 Divakar Sharma
Divakar Sharma Deepa Bisht
Deepa Bisht- Department of Biochemistry, National JALMA Institute for Leprosy and Other Mycobacterial Diseases, Agra, India
Drug resistance in tuberculosis predominantly, mono-resistance, multi drug resistance, extensively drug resistance and totally drug resistance have emerged as a major problem in the chemotherapy of tuberculosis. Failures of first and second line anti-tuberculosis drugs treatment leads to emergence of resistant Mycobacterium tuberculosis. Few genes are reported as the principal targets of the resistance and apart from the primary targets many explanations have been proposed for drug resistance but still some resistance mechanisms are unknown. As proteins involved in most of the biological processes, these are potentially explored the unknown mechanism of drug resistance and attractive targets for diagnostics/future therapeutics against drug resistance. In last decade a panel of studies on expression proteomics of drug resistant M. tuberculosis isolates reported the differential expression of uncharacterized proteins and suggested these might be involved in resistance. Here we emphasize that detailed bioinformatics analysis (like molecular docking, pupylation, and proteins-proteins interaction) of these uncharacterized and hypothetical proteins might predict their interactive partners (other proteins) which are involved in various pathways of M. tuberculosis system biology and might give a clue for novel mechanism of drug resistance or future drug targets. In future these uncharacterized targets might be open the new resistance mechanism and used as potential drug targets against drug resistant tuberculosis.
Introduction
Current Scenario
Tuberculosis (TB) still remains one of the deadliest infectious diseases worldwide which is caused by Mycobacterium tuberculosis. WHO reported 10.4 million people became ill and that 1.8 million died from TB (WHO, 2016). For controlling this situation the available current tools are vaccine diagnostics and drugs. Over the past 50 years, the Mycobacterium bovis bacille Calmette–Guérin (BCG) vaccine against TB has maintained its position as the world’s most widely used vaccine, despite showing highly variable efficacy (0–80%) in different trials (Andersen and Doherty, 2005).
Sputum smear microscopy is the most common TB diagnostic method worldwide. However, culture remains the gold standard and the use of rapid molecular testing like line probe assay (LPA) is increasing for detection of drug resistant M. tuberculosis strains. Recently in India, Revised National TB Control Program (RNTCP) has approved a study for the Validation of second line LPA for detecting resistance to fluoroquinolones, aminoglycosides (kanamycin, amikacin), and cyclic peptides (capreomycin). First and second line drugs are the effective and necessary component of short course chemotherapy. DOTS and DOTS plus program have reduced the incidence of TB caused by susceptible strain but emergence of multidrug-resistant tuberculosis (MDR-TB), extensively drug resistant tuberculosis (XDR-TB), and totally drug resistant tuberculosis (TDR-TB) have worsened the situation and became a major threat to public health. Current tools (vaccines, diagnostics, and therapeutics) are unable to offer the complete protection against these deadly drug resistant situations.
