
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 May 2017
Sec. Plant Pathogen Interactions
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.00720
 Amir Ktari1
Amir Ktari1 Abdellatif Gueddou1
Abdellatif Gueddou1 Imen Nouioui1
Imen Nouioui1 Guylaine Miotello2
Guylaine Miotello2 Indrani Sarkar3
Indrani Sarkar3 Faten Ghodhbane-Gtari1
Faten Ghodhbane-Gtari1 Arnab Sen3
Arnab Sen3 Jean Armengaud2
Jean Armengaud2 Maher Gtari1*
Maher Gtari1*Molecular signaling networks in the actinorhizal rhizosphere select host-compatible Frankia strains, trigger the infection process and eventually the genesis of nitrogen-fixing nodules. The molecular triggers involved remain difficult to ascertain. Root exudates (RE) are highly dynamic substrates that play key roles in establishing the rhizosphere microbiome. RE are known to induce the secretion by rhizobia of Nod factors, polysaccharides, and other proteins in the case of legume symbiosis. Next-generation proteomic approach was here used to decipher the key bacterial signals matching the first-step recognition of host plant stimuli upon treatment of Frankia coriariae strain BMG5.1 with RE derived from compatible (Coriaria myrtifolia), incompatible (Alnus glutinosa), and non-actinorhizal (Cucumis melo) host plants. The Frankia proteome dynamics were mainly driven by host compatibility. Both metabolism and signal transduction were the dominant activities for BMG5.1 under the different RE conditions tested. A second set of proteins that were solely induced by C. myrtifolia RE and were mainly linked to cell wall remodeling, signal transduction and host signal processing activities. These proteins may footprint early steps in receptive recognition of host stimuli before subsequent events of symbiotic recruitment.
The cohabitation of plant roots and soil microbes has been shaped by a long and complex coevolutionary process (Morgan et al., 2005), leading to specialized and durable interactions ranging from cheating to altruism (Mendes et al., 2011). The multiple plant-microbe interactions taking place in the rhizosphere are mediated through a dynamic network, via molecular signals secreted as a response to attack in the case of disease, or as a developmental root structure such as in rhizobial and arbuscular mycorrhizal symbiosis (Oldroyd, 2013). In addition to their nutritional value for almost all rhizosphere microbes, root exudates (RE) contain chemoattractants, repellents and/or defensins that play key roles in priming and sustaining molecular dialogs with beneficial and pathogenic microbes (Bais et al., 2006). Frankia are nitrogen-fixing soil actinobacteria that are best known for their symbiotic lifestyle with a wide range of dicotyledonous host plants collectively designated as actinorhizal plants (Gtari et al., 2013). These latter are pioneering species that enrich poorly fertile or new soils with nitrogen and organic materials and thus are important plants and shrubs in forestry, agroforestry, and for soil reclamation (Benson and Silvester, 1993). The molecular signaling network leading to infection/nodulation in the nitrogen-fixing symbiosis has been well documented in the case of legume-rhizobium associations. Lipo-chitooligosaccharide or Nod factors are the key signals secreted in response to host plant flavonoids perceived by the compatible rhizobia (Lerouge et al., 1990). These rhizobial Nod factors are sensed by the host plant via LysM-receptor-like kinases (LysM-RLKs), triggering a signal transduction cascade leading to invasion of root cortical cells and, further, to the genesis of functional nodules (Oldroyd, 2013). This host signal transduction cascade has been shown to be common in rhizobial, arbuscular mycorrhizal and also actinorhizal symbiosis (Oldroyd, 2013; Svistoonoff et al., 2013). From the microbial viewpoint of the symbiosis, the analysis of several Frankia genomes has failed to reveal the presence of these common canonical nodABC genes (nodA-acyl transferase, nodB-chitin deacetylase, nodC-chitin synthase) (Tisa et al., 2016), as has also been the case for several photosynthetic (Giraud et al., 2007) and non-photosynthetic (Miché et al., 2010) bradyrhizobia. While exceptions exist with two Candidatus Frankia genomes generated from nodule metagenomes and where canonical nodABC genes and sulfotransferase gene nodH have been reported (Persson et al., 2015; Nguyen et al., 2016). The absence of reliable methods for the genetic manipulation of Frankia is an obstacle to the identification of such equivalent and elusive microbial signaling molecules in Frankia-actinorhizal symbiosis (Benson et al., 2011).
Proteins are involved in the organization of most biochemical reactions, ranging from metabolism to signal processing. The majority of such proteins operate within large multimeric complexes, through biological networks (Schmidt et al., 2014). Extracellular and surface-associated proteins reflect the physiological state of an organism under a given condition and also indicate the systematic interaction between the organism and its biotic and abiotic environment (Armengaud et al., 2012). Study of the whole proteome is of the utmost importance in gaining a deeper knowledge of the molecular processes regulating the cellular physiology of an organism (Aebersold and Mann, 2003). It has been shown that besides Nod factors and surface polysaccharides, proteins transported through general or host-targeting secretion systems are determinants in establishing and maintaining functional nitrogen-fixing symbiosis for both rhizobia (Fauvart and Michiels, 2008) and mycorrhizal fungi (Doré et al., 2015; Wagner et al., 2015). Previous functional analyses through transcriptomics and proteomics have been limited to comparisons of gene expression patterns between Frankia in root nodules and free living cultures (Mastronunzio et al., 2009; Alloisio et al., 2010; Mastronunzio and Benson, 2010), and culture transitions between nitrogen-fixing and non nitrogen-fixing conditions (Alloisio et al., 2007; Bickhart and Benson, 2011; Udwary et al., 2011).
Frankia coriariae strain BMG5.1 was isolated from root nodules of Coriaria japonica (Gtari et al., 2015; Nouioui et al., 2017). As a member of cluster 2, the host range of this strain is limited to members of the Coriariaceae, Datiscaceae, Rosaceae, and Ceanothus. Using a high-throughput shotgun proteomic approach, we report here the abundance of Frankia coriariae proteins matching first-step receptive recognition and response to host plant stimuli. Symbiotic proteins were identified upon comparative proteomic analysis for the strain grown in media matching compatible, incompatible, and non-host-plant signaling systems, corresponding, respectively, to Coriaria myrtifolia, Alnus glutinosa, and Cucumis melo RE.
Coriaria myrtifolia represents the compatible actinorhizal host of Frankia coriariae strain BMG5.1, whereas A. glutinosa is an incompatible actinorhizal host for this strain. Cucumis melo was used in this study because it is taxonomically the closest representative to Coriaria for which the whole genome sequence is available. Seeds were incubated overnight in sterile tap water, surface-sterilized with 30% hydrogen peroxide for 30 min and washed several times with sterilized, distilled water. Seeds were then germinated on watered sterile sand at 28°C with a 16-h light period. Depending on the species, 5–15 days after germination, seedlings were aseptically transplanted into a Magenta GA-7 box containing 100 ml Broughton and Dilworth solution supplied with a nitrogen source, i.e., 5 mM KNO3 (BD+N) (Broughton and Dilworth, 1971). After 15 days' growth, BD+N medium was replaced by BD medium without nitrogen source (BD-N) and RE were collected after 3–4 weeks of plant growth, filter sterilized through a 0.22 μm polycarbonate membrane and freshly added to BMG5.1 cultures.
