 Fabio Castillo
Fabio Castillo Amal Benmohamed
Amal Benmohamed George Szatmari
George Szatmari- Département de Microbiologie, Infectiologie et Immunologie, Université de Montréal, Montréal, QC, Canada
The separation and segregation of newly replicated bacterial chromosomes can be constrained by the formation of circular chromosome dimers caused by crossing over during homologous recombination events. In Escherichia coli and most bacteria, dimers are resolved to monomers by site-specific recombination, a process performed by two Chromosomally Encoded tyrosine Recombinases (XerC and XerD). XerCD recombinases act at a 28 bp recombination site dif, which is located at the replication terminus region of the chromosome. The septal protein FtsK controls the initiation of the dimer resolution reaction, so that recombination occurs at the right time (immediately prior to cell division) and at the right place (cell division septum). XerCD and FtsK have been detected in nearly all sequenced eubacterial genomes including Proteobacteria, Archaea, and Firmicutes. However, in Streptococci and Lactococci, an alternative system has been found, composed of a single recombinase (XerS) genetically linked to an atypical 31 bp recombination site (difSL). A similar recombination system has also been found in 𝜀-proteobacteria such as Campylobacter and Helicobacter, where a single recombinase (XerH) acts at a resolution site called difH. Most Archaea contain a recombinase called XerA that acts on a highly conserved 28 bp sequence dif, which appears to act independently of FtsK. Additionally, several mobile elements have been found to exploit the dif/Xer system to integrate their genomes into the host chromosome in Vibrio cholerae, Neisseria gonorrhoeae, and Enterobacter cloacae. This review highlights the versatility of dif/Xer recombinase systems in prokaryotes and summarizes our current understanding of homologs of dif/Xer machineries.
Introduction
Bacteria and archaea have developed a variety of well-regulated and coordinated mechanisms of replication and segregation of their genomes that ensure the genetic material is transmitted faithfully to the daughter cells, despite the absence of temporal separation between DNA synthesis, chromosome separation and cell division (Reyes-Lamothe et al., 2012). However, the circular state of their chromosomes and plasmids constitutes a constant threat to genome stability and proper segregation because of dimer formation during recombinational exchanges between sister chromatids. These rearrangements can combine their genomes into larger molecules, compromising an equal distribution of the genetic material to the daughter cells (Barre et al., 2001; Yates et al., 2003; Massey et al., 2004; Grainge et al., 2007). This topological problem was fully addressed in 1981, when Austin et al. (1981) demonstrated that the stable inheritance of the prophage P1 was due to site-specific recombination (SSR), a specialized system that catalyzes DNA exchange between two defined DNA sequences, and which plays a major role in dimer resolution by converting multimeric forms to the monomeric forms. Later studies performed with the plasmid ColE1 connected SSR with plasmid monomerization and stability (Summers and Sherratt, 1984). Colloms et al. (1990), the site-specific recombinase (XerC) was identified as the first protein responsible for SSR on cer, followed by the identification of a second recombinase, XerD required for this reaction (Blakely et al., 1993).
The Disadvantage of Having Circular DNA
In most bacteria and some archaea, replication begins at a single origin of replication oriC at which DnaA binds and stimulates the assembly of the replisome (Jha et al., 2016). Replication forks then proceed bi-directionally until the two replication forks meet in an antipodal terminus region flanked by ter sequences. These sequences in conjunction with the replication terminator protein (Tus) stop the replication forks to synchronize their arrival at the same time and place (Elshenawy et al., 2015; Gowrishankar, 2015; Pandey et al., 2015). However, chromosome replication is not a continuous process and is continuously halted by different types of DNA lesions such as UV irradiation, free radicals, genotoxic agents, DNA replication errors, transcription-replication conflicts, tightly bound protein-DNA complexes, or RNA secondary structures (Kuzminov, 1999; Tehranchi et al., 2010; Ayora et al., 2011; Merrikh et al., 2012; Gowrishankar, 2015). To maintain their genomic integrity, bacteria have developed several and sophisticated mechanisms to minimize the frequency of these DNA lesions before the occurrence of replication. The initial barrier against deleterious DNA modifications is carried out by specialized mechanisms, each one required for a given type of lesion, such as proofreading, direct reversal of DNA damage, base excision repair, nucleotide excision repair and mismatch repair (Lenhart et al., 2012; Kisker et al., 2013; Groothuizen and Sixma, 2015; van der Veen and Tang, 2015). Additional groups of mechanisms are responsible for avoiding transcription-replication encounters, equally lethal for bacteria, such as the coordination of temporal and spatial gene activation and co-orientation, modulators of RNA polymerases (RNAPs) and replicative accessory helicases (Pomerantz and O’Donnell, 2010; Merrikh et al., 2012; Bruning et al., 2015; Roghanian et al., 2015). Nonetheless, it is unavoidable that some of this DNA damage or conflicts will escape the initial barrier and interfere with replication fork migration, leading to the eventual inactivation of the replication machinery and formation of double-strand breaks (DSBs), interstrand cross-links and single-stranded gaps (SSG). These represent critical forms of DNA damage that must be removed for chromosome replication and transcription to proceed (Heller and Marians, 2006; Yeeles et al., 2013). Therefore, a second barrier of repair is called into play to cope with these “evasive” damages. This second barrier is preferentially carried out by the homologous recombination repair system (HR). Estimates indicate that HR repair is required in almost every cycle of replication (Cox et al., 2000; Vos and Didelot, 2009; Darmon et al., 2014). In fact, the HR system is now not only considered as a functional mechanism for generating genetic diversity but also as a decisive factor in DNA repair, the latter being the primary role of this system in the maintenance of the genome and the main source of dimer events (Rocha et al., 2005). Thus, HR plays a central role in removal and/or repair of DNA damage and rescue and/or re-assembling of replication forks that have been broken or stalled (Heller and Marians, 2006; Costes and Lambert, 2012). In the traditional HR system in Escherichia coli, its mode of action consists of a multistep process of breakage and rejoining of homologous sequences (one old and one newly synthesized DNA strand). It initially involves (1) recognition of the DNA lesion by the complexes RecBCD or RecFOR, depending on the type of DNA lesion; (2) formation of 3′-ssDNA overhangs processed by the exo and endonuclease activity of the Rec proteins, and subsequent coating by RecA; (3) strand invasion of the 3-terminal ssDNA into the homologous duplex DNA molecule and search of the complementary strand; (4) formation of a D-loop intermediate, transformation into a branched intermediate and Holliday junction (HJ) formation and (5) completion of the recombination process by resolution of the HJ, catalyzed by the systems RuvABC or RecG (Chen et al., 2008; Handa et al., 2009; Zhang et al., 2010). HJ resolution can result in two alternative products;
• ‘crossover’ or spliced products; where reassortment of the flanking genes of the cleavage site has occurred, obtaining one different genotype at one side compared to the former DNA duplex, and therefore, long range of genetic exchange.
• ‘non-crossover’ or patch products where the flanking regions were not exchanged, and instead, the resulting DNA duplex contain a ‘patch’ of hybrid DNA with a shorter range of genetic exchange (Cromie and Leach, 2000; Lilley and White, 2001).
Because circular chromosomes do not have “ends,” they are vulnerable to concatenation during formation of an odd number of crossover events. Thus, swapping DNA flanking regions tangles the sister chromatids and forms larger ring chromosomes that compromise cellular division (Figure 1) (Steiner and Kuempel, 1998; Barre et al., 2001). To ensure proper chromosomal segregation, bacteria and archaea have overcome these major threats by two broad mechanisms. One is to minimize the formation of crossing-over events, and the other is to solve dimer formation by performing an additional DNA exchange, immediately prior to cell division, at a specific region called dif (Deletion-Induced Filamentation).
