of this prophage sequence element and contig 3 (directly after the WOcauB2 gp45 paralog), there is a gene sequence matching the B3gp45 gene protein (see below) which has no paralog in the WOcauB2 genome. Repeated efforts to close a predicted gap between contigs 2 and 3 failed; it is, thus, unclear if there is a WOcauB2 gene protein 24 paralog within the genome of sr1WOdamA1 or not. The extreme 5′ end of contig 3 contains what we propose here is a 217-nucleotide transposable element sequence with all the features of a Miniature Inverted-repeat Transposable Element (MITE), including 24-nucleotide inverted terminal repeats (ITRs), as well as a 9-nucleotide target-site duplication (Delihas, 2011). This MITE—named here as wDam-MITE-1—could be the beginning of a stretch of repetitive DNA lying between the head and base-plate and tail modules (i.e., contigs 2 and 3) of the sr1WOdamA1 genome that was too long for our PCR bridging efforts to gap-close.
BLASTn and tBLASTx homology searches of the shot-gun sequence data using the 16,703 nucleotide sequence reported to be upstream of WOcauB3 did not identify any significant sequence matches. However, four shot-gun sequence contigs (representing 5.265 kb of unique DNA) showed significant levels of homology with the upstream sequence of WOcauB2. As shown in Table 1, these four contigs show high levels of sequence identity with six of the 16 genes found immediately upstream of the WOcauB2 and WOri relic serine recombinase prophage sequences. Importantly, the gene sequences in these contigs correspond to four of the five gene sequences that are closest to the WOcauB2 and WOri relic integration sites. Hence, our 454 sequence run recovered DNA sequence matching the 4,239 bp immediately upstream of the WOcauB2 prophage and the 3,769 bp sequence immediately upstream of the WOri relic prophage (Table 1). In contrast with the other four genes immediately upstream of the WOcauB2 and WOri relic prophages, BLAST searches revealed that the gene for which we did not recover a paralog (corresponding to ACN95502 in wRi and BAH22204 in wCau) is incompletely conserved among the Wolbachia strains. The absence of this gene from the shot-gun sequence reads could be a consequence of its absence from the wDam genome. Sequence comparisons made between the wDam FtsZ gene contig recovered from our shot-gun sequence reads and the previous report confirmed that the FtsZ-positive BACs used in this study were of the same origin as the FtsZ gene first reported in 2010 (Crainey et al., 2010a). The 454 sequence contig showed >99% sequence identity with the previously published FtsZ (FN563974) sequence.
PCR screening for sr1WOdamA1 gene sequences within the seven FtsZ-positive BACs used in the original shot-gun sequence run, identified three colonies as containing sr1WOdamA1 phage sequences. Consistent with the notion that a full-length sr1WOdamA1 prophage element occurs immediately adjacent to the FtsZ gene of wDam, one FtsZ-positive BAC was confirmed (by two independent PCR reactions) as containing the sr1WOdamA1 serine recombinase gene (i.e., a WOcauB2 gp1 paralog) and also B2gp33 and B2gp34 paralogs (by two independent PCR reactions). As previous analysis of BAC clones from the S. squamosum E BAC library used in this study suggested that the average BAC contains around 128 kb of cloned DNA and the sr1WOdamA1 genomic sequence is over 32 kb, this strongly suggests that the sr1WOdamA1 prophage has integrated within 100 kb of the wDam FtsZ gene. Unexpectedly, two BACs tested positive (in two independent PCR tests) for just the tail-end of sr1WOdamA1 (i.e., the gp31 and gp32 genes).
The three contigs recovered in this work can be seen to correspond, roughly, to three distinct (and non-overlapping) WO modules (1–3), that can be considered, when compared to the WOcauB2 reference genome, collectively to make-up a near complete sr1WO genome. wDam WO contig 1, contains a replication and repair module thus far only associated with WOs that contain sr1 recombinases (sr1RRM); the wDam WO contig 2 contains a head and base-plate module and some “virulence” genes, and wDam WO contig 3 is a tail module (Figure 2). While the exact relationship between these wDam WO prophage sequences is presently unclear, the gene sequence order and orientation within these contigs can be seen to be almost identical to those reported for the WOcauB2 and WOcauB3 phage genomes. Although we acknowledge that alternative explanations for our results may exist (see Section Discussion), we believe that the most parsimonious explanation is that all three of the wDam WO contigs reported here derive from the same sr1WO genome, which we are proposing be named as sr1WOdamA1. In relation to the reference genome WOcauB2, the sr1WOdamA1 appears to be missing nine phage gene sequences corresponding to WOcauB2 gp24 and gp34–41 (Figure 2), but to contain paralogs for all other WOcauB2 genome sequences.