Frankia coriariae strain BMG5.1 was grown and maintained in BD-N supplemented with 2.5 mM pyruvate as a carbon source, at 28°C. Ten-day-old cultures were supplemented with one volume (vol/vol) of filter sterilized RE from each plant species and incubated for an additional 5 days. All experiments were performed to obtain three biological replicates. Total cellular protein was measured using the BCA method (Smith et al., 1985). Cellular respiration was assessed using an INT (2-(p- Iodophenyl)-3(p- nitrophenyl)-5- phenyl tetrazolium chloride) reduction assay (IRA) (Prin et al., 1990).
For proteomic analysis, cells were harvested by centrifugation at 13,000 × g for 10 min, resuspended in 90 μl lithium dodecyl sulfate β-mercaptoethanol protein gel sample buffer (Invitrogen, Carlsbad, CA, USA) and incubated at 99°C for 5 min as indicated previously (Hartmann et al., 2014). Proteins from 10 ml culture supernatants were precipitated with trichloroacetic acid. The resulting pellets were resuspended in 50 μl lithium dodecyl sulfate β-mercaptoethanol protein gel sample buffer (Invitrogen, Carlsbad, CA, USA) and heated at 99°C for 5 min. The samples were briefly centrifuged to remove large aggregates before SDS-polyacrylamide gel electrophoresis analysis on 10% Bis-TrisNuPAGE gels (Invitrogen). Volumes of 20 μl of either cellular proteomes corresponding to 60 μg of total proteins, or exoproteome corresponding to a few μg of secreted proteins, were loaded per well. Sodium dodecyl sulfate-PAGE was carried out in 1 × 3-(N-morpholino) propane sulfonic acid solution (Invitrogen) on an XCellSureLock Mini-cell (Invitrogen) under a constant voltage of 200 V for 5 min. Gels were stained for 10 min with SimplyBlue SafeStain, a ready-to-use Coomassie G-250 stain (Invitrogen), and then destained overnight with milliQ water. Polyacrylamide gel bands (equivalent in volume to 100 μl) comprising the entire cellular proteome or exoproteome—one band per entire proteome—were cut and processed for in-gel proteolysis with Sequencing Grade Trypsin (Roche, Meylan, France) in the presence of 0.01% Protease Max reagent (Promega, Madison, WI, USA), as described previously (Clair et al., 2010). Three independent biological replicates were analyzed per treatment.
Peptide digests were resolved on an Ultimate 3000 LC system (Thermo-Scientific, Villebon-sur-Yvette, France) before MS/MS measurements performed with a Q-Exactive HF tandem mass spectrometer incorporating an ultra-high-field Orbitrap analyzer (Thermo-Scientific). Conditions for operating the LC system were as described previously (Dedieu et al., 2011). The Q-Exactive HF tandem mass spectrometer was operated in data-dependent mode comprising a scan cycle initiated with a full scan of peptide ions in the ultra-high-field Orbitrap analyzer, followed by selection of the precursor, high energy collisional dissociation and MS/MS scans on the 20 most abundant precursor ions (Top20). Full scan mass spectra were acquired with an Automatic Gain Control Target set at 3 × 10E6 ions from m/z 350 to 1,800 at a resolution of 60,000. MS/MS scan was initiated at a resolution of 15,000 when the AGC target reached 10E5 ions with a threshold intensity of 83,000 and potential charge states of 2+ and 3+. A 10 s dynamic exclusion was activated throughout the gradient. MS/MS spectra were processed and interpreted with the MASCOT 2.3.02 search engine (Matrix Science, London, UK), with standard parameters as indicated previously (Hartmann and Armengaud, 2014). This analysis was performed against two complementary databases: (i) a complete list of annotated CDS from the draft genome of BMG5.1 (GenBank/EMBL/DDBJ accession number JWIO00000000.1) comprising 4,175 protein sequences, and (ii) a complete list of annotated CDS from the finished genome of Dg1, the Frankia symbiont of Datisca glomerata (GenBank/EMBL/DDBJ accession number CP002801.1) comprising 4,099 protein sequences. Peptide matches with a score above their peptidic identity threshold were filtered at P < 0.05. A protein was only validated when at least two peptides had been assigned to that protein according to the principle of parsimony. Using a previously described approach (Liu et al., 2004; Zivanovic et al., 2009), protein abundance was evaluated by shotgun analysis using MS/MS spectral counts. Normalized spectral count abundance factors were calculated as described by Paoletti et al. (2006) and presented as percentages of total signal.
To study the proteome and exoproteome of BMG5.1, self-customized Perl, Python, and R programs were adopted. STRING server version 10.0 (Szklarczyk et al., 2014) was utilized to obtain knowledge about the biological interactions between the different sets of proteins. Proteins of BMG 5.1 were further distributed among the functional Clusters of Orthologous Groups (COG) categories, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway categories were viewed for biochemical features and potential links to life and environment.
CodonW (Peden, 2005) was used to calculate diverse codon and amino acid usage indices such as GC (total guanine and cytosine content of a genome), GC3 (total content of guanine and cytosine present at the third position of the synonymous codons), Enc (effective number of codons), CAI (codon adaptation index; a direct measurement of the expression level of protein encoded by a particular gene), Fop (frequency of optimal codons), RSCU (relative synonymous codon usage), aromaticity, and hydrophobicity (Sen et al., 2008; Roy et al., 2015). These parameters can help us by providing a plethora of information regarding the codon bias nature of symbiosis-related genes and its implication in the lifestyle of the strain studied under different conditions. To further analyze the amino acid usage pattern of the core genes, aromaticity, gravy score, and protein biosynthetic cost were correlated with each other. The tRNA adaptation index (tAI) (dos Reis et al., 2004) was calculated via tRNAscan-SE and codonR software. The tAI of a gene predicts the usage of those codons that perfectly match with anti-codons of major isoaccepting tRNAs present in the studied organism. To assume the biosynthetic energy cost of each protein of BMG5.1, DAMBE software (Xia and Xie, 2001) was used, which calculates the protein energy cost based on the algorithm proposed by Akashi and Gojobori (2002). Pfam server (http://pfam.xfam.org/) was used to predict the protein domains present in the particular protein sets expressed under different RE treatments.
The mass spectrometry proteomic data were deposited at the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaíno et al., 2016) with the data set identifiers PXD005979 and DOI 10.6019/PXD005979 (cellular proteome data), and PXD005980 and DOI 10.6019/PXD005980 (exoproteome data).
A total of 149,629 and 144,213 MS/MS spectra were assigned on the basis of the BMG5.1 and Dg1 databases, respectively. As expected, the highest assignment rate was obtained with the genome of the strain used in this study. However, some additional spectra could be assigned using the Dg1 database because of missing gene sequences or annotations in the first genome. Indeed, the number of unique peptide sequences was 6,627 peptides when the BMG5.1 database was used, while 5,879 peptides were found against the Dg1 database. When merging both data sets, the additional Dg1 database search allowed the detection of 918 additional unique peptide sequences. The whole set of peptides indicated the presence of 1,330 proteins identified with at least two peptide sequences and 572 further protein candidates detected with only one peptide sequence (Supplementary Tables S1, S2). The high-throughput approach used here, which relies on one of the most recent tandem mass spectrometers, allows a deep characterization of bacterial proteins. An average of 21% of each protein sequence was covered by certified tryptic peptides, and a high redundancy in terms of spectral counts was obtained with an average of 166 MS/MS spectra recorded per protein. The abundances of the proteins were assessed by their spectral counts in the different replicates and conditions. Among the proteins specifically detected using the Dg1 database, and thus wrongly annotated in the BMG5.1 database (Supplementary Tables S1, S2), we noted the presence of high abundance proteins: three transcriptional regulators (KLL11059.1; WP_013874517.1; KLL10836.1) and several hypothetical proteins (WP_043603927.1; WP_050803503.1; WP_050803326.1; WP_050803366.1; WP_013875127.1). Remarkably, the DNA-directed RNA polymerase subunit beta (WP_013875121.1) and ATP-dependent Clp protease ATP-binding protein (WP_013875507.1) sequences were well covered with our proteogenomic approach based on the use of the Dg1 closely related genome information, with 60 and 36 different peptides, respectively. Table 1 summarizes the proteomic data predicted from genome sequences and experimentally determined in this study.