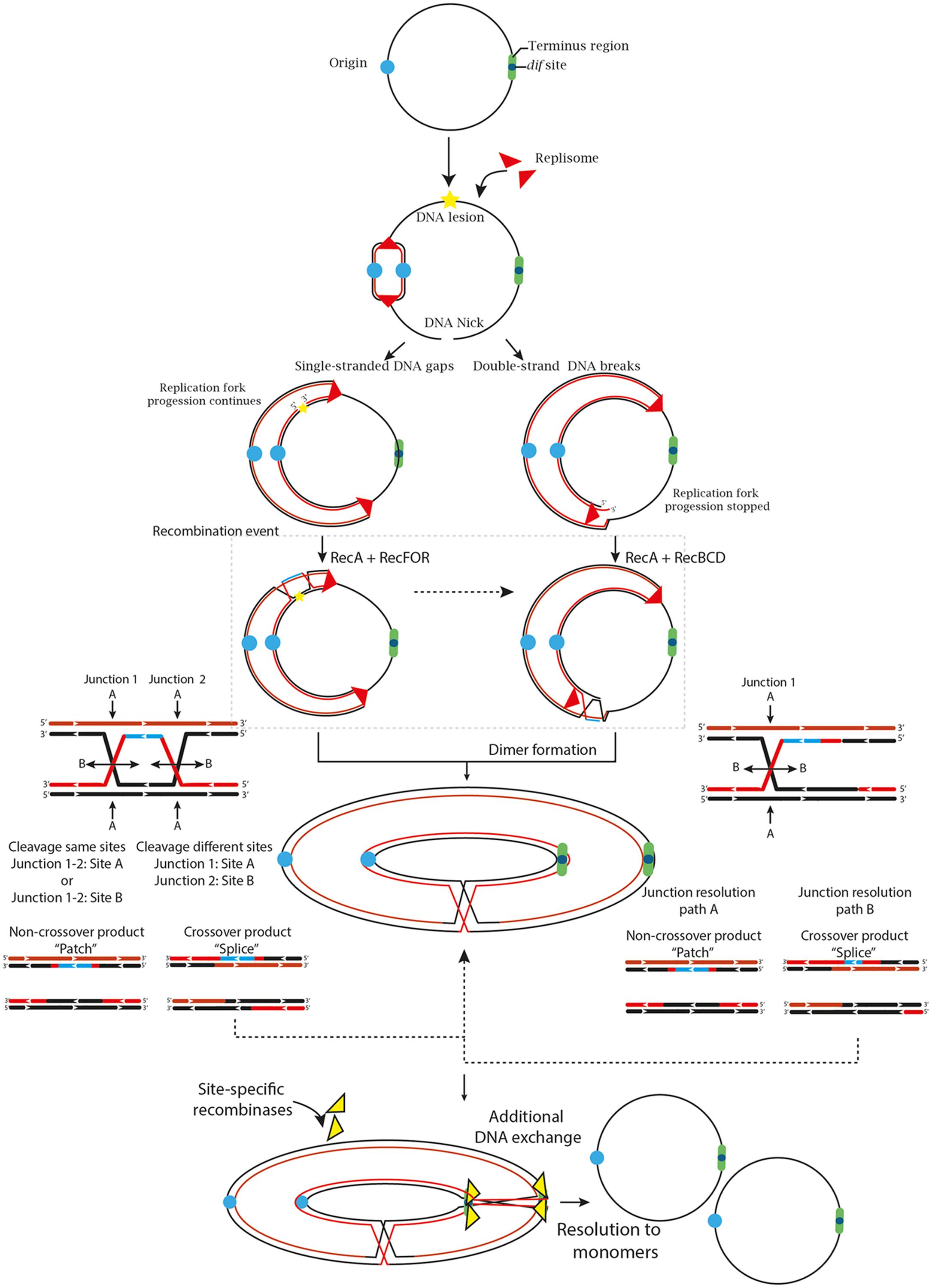
FIGURE 1. The two primary and most generalized pathways to solve chromosomal dimers generated by RecA-dependent repair or stalled replication forks. If the fork encounters a non-coding lesion (oxidative damage, pyrimidine dimer or an abasic site) depicted with a yellow star, it usually generates SSG (left path), although it can also lead to DSBs ends during repair by the RecFOR system. The pathways can also diverge on the location of the DNA lesion. If the lesion is located on the lagging strand template, the replisome will be able to bypass the lesion by blocking ‘Discontinuous DNA synthesis’ and then resume it downstream of the lesion, leaving a gap that would be repaired by the RecFOR system. On the other hand, a lesion on the leading strand template might transiently stop the replisome, cause its dissociation and then, bind further downstream to a new leading-strand primer, although these mechanisms are still under debate (Costes and Lambert, 2012; Yeeles et al., 2013). Alternatively, when the fork encounters a nick in the template strand (unrepaired SSG) or some cases of replication fork collapse, a DSB is generated (Right path). The DSBs are processed by the RecBCD complex that catalyzes the reattachment of the damaged DNA to the sister DNA duplex, forming a D-loop structure and eventual recognition by the replication-restart PriA protein that directs replisome assembly and resumption of the replication process in an origin-independent manner. However, odd numbers of crossover events generate dimer products (Yeeles and Dillingham, 2010; Azeroglu et al., 2016; Ðermić, 2015). Thus, if the resolution of the HJ occurs in the same sites, it will generate monomeric chromosome (Non-crossover products). In contrast, if the resolution takes place in different sites, it will generate chromosome dimers (Crossover products) (Carrasco et al., 2004; Ayora et al., 2011).
Avoiding Dimer Formation
One way to avoid dimer formation, as simple as it sounds, is to decrease the likelihood of dimer formation. This, however, is an intricate process of coordination and selection of the right enzymes at the right moment. Therefore, if the resolution of HJ intermediates by endonucleolytic cleavage can only result in crossover or non-crossover products, the likelihood of obtaining one or another is 50%. However, minimizing crossover events during homologous recombination repair seems to be the rule rather than the exception in organisms with circular chromosomes (Martini et al., 2006). In E. coli, homologous recombination repair is processed by two predominant recombinational pathways; The RecBCD pathway associated with DSB repair, replication fork collapse, replication fork reversal and replication fork arrest, and the RecF pathway, which is mostly involved in the repair of SSG, and under certain conditions, can also repair DSBs (Labib and Hodgson, 2007; Johnston et al., 2015). Both mechanisms lead to the formation of HJs that are mostly resolved by the RuvABC complex in E. coli or RecU in Firmicutes and Mollicutes (Carrasco et al., 2004). Deletion of the genes of the RuvABC complex eliminates non-crossover formation bias, supporting the idea that bias formation mostly depends on the action of Ruv proteins more than Rec proteins (Cromie and Leach, 2000). This idea was initially discussed by Van Gool et al. (1999) who demonstrated that crossover and non-crossover products are not random and, conversely, they are influenced by the positioning and orientation of the resolvasome on the HJ intermediate, which in turn directs RuvC strand cleavage direction. Additionally, topological conditions such as DNA supercoiling, DNA catenation, adjacent HJ intermediates or the presence of double HJ intermediates can also influence assembly of the RuvABC complex on the HJ and indirectly affect resolution (Van Gool et al., 1999). Subsequently, Cromie and Leach (2000) showed that RuvABC positioning may depend on the nature of the substrate caused by the type of DNA lesion, thus DSBs are more likely to result in crossover products (Frequently processed by RecBCD) whereas SSG are more likely to result in non-crossover products (frequently processed by RecF). Although, some fractions of SSG can cause DSBs (Spies and Kowalczykowski, 2005). However, the specific causes of crossover or non-crossover formation are still under debate, and different reactions cannot be completely dismissed. A clear example of this is the fact that RecBCD as well as RecF are not restricted to DSBs and SSG respectively and, on the contrary, the both may have interchangeable functions (Pagès, 2016). That would explain why some replication fork arrests generate non-crossover products even if they are mostly processed by the RecBCD pathway (Michel et al., 2000), or why RecF contributes almost equally to dimeric chromosome formation in E. coli despite the fact that it is responsible for SSG resolution (Barre et al., 2001).
