 John Vollmers
John Vollmers Martinique Frentrup
Martinique Frentrup Patrick Rast
Patrick Rast Christian Jogler
Christian Jogler Anne-Kristin Kaster
Anne-Kristin Kaster- 1Leibniz Institute DSMZ—German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany
- 2Department of Microbiology, Institute for Water and Wetland Research, Faculty of Science, Radboud University, Nijmegen, Netherlands
The kelp forest of the Pacific temperate rocky marine coastline of Monterey Bay in California is a dominant habitat for large brown macro-algae in the order of Laminariales. It is probably one of the most species-rich, structurally complex and productive ecosystems in temperate waters and well-studied in terms of trophic ecology. However, still little is known about the microorganisms thriving in this habitat. A growing body of evidence suggests that bacteria associated with macro-algae represent a huge and largely untapped resource of natural products with chemical structures that have been optimized by evolution for biological and ecological purposes. Those microorganisms are most likely attracted by algae through secretion of specific carbohydrates and proteins that trigger them to attach to the algal surface and to form biofilms. The algae might then employ those bacteria as biofouling control, using their antimicrobial secondary metabolites to defeat other bacteria or eukaryotes. We here analyzed biofilm samples from the brown macro-algae Macrocystis pyrifera sampled in November 2014 in the kelp forest of Monterey Bay by a metagenomic shotgun and amplicon sequencing approach, focusing on Planctomycetes and Verrucomicrobia from the PVC superphylum. Although not very abundant, we were able to find novel Planctomycetal and Verrucomicrobial species by an innovative binning approach. All identified species harbor secondary metabolite related gene clusters, contributing to our hypothesis that through inter-species interaction, microorganisms might have a substantial effect on kelp forest wellbeing and/or disease-development.
Introduction
Submarine kelp forests are one of the most species-rich, structurally complex, and productive ecosystems in temperate waters. They are extremely well studied in terms of trophic ecology (Dayton and Tegner, 1984; Steneck et al., 2002; Estes et al., 2004; Graham, 2004) and provide habitat and nutrition to diverse communities extending from microorganisms to mammals. Kelp forests are highly diverse at the phylum level (Steneck et al., 2002; Graham, 2004) and—like tropical rain forests—important habitats and hot spots of biological diversity. Given that individual kelp algae can grow up to 50 cm in length every day, kelp forests are also mayor players in CO2 fixation in temperate waters (Foster et al., 2012). The so-called giant kelp, Macrocystis pyrifera, dominates these ecosystems along the temperate west coasts of North America (Dayton, 1985; Foster and Schiel, 1985; Delille and Perret, 1991; Graham et al., 2007). Along the central coast of California the kelp forest is of tremendous importance for coastal biodiversity, productivity, and the human economy and kelp-surface-associated bacteria are believed to be important players in carbon and nitrogen turnover in this food web (Linley and Field, 1982; Graham, 2004). While this habitat has been intensively researched for decades, still little is known about the microorganisms associated and interacting with the kelp, as only recently molecular techniques have become available to study biofilm species composition and abundance (Bengtsson et al., 2010, 2011, 2012; Hollants et al., 2013; Michelou et al., 2013).
Bacteria-algae interactions include symbiotic and parasitic relationships and mainly depend on environmental parameters, such as the availability of inorganic nutrients and organic matter. In eutrophic coastal marine systems like the Monterey Bay kelp forest rapid bacterial biofilm colonization takes place. Heterotrophic microorganisms are most likely attracted by the macro-algae through secretion of specific carbohydrates and proteins that trigger them to attach to their surface and to form biofilms, while degrading complex algal polysaccharides (Bengtsson et al., 2011, 2012; Jeske et al., 2013; Wegner et al., 2013). Kelp exudates may shape bacterial community composition, and create communities that are kelp-specific rather than randomly assembled from the surrounding seawater (Taylor et al., 2004; Longford et al., 2007; Reis et al., 2009). A study by Michelou et al. (2013) with samples taken from Monterey Bay in the months March and May in 2010 using 454-tag pyrosequencing of 16S rRNA genes showed that bacterial community structure and membership correlated with the kelp surface serving as host, and varied over time. M. pyrifera surface was enriched with Rhodobacteraceae, Sphingomonadaceae (Alphaproteobacteria), Flavobacteraceae, Saprospiraceae (Bacteroidetes) families and unclassified Gammaproteobacteria. Interestingly, sequences from the phyla Verrucomicrobia and Planctomycetes were detected, although not in high abundance. Several taxa were highly similar to other bacteria known to either prevent the colonization of eukaryotic larvae or exhibit antibacterial activities, which holds true for Verrucomicrobia and Planctomycetes (Wagner and Horn, 2006; Rao et al., 2007).
Studies of biofilms from the kelp algae Laminaria hyperborea collected along the west coast of Norway showed Verrucomicrobia and Planctomycetes to be even among the most frequently detected lineages (Bengtsson and Øvreås, 2010). However, biofilm composition was subject to seasonal variations (Bengtsson et al., 2010) and due to the dominance of few abundant Operational Taxonomic Units (OTUs), the kelp surface was characterized as low-diversity habitat (Bengtsson et al., 2012). Given the slow growth of Planctomycetal species (Fuerst, 2013) their abundance in such habitats, that are packed with carbon sources in contrast to the largely oligotrophic surrounding water, appears counter intuitive (Lage and Bondoso, 2014). Most other heterotrophs that dwell in such ecological niches divide much faster (for example 1.2–6.3 h for Roseobacter species (Christie-Oleza et al., 2012; Hahnke et al., 2013) and should generally outcompete slowly growing competitors. However, the interactions with the algae might involve the production of various secondary metabolites that are antimicrobial (defense against other, faster growing, heterotrophic bacteria) or algicidal (to destroy other eukaryotes like algae, diatoms or cyanobacteria for scavenging), and algae use those prokaryotic species as biofouling control (Zheng et al., 2005; Goecke et al., 2010). Those inter-species interactions of algae and bacteria and their resulting natural products are however not well understood (Estes et al., 2004).
We here analyzed for the first time a biofilm sample from M. pyrifera of the Monterey Bay kelp forest by a metagenomic shotgun and amplicon sequencing approach with a focus on the PVC superphylum (Figure 1). This study reports an in-depth description of the diversity and phylogenetic association of the microbial communities associated with M. pyrifera. Through inter-species interactions Planctomycetes and Verrucomicrobia might have a substantial effect on kelp forest wellbeing or disease-development, providing a foundation for understanding the microbial ecology of kelp forests.
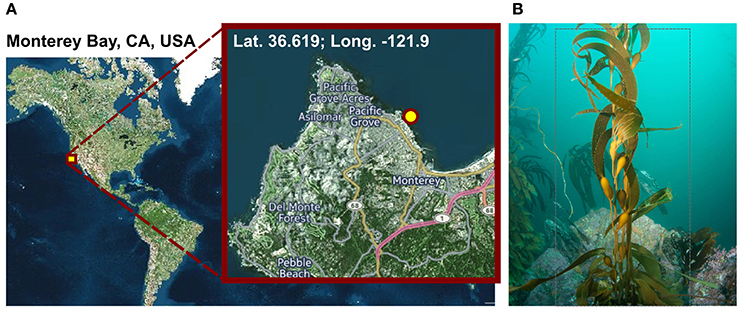
Figure 1. Sampling. (A) Geographic position of the sampling site. Visualization was done with cartoDB (https://www.carto.com). (B) Macrocystis pyrifera (kelp) specimen photographed at the sampling location during sampling in November 2014 at a water depth of 6 m and a water temperature of 12°C.
Materials and Methods
Sampling
Macrocystis pyrifera was collected in 6 m water depth at a temperature of 12°C from the kelp forest near the Monterey Bay Aquarium, California, USA (lat. 36.619; long. −121.901) in November 2014 (Figure 1). Samples were stored in sterile Artificial Sea Water (ASW; 0.8 M NaCl, 0.06 M Na2SO4, 0.1 M MgCl2× 6 H2O, 19.5 mM CaCl2 × 2 H2O, 4.6 mM NaHCO3, 18.5 mM KCl, 1.6 mM KBr, 0.08 mM SrCl2 × 6 H2O and 0.14 mM NaF) and shipped on ice to Germany the same day. Upon arrival, the algae were cut into several 5 cm2 pieces and its biofilm was partially scraped off into 20 ml fresh ASW using a sterile scalpel, in order to achieve a partial enrichment of biofilm associated bacteria and to circumvent extracting eukaryotic cell material. However, since the biofilm was found to be thin and not always clearly visible, original kelp pieces were also retained for subsequent DNA extractions. Kelp pieces and scraped-off biofilm were stored separately in fresh ASW at −20°C until further processing.