Mystery behind the Drug Resistance
Usually interrupted anti-TB drugs treatment (first and second line anti-mycobacterial drugs) leads to emergence of drug resistant M. tuberculosis strains. Probably these resistant M. tuberculosis strains can resist antibiotic actions by the series of mechanisms such as: mutations in target genes (Beauclerk and Cundliffe, 1987), enzymatic inactivation of antibiotic molecules (Welch et al., 2005), over expression of novel efflux pumps and porin alterations in the cell wall (Magnet et al., 2001; Nikaido, 2003), trapping of drugs and the over expression of proteins involved in neutralizing the effect of drugs (Magnet et al., 2003; Kumar et al., 2013; Lata et al., 2015a; Sharma et al., 2015a, 2016b). Genes involved in these drug resistance mechanisms are tabulated in Table 1 (adopted from Zhang and Yew, 2009 and updated). Figure 1 is the schematic diagram which showed the potential mechanism (s) of action of first line (Rifampicin-inhibition of RNA synthesis, Isoniazid-inhibition of mycolic acid biosynthesis, Ethambutol-inhibition of arabinogalactan synthesis, Pyrazinamide-depletion of membrane energy, and Streptomycin-inhibition of protein synthesis) and second line of anti-TB drugs (Amikacin, Kanamycin, Capreomycin-inhibition of protein synthesis and Quinolones-inhibition of DNA gyrase) and the potential mechanism (s) of drug resistance, respectively. These mechanisms of action of drugs were also tabulated in Table 1. Usually 36–95% resistances in M. tuberculosis were contributed by mutations in the target genes, however, remaining 5–64% does not have these mutations and signifying the contribution of some other resistance mechanism (s). Research through expression proteomics (2D gel electrophoresis) and bioinformatic tools (like molecular docking, pupylation, and proteins-proteins interaction) explored the other novel mechanisms of drug resistance were accumulated in last decade and still underway. Patch dock and fire dock (molecular docking) predicted, drug binds to the conserved domain of the hypothetical proteins and suggested that overexpression of these proteins of undefined role might be neutralize/modulate the effects of drugs (Sharma et al., 2015a, 2016b). Pupylation is a post translational modification through which small disordered protein Pup is conjugated to lysine residues of proteins marking them for proteasomal degradation. GPS-PUP (pupylation) predicted, that neutralized/modulated adduct (drug-protein complex) might be degraded by proteasome machinery complex {turnover of the proteins} (Sharma et al., 2015a, 2016b). As modification with pup is reversible, pupylation is also likely to have a regulatory role. Pup-proteasome system controlled by pupylation contributes to the virulence/survival strategy of M. tuberculosis in the host and makes the bacteria more resistant to various stresses. STRING-10 (Proteins-proteins interaction) predicted the potential interactive partners and suggested the metabolic pathways involved in resistance (Sharma et al., 2016a,b). Rv0148 (hypothetical protein/putative short-chain type dehydrogenase/reductase) was overexpressed in aminoglycosides resistant M. tuberculosis and had conserved SDR domain. We found that aminoglycosides binds to SDR domains and might be neutralized the drug effect (Sharma et al., 2015a). Further we characterized it and found that overexpression of Rv0148 involved in shift in MIC of aminoglycosides in recombinant E. coli (Sharma et al., 2015b). These findings might be used in the development of newer therapeutic agents or molecular markers which can directly be targeted to a gene/protein responsible for resistance. In this work we discuss the probable involvement of uncharacterized/hypothetical proteins (differential expression in drug resistance studies were previously reported) in drug resistance via bioinformatics analysis (like molecular docking, pupylation, and proteins-proteins interaction) and suggested that these might be explore novel mechanism of drug resistance or used as potential future drug target/diagnostics against drug resistance.
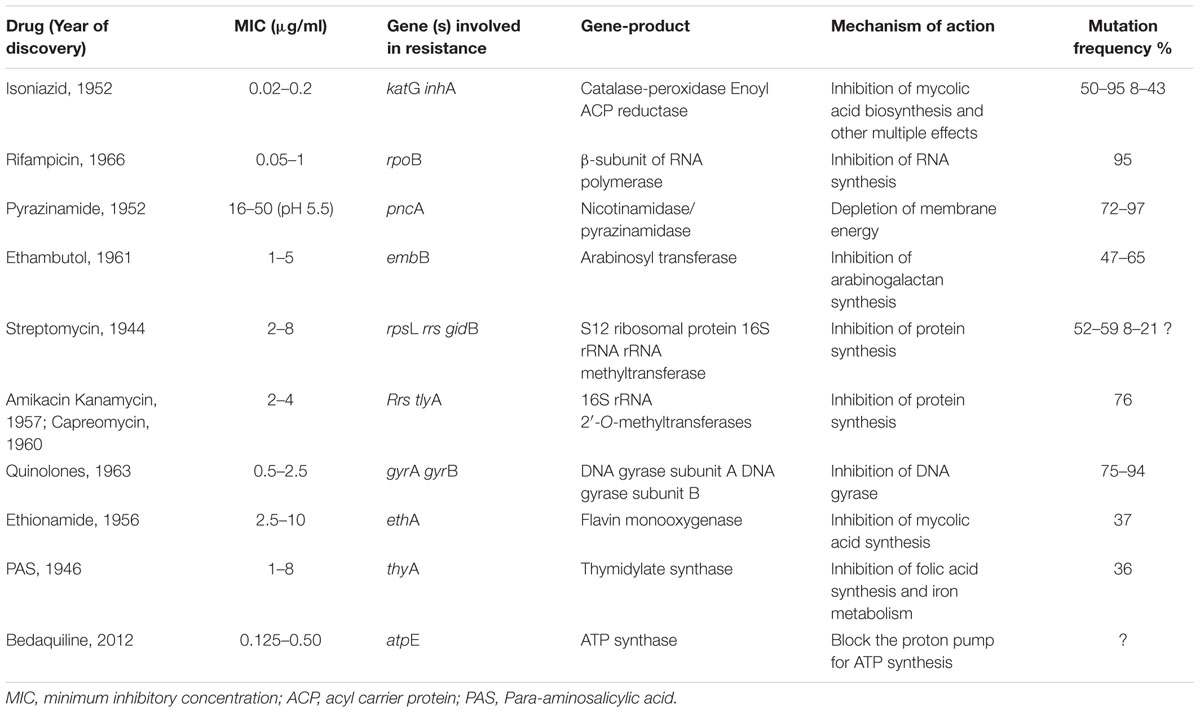
TABLE 1. Mechanisms of genes involved in drug resistance in Mycobacterium tuberculosis.
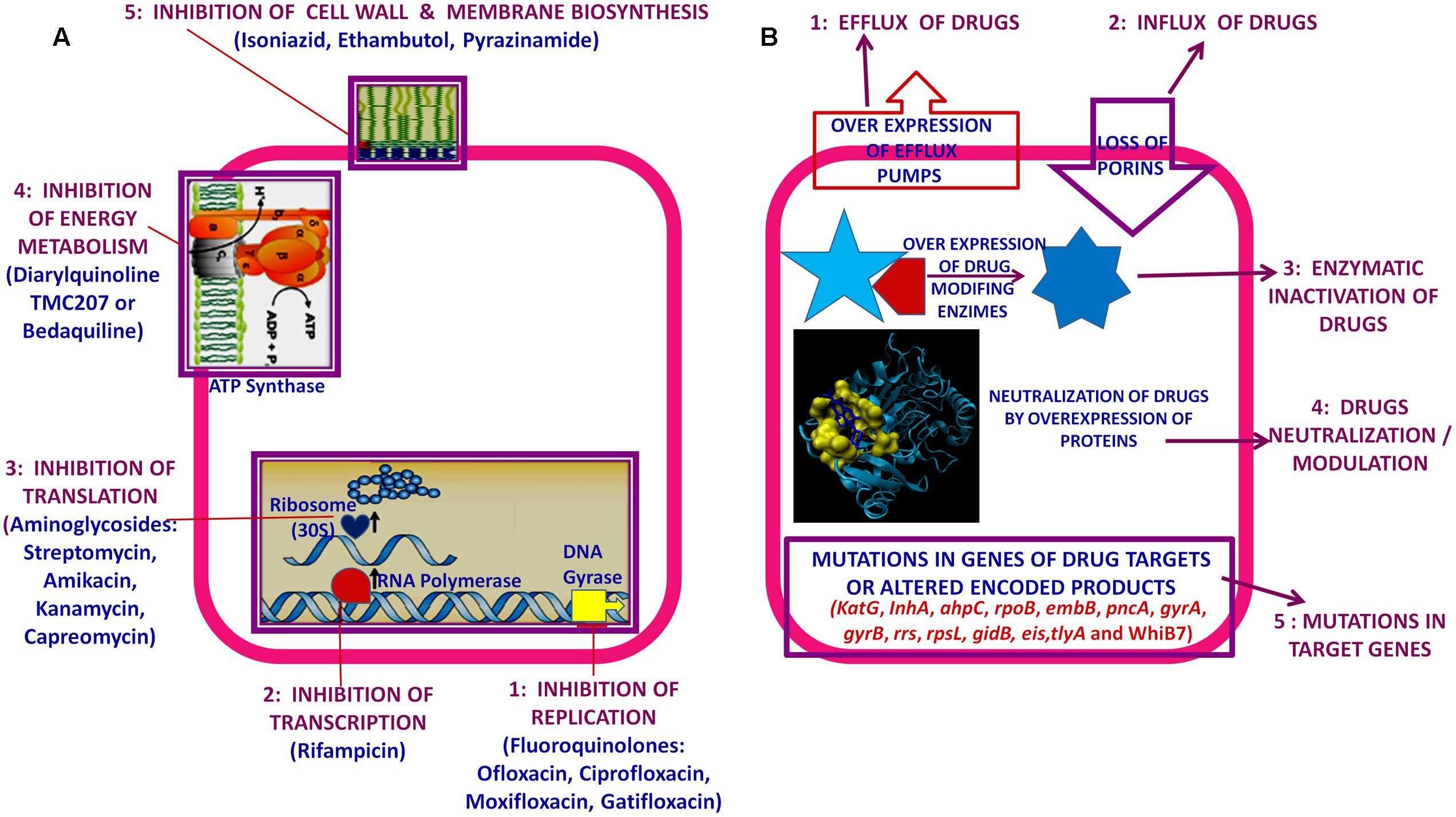
FIGURE 1. Schematic diagram showed: (A) Potential mechanism (s) of action of first and second line of anti-TB drugs. (B) Potential mechanism (s) of drug resistance in Mycobacterium tuberculosis.
Expression Proteomics and Bioinformatics Approaches: A Way for Exploring the Mystery of Drug Resistance