Interestingly, the fact that DNA lesions and transcription-replication conflicts are more abundant in the leading strand than in the lagging strand in E. coli, and that these lesions usually generate non-crossover products, reinforces the idea that organisms with circular chromosomes favored a system that minimizes dimer formation during HR repair completion (Cromie and Leach, 2000; Fijalkowska et al., 2012; Merrikh et al., 2012). These biased reactions have also been detected in other microorganisms such as B. subtilis where the resolvase RecU biases homologous recombination toward non-crossover products (Carrasco et al., 2004). Despite this non-crossover preference by HR system in recA+ cells, dimer formation still occurs reaching 10–15% of the growing cells (Carnoy and Roten, 2009).
Coping With Dimers
It is clear that dimer formation is regarded as a negative outcome that must be solved. Despite this, Mazin et al. (1996) proposed that under certain conditions of selective stress, plasmid dimerization could confer an advantage for the selection of adaptive mutations due to rapid accumulation and selection of plasmids carrying a specific mutation and subsequent segregation to the daughter cells. Berza et al. (2013) also reported that plasmid dimerization greatly increased synthesis of a foreign protein and that plasmid content is unaffected by dimer formation showing some advantages for transcriptional events. However, these benefits were only considered for plasmids. Regarding circular chromosomes, dimerization must be resolved by the action of site-specific recombinases (SSRs), which are enzymes that are responsible for breaking and rejoining specific sites without requiring DNA synthesis or high energy cofactors (Warren et al., 2013). The relevance of this system for proper chromosome segregation is supported by the high degree of conservation in Bacteria and Archaea. The Xer complex is considered one of the most conserved structural features in cells containing circular chromosomes, as well as RecA and FtsK enzymes (Carnoy and Roten, 2009; Kono et al., 2011; Diagne et al., 2014). The SSRs act on specific short DNA sequences, called recombination sites, where DNA exchange occurs in three different types of DNA rearrangements; deletion (divided into excision or resolution), insertion, or inversion. All these processes depend on the orientation and direction of the two recombination sites (Grindley et al., 2006) (Figure 1). All known site-specific recombinases are classified into two unrelated families, tyrosine–type or serine-type recombinases (Tyr or Ser) based on the amino acid residue that forms a covalent linkage between the protein and a phosphate at the DNA cleavage site (Hirano et al., 2011). Serine recombinases, often referred to as the resolvase/invertase family, act on a recombination site with just 2 bp separating the cleavage sites on top and bottom strands and the cleavages occur simultaneously to create a double strand break, while tyrosine recombinases, often referred to as the λ integrase family enzymes perform a two-step cleavage and rejoining process where the cleavage sites are separated by 6–11 bp. Each recombinase family possesses a distinct mechanism. Tyrosine recombinases are divided according to the recombination directionality; unidirectional or bidirectional recombinases. Whereas serine recombinases are divided according to their size; small or large recombinases (Grindley et al., 2006; Brown et al., 2011).
The chromosome dimer resolution (CDR) process and heritable stability were originally elucidated in E. coli (Stirling et al., 1988a; Colloms et al., 1990; Blakely et al., 1993; Sherratt et al., 1995, 2004), where two paralogous site-specific tyrosine recombinases, XerC (298 aa) and XerD (298 aa) (Chromosomally Encoded Recombinases) were shown to act on a 28 bp DNA sequence (dif site), located in the ter region. The synaptic XerCD/dif complex consists of two XerC and two XerD subunits respectively bound to two dif sites (Figure 2B). Limited structural information of some tyrosine recombinases have revealed a conserved catalytic domain fold (Swalla et al., 2003), facilitating the analysis of experimental data and allowing the development of a general model for Xer recombinases (Meinke et al., 2016) consisting of; XerD (Subramanya et al., 1997), XerA (Hwa Jo et al., 2016), XerH (Bebel et al., 2016) and other related tyrosine recombinases like Cre (Gopaul and Van Duyne, 1999; Martin et al., 2002), HP1 integrase (Hickman et al., 1997), FLP (Chen et al., 2000) and λ integrase (Kwon et al., 1997; Biswas et al., 2005). The E. coli dif site is divided into two 11 bp half-sites that share partial dyad symmetry linked by a 6 bp central region that defines the positions of strand cleavage and exchange (Kuempel et al., 1991). The initial step of SSR during dimer resolution requires the formation of a synaptic complex consisting of a tetrameric protein/DNA complex (four protomers of tyrosine recombinases and two recombination site duplexes). Once the synaptic complex is formed, two opposing and activated protomers cleave the DNA strand of each recombination site duplex. This occurs when the hydroxyl group of the nucleophilic tyrosine attacks the scissile phosphate in the central region to form a 3′ phosphotyrosyl intermediate and a 5′-hydroxyl end. This intermediate conserves the energy from the phosphodiester bond cleavage to perform the first strand exchange. The recently formed 5′- hydroxyl attacks the 3′ phosphotyrosyl linkage on the partner site to reseal the strand breaks creating a HJ intermediate (Warren et al., 2013). HJ formation and isomerization activates the second pair of subunits bound to the other half of the recombination sites and inactivates the first pair of subunits. The second pair of subunits then cleaves, exchanges and rejoins the second pair of strands by the same mechanism just described; this second cleavage allows the resolution of HJ-intermediate and results in the recombinant DNA (Figure 2B) (Rajeev et al., 2009; Meinke et al., 2016). This process implies that the specific pairs of recombinases and/or active sites are continuously switched on and off to synchronize when and how recombination occurs, this coordination depends on allosteric interactions between the recombinases and external factors imposed on the synaptic complex (Hallet et al., 1999; Aussel et al., 2002; Vanhooff et al., 2009). In the XerCD/dif system, XerC normally initiates catalysis of one pair of DNA strands to form the HJ-intermediate without a subsequent resolution by XerD. Therefore, the HJs are rapidly converted back to the original DNA rearrangement. This XerC-first interaction is functionally active during the integration of certain bacteriophages that utilize Xer recombination to integrate their genomes into their host dif sites or in the resolution of plasmid multimers. In contrast, during chromosomal dimer resolution, pre-synapsed XerCD/dif complexes favor XerD activation by the FtsK protein to mediate the first strand exchange, generating a transient (XerD-HJ) intermediate, subsequent isomerization forms a XerC-HJ intermediate that is rapidly resolved to recombinant DNA by XerC, see below and Figures 2A, 3A (Zawadzki et al., 2013; Diagne et al., 2014).