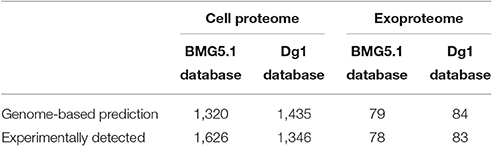
Table 1. Protein abundance, as predicted from Dg1 and BMG5.1 genome sequences and detected experimentally for Frankia coriariae strain BMG5.1.
The most abundant proteins of the BMG5.1 cellular proteome comprise housekeeping proteins involved in carbohydrate metabolism, nitrogen fixation and assimilation, and DNA replication, repair, transcription and translation (Figure 1). Proteins encoded by genes with unassigned functions were also recovered using our high-throughput proteomic analysis. An overall codon usage and amino acid usage study of these MS/MS-detected proteins revealed a high positive correlation (p < 0.001) between GC and CAI whereas a statistically significant negative correlation was obtained between GC and Enc (p < 0.001). On further analysis, we found a positive correlation between CAI and aromaticity as well as aromaticity and energy cost. A similar positive correlation was obtained between CAI, Fop, and tAI (Figure 2).
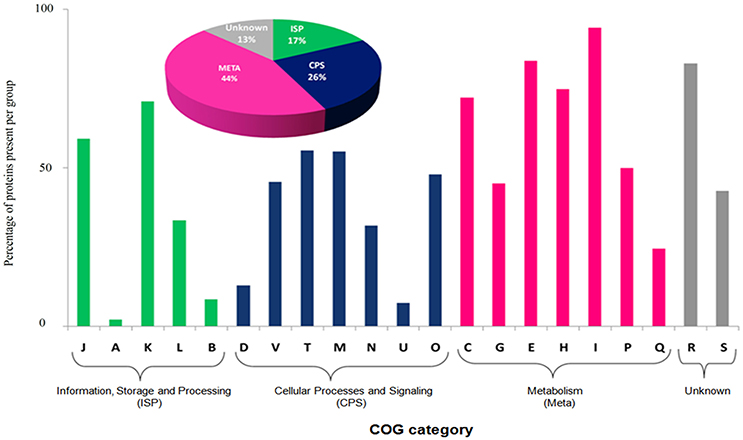
Figure 1. Functional classification based on Clusters of Orthologous Groups (COG) of BMG5.1 proteome. Proteins were distributed among the COG categories on the basis of percentage protein abundance. J, Translation, ribosomal structure and biogenesis; A, RNA processing and modification; K, Transcription; L, Replication; recombination and repair; B, Chromatin structure and dynamics; D, Cell cycle control, cell division, chromosome partitioning; V, Defence mechanisms; T, Signal transduction mechanisms; M, Cell wall/membrane biogenesis; N, Cell motility; U, Intracellular trafficking, secretion, and vesicular transport; O, Posttranslational modification, protein turnover, chaperones; C, Energy production and conversion; G, Carbohydrate transport and metabolism; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport and catabolism, R, General function prediction only; S, Function unknown.
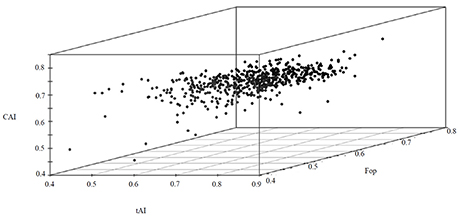
Figure 2. A 3D plot of CAI, Fop and tAI values for the BMG5.1 proteome, revealing the positive correlation between the three parameters.
The BMG5.1 exoproteome (Supplementary Tables S3, S4) contains a total of 98 proteins. Of these, 70 are shared with the cellular proteome, although more abundant in the exoproteome, while 28 are unique to the secreted proteome, mainly involved in essential physiological activities such as transcriptional regulation, ribosome formation, RNA degradation, oxidative phosphorylation, glycolysis/gluconeogenesis, and transport systems (Supplementary Table S5). Nitrogen fixation-related proteins were not detected in the exoproteome fraction. Thus, the exoproteome contains proteins that are in particular required for regulating the most essential biochemical pathways related to the existence of the organism itself. Cello server (Yu et al., 2006) queried with this list of exoproteins predicted preferentially cytoplasmic (74%), membrane-bound (13%), and extracellular (13%) proteins. It is worth noting that an ATP-dependent protease and a proteasome have been detected systematically in BMG5.1 culture supernatants. The COG category analysis detected “metabolism” as major functional category (Figure 3). The KEGG pathways are, broadly, carbohydrate metabolism, ribosome formation, amino acid metabolism, and signal transduction.
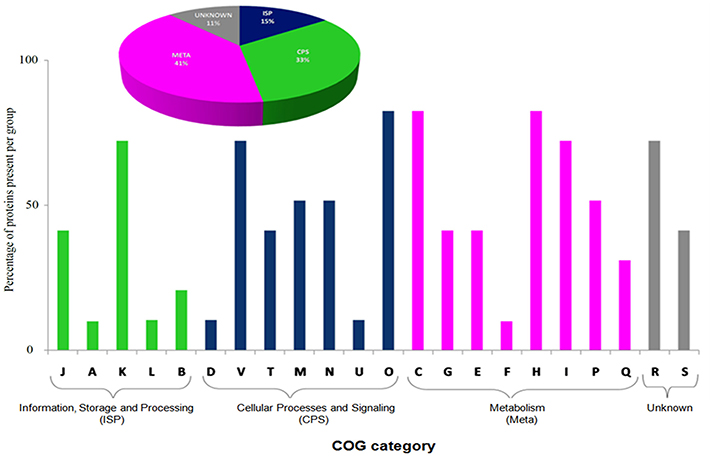
Figure 3. Functional Clusters of Orthologous Groups (COG) classification of the BMG5.1 exoproteome, showing “metabolism” as the major category. The plot was generated on the basis of percentage of protein abundance in the different COG categories. J, Translation, ribosomal structure, and biogenesis; A, RNA processing and modification; K, Transcription; L, Replication, recombination, and repair; B, Chromatin structure and dynamics; D, Cell cycle control, cell division, chromosome partitioning; V, Defence mechanisms; T, Signal transduction mechanisms; M, Cell wall/membrane biogenesis; N, Cell motility; U, Intracellular trafficking, secretion, and vesicular transport; O, Posttranslational modification, protein turnover, chaperones; C, Energy production and conversion; G, Carbohydrate transport and metabolism; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport, and catabolism; R, General function prediction only; S, Function unknown.
A detailed codon usage analysis of exoproteome-encoding genes revealed that they preferentially favor the use of GC-enriched codons. Negative correlation between ENc and GC3 verified GC compositional constraint as a major factor driving codon usage. However, significant positive correlation between tAI, Fop, and CAI (p < 0.001) indicates the favored utilization as well as the translational adaptability of major optimal codons. The energy cost of these proteins showed a significant positive correlation with aromaticity and tAI (p < 0.001). This result indicates the favored usage of costly aromatic amino acids in these proteins. Thus, the exoproteome of BMG5.1 is composed of costly aromatic amino acids, highlighting the high relevance of these amino acids in symbiotic association and for nitrogen fixation. Proteins that were detected in the exoproteome but not in the cellular proteome (n = 28) were further analyzed for aromatic amino acid counts, which were found to be lower than for the proteins that were shared with cell proteins.