Advancement in proteomics has explored the mystery behind any complex phenotypes like drug resistance. As proteins manifest most of the biological processes, these are attractive targets for exploring new mechanisms of the drug resistance. Expression proteomics (2-DE coupled with MALDI-TOF-MS) and bioinformatic tools {patch dock and fire dock for molecular docking, GPS-PUP for pupylation, and STRING-10 for protein-protein interaction} have now emerged as major analytical tools for identification and characterization of expression proteome (proteins and its species) (Sharma et al., 2015a; Sharma and Bisht, 2016). In the last decade (2006–2016) few reports on expression proteome of drugs resistant M. tuberculosis existed (Jiang et al., 2007; Sharma et al., 2010, 2014, 2015b, 2016b; Kumar et al., 2013; Lata et al., 2015a,b; Singh et al., 2015; Sharma and Bisht, 2017) and suggested that differential expression of functionally known and unknown proteins and their protein-protein interaction might be involved in resistance and could be explore the novel mechanism of resistance.
Hypothetical Proteins and Proteins of Unknown Function: Potential Targets of Drug Resistance or Novel Resistance Mechanisms
Since last decade, few proteomics and bioinformatics studies of drug resistant M. tuberculosis have been accumulated and reported the differential expression of a panel of uncharacterized (proteins of unknown function) and hypothetical proteins. Through in silico/bioinformatic (Interproscan and molecular docking) analysis they showed that drugs binds to the conserved domains of hypothetical proteins/uncharacterized proteins and suggested that the over expression of a panel of uncharacterized and hypothetical proteins might neutralize/compensate the effect of drugs. Sharma et al. (2015b, 2016a) reported that inducible over expression of cloned known and unknown proteins (Rv0148 and Rv3841) in E. coli makes it two- to threefolds more resistant under drug pressure. Here we emphasize that detailed bioinformatics analysis (like protein-protein interaction) of these uncharacterized and hypothetical proteins might predict their interactive partners (other proteins) which are involved in various pathways of M. tuberculosis system biology (exploring/deciphering the M. tuberculosis network biology through in silico/holistic approaches) and might give a clue for novel mechanism of drug resistance or future target. Research in this direction could prevent the emergence of MDR-TB, XDR-TB, and TDR-TB situation and also these targets may be used to discover the new drug entities as potential drug candidates against drug resistant tuberculosis.