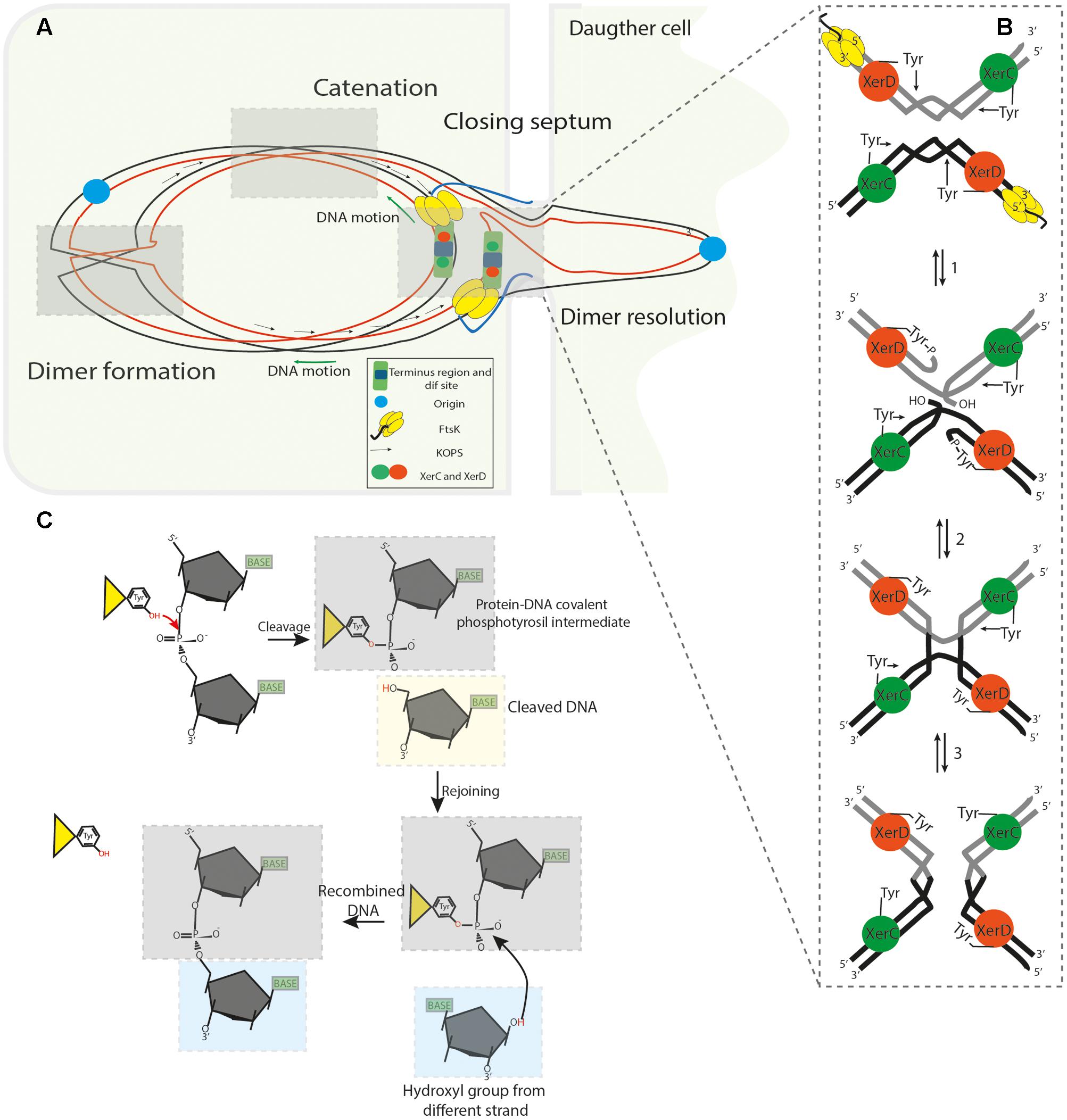
FIGURE 2. (A) Segregation of the sister chromatids during chromosome dimer resolution in E. coli. The illustration depicts the central part of a dividing cell in the final steps of chromosome segregation. The closing division septum, the motor domain αβ of FtsKC (yellow hexameric ring), the unstructured linker domain FtsKL (Blue ribbon), the KOPS sequences and the XerCD/dif synaptic complex are indicated. Concatenation prevents proper migration of the nascent chain of DNA; the origin regions move toward their respective cell poles, but the rest of the knotted DNA is stretched across and behind the septum. (B) FtsKC loads onto the KOPS sequences in an oriented manner and translocates toward XerCD/dif complexes. FtsK translocation allows it to reach the XerCD/dif complexes and bring them into proximity; as a consequence, the γ-subdomain of the FtsKC region activates XerD (Orange sphere) to perform the first strand cleavage. Then, XerC (Green sphere) mediates the second strand cleavage, allowing separation of the sister chromatids from each other. (C) Illustration of the SSR mechanism used by tyrosine recombinases: The OH group of the active residue tyrosine attacks the scissile phosphate forming a 3′-covalent phosphotyrosyl enzyme–DNA covalent intermediate and a free 5′-hydroxyl end. The covalent intermediate is attacked in turn by the other 5′- end to reverse the cleavage reaction and obtain a recombinant product.
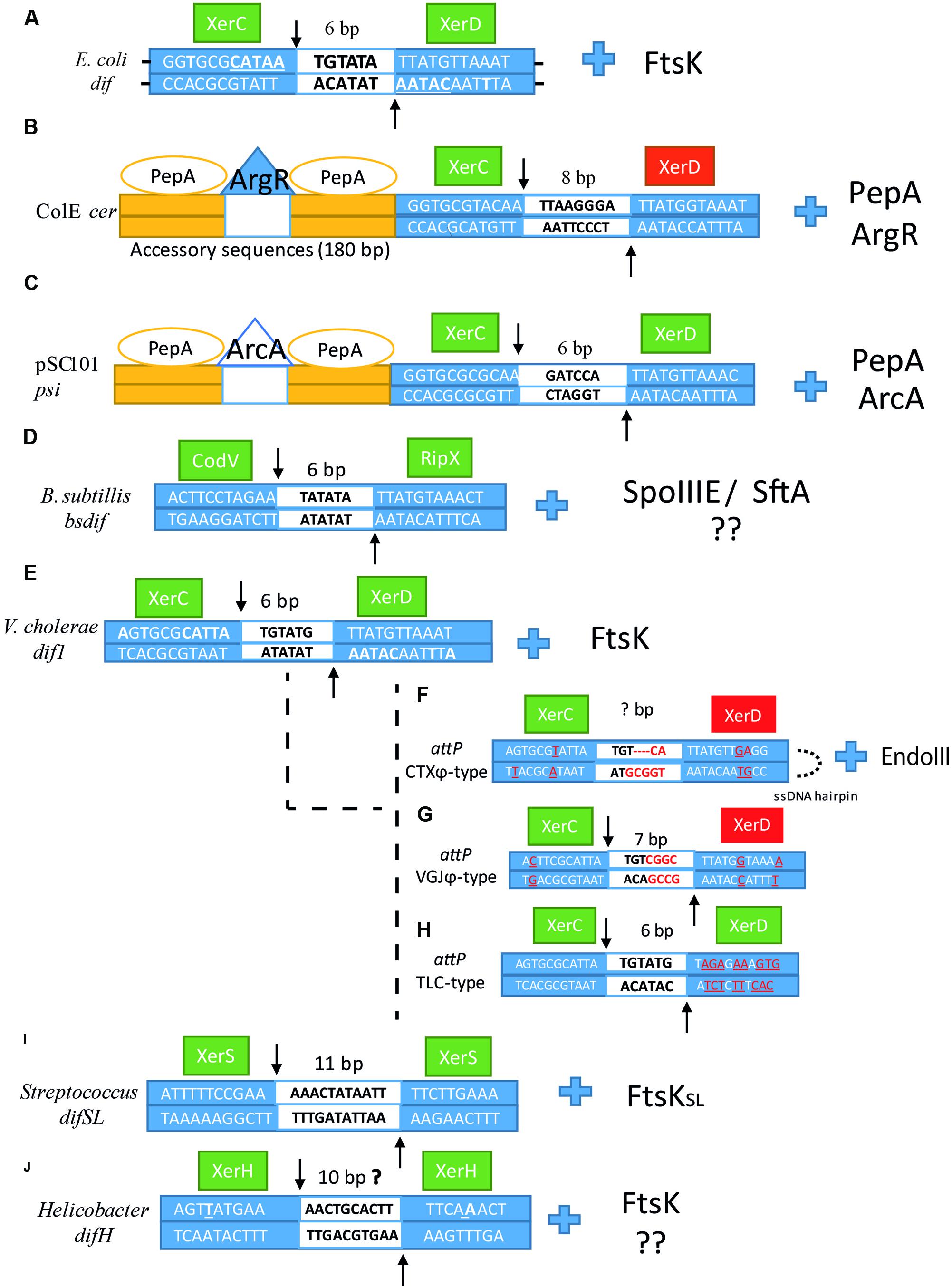
FIGURE 3. Sequence alignment of (A) dif, (B) cer, (C) psi, (D) Bsdif, (E) dif1, (F) attPCTX, (G) attPV GJ, (H) attPTLC, (I) difSL and (J) difH. Dyad bases in each arm are underlined in dif. XerC and XerD cleavage points are indicated by arrows, the central region is depicted in the middle of the sequence as a white box with the number of base pair corresponding to each dif site above. Left and right arms are depicted as blue boxes with their corresponding sequences for each dif site. The catalytic unit is depicted as a green box, whereas the inactivated unit is depicted as a red box. For (E–H), bases that differ from dif1 in V. cholerae are underlined and colored in red. Next to each dif alignment is the corresponding accessory protein that coordinates/activates dimer resolution.