Strain BMG5.1 grows better in the presence of C. myrtifolia RE than in BD-N medium, followed by A. glutinosa then C. melo RE, as shown by total cellular protein yield and cell respiration (IRA) assays, the two reliable indicators of biomass production and metabolic activities (Figure 4).
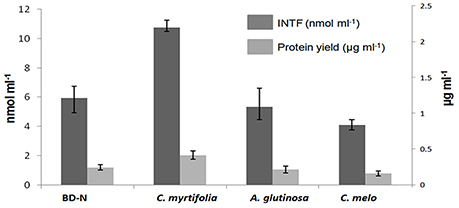
Figure 4. The effect of C. myrtifolia, A. glutinosa, and C. melo root exudates on Frankia coriariae BMG5.1 growth yield, as determined by protein yield and respiration assay using a spectrophotometric measurement of the reduction of 2-(p-iodophenyl)3-(p-nitrophenyl)-5-phenyl-tetrazolium chloride (INT) to iodo-nitrophenyl formazan (INTF). Strain BMG5.1 was grown in presence of filter sterilized root exudates from the indicated plant species. BD-N (nitrogen-free Broughton and Dilworth medium) was used as control.
To check potential interference of plant proteins derived from RE, we first interpreted MS/MS spectra for the identification of peptides derived from C. melo RE in comparison with the exoproteome of BMG5.1 grown in C. melo RE. The proteins obtained by acid precipitation from 40 ml RE were quantitatively insignificant, as verified by SDS-PAGE and by the very low level of MS/MS spectra recorded by our shotgun procedure (n = 373), which corresponded to 77 peptide sequences. The average MASCOT score of proteins from C. melo RE is 28.9, a very low value compared to the score for strain BMG5.1 grown in C. melo RE, 45.6, thus indicating a rather low confidence for exudates spectrum assignment. Further interpretation of BMG5.1 culture supernatants produced in the presence of C. melo RE against Frankia (Supplementary Table S1) and C. melo databases indicated that MS/MS spectra allocations wholly derived from Frankia peptides.
Comparison of BMG5.1 proteomes in BD-N medium with those obtained in the presence of all three RE revealed a total of 404 common proteins in the cellular proteome of BMG5.1 (while the number was 376 in Dg1 with the same conditions). When the RE were used individually, these counts were reduced to 371 (BMG5.1) and 343 (Dg1) in A. glutinosa RE, whereas and 308 (BMG5.1) and 292 (Dg1) in C. melo RE. However, as expected, the proteome size was higher (470 for BMG5.1; 438 for Dg1) using RE of C. myrtifolia, which is the natural host of the BMG5.1 strain.
These differentially expressed protein sets did not reveal a significant variation with respect to protein domains. Among the 404 proteins of BMG5.1 expressed under the influence of cumulative REs (which henceforth will be called as cREEP), we found a total of 37 functional domains. Of these 37 domains, there were four that were the most prevalent within the majority of the cREEP.
These four domains were PAS (pfam00989, pfam13426, pfam08447, pfam08448), GGDEF (pfam00990), HisKA (pfam00512, pfam07568, pfam07730), and cytochrome p450 (pfam00067), associated with bacterial signal transduction. Other domains were found to be involved in metabolism, especially carbohydrate metabolism. Some examples of these domains, present at moderate levels among the cREEP (supplementary Table S6), are Gp_dh_N (glyceraldehyde 3-phosphate dehydrogenase, NAD binding domain) (pfam00044), Gln-synt_C (glutamine synthetase, catalytic domain) (pfam00120), PGI (phosphoglucose isomerase) (pfam00342), 6PGD (6-phosphogluconate dehydrogenase, C-terminal domain) (pfam 00393), PGM_PMM_IV (phosphoglucomutase/phosphomannomutase, C-terminal domain) (pfam00408), and UDPG_MGDP_dh (UDP-glucose/GDP-mannose dehydrogenase family, central domain) (pfam00984).
The clustering coefficients and P-values obtained from biological interaction study of these cREEPs clearly indicate that they are biologically connected and the clustering coefficients were statistically supported. The functional COG category analysis of these proteins showed “metabolism” and “cellular process & signaling” to be the major functional groups. The overall unique and shared proteins of the cellular proteome and exoproteome expressed under different root exudates and BD-N are illustrated in a Venn diagram in Figure 5.
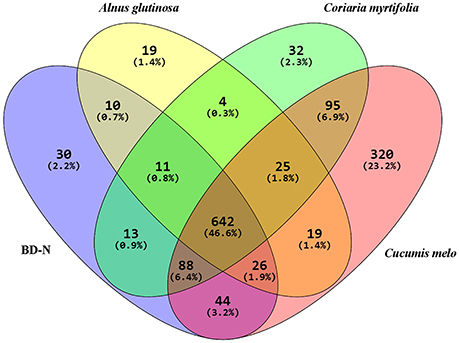
Figure 5. Venn Diagram showing the distribution of BMG5.1 proteins grown in BD-N (nitrogen-free Broughton and Dilworth medium) and in the presence of root exudates from the indicated plant species. A total of 642 proteins were detected under all conditions. These proteins were found to be mostly housekeeping proteins involved in metabolism and signal transduction.
Compared with the BMG5.1 proteome in BD-N medium, 83 unique proteins were induced for the BMG5.1 proteome in the presence of C. myrtifolia RE (Table 2). Among the above-mentioned proteins, 46 and 24 were shared with the BMG5.1 proteome in the presence of A. glutinosa and C. melo RE, respectively, while 32 were unique to C. myrtifolia RE. Cello server predicted the cellular localization of these 32 C. myrtifolia RE unique proteins to be cytoplasmic (55%), membrane bound (25%), and extracellular (20%). No signal peptide was detected among these proteins. Positive correlation among CAI, tAI and Fop of these protein-coding-genes also implies maximum usage of translationally optimal codons. Figure 6 illustrates the main alterations of BMG5.1 cellular processing when grown in C. myrtifolia RE.

Table 2. Proteins induced in Frankia strain BMG5.1 grown in root exudates.
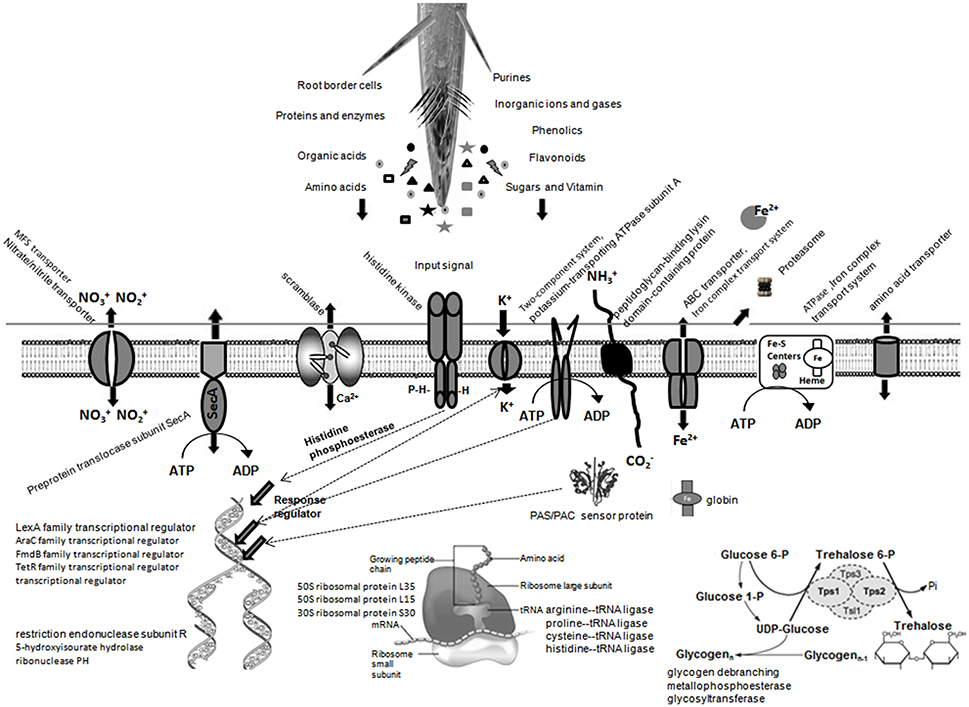
Figure 6. A predictive overview of the main alterations to strain BMG5.1 cellular processing in the presence of C. myrtifolia root exudates as expected from COG and KEGG functional assignments of the induced proteins.