In WOcauB2, and the other serine recombinase phage genomes that have paralogs, the genes gp34–41 (and their paralogs) have been ascribed tail functions (Figure 2 and Table 2). As this deletion begins following a 3′-truncated gp33 paralog, our findings indicate that sr1WOdamA1 lacks paralogs for these sequences because of a recent deletion and, thus, that sr1WOdamA1 is missing genes that its progenitor contained. As, however, this part of the tail region is highly variable among WOs and some, including other sr1WO group bacteriophages like WOvitA2 (Figure 2), lack recognizable tail modules, it is possible that sr1WOdamA1 and/or its progenitor retained active functions in the absence of a gp34–41 section. On the other hand, the absence of a WOcauB2 gp24 paralog from the sr1WOdamA1 genome, a well conserved gene component of the highly preserved phage base-plate region, is likely to render sr1WOdamA1 immobile. Although our failure to detect this gene could be an artifact of its expected location occurring in the break between two of our three sr1WOdamA1 contigs (Figure 2 and above), we believe that it is more likely that the gene has been disrupted by a transposable element integration. Our analysis identified a wDam-MITE-1 transposable element sequence at the extreme 5′-end of contig 3, immediately downstream of sr1WOdamA1's WOcauB2 gp25 gene protein paralog (where the WOcauB2 gp24 gene should occur). Because of this and because there was no trace of a WOcauB2 gp24 paralog detected from our initial shot-gun sequence run, we believe that the sr1WOdamA1 genome reported here is probably a dysfunctional prophage relic.
As shown in Table 2, most BLAST searches with the wDam WO prophage sequences recovered in this work, returned best match paralogous sequences from the genomes of other sr1WO group prophages. In every search performed with our 36 predicted sr1WOdamA1 gene sequences, the paralogous sequence matches listed in Table 2 were among the top ten closest matches in the non-redundant NCBI sequence repository. In addition to this, every search returned a best match sequence annotated as deriving from a Wolbachia genome or WO. Moreover, when the search results returned five or more significant matches, the top five hits were always annotated as not just deriving from a Wolbachia or WO genome but to be also sourced from an arthropod.
Figure 1 presents a phylogenetic tree constructed using WO recombinase genes and shows that the wDam WO phage sequence element recovered in contig 1 belongs to the same group of serine recombinase WOs to which WOcauB2 and WOcauB3 belong, and that was previously identified in the analysis of both Kent et al. (2011a) and Wang et al. (2013). As mentioned above, in addition to sharing closely-related recombinase genes, this group of five phages (WOcauB2, WOcauB3, WOvitA2, WOri relic, and now sr1WOdamA1) share an sr1RRM (spanning around 11 kb), which is not found in other (unrelated) WOs and corresponds almost exactly with contig 1 (Figure 2). Gene order and orientation is near perfectly conserved in the sr1RRM, with nine conserved WOcauB2 spanning gp1–6 paralog sequences recognizable in sr1WOdamA1 and all other serine recombinase WOs (Table 2 and Figure 2). Our first six predicted gene sequences have clear paralogs (appearing in the same order and orientation) and the last two gene sequences of the module (including a Holliday junction recombinase) have clear paralogs in all five phage genomes (Table 1 and Figure 2).
As shown in Table 2, BLAST searches (against the NCBI's entire non-redundant sequence database) with the 12 wDam WO contig gene sequences from this module returned best match sequences deriving from another serine recombinase WO eight times. In most cases the wDam WO-predicted gene sequences share similar levels of identity (>90%) with the other predicted phage gene sequences. For five out of eight of these genes, all differences between the sequences and their closest ones in the database are attributable to nucleotide substitutions, suggesting that these genes have been the subject of point mutation-based evolution. Indications of recombination, however, can be seen in Table 2. For example, most WOvitA2, WOri relic, WOcauB2, and WOcauB3 genes show similar levels of divergence from their sr1WOdamA1 paralogs, but the WOvitA2 B2gp3 paralog is markedly closer than the others. Similar signs of point-mutation-based WO evolution and of between-WO gene recombination are also evident from the BLAST search returns of wDam WO prophage head base-plate gene sequences (contig 2) and tail module gene sequences (contig 3) (Table 2).