DNA Extraction
In order to ensure a comprehensive representation of the kelp biofilm community, while minimizing eukaryotic DNA contamination from the algae itself and to enable a differential coverage binning approach, two different extraction methods were used to obtain DNA from kelp biofilm, resulting in DNA extracts A and B, respectively. Both extracts originated from the same kelp stipe, but from different blades. (Extract A) Sub-segments of one 5 cm2 kelp piece and 1.5 ml of scraped-off biofilm suspension were combined and subjected to pulse vortexing as well as 5 min of vigorous shaking in order to detach and capture tenacious biofilm community members which may not have been efficiently scraped off. Eukaryotic cells were then removed from the suspension via gravity flow filtration using a polycarbonate filter with 10 μm pore size (Celltrics filter, Partec, Münster, Germany). (Extract B) In order to minimize carry-over of eukaryotic cell fragments and to protect sensitive community members from shearing forces, 2 ml of undisturbed scraped-off biofilm suspension were carefully transferred to a new microcentrifuge tube without including the precipitate of residual algae fragments which formed by natural sedimentation. Subsequently, all DNA extraction steps were identical for both approaches, beginning with a two-step protocol adopted from Ferrera et al. (2010). Microbial cells were harvested by centrifugation (40 min, 16,000 g, 4°C) and re-suspended in 950 μl lysis buffer (40 mM EDTA, 50 mM Tris-HCl, 0.75 M sucrose). The cell suspensions were enzymatically lysed under slight shaking for 45 min at 37°C using 1 mg/ml lysozyme (final conc.), followed by chemical lysis under slight shaking for 60 min at 55°C using 1 mg/ml SDS and 0.2 mg/ml proteinase K (final conc., respectively). Two rounds of phenol:chloroform:isopentanol [25:24:1; v:v:v] extractions and two rounds of chloroform:isopentanol [24:1; v:v] extractions were performed. DNA of each sample was precipitated for 12 h at −20°C, using 0.3 M sodium acetate (final conc.) and 1 volume of ice-cold isopropanol, washed twice with 70% ethanol and re-suspended in 20 μl of nuclease free water. DNA purity was verified photometrically using a Nanodrop 2,000 Spectrometer (Thermo Fischer Scientific, Wilmington, USA), while DNA yield was determined using a Qubit® 1.0 Fluorometer with an dsDNA HS Assay Kit (Life Technologies, Darmstadt, Germany).
Metagenomic Shotgun Library Preparation
Separate shotgun libraries, KelpA and KelpB, were produced for DNA extracts A and B, respectively. DNA was sheared using a Covaris® S220 sonication device (Covaris Inc; Massachusetts, USA; 55.5 μl shearing volume, 175 W Peak Incident Power, 5% Duty factor, 200 burst cycles, 55 s treatment time). Sequencing libraries were prepared using the NEBnext® Ultra™ DNA Library Preparation Kit for Illumina® (New England Biolabs, Frankfurt, Germany) as per the manufacturer's instructions. Due to a high concentration of inhibitory contaminants, such as algal osmolytes released during sample treatment, extract A had to be diluted prior to library preparation, limiting the respective amount of input DNA to 5 ng. Therefore, 12 cycles of enrichment PCR were performed during the preparation of library KelpA. Since extract B was free of such contaminants, the full DNA yield of 105 ng could be utilized for preparation of library KelpB, necessitating only 6 cycles of enrichment PCR.
Amplicon Library Preparation
An aliquot of DNA extract A was diluted to 1 ng/μl with PCR-grade water and subjected to Whole Genome Amplification (WGA) using the illustra™ GenomiPhi™ V2 DNA amplification Kit (GE Healthcare) and a Veriti 96-Well thermal cycler (Applied Biosystems). In order to reduce stochastic amplification bias, 10 independent amplification reactions were performed, using 1 μl of diluted DNA in a 20 μl reaction volume each, and subsequently pooled. WGA products were quantified using a Qubit® 3.0 Fluorometer with the Qubit® dsDNA BR Assay Kit (Life Technologies, Darmstadt, Germany). The V3 region of the 16S rRNA gene was amplified from the pooled WGA products via two subsequent PCR steps. Both reactions were performed in 50 μl reaction volumes consisting of 0.02 U/μl Q5® High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA), 1 × Q5 high GC enhancer, 1 × Q5 reaction buffer and 200 μM dNTPs. While the applied temperatures varied, both PCR programs consisted of 10 amplification cycles, with 1 min each for denaturing, annealing and elongation, as well as a preliminary denaturing step and a final elongation step of 5 min each. Reactions were performed in triplicates, which were subsequently pooled in order to reduce stochastic amplification bias. The first pre-amplification step was performed using 0.1 μM each of the V3 region specific forward primer 341f (5'- CCT ACG GGW GGC WGC AG-3') and the universal reverse primer uni515r (5'- CCG CGG CTG CTG GCA C-3') modified from 341f and 518r by Muyzer et al. (1993), respectively. Three hundred nano grams of WGA product was used as template. Cycling temperatures were 94°C for denaturing, 63°C for annealing and 72°C for elongation. Illumina® sequencing adapters and barcodes were added during the second amplification step following the pooling and purification of the pre-amplification products. For this step, 0.2 μM each of the extended V3 region primers “V3 fwd” (5′- AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GCT CTT CCG ATC TCC TAC GGG WGG CWG CAG -3′) and “index 34 V3 rev” (5′- CAA GCA GAA GAC GGC ATA CGA GAT TAT TCG GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC TCC GCG GCT GCT GGC AC −3′) were used, both of which were modified from Bartram et al. (2011). Fifteen microliters of pre-amplification product was used as template. Denaturing, annealing and elongation temperatures were 98, 65, and 72°C, respectively. PCR products were separated by electrophoresis using 2% agarose gels in 1 × Tris-Acetate-EDTA (TAE) buffer. The gel was stained with SYBR® gold (Thermo Fisher) for 1 h and inspected under UV light. Amplicon bands (~300 bp sized fragments) were excised and extracted using a spin column based approach (NucleoSpin Gel and PCR Clean-up; Macherey-Nagel).
Sequencing and Read Processing
All libraries were sequenced on an Illumina® MiSeq using v3 chemistry, 301 cycles per read and paired end settings. Raw sequences were subjected to adapter clipping and quality trimming using Trimmomatic v.0.36 (Bolger et al., 2014) with the following arguments: “LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:105.” For preliminary metagenome assemblies, only the resulting read pairs were used, excluding orphans. For subsequent mapping and reassembly steps, as well as amplicon analyses, overlapping read pairs were identified and merged using FLASH v.1.2.11 (Magoč and Salzberg, 2011). Due to the short length of the amplicon inserts, amplicon read pairs overlapped in their entire sequence length. Therefore, merging resulted in high confidence consensus sequences which minimized the influence of random sequencing errors. Merged amplicon reads were furthermore analyzed and filtered based on the presence of the employed V3 region specific forward and reverse primer sequences, which were subsequently clipped from the reads.
Metagenome Shotgun Assembly
Processed reads were pooled into a combined dataset KelpAB and assembled using IDBA-UD v.1.1.1 (Peng et al., 2012). For reference, additional individual assemblies were performed for each dataset, separately. In order to take optimal advantage of the longer MiSeq sequencing read lengths for correctly resolving repetitive regions, the source code was slightly altered to allow k-mer lengths of up to 251 bp, according to guidelines provided by the developers (https://github.com/loneknightpy/idba). For the preliminary metagenome shotgun assemblies, only paired reads were used, as IDBA_UD is not optimized for unpaired or merged reads.
Binning and Reassembly
Initial binning was performed using Maxbin v.2.1.1 (Wu et al., 2016) yielding 136 bins (Supplementary Table 1). After analysis and classification of these preliminary bins using CheckM v.1.0.4 (Parks et al., 2015), 8 Planctomycetal bins and 2 Verrucomicrobial bins were selected for further processing, in order to improve the assembly and remove residual contaminating genome fragments (Table 1). Putative contaminants were identified by taxonomically classifying the scaffolds of each bin using the Least Common Ancestor (LCA) approach implemented in MEGAN5 (Huson and Weber, 2013). Classifications were based on blastx and blastn comparisons against the NCBI nt and nr databases respectively, using an evalue cutoff of 1e-20. Only scaffolds that could be unambiguously assigned to any other phyla than the expected Planctomycetes or Verrucomicrobia were removed as putative contaminants. Subsequently, the average read coverage of each metagenomic scaffold was determined by mapping the sequencing datasets KelpA and KelpB individually against the combined assembly (KelpAB) using bowtie2 (Langmead and Salzberg, 2012). This information was used to analyze the coverage distribution within each of the selected bins. All outliers, consisting of scaffolds with coverage values larger than three-fold the median of the respective dataset and bin, were removed. Furthermore, bins were examined for sets of scaffolds displaying inverse relative coverage profiles when comparing KelpA and KelpB (e.g., displaying high abundance in KelpA but low abundance in KelpB or vice versa) and divided into appropriate sub-bins if applicable. In order to enable a targeted re-assembly, the corresponding sequencing reads were extracted from datasets KelpA and KelpB based on bowtie2 mappings for each of the hereby pre-filtered bins. To maximize sequence information and coverage, orphaned reads and merged read pairs were included in the analyses. To ensure high specificity, the “end-to-end” setting of bowtie2 was employed accepting only reads aligning in the entirety of their length. Furthermore, read pairs were only accepted if both reads mapped to the respective bin. The resulting read subsets were individually reassembled using SPAdes v.3.7.1 (Bankevich et al., 2012) and utilizing the scaffolds of the respective purified preliminary bin as “untrusted reference.” For final purification, new coverage profiles were calculated based on bowtie2 mappings and analyzed in order to identify scaffolds displaying differential coverage between datasets KelpA and KelpB, indicating putative contaminating genome fragments in each bin. To this end, the coverage values for each bin and dataset were converted into z-scores representing the respective deviation from the mean coverage of each analyzed bin. Putative contaminants were identified as scaffolds displaying differences above a specified cutoff between the z-scores for datasets KelpA and KelpB. Z-scores were recalculated for the remaining scaffolds, each time a putatively contaminating scaffold was removed. This process was performed iteratively beginning at a z-score cutoff of 4, decreasing the cutoff by 1 each time no further contaminants could be identified, until a final cutoff value of 2 was reached (corresponding to a coverage difference of 2 x the standard deviation for each dataset). The final, processed bins were re-examined using CheckM with appropriate phylum specific marker sets for Planctomycetes or Verrucomicrobia (Table 1).