The main characteristic of the BMG5.1 shotgun proteome obtained in the present study is its adaptation to the costly aromatic amino acids, which may be of relevance for bacteria with a particular lifestyle in a nitrogen-fixing symbiotic association (Prasad et al., 2000; Randhawa and Hassani, 2002).
As previously reported for Frankia casuarinae (Mastronunzio et al., 2008, 2009), the BMG5.1 exoproteome was determined to be tiny, with few secreted proteins. The presence of extensive cell lysis activity was attributed as responsible for the existence of several cellular proteins in the secreted fraction (Mastronunzio et al., 2009). The total absence of signal peptide proteins and the detection of an ATP-dependent protease and a proteasome at higher level in BMG5.1 supernatant, further strengthen this hypothesis. It has been shown that some ATP-dependent proteases are essential elements of the complex regulatory networks enabling the viability of cells under adverse environmental conditions (Gottesman, 1996).
COG analysis attributed most of the secreted proteins to metabolism, especially carbohydrate and amino acid metabolism and transport, as a major functional category. This finding fully supports the findings of Udvardi and Poole (2013), who reported that nitrogen-fixing associations require smooth integration between the bacterial and plant “metabolism–transportation system” for fluid exchange of the metabolic products across the plant and bacterial membranes.
RE derived from BMG5.1-compatible (C. myrtifolia), BMG5.1-incompatible (A. glutinosa), and the non-actinorhizal (C. melo) host plants differ both qualitatively and quantitatively (Badri and Vivanco, 2009). RE contain high molecular weight components such as mucilage (polysaccharides) and proteins, and low molecular weight components such as amino acids, organic acids, sugars, and other secondary metabolites (Badri and Vivanco, 2009), which, besides their nutritional value, act as chelators of poorly soluble mineral nutrients and as chemoattractant signals to microbes (Haichar et al., 2014). Thus, Frankia growth depends on the ability of a given strain to assimilate nutrients, to tolerate toxic compounds, and to further interact with signaling molecules of a given RE (Turner et al., 2013). A study based on a plant-trapping assay demonstrated the occurrence of Frankia in the host and non-host rhizosphere (Smolander and Sarsa, 1990), with a rapid fall in its infective units outside its compatible host area (Benson and Silvester, 1993). Here we showed that RE-induced proteins share some functional domains essential for signaling mechanisms. For instance, GGDEF, HisKA, and PAS domains are associated with bacterial two-component signaling systems (Sarkar et al., 2016). These domains comprise the periplasmic sensory domains with important enzymatic functionalities. They remain associated with the diguanylate cyclase/phosphodiesterase activity of bacteria, which catalyze the synthesis and hydrolysis of the cyclic diguanylate (c-di GMP). This further helps regulation of extracellular polysaccharide formation, the cell development program, and other signaling cascade mechanisms (Galperin, 2004). Among the moderately present domains, Gp_dh_N, Gln-synt_C, PGI, 6PGD, PGM_PMM_IV, and UDPG_MGDP_dh together help in bacterial metabolism, especially carbohydrate metabolism (Saier and Reizer, 1992; Postma et al., 1993; Saier et al., 1995), and may act to bind the bacterial cells to those of the host (Chagnot et al., 2013). Thus, both metabolism and signal transduction are the dominant activities for BMG5.1 under the different RE conditions tested.
The RE-induced proteins represent an overview of the deeply affected metabolic pathways footprinting shifts of the Frankia strain from aposymbiotic to rhizosphere, and then from rhizosphere to a symbiotic lifestyle. This indicates (i) adaptation to rhizosphere colonization, which was also induced by the A. glutinosa and C. melo RE, and (ii) early receptive steps as recognition of host stimuli prior to triggering the root infection and nodulation processes, which was obtained only with C. myrtifolia RE.
The induced proteins matching rhizosphere colonization involve 31 and 15 items with assigned and unassigned biochemical features, respectively. On the first line of induced proteins are those related to protein synthesis, such as DNA transcriptional regulator [TetR family transcriptional regulator (KLL12053.1), ribosomal structure and biogenesis (WP_013874486.1/KLL10220.1; WP_047221577.1; KLL12035.1), ribonuclease PH (WP_013872713.1)], transporters and secretion systems, ATP-binding cassette (ABC) transporter (WP_013872693.1), major facilitator superfamily (MFS) transporter (KLL10396.1), SecA or type II secretory pathway (KLL10487.1) and ATPase transporters (WP_043607340.1), for diverse metabolites ranging from ions (nitrate/nitrite, iron, sulfur and potassium) to amino acids and proteins.
Amino acid-related activities supporting this active transcription and protein synthesis are also detected. A particular “loading” of tRNA with arginine, proline, cysteine, and histine affinity amino acids–tRNA ligase (WP_043602701.1; WP_013875400.1; WP_013875269.1) was noted in this study for actinorhizal RE. DNA replication, recombination and repair proteins (KLL12894.1KLL13116.1); restriction endonuclease subunit R (KLL09521.1); 5-hydroxyisourate hydrolase (KLL11137.1) may protect against cytotoxicity-generating DNA damage produced during the early step of plant defense toward microbial colonization (Baker and Orlandi, 1995; Bagnarol et al., 2007) or drastic adaptation of metabolism (Clair et al., 2012). Aldo/keto reductase (WP_050803659.1/KLL11374.1), which is an NADPH-dependent oxidoreductase, may help further detoxification of a wide range of aldehydes and ketones (Barski et al., 2008). Carbon-rich RE released by plants make the rhizosphere extremely nutrient-rich relative to the bulk soil environment (van Veen et al., 1997). Thus, the short-chain dehydrogenase/reductase (SDR) superfamily of proteins (WP_050803541.1) induced here, known as tyrosine-dependent oxidoreductases, are suggested to confer the ability to metabolize a wide range of carbon substrates that contribute to cell survival and proliferation in the nutritionally complex soil or rhizosphere environment (Jacob et al., 2008).