In addition to the 36 wDam prophage genes with paralogs in the WOcauB2 genome that were identified from the three prophage sequence elements recovered in this work, an SpvB-like protein was observed to occur at the terminal end of the wDam WO prophage tail module element (contig 3). BLASTn searches with the last 377 nucleotides of contig 3, best match the first 378 nucleotides of the WOcauB3 gp45 protein which is annotated as coding for an SpvB-motif protein (BAH22314). The two sequences share 83% identity (315/378). The second-best (and only other significant) match is with the first 378 nucleotides of the Wolbachia phage wNo_WO4 “SpvB and TcdB toxin domain protein” (AGJ99401), which shares 79% identity (299/378). BLASTx searches also provided best matches with these gene sequences (83 and 85% identity across 110 residues, respectively), as well as support for this gene having an insecticidal toxin function. Thus, while there are presently no other close relatives to these proteins in the NCBI database, the next 17 best matches are all with bacterial proteins, which share between 50 and 60% amino acid level identity and have similar properties to those predicted for the BAH22314 and AGJ99401 proteins. All 10 of these hits that have functional annotation, are described as SpvB proteins and/or toxins or “insecticidal toxins.” As in the WOcauB3 genome, the SpvB-like protein appears to occur at the terminal tail end of the wDam WO tail module contig.
In previous work we reported a novel Wolbachia FtsZ cell-division gene sequence and showed the genome of this Wolbachia to be well represented in a BAC library prepared from S. squamosum E blackfly larvae (Crainey et al., 2010a,b). In this work we have taken the first step toward characterizing this bacterium's genome and have provided evidence that it harbors Wolbachia prophage sequence elements close to its FtsZ cell-division gene. Following gap-closing PCRs, we have resolved >32 kb of WO prophage sequence elements, corresponding to three distinct WO functional modules, namely, an sr1RRM, a head and base-plate and a tail module. Although alternative explanations may exist (see below) as to why we recovered these three WO sequences from the pool of FtsZ-gene positive BACs sequenced, the most parsimonious explanation is that they all derive from a single WO prophage genome that occurs close to the FtsZ cell-division gene.
Alternative explanations may include, for instance, the generation of chimeric BAC clones, created during the cloning process by ligating WO and wDam genomic fragments (with different origins and which do not occur close together in the wDam genome in nature). This would have required these sequences co-incidentally being cloned into the same BAC vector. However, there are good reasons to doubt such an explanation. Firstly, BAC libraries have been widely used in genome research for over 30 years and reports of such chimeric ligation being generated by the cloning process are extremely rare. Second, our former characterization of the BAC library used for this work suggests that wDam DNA represents only about 1% of the total cloned DNA (Crainey et al., 2010a). Hence, one would expect that there is about a 99% chance that any randomly created BAC chimera including a wDam genomic fragment would be composed of wDam and non-wDam DNA (i.e., most likely S. squamosum genomic DNA). The PCRs we did on our BAC clones showed that three of our FtsZ-positive BACs contain both wDam genomic DNA and WO phage sequences; thus, this would require three such events to have occurred (each with a 1% chance) ignoring that the cloning process only very rarely creates chimeric BAC clones. Therefore, the possibility that our results are explained by such a phenomenon is in the order of one in a million. We are, therefore, reasonably confident that all the wDam WO sequences recovered are from the wDam genome and thus of prophage origin. As a result, we are tentatively proposing that they are all from the same WO prophage genome, named here sr1WOdamA1. This sr1WOdamA1 is most similar to the Wolbachia phage from the almond moth Cadra cautella, WOcauB2 (Tanaka et al., 2009). Several similarities exist between our sequences and other WOs, as well as unique aspects which we discuss in relation to phage evolution, Wolbachia-based disease control programmes and the relevance of sr1WOdamA1 to onchocerciasis epidemiology below.