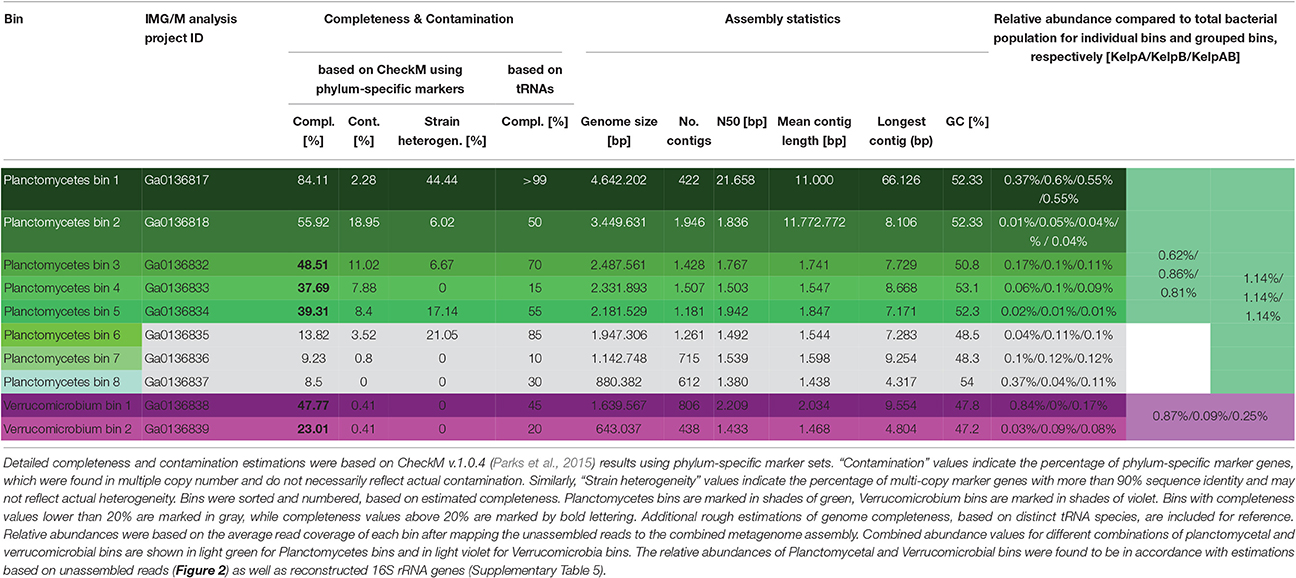
Table 1. General statistics of Planctomycetes and Verrucomicrobia bins.
Annotation and Functional Analyses
Gene calling and annotation was performed for processed bins using the Prokka pipeline v.1.12 (Seemann, 2014). Additionally, genes were annotated with SEED categories, based on blastp alignments against the NCBI nr database and subsequent LCA analyses using MEGAN5. AntiSMASH v.3.0 was employed to identify putative secondary metabolite gene clusters (Weber et al., 2015).
Analyses of Shotgun Metagenomic 16S rRNA Genes
RNAmmer v.1.2 was used to identify almost full-length (>1,100 bp) 16S rRNA gene sequences in the assembled metagenomes (Lagesen et al., 2007). However, due to the highly conserved nature of 16S rRNA genes, such sequences are likely to represent chimeras of different related strains when obtained by metagenome assembly. Therefore, the dedicated software tool EMIRGE (Miller et al., 2011) for reconstructing 16S rRNA genes from short read metagenomic data, was additionally employed for verification purposes. The resulting 16S rRNA sequences were aligned to the SILVA reference database and taxonomically classified using SINA (Pruesse et al., 2012; Quast et al., 2013; Yilmaz et al., 2014). Additionally, the sequences were phylogenetically clustered using a neighbor joining approach with 1,000 bootstrap iterations as implemented in ARB v.6.0.2 (Ludwig et al., 2004). A combination of phylogenetic, Multi Locus Sequence Analyses (MLSA) and coverage analyses was used to identify unambiguous connections between metagenomic bins and 16S rRNA gene sequences. Classification and relative abundances were visualized using the software tool Krona (Ondov et al., 2011).
Analyses of 16S rRNA Amplicon Sequences
The low sequence length and high throughput of the amplicon sequencing data required a slightly different analyses pipeline than the almost full-length sequences reconstructed from the shotgun metagenome datasets. However, the same reference database as well as general taxonomic classification framework was used. Classification of processed amplicon reads was performed using the SILVAngs service platform and associated analysis pipeline (Quast et al., 2013; Yilmaz et al., 2014). Reads were uploaded as suggested in the SILVAngs user-guide and processed by the SILVAngs software according to the recommended protocol, including sequence alignment with the SINA aligner (Pruesse et al., 2012). During the process, a de-replication step, eliminating 100% identical reads, as well as OTU definition and clustering was performed. OTUs were classified by a local BLAST search using blastn with default parameters in accordance to the non-redundant version of the SILVA SSU Ref database. Classification and relative abundance of defined OTUs was visualized using the implemented software tool Krona (Ondov et al., 2011).
Multi Locus Sequence Analyses (MLSA)
Proteinortho5 (Lechner et al., 2011) was used to detect groups of orthologous genes shared between selected reference genomes and processed metagenomic bins. Reference genomes were obtained from NCBI and IMG/M. For each set of bins and comparison genomes, the respective “unique core genome” was determined as the list of gene products universally present in single copy. The size and composition of the resulting unique core genome set is strongly dependent on the reference genomes and the completeness of the analyzed bins. Therefore, separate analyses were performed for each processed metagenomic bin as well as combinations of bins associated with the same phylum. The resulting unique core genome gene products were individually aligned using MUSCLE v.3.8.31 (Edgar, 2004) and subsequently concatenated. Regions that could not be aligned were filtered from the alignments using Gblocks v.0.91b (Castresana, 2000). The filtered alignments were clustered using the neighbor joining algorithm with 1,000 bootstrap permutations, in order to reliably place each bin into a phylogenetic context.
Accession Numbers
Metagenome data and sequences of the 8 Planctomycetal and 2 Verrucomicrobial bins were deposited with the Integrated Microbial Genomes (IMG) database. KelpAB (coassembly): Ga0136809, KelpA (single assembly): Ga0136854, KelpB (single assembly): Ga0136855; Planctomycetal bin 1: Ga0136817, Planctomycetal bin 2: Ga0136818, Planctomycetal bin 3: Ga0136832, Planctomycetal bin 4: Ga0136833, Planctomycetal bin 5: Ga0136834, Planctomycetal bin 6: Ga0136835, Planctomycetal bin 7: Ga0136836, Planctomycetal bin 8: Ga0136837; Verrucomicrobial bin 1: Ga0136838, Verrucomicrobial bin 2: Ga0136839.