We identified, in the category of proteins suspected to be symbiotically related, 21 and 11 proteins with assigned and unassigned biochemical features, respectively. Proteins involved in the remodeling of cell surface structures are abundantly modulated. Beauchemin et al. (2012) also reported that among different RE tested, only C. cunninghamiana was able to alter bacterial surface properties at the fatty acid and carbohydrate levels, leading to “curling” on its compatible F. casuarinae strain CcI3 hyphae. In fact, microbial membrane proteins play a predominant role in bacterial auto-aggregation and host cell adhesion that is well known both for bacterial survival and the host plant colonization process (Bogino et al., 2013). Lipid metabolism proteins included cyclopropane-fatty-acyl-phospholipid synthase (KLL12366.1), which is implicated in membrane modifications (Tiricz et al., 2013), and 3-oxoacyl ACP synthase (KLL12268.1) that contributes to the biosynthesis and transfer of fatty acyl residues, which serve as acyl donors in different acyl transfers during lipid biosynthesis reactions including phospholipids (Geiger and López-Lara, 2002). Scramblase (WP_013875539.1), a calcium activation protein, is implicated in membrane depolarization through passive translocation of phospholipids between the two sides of the membrane.
Besides HisKA, and PAS sensors discussed above, a GlcNAc-PI de-N-acetylase (KLL11416.1), also named N-acetylglucosaminylphosphatidylinositol deacetylase was also induced. It catalyzes the production of peptidoglycans that contain a backbone of N-acetylglucosamine (GlcNAc) residues (Gust et al., 2012). These GlcNAc residues serve as sensing ligands to plant LysM receptor involved in early plant-microbe interactions (Rey et al., 2013). Peptidoglycan-binding lysin domain-containing protein (WP_013872588.1) may contain beside the truly secreted proteins, outer-membrane proteins, lipoproteins or proteins bound to the cell wall in a non-covalent manner (Buist et al., 2008).
In conclusion, using next-generation proteomic analysis, an overview is provided here of proteome shotgun footprinting of metabolic alterations in response to RE stimuli, which orchestrate the first steps in the recruitment of Frankia by compatible host plants through (i) rhizosphere colonization and then (ii) early positive perception triggering root infection and subsequent nodulation processes. In the absence of reliable methods to manipulate the genes of interest for Frankia strains, comparative approaches based on high-throughput gene-expression analysis after plant and bacterial stimuli provide an alternative basis for elucidating the elusive bacterial triggers in actinorhizal symbiosis.
MG conceived and designed the study. AK, AG, IN, and FG performed root exudates production and Frankia treatments. JA and GM performed Nano liquid chromatography–MS/MS and identified the proteins. AS and IS performed bioinformatics analyses. MG, AS, and JA wrote the manuscript. All listed authors have made a substantial contribution to the work, and have approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Laboratoire Microorganismes & Biomolécules Actives, Université Tunis El-Manar, Tunisia (grant LR03ES03). JA acknowledges long-term support from the Commissariat à l'Energie Atomique et aux Energies Alternatives. AS acknowledges DBT, Government of Bengal (Grant no. 206/Bt (Estt)/RD-22/2014). IS acknowledges the receipt of a UGC-BSR fellowship.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00720/full#supplementary-material
Aebersold, R., and Mann, M. (2003). Mass spectrometry-based proteomics. Nature 422, 198–207. doi: 10.1038/nature01511
Akashi, H., and Gojobori, T. (2002). Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 99, 3695–3700. doi: 10.1073/pnas.062526999
Alloisio, N., Félix, S., Maréchal, J., Pujic, P., Rouy, Z., Vallenet, D., et al. (2007). Frankia alni proteome under nitrogen-fixing and nitrogen-replete conditions. Physiol. Plant. 130, 440–453. doi: 10.1111/j.1399-3054.2007.00859.x
Alloisio, N., Queiroux, C., Fournier, P., Pujic, P., Normand, P., Vallenet, D., et al. (2010). The Frankia alni symbiotic transcriptome. Mol. Plant Microbe Interact. 23, 593–607. doi: 10.1094/MPMI-23-5-0593
Armengaud, J., Christie-Oleza, J. A., Clair, G., Malard, V., and Duport, C. (2012). Exoproteomics: exploring the world around biological systems. Expert Rev. Proteomics 9, 561–575. doi: 10.1586/epr.12.52
Badri, D. V., and Vivanco, J. M. (2009). Regulation and function of root exudates. Plant Cell Environ. 32, 666–681. doi: 10.1111/j.1365-3040.2008.01926.x
Bagnarol, E., Popovici, J., Alloisio, N., Maréchal, J., Pujic, P., Normand, P., et al. (2007). Differential Frankia protein patterns induced by phenolic extracts from Myricaceae seeds. Physiol. Plant. 130, 380–390. doi: 10.1111/j.1399-3054.2007.00875.x
Bais, H. P., Weir, T. L., Perry, L. G., Gilroy, S., and Vivanco, J. M. (2006). The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 57, 233–266. doi: 10.1146/annurev.arplant.57.032905.105159
Baker, C. J., and Orlandi, E. W. (1995). Active oxygen in plant pathogenesis. Annu. Rev. Phytopathol. 33, 299–321. doi: 10.1146/annurev.py.33.090195.001503
Barski, O. A., Tipparaju, S. M., and Bhatnagar, A. (2008). The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab. Rev. 40, 553–624. doi: 10.1080/03602530802431439
Beauchemin, N. J., Furnholm, T., Lavenus, J., Svistoonoff, S., Doumas, P., Bogusz, D., et al. (2012). Casuarina root exudates alter the physiology, surface properties, and plant infectivity of Frankia sp. strain CcI3. Appl. Environ. Microbiol. 78, 575–580. doi: 10.1128/AEM.06183-11
Benson, D. R., Brooks, J. M., Huang, Y., Bickhart, D. M., and Mastronunzio, J. E. (2011). The biology of Frankia sp. strains in the post-genome era. Mol. Plant Microbe Interact. 24, 1310–1316. doi: 10.1094/MPMI-06-11-0150
Benson, D. R., and Silvester, W. B. (1993). Biology of Frankia strains, actinomycete symbionts of actinorhizal plants. Microbiol. Rev. 57, 293–319.