Establishing Rules for Dimer Resolution
One of the fundamental questions about SSR at dif concerns how the system is controlled to ensure a proper CDR into monomers in the right place and at the right time without promoting the reverse reaction, which would generate dimers from monomers. It is understood that Xer-mediated recombination mostly depends on an active HR system because it is the major process that provides concatenated chromosomes. However, catenation problems caused by replication may require Xer-recombination system as well (Recchia et al., 1999; Grainge et al., 2007; Midonet et al., 2014). This reaction occurs at two polarized and specific regions of ∼10 kb at either side of dif called DAZ (dif Activation Zone), where oppositely oriented KOPS (FtsK Orienting Polar Sequences) converge and guide FtsK DNA translocation toward the dif locus (Pérals et al., 2000). This directional control is achieved by the interaction between the Xer recombinase system and the C-terminal domain of FtsK (Filamentous Temperature-Sensitive cell division protein K), a large division septum-associated DNA translocase, which coordinates chromosome segregation and cell division when chromosome organization has been affected (e.g., chromosome dimer formation, decatenation or delayed replication) (Löwe et al., 2008; Graham et al., 2009; Grainge et al., 2011; Nolivos et al., 2012; Bisicchia et al., 2013; Besprozvannaya and Burton, 2014; Diagne et al., 2014). FtsK was initially documented in 1995 due to observations in E. coli TOE44 (AB2497 ftsK44) mutant cells and their ability to form long chains of cells due to a single substitution of one amino acid in the N-terminal domain (FtsKN), by then, FtsK was thought to participate in septum formation as a peptidoglycan-modifying enzyme. Then, Yu et al. (1998) demonstrated that inactivation of the C-terminal region of FtsK affected normal chromosome segregation due to the formation of long chains of cells and detected abnormal DNA distribution in some ftsK1::cat minB double mutants of E. coli minicells. Finally, in Steiner et al. (1999) discovered that SSR at dif requires FtsKC and thus, CDR only occurs in its presence. The ∼1329 aa FtsK protein can be divided into three domains; The ∼279 aa N-terminal domain (FtsKN) is responsible for attachment of the protein to the membrane by four transmembrane segments and interaction with other proteins of the division septum such as FtsZ (Berezuk et al., 2014). The linker domain FtsKL, not commonly conserved in FtsK homologs, is primarily composed of proline-glutamine residues (Dubarry et al., 2010). Its length and composition varies between species, being ∼650 amino acids long in E. coli and most of Proteobacteria, ∼200 aa long in Vibrios (Val et al., 2008) or ∼125 aa in Pseudomonas (Massey et al., 2006). Experiments performed by Bigot et al. (2004) demonstrated that ftsKL mutations increased filamentation phenotypes even higher than xer mutants and that this filamentous formation did not correspond to problems in CDR. On the contrary, it was thought to be due to a deficient positioning of the protein, reducing the possibility of contact between FtsKC and the DAZ region of the chromosome. Subsequently, Dubarry et al. (2010) revealed that different parts of the linker domain interact with other proteins of the divisome such as FtsQ, L, I and Z and these interactions help to stabilize the whole divisome at the site of septation. Interestingly, they also suggested that FtsKL domain may stop or slow down cell division during dimer resolution because of the destabilization of the divisome components when FtsKC has been pulled by the DNA during translocation, this force can separate FtsZ and delay septum constriction. There is also a proportional relation between the glutamine-proline concentration and its length, where the longest linkers are usually richer in these residues (Bigot et al., 2007). Following the linker, the highly conserved ∼500 aa C-terminal domain (FtsKC), usually referred as the motor of the FtsK, is comprised of three separated subdomains called α, β and γ. Structural studies of the translocation module FtsKαβ of Pseudomonas aeruginosa demonstrated that it assembles as a hexameric ring around double-stranded DNA forming a central channel of 30 Å in diameter, where double stranded DNA (dsDNA) passes through (Massey et al., 2006; Demarre et al., 2013). Later structural studies of the orientation module FtsKγ of P. aeruginosa and E. coli demonstrated that six γ subdomains are loosely attached by a short linker of 10 aa to the hexameric ring FtsKαβ (Sivanathan et al., 2006; Löwe et al., 2008). The FtsKαβ subdomains are responsible for the ATP hydrolysis-dependent DNA translocation of the protein. The 68 aa FtsKγ subdomain is a helix-wing-helix domain that performs two main functions. The first role of this subdomain is to recognize the 8 bp KOPS sites and then directs FtsK translocation toward the dif site located within the ter region, at which, if concatenation occurs, two dif sites will be brought together to form the synaptic complex XerCD/dif (Figure 2A). KOPS are over-represented on the leading strand of replication where their concentration gradually increases as dif is reached; indeed, more than 90% of KOPS sequences nearby dif are located on the leading strand (Nolivos et al., 2012); giving a possible estimate of 34 KOPS motifs located in the DAZ region (Lee et al., 2014) with a frequency of 1 motif every 13Kb (Besprozvannaya and Burton, 2014). The second main function of the FtsKγ subdomain is to activate the XerD catalytic function to generate the first HJ and subsequent dimer resolution (Grainge et al., 2011; Lee et al., 2014; Keller et al., 2016). How FtsK locates and assembles to initiate translocation in the correct KOPS sequence is still arguable, for this reason two models have been proposed; the loading model and the target search model, recently reviewed by Besprozvannaya and Burton (2014). New evidence strongly suggests that FtsK acts in a 350-kb region around dif that covers 7% of the genome where monomers of FtsKC assemble exclusively at KOPS motifs as described by the loading model. An initial interaction of a single monomer of FtsKγ will trigger a rapid and stepwise formation of the hexameric ring under high concentration of FtsK (Graham et al., 2010; May et al., 2015). It is likely that FtsKγ assembles quickly and binds to KOPS as a trimer initially, with three FtsKγ modules interacting with consecutive GGG, NA, and GGG bases and then it hexamerizes gradually (Löwe et al., 2008). Once a KOPS motif is detected, allosteric modifications occur leading to hexamerization of FtsKαβ, which alters the angular conformation of FtsKγ on the DNA affecting KOPS recognition, and activates FtsKαβ ATP hydrolysis. As a consequence, FtsK is no longer able to recognize subsequent KOPS motifs during translocation, unless FtsK migration is impaired, and KOPS recognition is obligated to restart (Sivanathan et al., 2006; Löwe et al., 2008; Crozat and Grainge, 2010; Lee et al., 2014). FtsK has been demonstrated to be the fastest known DNA translocase, reaching levels of 17.5 ± 3.5 kb/s at 37°C or even faster with a striking stall force and a slight supercoiling induction, 1 positive supercoil per every 150 bp translocated (Saleh et al., 2005; Graham et al., 2009; Lee et al., 2012). It has also demonstrated a striking capacity to displace, evict or bypass different obstacles, especially proteins bound to the DNA such as RNA polymerases (Lee et al., 2014). However, FtsK acts differently upon collision with RecBCD and XerD-XerCD/dif complex proteins. When FtsK collides with XerCD/dif, in a synapsed form, it activates XerD to create the XerD-HJ transient intermediate (structural rearrangements increase the distance between dif sites from about 53 to 67 A°) (Zawadzki et al., 2013; Bebel et al., 2016) followed by a rapid dissociation from the DNA (dissociation takes to 0.5–1 s). Cleverly, May et al. (2015) demonstrated that recombination of the synaptic complex XerCD/dif takes 1 s longer than the FtsK dissociation time. Therefore, they suggested that this time span can provide a regulatory control for dimer resolution because concatenated chromosomes will reform XerCD/dif synaptic complexes every time that resolution failed. Thus, multiple sets of FtsK hexamers colliding multiple times against XerCD/dif synaptic complexes will increase the likelihood of generating recombinant products. This regulatory mechanism ensures monomeric products are formed during translocation of impaired DNA.