Traditional phylogenetic classification of Wolbachia phages has focused on the minor capsid or “orf7” gene (Bordenstein and Wernegreen, 2004; Gavotte et al., 2007; Chafee et al., 2010). More recently, however, WO researchers have begun performing phylogenetic analysis on the integrase/recombinase genes of WOs (Kent et al., 2011a; Wang et al., 2013). To classify our novel wDamWO prophage sequence elements, we used both approaches (Figures 1, 3). Using the serine recombinase gene of the wDam WO sr1RRM prophage sequence module, our analysis resulted in the same four serine phylogenetic groupings generated by Kent et al. (2011a) and Wang et al. (2013), and showed that this wDam WO prophage sequence element, at least, belongs to a cluster of four other serine recombinase phages (sr1WOs) that share several structural features (Figures 1, 2). Phylogenetic analysis with the minor capsid gene from the wDam WO head and base-plate module prophage sequence element (Figure 3), by contrast, did not share the same degree of congruence with WO structural features or agree well with the phylogeny constructed using the serine recombinase genes. Supporting the notion that our wDam WO sr1RMM prophage element and our wDam WO head and base-plate modules derive from the same (sr1WOdamA1) genome, the minor capsid gene phylogeny shown in Figure 3, clustered the wDam prophage minor capsid gene in a bootstrap-supported monophyletic group with the genes of three other sr1 group WOs. This group, however, also contained two non-sr1 group WOs and excluded the sr1group WOvitA2 bacteriophage, supporting previous reports that this gene has been exchanged between WO families via recombination and suggesting that this is not a reliable gene for WO classification. As the conservation of the sr1RRM probably reflects a fundamental difference in phage life-cycle and serine recombinase-based phylogeny grouped all of the WOs that share this feature together, we think that classifying and naming our phage based on this feature (rather than by its minor capsid protein) has more biological meaning.
With the inclusion of sr1WOdamA1 in the sr1WO group, the latter can be considered as, currently, having five members, namely sr1WOdamA1, sr1WOvitA2, WOcauB2, WOcauB3, and WOri relic (Figures 1, 2). While we have been unable to resolve completely the modular organization of sr1WOdamA1 recorded here, we have been able to resolve most of its within-modular structure, and from this it is apparent that gene sequence identity, gene sequence order and orientation are all well preserved among this group (Figure 2 and Table 2). Our results do, however, suggest that modular architecture of sr1WO group bacteriophages may vary as for many other WO families (Klasson et al., 2008; Kent et al., 2011a). Thus, while the occurrence of the B2gp13 homolog gene sequence—corresponding to the first portion of the gene—at the end of contig 1, and the occurrence of the B2gp13 homolog gene sequence—corresponding to the end of the gene—in contig 2, strongly suggest that they are from the same WO genome, the fact that we were unable to bridge the gap by PCR suggests that they may not be orientated in the same way as the WOcauB2, WOcauB3, and WOvitA2 genomes (Figure 2). Consistent with the idea of variant modular architectures occurring within the sr1WO group, the “terminal” end of the WOri relic (like the end of the sr1WOdamA1 contig 1) corresponds to the 5′-end of a B2gp13 paralog (Klasson et al., 2009). The sr1RRM of the sr1WO phage group may have become separated from the head and base-plate modules (and therefore not occur in sr1WOdamA1 as they do in WOcauB2, WOcauB3, and WOvitA2 genomes) in the progenitors of this relic and the sr1WOdamA1 prophage. A variant modular organization of sr1WOdamA1 may also help to explain the non-joining of contigs 2 and 3 (and thus the sr1WOdamA1 head and tail modules).
In addition to shared sequence and structural features, some of the sr1WO bacteriophages also share a common integration preference. The occurrence of the sr1WOdamA1 genome within BACs that contain four of the five genes immediately upstream of two other sr1 group prophages (WOcauB2 and a WOri relic) suggest that the sr1WOdamA1 prophage belongs to a group of Wolbachia prophages with a target site preference. This observation and the fact that most of the sr1RRM genes do not have clear paralogs in the genomes of other Wolbachia phages, suggest that the sr1RRM prophage may be involved in a common and targeted genomic integration method. However, it should be noted that while the WOri relic prophage, WOcauB2 and sr1WOdamA1 all appear to have integrated close to the Wolbachia FtsZ gene, the WOcauB3 appears to have integrated at a different genomic location (Klasson et al., 2008; Tanaka et al., 2009). As more serine recombinase Wolbachia phages have their genome sequences and integration sites resolved, the nature of this apparent genomic targeting will become better understood. Our observation that the tail end of sr1WOdamA1 appears to be closer to the FtsZ gene than the recombinase gene, suggests that the integration process may not require the bacteriophage to be integrated in a fixed orientation and highlights how little is presently known about the process by which Wolbachia phages integrate into their host Wolbachia genomes, with the data presented here contributing substantially to the current knowledge base.