Bickhart, D. M., and Benson, D. R. (2011). Transcriptomes of Frankia sp. strain CcI3 in growth transitions. BMC Microbiol. 11:192. doi: 10.1186/1471-2180-11-192
Bogino, P. C., Oliva, M. D. L. M., Sorroche, F. G., and Giordano, W. (2013). The role of bacterial biofilms and surface components in plant-bacterial associations. Int. J. Mol. Sci. 14, 15838–15859. doi: 10.3390/ijms140815838
Broughton, W. J., and Dilworth, M. J. (1971). Control of leghaemoglobin synthesis in snake beans. Biochem. J. 125, 1075–1080. doi: 10.1042/bj1251075
Buist, G., Steen, A., Kok, J., and Kuipers, O. P. (2008). LysM, a widely distributed protein motif for binding to (peptido)glycans. Mol. Microbiol. 68, 838–847. doi: 10.1111/j.1365-2958.2008.06211.x
Chagnot, C., Zorgani, M. A., Astruc, T., and Desvaux, M. (2013). Proteinaceous determinants of surface colonization in bacteria: bacterial adhesion and biofilm formation from a protein secretion perspective. Front. Microbiol. 4:303. doi: 10.3389/fmicb.2013.00303
Clair, G., Roussi, S., Armengaud, J., and Duport, C. (2010). Expanding the known repertoire of virulence factors produced by Bacillus cereus through early secretome profiling in three redox conditions. Mol. Cell. Proteomics 9, 1486–1498. doi: 10.1074/mcp.M000027-MCP201
Clair, G., Armengaud, J., and Duport, C. (2012). Restricting fermentative potential by proteome remodeling an adaptive strategy evidenced in Bacillus cereus. Mol. Cell. Proteomics 11, M111–013102. doi: 10.1074/mcp.M111.013102
Dedieu, A., Gaillard, J. C., Pourcher, T., Darrouzet, E., and Armengaud, J. (2011). Revisiting iodination sites in thyroglobulin with an organ-oriented shotgun strategy. J. Biol. Chem. 286, 259–269. doi: 10.1074/jbc.M110.159483
Doré, J., Perraud, M., Dieryckx, C., Kohler, A., Morin, E., Henrissat, B., et al. (2015). Comparative genomics, proteomics and transcriptomics give new insight into the exoproteome of the basidiomycete Hebeloma cylindrosporum and its involvement in ectomycorrhizal symbiosis. New Phytol. 208, 1169–1187. doi: 10.1111/nph.13546
dos Reis, M., Savva, R., and Wernisch, L. (2004). Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 32, 5036–5044. doi: 10.1093/nar/gkh834
Fauvart, M., and Michiels, J. (2008). Rhizobial secreted proteins as determinants of host specificity in the rhizobium-legume symbiosis. FEMS Microbiol. Lett. 285, 1–9. doi: 10.1111/j.1574-6968.2008.01254.x
Galperin, M. Y. (2004). Bacterial signal transduction network in a genomic perspective. Environ. Microbiol. 6, 552–567. doi: 10.1111/j.1462-2920.2004.00633.x
Geiger, O., and López-Lara, I. M. (2002). Rhizobial acyl carrier proteins and their roles in the formation of bacterial cell-surface components that are required for the development of nitrogen-fixing root nodules on legume hosts. FEMS Microbial lett 208, 153–162. doi: 10.1111/j.1574-6968.2002.tb11075.x
Giraud, E., Moulin, L., Vallenet, D., Barbe, V., Cytryn, E., Avarre, J. C., et al. (2007). Legumes symbioses: absence of Nod genes in photosynthetic bradyrhizobia. Science 316, 1307–1312. doi: 10.1126/science.1139548
Gottesman, S. (1996). Proteases and their targets in Escherichia coli 1. Annu. Rev. Genet. 30, 465–506. doi: 10.1146/annurev.genet.30.1.465
Gtari, M., Ghodhbane-Gtari, F., Nouioui, I., Ktari, A., Hezbri, K., Mimouni, W., et al. (2015). Cultivating the uncultured: growing the recalcitrant cluster-2 Frankia strains. Sci. Rep. 5:13112. doi: 10.1038/srep13112
Gtari, M., Tisa, L. S., and Normand, P. (2013). “Diversity of Frankia strains, actinobacterial symbionts of actinorhizal plants,” in Symbiotic Endophytes, ed R. Aroca (Berlin; Heidelberg: Springer), 123–148.
Gust, A. A., Willmann, R., Desaki, Y., Grabherr, H. M., and Nurnberger, T. (2012). Plant LysM proteins: modules mediating symbiosis and immunity. Trends Plant Sci. 17, 495–502. doi: 10.1016/j.tplants.2012.04.003
Haichar, F. Z., Santaella, C., Heulin, T., and Achouak, W. (2014). Root exudates mediated interactions belowground. Soil Biol. Biochem. 77, 69–80. doi: 10.1016/j.soilbio.2014.06.017
Hartmann, E. M., Allain, F., Gaillard, J. C., Pible, O., and Armengaud, J. (2014). “Taking the shortcut for high-throughput shotgun proteomic analysis of bacteria,” in Host-Bacteria Interactions: Methods and Protocols, eds A. C. Vergunst, and D. O'Callaghan (New York, NY: Springer), 275–285.
Hartmann, E. M., and Armengaud, J. (2014). N-terminomics and proteogenomics, getting off to a good start. Proteomics 14, 2637–2646. doi: 10.1002/pmic.201400157
Jacob, A. I., Adham, S. A., Capstick, D. S., Clark, S. R., Spence, T., and Charles, T. C. (2008). Mutational analysis of the Sinorhizobium meliloti short-chain dehydrogenase/reductase family reveals substantial contribution to symbiosis and catabolic diversity. Mol. Plant Microbe Interact. 21, 979–987. doi: 10.1094/MPMI-21-7-0979
Lerouge, P., Roche, P., Faucher, C., Maillet, F., Truchet, G., Promé, J. C., et al. (1990). Symbiotic host-specificity of Rhizobium meliloti is determined by a sulphated and acylated glucosamine oligosaccharide signal. Nature 344, 781–784. doi: 10.1038/344781a0
Liu, T., Qian, W. J., Strittmatter, E. F., Camp, D. G., Anderson, G. A., Thrall, B. D., et al. (2004). High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal. Chem. 76, 5345–5353. doi: 10.1021/ac049485q
Mastronunzio, J. E., and Benson, D. R. (2010). Wild nodules can be broken: proteomics of Frankia in field-collected root nodules. Symbiosis 50, 13–26. doi: 10.1007/s13199-009-0030-1
Mastronunzio, J. E., Huang, Y., and Benson, D. R. (2009). Diminished exoproteome of Frankia spp. in culture and symbiosis. Appl. Environ. Microbiol. 75, 6721–6728. doi: 10.1128/AEM.01559-09
Mastronunzio, J. E., Tisa, L. S., Normand, P., and Benson, D. R. (2008). Comparative secretome analysis suggests low plant cell wall degrading capacity in Frankia symbionts. BMC Genomics 9:47. doi: 10.1186/1471-2164-9-47
Mendes, R., Kruijt, M., de Bruijn, I., Dekkers, E., van der Voort, M., Schneider, J. H., et al. (2011). Deciphering the rhizosphere microbiome for disease suppressive bacteria. Science 332, 1097–1100. doi: 10.1126/science.1203980
Miché, L., Moulin, L., Chaintreuil, C., Contreras-Jimenez, J. L., Munive-Hernández, J. A., Del Carmen Villegas-Hernandez, M., et al. (2010). Diversity analyses of Aeschynomene symbionts in Tropical Africa and Central America reveal that nod-independent stem nodulation is not restricted to photosynthetic bradyrhizobia. Environ. Microbiol. 12, 2152–2164. doi: 10.1111/j.1462-2920.2009.02090.x
Morgan, J. A. W., Bending, G. D., and White, P. J. (2005). Biological costs and benefitsto plant–microbe interactions in the rhizosphere. J. Exp. Bot. 56, 1729–1739. doi: 10.1093/jxb/eri205
Nguyen, T. V., Wibberg, D., Battenberg, K., Blom, J., Vanden Heuvel, B., Berry, A. M., et al. (2016). An assemblage of Frankia Cluster II strains from California contains the canonical nod genes and also the sulfotransferase gene nodH. BMC Genomics 17:796. doi: 10.1186/s12864-016-3140-1
Nouioui, I., Ghodhbane-Gtari, F., Rohde, M., Klenk, H.-P., and Gtari, M. (2017). Frankia coriariae sp. nov., an infective and effective microsymbiont isolated from Coriaria japonica. Int. J. Syst. Evol. Microbiol. doi: 10.1099/ijsem.0.001797. [Epub ahead of print].
Oldroyd, G. E. (2013). Speak, friend, and enter: signaling systems that promote beneficial symbiotic associations in plants. Nature Rev. Microbiol. 11, 252–263. doi: 10.1038/nrmicro2990
Paoletti, A. C., Parmely, T. J., Tomomori-Sato, C., Sato, S., Zhu, D., Conaway, R. C., et al. (2006). Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. U.S.A. 103, 18928–18933. doi: 10.1073/pnas.0606379103
Persson, T., Battenberg, K., Demina, I. V., Vigil-Stenman, T., Heuvel, B. V., Pujic, P., et al. (2015). Candidatus Frankia datiscae Dg1, the actinobacterial microsymbiont of Datisca glomerata, expresses the canonical nod genes nodABC in symbiosis with its host plant. PLoS ONE 10:e0127630. doi: 10.1371/journal.pone.0127630
Postma, P. W., Lengeler, J. W., and Jacobson, G. R. (1993). Phosphoenolpyruvate: carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57, 543–594.