Alternative dif/Xer Resolution in Prokaryotes
Plasmid Resolution: Multicopy Plasmid ColE1 and Accessory Proteins
Plasmid dimerization and eventual multimerization has been termed as the “Dimer catastrophe” due to its deleterious effect in cell populations (Summers et al., 1993; Field and Summers, 2011). Dimer catastrophe represents two major problems in bacteria; (1) unequal plasmid distribution among populations, in particular, multicopy plasmids that are more vulnerable to plasmid loss and (2) metabolic burden caused by the rapid accumulation of dimers into the host (Field and Summers, 2011; Million-Weaver and Camps, 2014). As mentioned previously, dimer resolution was originally elucidated in ColE1, resulting in the first functional characterization of XerC and subsequent identification of XerD by sequence homology to XerC (Blakely et al., 1993). These discoveries constituted a new approach for site-specific recombinases and their role in dimer resolution. Subsequent investigations led to the identification of SSR enzymes involved in dimer resolution in other plasmids of E. coli, and other bacteria. Current estimates have identified more than 1300 tyrosine recombinases where many of them are associated with other host proteins to regulate their activity, directionality, or processivity (Meinke et al., 2016). Large plasmids usually carry their own recombinase machineries adjacent to the recombination site. Whereas, small plasmids, like those in the ColE1 family, use the chromosomally encoded dimer resolution system of their host (Sengupta and Austin, 2011; Crozat et al., 2014). For ColE1 resolution, XerC/D proteins act on a specific site called cer (Figure 3B), a non-codifying region of 280 bp where two additional proteins act with XerCD to catalyze SSR reactions: the arginine repressor (ArgR) (an arginine-dependent DNA binding protein originally called XerA) (Stirling et al., 1988a,b), and aminopeptidase A (PepA) (a bifunctional transcriptional regulatory protein that reacts to environmental signals, which was originally called XerB) (Stirling et al., 1989). SSR in cer is catalyzed by XerC within a sequence of 30 bp composed of two 11 bp half sides and a central region of 8 bp. XerC and XerD bind to the left and right halves cooperatively and respectively. Strand exchanges are catalyzed by XerC to form a HJ intermediate that is eventually resolved by an uncharacterized cellular HJ resolvase to generate a recombinant product (Colloms et al., 1996; Cornet et al., 1997). The cer site is comprised of a 30 bp core recombination site and two accessory DNA sequences of ∼180 bp in length, in which one or two hexamers of PepA and one hexamer of ArgR control the reaction (Colloms, 2013). The two accessory proteins are necessary for recombination, since in their absence, plasmid dimer resolution cannot be completed. However, at abnormal high concentrations of PepA, recombination in vitro can proceed without the help of ArgR (Reijns et al., 2005; Sénéchal et al., 2010). This is also seen at the recombination site psi of plasmid pSC101 which requires XerC, XerD and PepA but not ArgR (psi dimer resolution requires another accessory protein called ArcA instead of ArgR, and the cleavage reaction is performed by XerC and XerD) (Cornet et al., 1994; Colloms et al., 1996). In cer, PepA and ArgR control recombination directionality so that dimers can only be converted into monomers and not the opposite reaction. Dimer resolution directionality caused by these two proteins involves the formation of two directly repeated cer sequences positioned in an antiparallel direction; this conformation is favored by negative supercoiling where the cer sequences are interwrapped three times around the proteins resulting in the formation of a right-handed synapse structure that brings the XerCD binding sites together. Sites in an inverted repeat position prevent right-handed formation; this ensures only dimer resolution occurs. Thus, XerC and XerD bind to the 30 bp cer synapse region and may interact with the N-terminal domains of the PepA hexamers. Whereas the ArgR protein, which is flanked by one or two PepA hexamers, might be involved in bending the DNA, tightening it and activating cer SSR by possible interaction with the C-terminal region of the Xer recombinases. Another possible function is to bring the two cer sites together and to allow PepA loading to form a nucleoprotein complex that promotes XerCD binding and recombination (Reijns et al., 2005; Sénéchal et al., 2010; Colloms, 2013). Additionally, cer also encodes for a 70 nt RNA fragment called Rcd that is only transcribed during dimer formation by the Pcer promoter and regulated in a sequence-specific manner by the FIS protein (Blaby and Summers, 2009). Rcd binds to the enzyme tryptophanase and induces a quiescent state by increasing indole production within the cell. The quiescent state permits the cell to arrest cell division and chromosomal replication but still be active metabolically. This process is thought to be part of a dimer formation checkpoint that allows the XerCD/cer system to resolve dimer formation during this pause (Field and Summers, 2011).