Results and Discussion
We here analyzed the biofilm community of M. pyrifera sampled from Monterey Bay kelp forest in November 2014 on multiple levels: Amplicon sequences, unassembled metagenomic shotgun reads, and assembled metagenome (Figure 2, Supplementary Figures 1, 2). Amplicon library sequencing yielded in 660.000 high quality reads in overlapping pairs, which could be merged into 330.000 consensus reads (Supplementary Figure 1A). Metagenomic shotgun sequencing yielded 36 mio raw reads per sequencing library. After quality trimming, 28 mio high quality reads were retained for library of KelpA and 34 mio reads for KelpB. In both cases, a large fraction of ~23 mio reads formed overlapping read pairs, which could be merged into longer consensus sequences prior to assembly (Supplementary Figure 1B). The amplicon library based approach indicated a bacterial biofilm community comprised predominantly of Proteobacteria and Bacteroidetes, with Planctomycetes and Verrucomicrobia representing lower but nonetheless substantial community fractions of 4% each (Figure 2A). This observation is in concurrence with a previous amplicon based analysis of M. pyrifera associated communities sampled during spring 2010. The overall trend of this community profile was furthermore confirmed by the Least Common Ancestor (LCA) taxonomic classification of a metagenomic shotgun sequencing dataset (KelpA) obtained from the same sample (Figure 2B), albeit with pronounced differences in the exact proportions of each bacterial fraction. Among the most noteworthy differences between the amplicon and the shotgun sequencing approach are the considerably lower fractions of Planctomycetes and Verrucomicrobia in the latter, comprising only 0.9 and 0.2% of the bacterial community, respectively. A cause for these discrepancies might be the bias introduced by the Phi29 based Whole Genome Amplification (Lasken, 2009) used in the preparation of the amplicon, but not the metagenomic shotgun library. However, since correct classification of short shotgun reads is highly dependent on the presence of closely related genome sequences within public databases, this divergence probably reflects the low representation of the PVC superphylum within currently sequenced genomes as well as the uniqueness of the Planctomycetes and Verrucomicrobia strains present in the here sampled habitat. The latter assumption is partially supported by the fact that relative abundances of the PVC superphylum diverge by a factor of 4.4–20% between amplicon and metagenomic shotgun data sets, while most other phyla and classes diverge only by a factor of 1.5–5%. The underrepresentation of PVC-classified reads, caused by the generally high variability of protein coding genes in combination with the low number of currently available PVC reference genomes also increases the likelihood of PVC-associated reads being classified as “unassigned bacteria.”
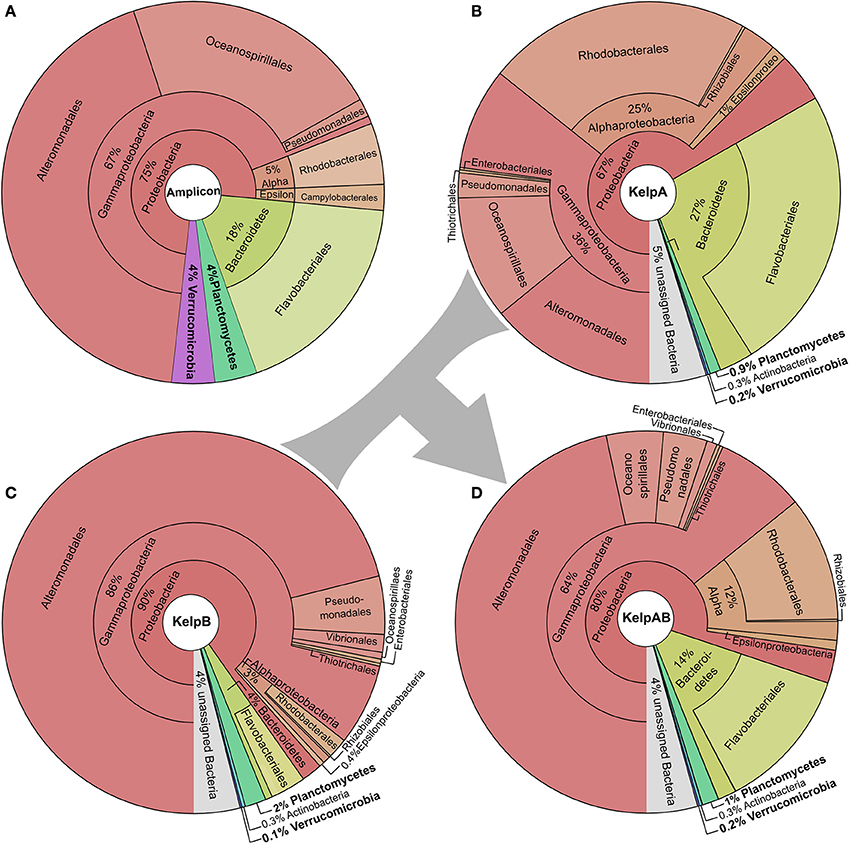
Figure 2. Taxonomic composition of the kelp biofilm community. Pie charts showing the relative abundances of sequences associated with bacterial taxa within each sequencing dataset down to order level. Visualizations were done using KRONA (Ondov et al., 2011). (A) 16S rRNA amplicon dataset. Relative abundances were determined using the SILVA NGS pipeline (Quast et al., 2013; Yilmaz et al., 2014). (B,C) Metagenomic shotgun libraries of KelpA & KelpB, respectively. KelpA (B) was derived from the same sample as the amplicon library, while KelpB (C) was derived from a separate sample obtained from a different leaf of the same Kelp specimen, using a slightly modified DNA extraction protocol. Relative abundances were determined based on sequencing read classifications obtained by using the Least Common Ancestor (LCA) approach implemented in MEGAN5 (Huson and Weber, 2013). In order to maximize coverage for low abundant community members, both datasets were treated as a composite dataset KelpAB (D) for assembly.
In the past, PVC community members have been often reported to be highly abundant and sometimes even dominant in kelp biofilm communities (Bengtsson and Øvreås, 2010; Michelou et al., 2013). However, the dominance of specific species or OTUs is often only temporary (Bengtsson et al., 2012; Michelou et al., 2013), e.g., in form of seasonal blooms. Metagenomics data are therefore likely not generally representative for the respective environment but only for the specific set of conditions during sampling. In addition, virus infections might change a microbial community within hours. Therefore, crucial members of communities may only be found in low abundance most of the time, in some cases even always (K-strategists). Such organisms may be much harder to isolate and analyze, but represent a huge hidden genomic potential, which still remains to be discovered. Unfortunately, whether trying to obtain isolates from environmental samples, or trying to reconstruct genomes from metagenomes (Albertsen et al., 2013; Sangwan et al., 2016) the focus is most often laid on the most abundant community members. As a result, low abundant and/or slowly growing species are likely to be underrepresented in public databases. With our innovative binning approach we here provide insights into the genomes of some low abundance but ubiquitous Planctomycetal and Verrucomicrobial species on algae and were able to generate a good draft genome of a novel Planctomycete which cannot be assigned to any of the known genera with cultured representatives.
Since binning efficiency is significantly improved by employing multiple related datasets, two metagenomic shotgun libraries were produced using slightly altered extraction protocols and different blade sections of the same kelp specimen, resulting in the datasets KelpA and KelpB. Similar to KelpA, KelpB was dominated by Proteobacteria, particularly Gammaproteobacteria, and contained only relatively small fractions of Planctomycetes and Verrucomicrobia (Figure 2C). However, both datasets differed in the exact proportions of the individual observed taxa, caused by a combination of community variability within kelp specimens as well as differences in extraction protocols. Differences include a lower relative abundance of Planctomycetes in KelpA compared to KelpB, while the opposite seems to be true for Verrucomicrobia, indicating that coverage differences between these datasets may be utilized to improve subsequent binning steps. The most pronounced differences were observed for the class Alphaproteobacteria and the phylum Bacteroidetes, which are present in considerably lower abundance in KelpB compared to KelpA by factors of ~8 and ~7, respectively. This indicates that intra-host variation and differences in extraction methods may greatly influence the community structure observed by metagenomic analyses, but can be utilized for differential coverage binning approaches. While both libraries confirm the dominance of Proteobacteria observed in the amplicon library, the observed fraction of Planctomycetes and Verrucomicrobia is considerably lower, representing only 0.1–2% of the bacterial community. In order to maximize sequencing coverage for low abundant community members present in both sequencing datasets, KelpA and KelpB were pooled into the combined dataset KelpAB (Figure 2D) and re-assembled, prior to binning.
Unfortunately, the preliminary bins obtained using Maxbin v.2.0 were of mixed quality, with a large degree of fragmentation as well as contamination in most bins (Supplementary Table 1). In order to obtain the most reliable and representative portrayal of contained PVC superphylum members, selected bins were rigorously filtered and processed using a stringent custom pipeline (for details please see Materials and Methods). This resulted in one almost complete and seven partial Planctomycetal and two partial Verrucomicrobial bins (Table 1). However, the final processed bins did not include 16S rRNA sequences, since those were predominantly encoded on small genome fragments, which did not include any additional markers. Due to the conserved nature of 16S rRNA genes, such fragments could therefore not be unambiguously binned based on sequence composition.
Instead, two complementary methods were employed to obtain 16S rRNA sequences from the original metagenomic datasets: Direct extraction of 16S rRNA gene sequences using the hidden Markov model approach of RNAmmer, and the mapping based reconstruction approach implemented by EMIRGE. However, since the resulting genes do not represent clonal sequences they should be regarded as consensus sequences, analog to so-called Operational Taxonomic Units (OTUs). After checking for potential chimeras using DECIPHER (Wright et al., 2012), taxonomic classification using the LCA-approach of SINA, and screening for PVC-associated genes, at least two distinct sequences, representing separate organisms, were identified for each, Verrucomicrobia and Planctomycetes. Neither of those sequences could be assigned to a currently cultivated species (Figures 3A,C) and were therefore considered novel. An overview of the total extracted and reconstructed 16S rRNA sequences, as well as their taxonomic annotation and relative abundances, is given in Supplementary Table 2.