Modular theory predicts that phages can exchange gene sequences freely across a broad range of ecological niches (Kent et al., 2011b; Metcalf and Bordenstein, 2012). It has been proposed, however, that the normal rules of modular evolution do not apply to WOs and that, while WOs can exchange gene sequences among themselves, they do not appear commonly to exchange genes with non-Wolbachia phages (Kent et al., 2011b; Metcalf and Bordenstein, 2012). The BLAST searches performed with each of the sr1WOdamA1 36 predicted gene sequences, returned a best match sequence deriving from a previously characterized WO sequence. In most cases the best match sequence was from another serine recombinase WO, suggesting that there may $$even be restriction of gene flow between WO subgroups (Kent et al., 2011b; Metcalf and Bordenstein, 2012). In line with previous analysis, however, these searches did provide clear evidence of genetic exchange between sr1WOdamA1 and other WO sequences that infect arthropod-infecting Wolbachia (Klasson et al., 2008; Kent et al., 2011b; Wang et al., 2013).
Although evidence of frequent Wolbachia phages horizontally transferring between strains has recently emerged, the evidence for this has been entirely based on the minor capsid protein and, thus, the structure and biology of the bacteriophages involved in these transfers have hitherto been completely unknown (Wang G. H. et al., 2016; Wang N. et al., 2016). In this study, we have isolated three novel WO prophage sequence elements (which probably all derive from the same WO genome) from the wDam Wolbachia genome. The wDam Wolbachia strain is the first from outside the A and B super clades to be shown to be infected with an sr1WO group bacteriophage (Crainey et al., 2010a; Kent et al., 2011a; Ellegaard et al., 2013). This has two important implications for Wolbachia-based disease control strategies. Firstly, it suggests that artificially-introduced Wolbachia, like those being used to control Aedes aegypti-transmitted dengue in Australia, Brazil and elsewhere, could themselves be infected by naturally occurring phages (Hoffmann et al., 2011, 2015; Caragata et al., 2016). Given the present plans to expand the use of Wolbachia-based disease control techniques and the possibility that phage integrations into artificial Wolbachia infections could impact on vector characters of epidemiological importance, this is not a trivial observation but one that may have wide-reaching consequences (Woolfit et al., 2013; Sutton et al., 2014; Jeffries and Walker, 2015). For example, although variant strains of wMel Wolbachia bacteria currently used for control appear to be near-identical in gene-coding regions, very minor differences in repeat-region sequences have a major impact on the longevity (and thus epidemiological importance) of Ae. aegypti (Woolfit et al., 2013). On the other hand, this observation suggests that if a WO can be manipulated to modify genetically Wolbachia (such as lambda, which is routinely used to infect E. coli), one phage could potentially be used to modify a broad range of Wolbachia stains (Tanaka et al., 2009; Kent and Bordenstein, 2010; Wang et al., 2013). In this context, our discovery that sr1WOdamA1 and WOcauB2 belong to a group of phages that may integrate into a single Wolbachia genomic site is particularly interesting as it suggests that they may be adapted to provide a genetic modification system for Wolbachia.
Because some Wolbachia strains seem to be able to change radically and spontaneously the way in which they infect their insect hosts (for example, from exerting a cytoplasmic incompatibility to male-killing), WOs have long been suspected as having an important influence on these characteristics (Kent and Bordenstein, 2010; Metcalf and Bordenstein, 2012). Thus far, however, precious little evidence has been uncovered to support this hypothesis. The existence of an SpvB-like protein at the extreme terminal end of the WOcauB3 phage is regarded as the best evidence yet that these bacteriophages could influence the insect host as this gene is believed to have insecticidal properties (Kent and Bordenstein, 2010; Metcalf and Bordenstein, 2012). The impact that wDam and sr1WOdamA1 might have on S. squamosum E is presently unknown (Crainey et al., 2010b), but the occurrence of an SpvB-like gene at the terminal end of the tail module of the WO prophage sequence elements isolated in this study suggests that the WOs of wDam could be influencing S. squamosum E biology.
Male-killing is a common form of reproductive parasitism induced by Wolbachia (Zug and Hammerstein, 2015). Selective expression of insecticidal proteins such as SpvB in a male insect environment could provide a molecular mechanism for such a phenomenon (Kent and Bordenstein, 2010; LePage and Bordenstein, 2013; Metcalf and Bordenstein, 2012). Although there are presently no reports of male-killing Wolbachia infections in the S. damnosum s.l. complex, which contains the most important vectors of human onchocerciasis in Africa (including S. squamosum E), efforts to get the species into laboratory colonies have repeatedly failed because the entire population has become female over time (Simmons and Edman, 1982; Raybould and Boakye, 1986; Crainey et al., 2017). If the Wolbachia infecting S. squamosum E is promoting its spread by male-killing, this could be expected to increase the proportion of female flies in the onchocerciasis foci where this species occurs, and this could, in turn, be expected to increase disease transmission in areas where such infections occur. Similarly, it would suggest that antibiotic treatment might aid getting this notoriously difficult species into laboratory colonies.