The Bacillus subtilis Model and the Effect of Two Translocases
The capacity to perform SSR to resolve chromosome dimers is highly distributed among bacteria and archaea. Thus, homologs of XerC and XerD have been sequenced in a variety of species (Wang et al., 2013; Crozat et al., 2014). In B. subtilis, two homologs of XerC and XerD called CodV and RipX perform dimer resolution at a 28 bp dif site (Bsdif) close to the terminus region (Figure 3D). The Bsdif region is comprised of two 11 bp half-sites with imperfect dyad symmetry where CodV and RipX bind simultaneously and a 6 bp central region where DNA exchange occurs. Both CodV and RipX share a 37 and 44% identity with the XerC and XerD respectively, and 39% between them (Sciochetti et al., 1999). CodV binds preferentially to the left half-site and preferentially cleaves the top strand whereas RipX is able to bind to both sides with preferential binding to the right-half-site and preferential cleavage of the bottom strand. Cleavage by CodV is more efficient than cleavage by RipX, which suggests that CodV performs the first strand cleavage followed by RipX in in vitro experiments (Sciochetti et al., 2001). Sciochetti et al. (1999) also demonstrated that RipX could interact effectively with the E. coli dif site, unlike CodV which showed a weaker interaction with this substrate. However, addition of XerC to RipX/difE.coli or XerD to CodV/difE. coli generated larger complex formation in gel retardation analysis, demonstrating protein-protein interactions between these four proteins, which confirms some conserved features of tyrosine recombinases among bacteria. This is supported by the fact that the right half-site presents highly conserved features with respect to other dif sites among some bacteria, whereas the left-half site is less conserved, which could explain why RipX can binddifE. coli (Sciochetti et al., 2001). In contrast to E. coli, the synaptic complex can be brought together by the action of two DNA translocases: the membrane-associated SpoIIIE protein (Stage III Sporulation Protein E) and the soluble SftA protein (Septum-associated FtsK-like Translocase of DNA). Both translocases harbor AAA+-ATPase and C-terminal domains with 56% of sequence similarity between them. SftA exhibits 50% identity with respect to the E. coli FtsKγ domain whereas SpoIIIE exhibits a 50% of similarity to the FtsKαβ subdomain and 42% of similarity to the FtsKγ subdomain of E. coli (Barre, 2007). The N-terminal domains of these proteins are more divergent; SpoIIIE and FtsK share 36% of identity to respect to the four transmembrane helix whereas SftA lacks the transmembrane spanning domain (Wu, 2009; Kaimer et al., 2011). N-terminal domain variations coincide with their different location and activation in the genome. FtsK and SpoIIIE share a similar mechanism to anchor to the inner membrane of the dividing cell by their N-terminal regions; this transmembrane interaction is possibly reinforced by interactions with FtsZ or other cell division components such as FtsA or ZapA (Dubarry et al., 2010). During vegetative growth, SpoIIIE shows two predominant states: a static phase, where SpoIIIE is assembled close to future sites of cellular septation, and a mobile phase, where SpoIIIE does not occupy a specific position. Once cellular division begins, the static phase takes place when SpoIIIE is recruited by FtsZ and other division machinery proteins and is escorted to the center of the division septum (Fiche et al., 2013). SpoIIIE remains in the invaginating septum and hexamerizes independently of the cell division stages (Vegetative, division and sporulation stages) and independently of DNA interaction (Cattoni et al., 2014), suggesting that SpoIIIE assembly may not be restricted to the presence of impaired DNA and on the contrary, may be involved in normal DNA segregation as demonstrated by (Fiche et al., 2013). Experiments using high-resolution microscopy revealed that under formation of asymmetric (sporulation) or symmetric (vegetative growth) septa, the SpoIIIE concentration increased 2.5-fold around the constricting septa, even without evident formation of the septa, indicating close interaction with other components of the division machinery, that in turn regulates its activity under specific conditions (Fiche et al., 2013). SftA can be localized either to the cell center or more frequently, to the forming division septum. Although SftA lacks an integral membrane domain, the FtsZ ring recruits the enzyme and attaches it to the division septum during the initiation of cellular division, which explains its localization through the cell cycle (Kaimer et al., 2009, 2011; Kaimer and Graumann, 2011). These patterns of localization suggest that both translocases (SftA and SpoIIIE) are present at the septum at various times of segregation and that they perform DNA migration independently of each other, although SftA is only involved in DNA cytokinesis in contrast to SpoIIIE that may be involved in cytokinesis and cell division processes (Kaimer et al., 2009). DNA translocation is initially carried out by SftA during septation, probably by recognition of the 8-nucleotide SRS motifs (SpoIIIE Recognition Sequences) which are similar to the E. coli KOPS sequences. The SRS motifs are mostly located on the leading strand (up to 85%), and direct translocation toward the Bsdif site (Besprozvannaya and Burton, 2014). Therefore, the primary function of SftA consists of moving chromosomal DNA until the ter regions are positioned at midcell and the origin regions migrate to each pole of the cells. The SftA may also be required for other proteins involved in cytokinesis and FtsZ positioning (Biller and Burkholder, 2009). SpoIIIE, the second translocase in B. subtilis, may take over DNA translocation working synergistically but not interchangeably with SftA, it can also function as a DNA segregation checkpoint preventing membrane fusion until chromosome segregation is completed (Kaimer et al., 2011; Fiche et al., 2013). Cattoni et al. (2014) suggested that SpoIIIE binds non-specifically to the DNA in a pre-formed hexameric open ring conformation and then searches for SRS motifs without hydrolysis of ATP. Similarly to FtsK proteins, SRS recognition by the SpoIIIEy domain triggers allosteric modifications that activate the ATPase activity of SpoIIIEαβ and therefore, DNA translocation (Besprozvannaya et al., 2013). Once it encounters SRS motifs, the hexameric ring changes to the closed and active form pumping the chromosome in an oriented manner by recognizing further SRS motifs and translocating it toward Bsdif (Cattoni et al., 2014). As mentioned before, SpoIIIE is actively expressed in all growing cells and is essential during sporulation to translocate the remaining DNA from the mother cell into the forespore compartment, and during vegetative growth to guarantee that concatenate formation or disrupted genomes will not affect normal cellular division. Moreover, SpoIIIE is also required for septal membrane fusion after completion of chromosome translocation. During sporulation, asymmetric septation encloses the DNA and traps 25–30% of one chromosome into the forespore. SpoIIIE pumps the remaining 70–75% by an analogous mechanism used by FtsK; the reaction only takes 20 min demonstrating its incredible speed (Demarre et al., 2013; Bose et al., 2016). The mechanism of SpoIIIE DNA translocation through the membrane is still unclear, since recent single molecule-imaging experiments still provide valid information for two main models; the paired DNA conducting channel model (Burton et al., 2010; Yen Shin et al., 2015) and the aqueous channel model (Fiche et al., 2013). This system of both translocases has been also detected in Staphylococcus aureus termed FtsK and SpoIIIE because of their amino acid homology to SpoIIIE and FtsK from B. subtilis and E. coli respectively. However, in contrast to B. subtilis system, in S. aureus both enzymes seems to present a redundant, although independent role in DNA segregation. Individual deletions of either FtsK or SpoIIIE did not exhibit major changes in chromosome segregation for S. aureus, however, when combined together they represented a major threat for S. aureus genome stability (Veiga and Pinho, 2016).
Consistent with their different roles, SftA and SpoIIIE do not colocalize during vegetative-replicative stages or sporulation. Thus, SftA in concert with FtsZ and division proteins moves chromosomal DNA away from the closing division septum. Then, upon septum closure, entrapped DNA is translocated through the SpoIIIE pore or channels into the correct compartment (either a forespore or a daughter cell). However, unlike FtsK that activates XerCD recombination reactions, neither SftA nor SpoIIIE directly activate CodV or RipX recombinases. In this case, SftA and SpoIIIE affect the CodV/RipX reaction by proper positioning of the ter region, but there is no evidence of direct interaction between these enzymes to date (Biller and Burkholder, 2009; Kaimer et al., 2011).
Multichromosome Bacteria and IMEX
Vibrio cholerae, as well as 10% of sequenced bacteria to date, possess a very distinct property among bacteria; it harbors more than one chromosome (Jha et al., 2012). One ancestral chromosome I (chrI) of 2.96 Mbp and one plasmid-derived chromosome II (chrII) or ‘chromid’ of 1.072 Mbp, encode 2,775 and 1,115 ORFs, respectively. ChrI contains most of the housekeeping genes whereas chrII contains essential genes specialized in adaptation to new environments or pathogenicity (Xu et al., 2003; Harrison et al., 2010; Kirkup et al., 2010; Val et al., 2016). Harboring two or more chromosomes have shown to be highly heritable among these bacteria, which suggests that multiple chromosomes offer a positive selective pressure to maintain them. One possible explanation is that multiple chromosomes might offer an advantageous feature against dimer formation. Val et al. (2008) showed that dimer formation increases exponentially in relation to the size of the replicons, thus, dividing a single replicon into two or more replicons may reduce this topological problem. However, genome size might not be relevant for the presence or absence of Xer/dif recombination machinery. Some large chromosomes do not require Xer/dif recombination machinery as in some Legionellales (genome size ranging from 2 to 5 Mb) whereas some small-sized chromosomes still require Xer/dif recombination machinery as demonstrated by some Rickettsiales (genome ranging from 0.85 to 1.52 Mb in size) (Carnoy and Roten, 2009).