Prasad, C. K., Vineetha, K. E., Hassani, R., Gupta, R., and Randhawa, G. S. (2000). Isolation and symbiotic characterization of aromatic amino acid auxotrophs of Sinorhizobium meliloti. Indian J. Exp. Biol. 38, 1041–1049.
Prin, Y., Neyra, M., and Diem, H. G. (1990). Estimation of Frankia growth using Bradford protein and INT reduction activity estimations: application to inoculum standardization. FEMS Microbiol. Lett. 69, 91–95. doi: 10.1111/j.1574-6968.1990.tb04181.x
Randhawa, G. S., and Hassani, R. (2002). Role of rhizobial biosynthetic pathways of amino acids, nucleotide bases and vitamins in symbiosis. Indian J. Exp. Biol. 40, 755–764.
Rey, T., Nars, A., Bonhomme, M., Bottin, A., Huguet, S., Balzergue, S., et al. (2013). NFP, a LysM protein controlling Nod factor perception, also intervenes in Medicago truncatula resistance to pathogens. New Phytol. 198, 875–886. doi: 10.1111/nph.12198
Roy, A., Mukhopadhyay, S., Sarkar, I., and Sen, A. (2015). Comparative investigation of the various determinants that influence the codon and amino acid usage patterns in the genus Bifidobacterium. World J. Microb. Biotechhnol. 31, 959–981. doi: 10.1007/s11274-015-1850-1
Saier, M. H., Chauvaux, S., Deutscher, J., Reizer, J., and Ye, J. J. (1995). Protein phosphorylation and regulation of carbon metabolism in Gram-negative versus Gram-positive bacteria. Trends Biochem. Sci. 20, 267–271. doi: 10.1016/S0968-0004(00)89041-6
Saier, M. H. Jr., and Reizer, J. (1992). Proposed uniform nomenclature for the proteins and protein domains of the bacterial phosphoenolpyruvate: sugar phosphotransferase system. J. Bacteriol. 174:1433. doi: 10.1128/jb.174.5.1433-1438.1992
Sarkar, I., Normand, P., Tisa, L. S., Gtari, M., Bothra, A., and Sen, A. (2016). Characterization of PAS domains in Frankia and selected Actinobacteria and their possible interaction with other co-domains for environmental adaptation. Symbiosis 70, 69–78. doi: 10.1007/s13199-016-0413-z
Schmidt, A., Forne, I., and Imhof, A. (2014). Bioinformatic analysis of proteomics data. BMC Syst. Biol. 8:s3. doi: 10.1186/1752-0509-8-s2-s3
Sen, A., Sur, S., Bothra, A. K., Benson, D. R., Normand, P., and Tisa, L. S. (2008). The implication of life style on codon usage patterns and predicted highly expressed genes for three Frankia genomes. Antonie Van Leeuwenhoek 93, 335–346. doi: 10.1007/s10482-007-9211-1
Smith, P., Krohn, R. I., Hermanson, G. T., Mallia, A. K., Gartner, F. H., Provenzano, M., et al. (1985). Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85. doi: 10.1016/0003-2697(85)90442-7
Smolander, A., and Sarsa, M. (1990). Frankia strains in soil under Betula pendula: behavior in soil and in pure culture. Plant Soil 122, 129–136. doi: 10.1007/BF02851920
Svistoonoff, S., Benabdoun, F. M., Nambiar-Veetil, M., Imanishi, L., Vaissayre, V., Cesari, S., et al. (2013). The independent acquisition of plant root nitrogen-fixing symbiosis in Fabids recruited the same genetic pathway for nodule organogenesis. PLoS ONE 8:e64515. doi: 10.1371/journal.pone.0064515
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., and Huerta-Cepas, J. (2014). STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi: 10.1093/nar/gku1003
Tiricz, H., Szűcs, A., Farkas, A., Pap, B., Lima, R. M., Maróti, G., et al. (2013). Antimicrobial nodule-specific cysteine-rich peptides induce membrane depolarization associated changes in the transcriptome of Sinorhizobium meliloti. Appl. Environ. Microbiol. 79, 6737–6746. doi: 10.1128/AEM.01791-13
Tisa, L. S., Oshone, R., Sarkar, I., Ktari, A., Sen, A., and Gtari, M. (2016). Genomic approaches toward understanding the actinorhizal symbiosis: an update on the status of the Frankia genomes. Symbiosis 70, 1–12. doi: 10.1007/s13199-016-0390-2
Turner, T. R., Ramakrishnan, K., Walshaw, J., Heavens, D., Alston, M., Swarbreck, D., et al. (2013). Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J. 7, 2248–2258. doi: 10.1038/ismej.2013.119
Udvardi, M., and Poole, P. S. (2013). Transport and metabolism in legume-rhizobia symbioses. Annu. Rev. Plant Biol. 64, 781–805. doi: 10.1146/annurev-arplant-050312-120235
Udwary, D. W., Gontang, E. A., Jones, A. C., Jones, C. S., Schultz, A. W., Winter, J. M., et al. (2011). Significant natural product biosynthetic potential of actinorhizal symbionts of the genus Frankia, as revealed by comparative genomic and proteomic analyses. Appl. Environ. Microbiol. 77, 3617–3625. doi: 10.1128/AEM.00038-11
van Veen, J. A., van Overbeek, L. S., and van Elsas, J. D. (1997). Fate and activity of microorganisms introduced into soil. Microbiol. Mol. Biol. Rev. 61, 121–135.
Vizcaíno, J. A., Csordas, A., del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., et al. (2016). 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 44, D447–D456. doi: 10.1093/nar/gkv1145
Wagner, K., Linde, J., Krause, K., Gube, M., Koestler, T., Sammer, D., et al. (2015). Tricholoma vaccinum host communication during ectomycorrhiza formation. FEMS Microbiol. Ecol. 91:fiv120. doi: 10.1093/femsec/fiv120
Xia, X., and Xie, Z. (2001). DAMBE: software package for data analysis in molecular biology and evolution. J. Hered. 92, 371–373. doi: 10.1093/jhered/92.4.371
Yu, C. S., Chen, Y. C., Lu, C. H., and Hwang, J. K. (2006). Prediction of protein subcellular localization. Proteins 64, 643–651. doi: 10.1002/prot.21018
Keywords: root exudates, Frankia, Proteogenome, symbiosis, signaling pathways
Citation: Ktari A, Gueddou A, Nouioui I, Miotello G, Sarkar I, Ghodhbane-Gtari F, Sen A, Armengaud J and Gtari M (2017) Host Plant Compatibility Shapes the Proteogenome of Frankia coriariae. Front. Microbiol. 8:720. doi: 10.3389/fmicb.2017.00720
Received: 10 January 2017; Accepted: 06 April 2017;
Published: 02 May 2017.
Edited by:
Benjamin Gourion, Laboratoire Interactions Plantes-Microorganismes (INRS), FranceReviewed by:
Hassen Gherbi, Institut de Recherche pour le Développement (IRD), FranceCopyright © 2017 Ktari, Gueddou, Nouioui, Miotello, Sarkar, Ghodhbane-Gtari, Sen, Armengaud and Gtari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maher Gtari, bWFoZXIuZ3RhcmlAZnN0LnJudS50bg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.