Conclusion and Future Perspective
Based on the evidence discussed above related to expression proteomics and bioinformatics studies of drug resistant M. tuberculosis have been reported the differential expression of uncharacterized and hypothetical proteins (Jiang et al., 2007; Sharma et al., 2010, 2014, 2015b, 2016b; Kumar et al., 2013; Lata et al., 2015a; Singh et al., 2015; Sharma and Bisht, 2017). Molecular docking analysis of these uncharacterized and hypothetical proteins suggested that their over expression might neutralize/compensate the effect of drugs (Sharma et al., 2010, 2015a, 2016b; Kumar et al., 2013; Lata et al., 2015a; Sharma and Bisht, 2017) which further proved by other reports and showed inducible over expression of cloned known and unknown proteins in E. coli which makes it few folds more resistant under drug pressure (Sharma et al., 2015b). Here we emphasized the detailed in silico analysis through patch dock, fire dock (molecular docking), GPS-PUP (pupylation), and STRING-10 (predicts the proteins-proteins interactions/interactome) of these uncharacterized and hypothetical proteins which might be involved in various pathways of M. tuberculosis system biology (Lata et al., 2015a; Sharma et al., 2015a, 2016b; Sharma and Bisht, 2017). Future research in this direction could be uncovering the novel mechanism of drug resistance or future target. Which ultimately prevent the emergence of deadly resistant strains of M. tuberculosis and could leads to discovery of the new drug entities against deadly drug resistant tuberculosis.
Author Contributions
DS design the concept and wrote the manuscript. DS and DB finalized the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Director, NJIL & OMD for the support. DS is ICMR-PDFs (ICMR, New Delhi).
References
Andersen, P., and Doherty, T. M. (2005). The success and failure of BCG—implications for a novel tuberculosis vaccine. Nat. Rev. Microbiol. 3, 656–662. doi: 10.1038/nrmicro1211
Beauclerk, A. A. D., and Cundliffe, E. (1987). Site of action of two ribosomal RNA methylases responsible for resistance to aminoglycoside. J. Mol. Biol. 193, 661–671. doi: 10.1016/0022-2836(87)90349-4
Jiang, X., Zhang, W., Gao, F., Huang, Y., Lv, C., and Wang, H. (2007). Comparison of the proteome of isoniazid-resistant and -susceptible strains of Mycobacterium tuberculosis. Microb. Drug Resist. 12, 231–238. doi: 10.1089/mdr.2006.12.231
Kumar, B., Sharma, D., Sharma, P., Katoch, V. M., Venkatesan, K., and Bisht, D. (2013). Proteomic analysis of Mycobacterium tuberculosis isolates resistant to kanamycin and amikacin. J. Proteomics 94, 68–77. doi: 10.1016/j.jprot.2013.08.025
Lata, M., Sharma, D., Deo, N., Tiwari, P. K., Bisht, D., and Venkatesan, K. (2015a). Proteomic analysis of ofloxacin-mono resistant Mycobacterium tuberculosis isolates. J. Proteomics 127, 114–121. doi: 10.1016/j.jprot.2015.07.031
Lata, M., Sharma, D., Kumar, B., Deo, N., Tiwari, P. K., Bisht, D., et al. (2015b). Proteome analysis of ofloxacin and moxifloxacin induced Mycobacterium tuberculosis isolates by proteomic approach. Protein Pept. Lett. 22, 362–371.
Magnet, S., Courvalin, P., and Lambert, T. (2001). Resistance modulation cell division type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii BM4454. Antimicrob. Agents Chemother. 45, 3375–3380. doi: 10.1128/AAC.45.12.3375-3380.2001
Magnet, S., Smith, T. A., Zheng, R., Nordmann, P., and Blanchard, J. S. (2003). Aminoglycosides resistance resulting from tight drug binding to an altered aminoglycosides acetyl transferase. Antomicrob. Agents Chemother. 47, 1577–1583.