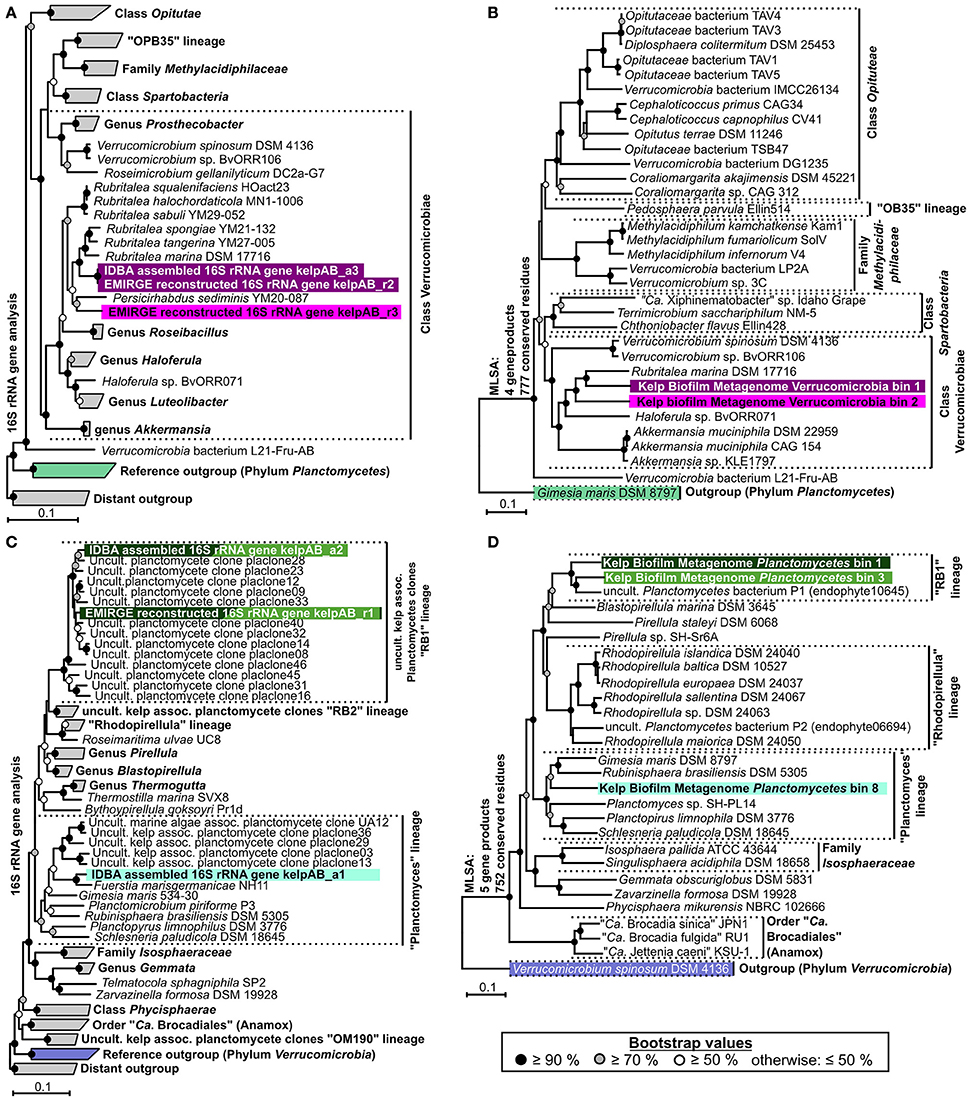
Figure 3. Phylogenetic analyses. (A,C) Neighbor joining trees showing the phylogenetic placement of ribosomal genes of Verrucomicrobia and Planctomycetes, respectively. Confidence values are based on 1,000 bootstrap permutations and indicated by appropriate symbols at each node. Ribosomal sequences and metagenomic bins obtained in this study are highlighted by different background colors representing putative different bacterial species & strains. Trees are based on aligned 16S rRNA genes >1,100 bp. For better presentability, outgroups, and several reference clades are shown in collapsed form. (B,D) Multi Locus Sequence Analyses (MLSA) based on unique core-genome gene products of Verrucomicrobial and Planctomycetal bins, respectively. The number of unique universally shared orthologs as well as the respective number of conserved amino acid positions considered for each analysis is indicated at the left of each tree. The exact unique core genome used for MLSA of different combinations of comparison genomes can be deduced from the results of the ortholog detection given in Supplementary Tables 3, 4. Due to the fragmented and incomplete nature of most metagenomic bins, relatively few universal orthologs could be considered in order to allow the simultaneous depiction of multiple representative bins. Higher phylogenetic resolutions can be found in the individual MLSA based phylogenies determined for each bin given in Supplementary Figure 3, which support the topologies depicted here. The MLSA results are in accordance with the observed 16S rRNA based phylogenies. Even though, due to strict binning parameters, none of the 16S rRNA genes depicted in (A,C) were included in any of the processed bins, each bin can be associated to a 16S rRNA gene based on phylogenetic placement as well as differential coverage information (Supplementary Table 5), as indicated by the respective background coloring (pink and purple = Verrucomicrobia, shades of green = Planctomycetes). Reference outgroups are marked in light violet (Verrucomicrobia) and light green (Planctomycetes)
In order to enable a similar phylogenetic placement of the processed metagenomic bins, a Multi Locus Sequence Analysis (MLSA) approach was employed (Figures 3B,D, Supplementary Figure 3). The unique core genome used for MLSA of different combinations of genomes can be deduced from the results of the ortholog detection given in Supplementary Tables 3, 4. Since the MLSA- and 16S rRNA based phylogenies were fully coherent with each other, the resulting phylogenetic information was used in combination with the respective abundance profiles to link 16S rRNA sequences with the most likely corresponding bins (Supplementary Table 5). The two obtained partial Verrucomicrobia bins, cluster within the Verrucomicrobiales, with bin 1 being more or less directly associated with the family Rubritaleaceae (closest sequenced relative: Rubritalea marina DSM 17716 with an MLSA identity of 79%), and bin 2 being positioned between the Rubritaleaceae and the Verrucomicrobiaceae (closest sequenced relatives: Rubritalea marina DSM 17716 and Haloferula sp. BvORR071 with MLSA identities of 76 and 69%, respectively; Figure 3B). A highly similar topology is shown for the respective 16S phylogenies (Figure 3A), with the identical sequences KelpAB_r2 and KelpAB_a3 corresponding to bin 1 (closest relative: Rubritalea marina DSM 17716 with 95% identity) and KelpAB_r3 corresponding to bin 2 (closest relative Persicirhabdus sedimis YM20-087 with 93% identity, situated between Rubritaleaceae and Verrucomicrobiaceae).
The Planctomycetal bins 1–7 show on MLSA level the closest similarity to an uncultured Planctomycete genome P1 (MLSA identity values ranging from 73 to 79%), which was reconstructed from a red algae endophyte metagenome and is not to be confused with the Rhodopirellula sp. P1 (Bengtsson and Øvreås, 2010). Despite their relation, the observed phylogenetic distances imply that the here presented bins and the published uncultured endophyte Planctomycete genome P1 represent different taxa, at least on species and likely on genus level. This is supported by Average Nucleotide Identity (ANI) values of <90%, as determined using the Pyani package (https://github.com/widdowquinn/pyani), a value well below the commonly used species thresholds. Interestingly, the uncultured endophyte genome P1 contains a 195 bp long 16S rRNA gene fragment. While this fragment is unfortunately too short to be included in a comprehensive 16S rRNA based phylogenetic tree, BLAST comparisons revealed ~95% sequence identity to the kelp biofilm sequences KelpAB_r1 and KelpAB_a2, which are directly associated with the uncultured kelp associated “RB1” lineage. Therefore, and because of the similar tree topologies of MLSA and 16S rRNA based approaches (Figures 3 C,D), the aforementioned bins and the endophyte genome P1 could be unambiguously assigned to the “RB1” lineage, which has not previously been associated with genomic sequences to this day. This interesting lineage was previously shown to be the most abundant Planctomycetal group within biofilm communities on the surface of the kelp Laminaria hyperborea. Its members are highly diverse as indicated by the large number of closely related but distinct 16S rRNA gene sequences found in the respective clone libraries (Figure 3C). Assuming a similar presence of multiple closely related strains of the “RB1” lineage within the biofilm community of M. pyrifera would explain the high number and fragmented nature of the respective obtained Planctomycetal bins. Furthermore, this would explain the divergence between the “RB1” associated 16S rRNA genes obtained by the mapping based reconstruction approach (KelpAB_r1) and by direct assembly (KelpAB_a2). Such a divergence was not observed for Verrucomicrobia, where the 16S rRNA gene sequence obtained by direct assembly (KelpAB_a3) was identical to its respective counterpart obtained by the mapping based approach (KelpAB_r2). As a consequence, all attempts to reconstruct further genomes of the “RB1” lineage or to obtain respective isolates should take potential strain heterogeneity into account and justifying the strict bin processing approach employed in this study (please see Materials and Methods).