In this study we have shown that the genome of a genetically-distinct Wolbachia named here as wDam harbors at least one serine recombinase Wolbachia prophage relic. Although the three prophage sequence elements we have characterized correspond to three distinct non-overlapping WO functional modules (and could in theory have multiple origins), we believe that they almost certainly all derive from a single WO genome that we have named sr1WOdamA1. Although this WO is unlikely to be active, its existence in the wDam genome implies that active, naturally occurring bacteriophages can infect a broad range of genetically diverse Wolbachia strains and that naturally occurring WOs could pose a greater risk to the artificial Wolbachia infections currently used for disease control than previously thought. The occurrence of an sr1RRM prophage sequence in the same BAC clones in which the FtsZ gene is found is consistent with the notion that at least some of the sr1WO group WOs, notably the WOcauB2 phage, may have a target site preference and could be used for targeted introduction of recombinant genes into Wolbachia genomes. The occurrence of an SpvB gene in the genome of the wDam WO prophage sequences suggests that these genes may be a more common feature of Wolbachia bacteriophages than hitherto realized, an observation consistent with previous proposals that WOs could be important drivers of Wolbachia reproductive parasitism and thus could be causing male-killing in the onchocerciasis S. damnosum s.l. species complex with implications for the laboratory colonization of vector species and the epidemiology of onchocerciasis.
RJP, MGB, and RAC secured financing for this work. JLC, RJP, MGB, and MDW contributed to the planning of the work. JLC performed the laboratory work, except for the Sanger sequencing which was done by CEG, JLC, JH, and CPMA performed the DNA sequence analysis. RJP, RAC, and PHLL collected S. squamosum E. MGB, RJP, RAC, and PHLL made substantial contributions to the interpretation and editing of the manuscript. All authors read and approved the final version of the manuscript.
PHLL, RAC, MDW, MGB, and RJP acknowledge financial support from The Wellcome Trust (grant 085133/Z/08/Z to MGB); PHLL received funding from a Junior Research Fellowship at Imperial College London. Presently PHLL is funded by a European Research Council Starting Grant [680088 SCHISTO_PERSIST], a Wellcome Trust ISSF Grant [105614/Z/14/Z] and a Lord Kelvin Adam Smith Leadership Fellowship. RJP acknowledges funding from the Medical Research Council (grant 77615). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful for the support provided in Ghana by Daniel A. Boakye, Mike Y. Osei-Atweneboana, and Anthony Tetteh-Kumah for the collection of the simuliid larval samples. We are also grateful to the Natural History Museum's DNA sequencing facility, and especially to Julia Llewellyn-Hughes and Lisa Smith, for assisting with the Sanger sequencing used in this study.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00852/full#supplementary-material
BAC, bacterial artificial chromosome; BLAST, Basic Local Alignment Search Tool; bp, base pair; FtsZ, Filamenting temperature-sensitive mutant Z; gp, gene product; ITR, inverted terminal repeat; kb, kilo base; MITE, Miniature Inverted-repeat Transposable Element; nts, nucleotides; PCR, polymerase chain reaction; pgp, paralogous gene product; SpvB-like protein, Salmonella virulence plasmid B protein homolog; sr1RRM, group 1 serine recombinase and replication module; TcdB toxin, Clostridium difficile toxin B; WO, Wolbachia bacteriophage.
Bordenstein, S. R., and Wernegreen, J. J. (2004). Bacteriophage flux in endosymbionts (Wolbachia): infection frequency, lateral transfer, and recombination rates. Mol. Biol. Evol. 21, 1981–1991. doi: 10.1093/molbev/msh211
Bourtzis, K., Dobson, S. L., Xi, Z., Rasgon, J. L., Calvitti, M., Moreira, L. A., et al. (2014). Harnessing mosquito-Wolbachia symbiosis for vector and disease control. Acta Trop. 132, S150–S163. doi: 10.1016/j.actatropica.2013.11.004
Caragata, E. P., Dutra, H. L., and Moreira, L. A. (2016). Exploiting intimate relationships: controlling mosquito-transmitted disease with Wolbachia. Trends Parasitol. 32, 207–218. doi: 10.1016/j.pt.2015.10.011
Chafee, M. E., Funk, D. J., Harrison, R. G., and Bordenstein, S. R. (2010). Lateral phage transfer in obligate intracellular bacteria (Wolbachia): verification from natural populations. Mol. Biol. Evol. 27, 501–505. doi: 10.1093/molbev/msp275
Crainey, J. L., Hurst, J., Wilson, M. D., Hall, A., and Post, R. J. (2010a). Construction and characterisation of a BAC library made from field specimens of the onchocerciasis vector Simulium squamosum (Diptera: Simuliidae). Genomics 96, 251–257. doi: 10.1016/j.ygeno.2010.06.004
Crainey, J. L., Medeiros, J. F., Pessoa, F. A. C., and Luz, S. L. B. (2017). “Onchocerciasis,” in Arthropod Borne Diseases, ed C. B. Macondes (Cham: Springer International Publishing), 383–403.