Nikaido, H. (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656. doi: 10.1128/MMBR.67.4.593-656.2003
Sharma, D., and Bisht, D. (2016). An efficient and rapid lipophilic proteins extraction from Mycobacterium tuberculosis H37Rv for two dimensional gel electrophoresis. Electrophoresis 37, 1187–1190. doi: 10.1002/elps.201600025
Sharma, D., and Bisht, D. (2017). Secretory proteome analysis of streptomycin resistant Mycobacterium tuberculosis clinical isolates. SLAS Discov. (in press). doi: 10.1177/2472555217698428
Sharma, D., Kumar, B., Lata, M., Joshi, B., Venkatesan, K., Shukla, S., et al. (2015a). Comparative proteomic analysis of aminoglycosides resistant and susceptible Mycobacterium tuberculosis clinical isolates for exploring potential drug targets. PLoS ONE 10:e0139414. doi: 10.1371/journal.pone.0139414
Sharma, D., Lata, M., Faheem, M., Khan, A. U., Joshi, B., Venkatesan, K., et al. (2015b). Cloning, expression and correlation of Rv0148 to amikacin & kanamycin resistance. Curr. Proteomics 12, 96–100. doi: 10.2174/157016461202150903113053
Sharma, D., Lata, M., Faheem, M., Khan, A. U., Joshi, B., Venkatesan, K., et al. (2016a). M. tuberculosis ferritin (Rv3841): potential involvement in Amikacin (AK) & Kanamycin (KM) resistance. Biochem. Biophys. Res. Commun. 478, 908–912. doi: 10.1016/j.bbrc.2016.08.049
Sharma, D., Lata, M., Singh, R., Deo, N., Venkatesan, K., and Bisht, D. (2016b). Cytosolic proteome profiling of aminoglycosides resistant Mycobacterium tuberculosis clinical isolates using MALDI-TOF/MS. Front. Microbiol. 7:1816.
Sharma, D., Shankar, H., Lata, M., Joshi, B., Venkatesan, K., and Bisht, D. (2014). Culture filtrate proteome analysis of aminoglycoside resistant clinical isolates of Mycobacterium tuberculosis. BMC Infect. Dis. 14(Suppl.):60. doi: 10.1186/1471-2334-14-S3-P60
Sharma, P., Kumar, B., Gupta, Y., Singhal, N., Katoch, V. M., Venkatesan, K., et al. (2010). Proteomic analysis of streptomycin resistant and sensitive clinical isolates of Mycobacterium tuberculosis. Proteome Sci. 8:59. doi: 10.1186/1477-5956-8-59
Singh, A., Gopinath, K., Sharma, P., Bisht, D., Sharma, P., Singh, N., et al. (2015). Comparative proteomic analysis of sequential isolates of Mycobacterium tuberculosis from a patient with pulmonary tuberculosis turning from drug sensitive to multidrug resistant. Indian J. Med. Res. 141, 27–45. doi: 10.4103/0971-5916.154492
Welch, K. T., Virga, K. G., Whittemore, N. A., Ozen, C., Wright, E., Brown, C. L., et al. (2005). Discovery of non-carbohydrate inhibitors of aminoglycoside-modifying enzymes. Bioorg. Med. Chem. 13, 6252–6363. doi: 10.1016/j.bmc.2005.06.059
WHO (2016). Global Tuberculosis Control 2016. Available at: http://www.who.int/tb/publications/global_report/en/
Keywords: M. tuberculosis, proteomics, bioinformatics, hypothetical proteins, mechanism of drug resistance
Citation: Sharma D and Bisht D (2017) M. tuberculosis Hypothetical Proteins and Proteins of Unknown Function: Hope for Exploring Novel Resistance Mechanisms as well as Future Target of Drug Resistance. Front. Microbiol. 8:465. doi: 10.3389/fmicb.2017.00465
Received: 06 January 2017; Accepted: 07 March 2017;
Published: 21 March 2017.
Edited by:
Santi M. Mandal, Indian Institute of Technology Kharagpur, IndiaReviewed by:
Noton Kumar Dutta, Johns Hopkins University, USAMairaj Ahmed Ansari, Rosalind Franklin University of Medicine and Science, USA
Copyright © 2017 Sharma and Bisht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Divakar Sharma, ZGl2YWthcnNoYXJtYTg4QGdtYWlsLmNvbQ==