To this day, no genomic sequence fragments or isolate cultures have been directly associated with the “RB1” lineage. The here presented Planctomycetal bins 1–7 represent multiple closely related strains of this lineage in various degrees of fragmentation and completeness (Table 1), providing an interesting glimpse into the “RB1” pan-genome. Of these bins, Planctomycetal bin 1 possessed the highest quality, with a presumed completeness of up to 84.11% or even >99% based on marker gene and tRNA counts, respectively. The degree of putative contamination, determined using standard checkM quality analyses workflows with Planctomycetes specific marker sets was ~2%. This is far below the average value for other published Planctomycetal bins, since equivalent analyses of Planctomycetes genomes frequently yield “contamination” values of more than 3% and up to 7% (Supplementary Table 6). That is why this bin can be considered “pure” to the greatest possible extent. A linear visualization of Planctomycetal bin 1 shows that the contained scaffolds possess orthologs to reference isolate Planctomycetes throughout the binned genome (Supplementary Figure 4), with the most orthologs found in the uncultured endophyte reference genome P1, confirming a low, if any, degree of contamination. Nonetheless, distinct genomic islands are also recognizable as short stretches with relatively few orthologs in reference genomes, illustrating the large potential for new genomic features of this lineage.
The presence of a “RB1” associated OTU was also confirmed by a similar, but different, sequence obtained by the mapping based 16S rRNA reconstruction approach (kelpAB_r1), showing 97% identity to KelpAB_a2 and 98% identity to RB1 associated clone “placlone40.” Members of the “RB1”-lineage have been previously shown to be abundant and highly diverse within plasmid clone libraries of kelp-associated communities (Bengtsson and Øvreås, 2010). This diversity may be reflected in the observed distinct clustering of the two “RB1”-associated 16S rRNA gene sequences (kelpAB_a2 & kelpAB_r1) obtained in this study using separate methods, indicating that they may actually be consensus sequences representing multiple diverse but closely related strains. Interestingly sequence kelpAB_a1 clusters more closely to Fuerstia marisgermanicae NH11 (96% identity), a recently discovered novel Planctomycetal species, than to related plasmid clones obtained from other kelp-associated communities (Kohn et al., 2016).
In addition to the “RB1” associated bins, the low abundant and highly fragmented bin 8 as well as the corresponding 16S rRNA sequence KelpAB_a1, could be associated with the “planctomyces lineage.” This lineage contains a deep branching sub-group which clusters relatively close to, but nonetheless distinctly apart from several planctomycetes strains such as Gimesia maris. Interestingly, this deep-branching sub-group was also observed by Bengtsson and Øvreås (2010) to co-occur with the “RB1” lineage within kelp biofilm communities of L. hyperborea in relatively low abundance. However, the insights into the genomic potential of this sub-group are limited by the small size and low completeness of Planctomycete bin 8 (Table 1). Fortunately, the recently isolated strain Fuersteria marisgermanica (Kohn et al., 2016) proved to be not only associated with the aforementioned sub-group, but also most closely related to the here presented bin 8. Genome analyses of this species may provide more detailed insights into the potential role of this sub-group within marine biofilm communities.
All plancomycetal bins, with the exception of bin 8, contained genes encoding for flagellum synthesis. No such genes were identified in the Verrucomicrobia bins, which is in concordance with the non-motile lifestyle of their most closely related cultured representatives Rubritalea marina and Persicirhabdus sediminis (Scheuermayer et al., 2006; Yoon et al., 2008). Additional specifically noteworthy features identified in the PVC bins are listed in Supplementary Table 7.
According to SEED annotations obtained via MEGAN5 analyses, a relatively high proportion of sulfur metabolism related genes was found in the Planctomycetal and Verrucomicrobial bins, which comprise a ~2–5 times higher fraction of the total SEED annotated genes compared to the total biofilm community (Figure 4; Supplementary Table 8). This observation is true for all of the processed PVC bins, including the almost-complete Planctomycetes bin 1, indicating that this is a general feature of the respective genomes and not biased by partial genome representation. The PVC-superphylum members might therefore represent the primary metabolizers of organic sulfur compounds within the kelp biofilm community. Such compounds are commonly produced in a large variety by marine macro algae (Sunda et al., 2002; Lage and Bondoso, 2014; Michel et al., 2006), and may therefore constitute key factors for the successful colonialization of, and adaptation to, algal surfaces. A high propensity for the utilization of sulfated glycopolymers has also been reported for several Planctomycetes and Verrucomicrobia cultures (Lage and Bondoso, 2011, 2012; Wegner et al., 2013; Spring et al., 2016). Furthermore, sulfated polysaccharides were the predominantly hydrolyzed glycopolymers during an unusual population spike of Verrucomicrobia observed at a Svalbard Fjord in 2008 (Cardman et al., 2014).
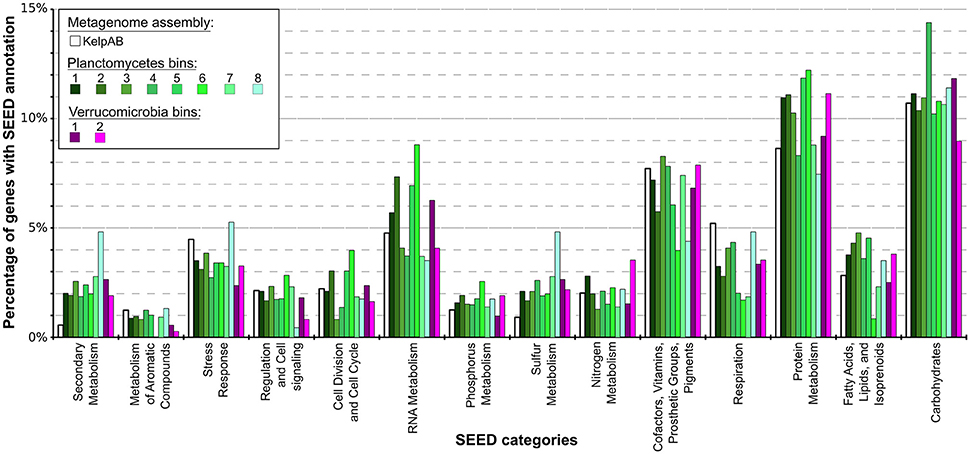
Figure 4. Distribution of SEED category annotations for the metagenomic assemblies as well as each bin. Relative fractions of selected SEED categories compared to the total number of SEED annotations in each assembly or bin are shown. ORF finding and gene prediction was performed using Prodigal (Hyatt et al., 2010). SEED annotations were obtained by aligning protein sequences against the NCBI nr database using Diamond (Buchfink et al., 2014) and analyzing the results using MEGAN5 (Huson and Weber, 2013). The Planctomycetal and Verrucomicrobial bins (shown in shades of green and violet, respectively) display two to four times higher proportions of gene products associated with secondary metabolism than average for the kelp biofilm community represented by the KelpAB metagenome (depicted in white), and a slightly higher fraction of genes associated with protein metabolism. Furthermore, genes associated with RNA metabolism are slightly more represented in Planctomycetal bins.
Other examples of nutrient scavenging which have been observed in different macroalgal associates include phosphorous and nitrogen utilization (Egan et al., 2013). Although a slight increase of genes encoding for these categories can be observed for several bins, including the almost complete Planctomycetes bin 1, this increase far less distinct compared to the aforementioned sulfur metabolism and not coherent for all PVC bins. The same holds true for genes involved in fatty acid, lipid and isoprenoid metabolism, which may be relevant for host-microbe interactions due to the production of polyunsaturated fatty acids (PUFAs) and terpenes frequently observed in marine macroalgae (Potin et al., 2002). Therefore, while it may be hypothesized that the PVC-superphylum members are also involved in the metabolisation of said nutrients and compounds in the kelp biofilm community, it is not conclusive whether they play a dominant role in this regard.
Genes encoding for respiration were found to be underrepresented in all of the PVC bins, indicating a relatively low metabolic activity. This might be contrasted by the comparatively high fraction of RNA metabolism related genes in several bins, which could be indicative for a complex regulatory network. These observations however do support the assumption that these genomes represent slow growing organisms, as frequently observed for PVC superphylum members.