Crainey, J. L., Wilson, M. D., and Post, R. J. (2010b). Phylogenetically distinct Wolbachia gene and pseudogene sequences obtained from the African onchocerciasis vector Simulium squamosum. Int. J. Parasitol. 40, 569–578. doi: 10.1016/j.ijpara.2009.10.017
Delihas, N. (2011). Impact of small repeat sequences on bacterial genome evolution. Genome Biol. Evol. 3, 959–973. doi: 10.1093/gbe/evr077
Echaubard, P., Duron, O., Agnew, P., Sidobre, C., Noël, V., Weill, M., et al. (2010). Rapid evolution of Wolbachia density in insecticide resistant Culex pipiens. Heredity 104, 15–19. doi: 10.1038/hdy.2009.100
Ellegaard, K. M., Klasson, L., Näslund, K., Bourtzis, K., and Andersson, S. G. (2013). Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet. 9:e1003381. doi: 10.1371/journal.pgen.1003381
Ewing, B., and Green, P. (1998). Base-calling of automated sequencer traces using PHRED II. Error probabilities. Genome Res. 8, 186–194. doi: 10.1101/gr.8.3.186
Ewing, B., Hillier, L., Wendl, M. C., and Green, P. (1998). Base-calling of automated sequencer traces using PHRED. I. Accuracy Assess. Genome Res. 8, 175–185. doi: 10.1101/gr.8.3.175
Felsenstein, J. (2002). PHYLIP (Phylogeny Inference Package). Version 3.6. Distributed by the Author. Department of Genetics, University of Washington, Seattle, U.S.A. Available online at: http://evolution.genetics.washington.edu/phylip.html
Gavotte, L., Henri, H., Stouthamer, R., Charif, D., Charlat, S., Boulétreau, M., et al. (2007). A survey of the bacteriophage WO in the endosymbiotic bacteria Wolbachia. Mol. Biol. Evol. 24, 427–435. doi: 10.1093/molbev/msl171
Hoffmann, A. A., Montgomery, B. L., Popovici, J., Iturbe-Ormaetxe, I., Johnson, P. H., Muzzi, F., et al. (2011). Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 476, 454–457. doi: 10.1038/nature10356
Hoffmann, A. A., Ross, P. A., and Rašić, G. (2015). Wolbachia strains for disease control: ecological and evolutionary considerations. Evol. Appl. 8, 751–768. doi: 10.1111/eva.12286
Jeffries, C. L., and Walker, T. (2015). The potential use of Wolbachia-based mosquito biocontrol strategies for Japanese encephalitis. PLoS Negl. Trop. Dis. 9:e0003576. doi: 10.1371/journal.pntd.0003576
Kent, B. N., and Bordenstein, S. R. (2010). Phage WO of Wolbachia: lambda of the endosymbiont world. Trends Microbiol. 18, 173–181. doi: 10.1016/j.tim.2009.12.011
Kent, B. N., Funkhouser, L. J., Setia, S., and Bordenstein, S. R. (2011a). Evolutionary genomics of a temperate bacteriophage in an obligate intracellular bacteria (Wolbachia). PLoS ONE 6:e24984. doi: 10.1371/journal.pone.0024984
Kent, B. N., Salichos, L., Gibbons, J. G., Rokas, A., Newton, I. L., Clark, M. E., et al. (2011b). Complete bacteriophage transfer in a bacterial endosymbiont (Wolbachia) determined by targeted genome capture. Genome Biol. Evol. 3, 209–218. doi: 10.1093/gbe/evr007
Klasson, L., Walker, T., Sebaihia, M., Sanders, M. J., Quail, M. A., Lord, A., et al. (2008). Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol. Biol. Evol. 25, 1877–1887. doi: 10.1093/molbev/msn133
Klasson, L., Westberg, J., Sapountzis, P., Naslund, K., Lutnaes, Y., Darby, A. C., et al. (2009). The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc. Natl. Acad. Sci. USA 106, 5725–5730. doi: 10.1073/pnas.0810753106
LePage, D., and Bordenstein, S. R. (2013). Wolbachia: can we save lives with a great pandemic? Trends Parasitol. 29, 385–393. doi: 10.1016/j.pt.2013.06.003
Metcalf, J. A., and Bordenstein, S. R. (2012). The complexity of virus systems: the case of endosymbionts. Curr. Opin. Microbiol. 15, 546–552. doi: 10.1016/j.mib.2012.04.010
Post, R. J., Crainey, J. L., Bivand, A., and Renz, A. (2009). Laser-assisted microdissection for the study of the ecology of parasites in their hosts. Mol. Ecol. Res. 9, 480–486. doi: 10.1111/j.1755-0998.2008.02437.x
Raybould, J. N., and Boakye, D. A. (1986). Temporary small-scale colonization of the Beffa form of the Simulium damnosum Theobald complex (Diptera: Simuliidae) in Africa. J. Am. Mosq. Control Assoc. 2, 14–17
Simmons, J. D., and Edman, L. (1982). Laboratory colonization of the human onchocerciasis vector Simulium damnosum complex (Diptera: Simuliidae), using an enclosed, gravity-trough rearing system. J. Med. Entomol. 19, 117–126. doi: 10.1093/jmedent/19.2.117
Slatko, B. E., Luck, A. N., Dobson, S. L., and Foster, J. M. (2014). Wolbachia endosymbionts and human disease control. Mol. Biochem. Parasitol. 195, 88–95. doi: 10.1016/j.molbiopara.2014.07.004
Sutton, E. R., Harris, S. R., Parkhill, J., and Sinkins, S. P. (2014). Comparative genome analysis of Wolbachia strain wAu. BMC Genomics 15:928. doi: 10.1186/1471-2164-15-928
Tanaka, K., Furukawa, S., Nikoh, N., Sasaki, T., and Fukatsu, T. (2009). Complete WO phage sequences reveal their dynamic evolutionary trajectories and putative functional elements required for integration into the Wolbachia genome. Appl. Environ. Microbiol. 75, 5676–5686. doi: 10.1128/AEM.01172-09
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Wang, G. H., Jia, L. Y., Xiao, J. H., and Huang, D. W. (2016). Discovery of a new Wolbachia supergroup in cave spider species and the lateral transfer of phage WO among distant hosts. Infect. Genet. Evol. 41, 1–7. doi: 10.1016/j.meegid.2016.03.015
Wang, G. H., Xiao, J. H., Xiong, T. L., Li, Z., Murphy, R. W., and Huang, D. W. (2013). High-efficiency thermal asymmetric interlaced PCR (hiTAIL-PCR) for determination of a highly degenerated prophage WO genome in a Wolbachia strain infecting a fig wasp species. Appl. Environ. Microbiol. 79, 7476–7478. doi: 10.1128/AEM.02261-13
Wang, N., Jia, S., Xu, H., Liu, Y., and Huang, D. (2016). Multiple horizontal transfers of bacteriophage WO and host Wolbachia in fig wasps in a closed community. Front. Microbiol. 7:136. doi: 10.3389/fmicb.2016.00136
Woolfit, M., Iturbe-Ormaetxe, I., Brownlie, J. C., Walker, T., Riegler, M., Seleznev, A., et al. (2013). Genomic evolution of the pathogenic Wolbachia strain, wMelPop. Genome Biol. Evol. 5, 2189–2204. doi: 10.1093/gbe/evt169
Keywords: Wolbachia, Wolbachia phages, serine recombinase, SpvB protein homolog, Simulium squamosum E, onchocerciasis
Citation: Crainey JL, Hurst J, Lamberton PHL, Cheke RA, Griffin CE, Wilson MD, de Araújo CPM, Basáñez M-G and Post RJ (2017) The Genomic Architecture of Novel Simulium damnosum Wolbachia Prophage Sequence Elements and Implications for Onchocerciasis Epidemiology. Front. Microbiol. 8:852. doi: 10.3389/fmicb.2017.00852
Received: 11 October 2016; Accepted: 26 April 2017;
Published: 29 May 2017.
Edited by:
Marina G. Kalyuzhanaya, San Diego State University, USAReviewed by:
Marla Trindade, University of the Western Cape, South AfricaCopyright © 2017 Crainey, Hurst, Lamberton, Cheke, Griffin, Wilson, de Araújo, Basáñez and Post. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María-Gloria Basáñez, bS5iYXNhbmV6QGltcGVyaWFsLmFjLnVr
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.