Interestingly, an outstanding large relative fraction of the respective encoding genes in all of the processed bins are associated with secondary metabolism (Figure 4; Supplementary Table 8). This fraction is ~4–9 times higher in the Planctomycetal and Verrucomicrobial bins compared to the total kelp biofilm community and found evidence for a relatively high potential for secondary metabolite production in those species. Putative gene clusters were identified by AntiSMASH v.3 in all of the processed bins, including several potential Polyketide Synthase (PKS) clusters (Figure 5). PKSs are enzyme complexes or multi-domain enzymes which perform a stepwise biosynthesis of complex organic compounds, so-called polyketides, with varying pharmaceutical properties such as antibiotic activity (Jenke-Kodama et al., 2005). Nutrient rich habitats like kelp forests attract other –more rapidly- growing- heterotrophic bacteria that should be able to outcompete slow-growers. As Verrucomicrobia and Planctomycetes are both known to be relatively slowly growing organisms, secondary metabolites may likely serve as inhibitory agents against more rapidly growing competitors within the biofilm community. This would help explain the persistence and even frequent dominance of PVC superphylum members in kelp-associated communities, despite the constant danger of being out-competed by rapidly growing Proteobacteria. We therefore hypothesize that Verrucomicrobia and Planctomycetes are attracted by kelp through secretion of specific carbohydrates that trigger them to attach to the algal surface and to form biofilms. The kelp might then employ them as biofouling control, using their antimicrobial secondary metabolites to defeat other bacteria or eukaryotes. This would be in accordance with the low abundancy but ubiquitous distribution of these species on marine algae, and shows that these phyla are very interesting for natural products production/pharmaceuticals. Only recently, Planctomycetes were demonstrated to produce small molecules with bioactivity and their antibiotic effects were recently demonstrated (Jeske et al., 2013).
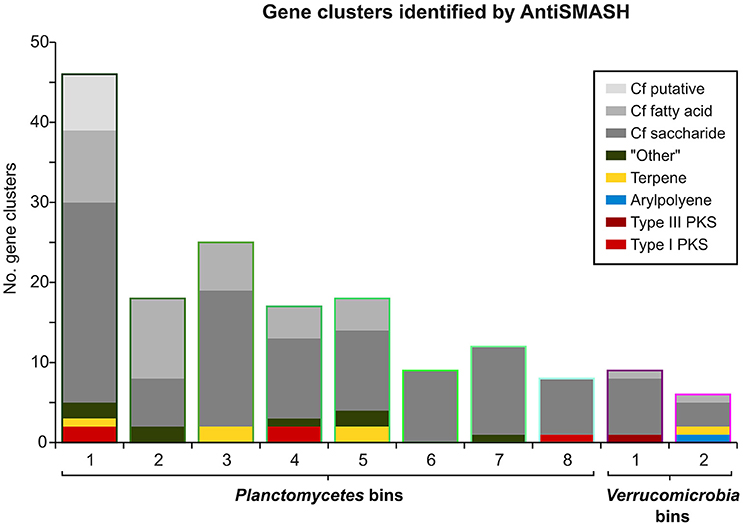
Figure 5. Gene Clusters identified by AntiSMASH. Bar heights indicate the total number of AntiSMASH hits identified in each bin. The respective relative fractions of different functional classes are indicated by distinct coloring.
Although several studies have reported the discovery of new bioactive compounds from marine organisms and more than 15,000 structurally diverse bioactive compounds were isolated during the past 30 years (Hu et al., 2015), only two new classes of antibiotics have been brought to market during this period of time (Coates et al., 2011). In the post-genomic era one has come to realize that naturally occurring reservoirs of genetic diversity contain vast, untapped potential for production of biologically active chemical compounds that needs to be made available for society with the emerge of new technologies. The PVC superphylum is known for its biotechnological and medical relevance (Wagner and Horn, 2006) and a promising resource for natural products, however due to the relatively low representation of these phyla in public sequencing databases there are still may gaps to fill by e.g., specific isolation attempts or combinations of single cell (Rinke et al., 2013; Kaster et al., 2014) and metagenomics. Our study provides a first glimpse into this microbial dark matter by using an innovative binning approach to reconstruct genomes of low abundant species, shedding light on Planctomycetes and Verrucomicrobia from biofilms of kelp algae.
Author Contributions
AK and JV designed the experiments, performed the data analyses and wrote the paper. MF and PR performed the experiments. CJ collected the samples by scuba diving.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Professor Alfred Spormann (Stanford University) and the staff of Hopkins Marine Station, especially Judy Thompson, for their generous help with the collection and shipment of the samples.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00472/full#supplementary-material
References
Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K. L., Tyson, G. W., and Nielsen, P. H. (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538. doi: 10.1038/nbt.2579
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bartram, A. K., Lynch, M. D., Stearns, J. C., Moreno-Hagelsieb, G., and Neufeld, J. D. (2011). Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl. Environ. Microbiol. 77, 3846–3852. doi: 10.1128/AEM.02772-10
Bengtsson, M. M., and Øvreås, L. (2010). Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiol. 10:261. doi: 10.1186/1471-2180-10-261
Bengtsson, M. M., Sjøtun, K., and Øvreås, L. (2010). Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea. Aquat. Microb. Ecol. 60, 71–83. doi: 10.3354/ame01409
Bengtsson, M. M., Sjøtun, K., Lanzén, A., and Øvreås, L. (2012). Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. ISME J. 6, 2188–2198. doi: 10.1038/ismej.2012.67
Bengtsson, M. M., Sjøtun, K., Storesund, J. E., and Øvreås, J. (2011). Utilization of kelp-derived carbon sources by kelp surface-associated bacteria. Aquat. Microb. Ecol. 62, 191–199. doi: 10.3354/ame01477
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cardman, Z., Arnosti, C., Durbin, A., Ziervogel, K., Cox, C., Steen, A., et al. (2014). Verrucomicrobia are candidates for polysaccharide-degrading bacterioplankton in an arctic fjord of Svalbard. Appl. Environ. Microbiol. 80, 3749–3756. doi: 10.1128/AEM.00899-14
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Christie-Oleza, J. A., Pi-a-Villalonga, J. M., Bosch, R., Nogales, B., and Armengaud, J. (2012). Comparative proteogenomics of twelve Roseobacter exoproteomes reveals different adaptive strategies among these marine bacteria. Mol. Cell. Proteomics 11:M111.013110. doi: 10.1074/mcp.M111.013110
Coates, A. R., Halls, G., and Hu, Y. (2011). Novel classes of antibiotics or more of the same? Br. J. Pharmacol. 163, 184–194. doi: 10.1111/j.1476-5381.2011.01250.x
Dayton, P. K. (1985). Ecology of kelp communities. Annu. Rev. Ecol. Syst. 16, 215–245. doi: 10.1146/annurev.es.16.110185.001243
Dayton, P. K., and Tegner, M. J. (1984). Catastrophic storms, el nino, and patch stability in a southern california kelp community. Science 224, 283–285.
Delille, D., and Perret, E. (1991). The influence of giant kelp Macrocystis pyrifera on the growth of subantarctic marine bacteria. J. Exp. Mar. Biol. Ecol. 153, 227–239. doi: 10.1016/0022-0981(91)90227-N
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Egan, S., Harder, T., Burke, C., Steinberg, P., Kjelleberg, S., and Thomas, T. (2013). The seaweed holobiont: understanding seaweed-bacteria interactions. FEMS Microbiol. Rev. 37, 462–476. doi: 10.1111/1574-6976.12011
Estes, J. A., Danner, E. M., Doak, D. F., Konar, B., Springer, A. M., Steinberg, P. D., et al. (2004). Complex trophic interactions in kelp forest ecosystems. Bull. Mar. Sci. 74, 621–638.
Ferrera, I., Massana, R., Balague, V., Pedros-Alio, C., Sanchez, O., and Mas, J. (2010). Evaluation of DNA extraction methods from complex phototrophic biofilms. Biofouling 26, 349–357. doi: 10.1080/08927011003605870
Foster, M. S., Reed, D. C., Carr, M. H., Dayton, P. K., Malone, D. P., Pearse, J. S., et al. (2012). Kelp forests in California. Smithson. Contrib. Mar. Sci. 39, 115–132.
Foster, M. S., and Schiel, D. R. (1985). Ecology of Giant Kelp Forests in California: A Community Profile. Moss Landing, CA: San Jose State University, Moss Landing Marine Labs.
Fuerst, J. A. (ed.). (2013). Planctomycetes: Cell Structure, Origins and Biology. Totowa, NJ: Humana.
Goecke, F., Labes, A., Wiese, J., and Imhoff, J. F. (2010). Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 409, 267–299. doi: 10.3354/meps08607
Graham, M. H. (2004). Effects of local deforestation on the diversity and structure of Southern California giant kelp forest food webs. Ecosystems 7, 341–357. doi: 10.1007/s10021-003-0245-6
Graham, M. H., Kinlan, B. P., Druehl, L. D., Garske, L. E., and Banks, S. (2007). Deep-water kelp refugia as potential hotspots of tropical marine diversity and productivity. Proc. Natl. Acad. Sci. U.S.A. 104, 16576–16580. doi: 10.1073/pnas.0704778104
Hahnke, S., Tindall, B. J., Schumann, P., Simon, M., and Brinkhoff, T. (2013). Pelagimonas varians gen. nov., sp. nov., isolated from the southern North Sea. Int. J. Syst. Evol. Microbiol. 63, 835–843. doi: 10.1099/ijs.0.040675-0
Hollants, J., Leliaert, F., De Clerck, O., and Willems, A. (2013). What we can learn from sushi: a review on seaweed-bacterial associations. FEMS Microbiol. Ecol. 83, 1–16. doi: 10.1111/j.1574-6941.2012.01446.x
Hu, Y., Chen, J., Hu, G., Yu, J., Zhu, X., Lin, Y., et al. (2015). Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 13, 202–221. doi: 10.3390/md13010202
Huson, D. H., and Weber, N. (2013). Microbial community analysis using MEGAN. Methods Enzymol. 531, 465–485. doi: 10.1016/B978-0-12-407863-5.00021-6
Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jenke-Kodama, H., Sandmann, A., Müller, R., and Dittmann, E. (2005). Evolutionary Implications of bacterial polyketide synthases. Mol. Biol. Evol. 22, 2027–2039. doi: 10.1093/molbev/msi193
Jeske, O., Jogler, M., Petersen, J., Sikorski, J., and Jogler, C. (2013). From genome mining to phenotypic microarrays: planctomycetes as source for novel bioactive molecules. Antonie van Leeuwenhoek 104, 551–567. doi: 10.1007/s10482-013-0007-1
Kaster, A. K., Mayer-Blackwell, K., Pasarelli, B., and Spormann, A. M. (2014). Single cell genomic study of Dehalococcoidetes species from deep-sea sediments of the Peruvian Margin. ISME J. 8, 1831–1842. doi: 10.1038/ismej.2014.24
Kohn, T., Heuer, A., Jogler, M., Vollmers, J., Boedeker, C., Bunk, B., et al. (2016). Fuerstia marisgermanicae gen. nov., sp. nov., an Unusual Member of the Phylum Planctomycetes from the German Wadden Sea. Front. Microbiol. 7:2079. doi: 10.3389/fmicb.2016.02079
Lage, O. M., and Bondoso, J. (2011). Planctomycetes diversity associated with macroalgae. FEMS Microbiol. Ecol. 78, 366–375. doi: 10.1111/j.1574-6941.2011.01168.x
Lage, O. M., and Bondoso, J. (2012). Bringing Planctomycetes into pure culture. Front. Microbiol. 3:405. doi: 10.3389/fmicb.2012.00405
Lage, O. M., and Bondoso, J. (2014). Planctomycetes and macroalgae, a striking association. Front. Microbiol. 5:267. doi: 10.3389/fmicb.2014.00267
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lasken, R. S. (2009). Genomic DNA amplification by the multiple displacement amplification (MDA) method. Biochem. Soc. Trans. 37, 450–453. doi: 10.1042/BST0370450
Lechner, M., Findeiss, S., Steiner, L., Marz, M., Stadler, P. F., and Prohaska, S. J. (2011). Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12, 124–124. doi: 10.1186/1471-2105-12-124
Linley, E. A. S., and Field, J. G. (1982). The nature and ecological significance of bacterial aggregation in a nearshore upwelling ecosystem. Estuarine Coastal Shelf Sci. 14, 1–11. doi: 10.1016/S0302-3524(82)80063-7
Longford, S. R., Tujula, N. A., Crocetti, G. R., Holmes, A. J., Holmstrom, C., Kjelleberg, S., et al. (2007). Comparisons of diversity of bacterial communities associated with three sessile marine eukaryotes. Aquat. Microb. Ecol. 48, 217–229. doi: 10.3354/ame048217
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Michel, G., Nyval-Collen, P., Barbeyron, T., Czjzek, M., and Helbert, W. (2006). Bioconversion of red seaweed galactans: a focus on bacterial agarases and carrageenases. Appl. Microbiol. Biotechnol. 71, 23–33. doi: 10.1007/s00253-006-0377-7
Michelou, V. K., Caporaso, J. G., Knight, R., and Palumbi, S. R. (2013). The ecology of microbial communities associated with Macrocystis pyrifera. PLoS ONE 8e67480. doi: 10.1371/annotation/48e29578-a073-42e7-bca4-2f96a5998374
Miller, C. S., Baker, B. J., Thomas, B. C., Singer, S. W., Banfield, J. F., Pace, N. R., et al. (2011). EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 12:R44. doi: 10.1186/gb-2011-12-5-r44
Muyzer, G., de Waal, E. C., and Uitterlinden, A. G. (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59, 695–700.
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12, 385–385. doi: 10.1186/1471-2105-12-385
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Potin, P., Bouarab, K., Salaün, J.-P., Pohnert, G., and Kloareg, B. (2002). Biotic interactions of marine algae. Curr. Opin. Plant Biol. 5, 308–317. doi: 10.1016/S1369-5266(02)00273-X
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rao, D., Webb, J. S., Holmstrom, C., Case, R., Low, A., Steinberg, P., et al. (2007). Low densities of epiphytic bacteria from the marine alga Ulva australis inhibit settlement of fouling organisms. Appl. Environ. Microbiol. 73, 7844–7852. doi: 10.1128/AEM.01543-07
Reis, A. M. M., Araújo S. D. Jr., Moura, R. L., Francini-Filho, R. B., Pappas G. Jr., Coelho, A. M. A., et al. (2009). Bacterial diversity associated with the Brazilian endemic reef coral Mussismilia braziliensis. J. Appl. Microbiol. 106, 1378–1387. doi: 10.1111/j.1365-2672.2008.04106.x
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J.-F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Sangwan, N., Xia, F., Gilbert, J. A., Ramette, A., Tiedje, J. M., Tyson, G. W., et al. (2016). Recovering complete and draft population genomes from metagenome datasets. Microbiome 4:8. doi: 10.1186/s40168-016-0154-5
Scheuermayer, M., Gulder, T. A. M., Bringmann, G., and Hentschel, U. (2006). Rubritalea marina gen. nov., sp. nov., a marine representative of the phylum “Verrucomicrobia,” isolated from a sponge (Porifera). Int. J. Syst. Evol. Microbiol. 56, 2119–2124. doi: 10.1099/ijs.0.64360-0
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Spring, S., Bunk, B., Spröer, C., Schumann, P., Rohde, M., Tindall, B. J., et al. (2016). Characterization of the first cultured representative of Verrucomicrobia subdivision 5 indicates the proposal of a novel phylum. ISME J. 10, 2801–2816. doi: 10.1038/ismej.2016.84
Steneck, R., Graham, M. H., Bourque, B. J., Corbett, D., Erlandson, J. M., Estes, J. A., et al. (2002). Kelp forest ecosystems: biodiversity, stability, resilience and future. Environ. Conserv. 29, 436–459 doi: 10.1017/s0376892902000322
Sunda, W., Kieber, D. J., Kiene, R. P., and Huntsman, S. (2002). An antioxidant function for DMSP and DMS in marine algae. Nature 418, 317–320. doi: 10.1038/nature00851
Taylor, M. W., Schupp, P. J., Dahllöf, I., Kjelleberg, S., and Steinberg, P. D. (2004). Host specificity in marine sponge-associated bacteria, and potential implications for marine microbial diversity. Environ. Microbiol. 6, 121–130. doi: 10.1046/j.1462-2920.2003.00545.x
Wagner, M., and Horn, M. (2006). The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr. Opin. Biotechnol. 17, 241–249. doi: 10.1016/j.copbio.2006.05.005
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Wegner, C. E., Richter-Heitmann, T., Klindworth, A., Klockow, C., Richter, M., Achstetter, T., et al. (2013). Expression of sulfatases in Rhodopirellula baltica and the diversity of sulfatases in the genus Rhodopirellula. Mar. Genomics 9, 51–61. doi: 10.1016/j.margen.2012.12.001
Wright, E. S., Yilmaz, L. S., and Noguera, D. R. (2012). DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl. Environ. Microbiol. 78, 717–725. doi: 10.1128/AEM.06516-11
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Yoon, J., Matsuo, Y., Adachi, K., Nozawa, M., Matsuda, S., Kasai, H., et al. (2008). Description of Persicirhabdus sediminis gen. nov., sp. nov., Roseibacillus ishigakijimensis gen. nov., sp. nov., Roseibacillus ponti sp. nov., Roseibacillus persicicus sp. nov., Luteolibacter pohnpeiensis gen. nov., sp. nov. and Luteolibacter algae sp. no. Int. J. Syst. Evol. Microbiol. 58, 998–1007. doi: 10.1099/ijs.0.65520-0
Keywords: algal biofilms, syntrophic interactions, biofouling control, natural product producers, genome binning
Citation: Vollmers J, Frentrup M, Rast P, Jogler C and Kaster A-K (2017) Untangling Genomes of Novel Planctomycetal and Verrucomicrobial Species from Monterey Bay Kelp Forest Metagenomes by Refined Binning. Front. Microbiol. 8:472. doi: 10.3389/fmicb.2017.00472
Received: 08 October 2016; Accepted: 07 March 2017;
Published: 29 March 2017.
Edited by:
Damien Paul Devos, University Pablo Olavide, SpainReviewed by:
Peter Dunfield, University of Calgary, CanadaJorge L. M. Rodrigues, University of California, Davis, USA
Copyright © 2017 Vollmers, Frentrup, Rast, Jogler and Kaster. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne-Kristin Kaster, YS5rYXN0ZXJAZHNtei5kZQ==