Homologs of XerC/XerD and FtsK have been characterized on chrI, referred as XerCV C and XerDV C with 53 and 68% of amino acid similarity to E. coli XerC and XerD, respectively (Huber and Waldor, 2002; McLeod and Waldor, 2004). Whereas chrII does not encode any Xer recombinase involved in dimer resolution. dif-like sequences are present in both chromosomes (dif1 and dif2) located near GC skew shift-points (Val et al., 2008; Kono et al., 2011). Interestingly, both dif sites differ from each other in their sequences, dif2 harbors five different nucleotides compared to dif1 and most α-proteobacterial dif sites, four of them in the central region, resembling dif-like plasmid composition (Kono et al., 2011). Dimer resolution in V. cholerae requires FtsKV C translocation by recognition of KOPS-like motifs (GGGNAGGG) in a similar way to that found in E. coli. Once the dif sites are brought together nearby, FtsKV C activates XerDV C, which is positioned to cleave the bottom strand, and perform the first strand cleavage. Then XerCV C cleaves the top strand and performs the second strand cleavage; these reactions are carried out on both chromosomes at their respective dif sites (Figure 3E) (Val et al., 2008). Additional studies demonstrated that E. coli FtsK was able to activate 50% of the XerCDV C synaptic complexes at dif1 whereas only 20% of XerCDV C were activated at dif2, suggesting that the dif2 recombination process requires more accurate interactions between the FtsK proteins and the XerCD complex (Val et al., 2008). An additional feature of multiple chromosomes is their capacity to synchronize replication termination at the same time despite their different sizes (Val et al., 2016). This capacity may confer an additional regulatory control against dimer formation due to the time-lapse between the replicated chromosomes and cellular division. Demarre et al. (2014) showed that terII sites (chrII) separate earlier than terI and that this early separation keeps terII sites at midcell by the macro domain MatP/matS organization system. This restriction during concatenation induces several collisions at midcell between terII sites, increasing the number of recombinational events and the likelihood of dimer resolution. It also ensures that ter sites of bacterial chromosomes remain exclusively in mid-cell to be processed by FtsK.
Although XerC and XerD recombinases normally perform dimer resolution, they are also exploited by other replicons such as plasmids, bacteriophages, and other integrative elements. Indeed, initial studies on plasmid stability in ColE1 and phage integration of bacteriophage λ led to the discovery of XerC and the mechanistic insights of the tyrosine family (Meinke et al., 2016). In V. cholerae, the causative agent of the potentially fatal human disease cholera, XerCV C and XerDV C are hijacked by some vibriophages to integrate their genomes into the chromosome. They are usually referred to as IMEX (Integrative Mobile Elements Exploiting Xer), and the best known ones are VGJϕ (Vibrio Guillermo Javier filamentous phage), TLCϕ (Toxic Linked Cryptic), and CTXϕ (Cholera Toxin Phage). CTXϕ is a lysogenic [(+)ssDNA] filamentous bacteriophage that encodes the A-B type enterotoxin CT in V. cholerae (Das, 2014). These three vibriophages harbor a particular attachment site (attP), a dif-like site that serves to classify the three different groups of IMEX, (CTXϕ-type, VGJϕ-type and TLCϕ-type) (Das et al., 2013). Although the components to integrate their genomes are very similar, their mechanisms of integration differ from one to the other and from their host strains. Direct ssDNA integration by CTXϕ-type phages is characterized by the formation of a ∼150 bp folded structure created by the intra-strand base pairing interaction between two palindromic attP sites (attP1 and attP2) separated by 90 nt on the ssDNA sequence (Figure 3F) (Das, 2014). The two overlap regions attP1 and attP2 reassemble the XerCV C side of dif1 and dif2 regions but differ from the XerD-side. This lack of homology between XerDV C recognition site and attPCTXϕ limits the catalytic reaction to XerCV C that catalyzes the complete reaction. An additional host factor called EndoIII participates in the directionality of the reaction, which blocks further rounds of strand cleavage by XerCV C causing its dissociation and therefore preventing CTXϕ excision (Bischerour et al., 2012). Although XerDV C is not involved in the catalytic reaction, it is still necessary for a successful integration, probably by its role in synaptic complex formation (Val et al., 2005). Once the integration is completed, host DNA replication proteins resolve the formed HJ intermediate and convert it to dsDNA. Prophage CTXϕ cannot be excised from its host since it loses the capacity to fold itself, which in turn prevents further base-pairing interactions between the attP sites, which ultimately abolishes the XerCV C catalytic reaction (Das, 2014). Interestingly, CTXϕ integration in El Tor strains is only found in chrI, and it is generally associated with two other vibriophages, TLCϕ and RS1 that enable CTXϕ integration in V. cholerae genome by reconstituting a functional dif site, and by promoting CTXϕ replication and transmission (Hassan et al., 2010). In the classical biotype strains, CTXϕ usually targets both chromosomes (Faruque and Mekalanos, 2012).
Similarly, to CTXϕ, VGJϕ integration uses the XerCV C catalytic reaction at the dif1 site, but unlike CTXϕ, it only harbors one dif-like attachment site (attPV GJϕ) of 29 bp that allows its integration into the chromosome as a dsDNA. The attP central region contains four different nucleotides close to the XerD binding side with respect to the central region of the dif1 site (Figure 3G). The lack of homology at the XerDV C central region side prevents XerDV C participation in the catalytic reaction. Once integrated, prophage VGJϕ acquires two attP sites (attPL and attPR), equally functional for the XerCV C excision reaction, in contrast to CTXϕ, where Xer recombinases can process VGJϕ excision from the host genome (Das et al., 2013). TLCϕ also depends on host encoded Xer recombinases for its integration. Its attPTLCϕ site possesses high homology with the XerCV C binding side and central region of dif1 whereas it is highly divergent from the XerDVC binding site (Figure 3H). The prophage form of TLCϕ is almost always linked to CTXϕ integration confirming the regular synergistic interactions found in most IMEX. Paradoxically, despite the lack of homology between the XerD binding sites of dif1 and attPTLCϕ, TLCϕ integration/excision is mediated by XerDV C and then completed by XerCV C resembling dimer resolution in bacteria, but independently of FtsK participation (Midonet et al., 2014).
IMEX are recombination platforms that permit bacteria to evolve and adapt through the acquisition and reordering of relevant genes. They have strengthened bacterial evolution, playing an important role in the rise of multidrug resistance, gene transfer mechanisms and virulence factors among clinically relevant bacteria (Fournes et al., 2016; Midonet and Barre, 2016). Besides the vibriophages just described above, some other relevant IMEX have been found; the gonococcal genomic island (GGI) related to pathogenic Neisseria species (Domínguez et al., 2011) and the EludIMEX-1 found in Enterobacter ludwigii (Antonelli et al., 2015). GGI is an unusually long IMEX (57 kb long) found in almost 80% of Neisseria gonorrheae strains and is involved in the expression of type IV secretion system (T4SS) genes (Christie et al., 2014). GGI carries a degenerate dif site called difGGI of 28 bp with a XerC-binding site and a central region homologous to the conserved Neisseria dif site (difNg) and a divergent XerD-binding site. GGI insertion into the Neisseria genome follows a CDR-like process where FtsK activates XerD to perform the first strand cleavage between difNg and difGGI followed by isomerisation of the synaptic complex and activation of XerC to perform the second strand cleavage, creating a GGI integrated form with two active Xer binding sites. Interestingly, the GGI synapse has given important clues about how IMEX might remain integrated in the host genome despite the presence of dif sites. Fournes et al. (2016) revealed by experiments in vitro that a trimeric form of the E. coli FtsK protein (t-FtsKαβγEc) was unable to activate XerCD recombination at one of the two dif sites (the difGGI site), in fact, the XerCD/difGGI complex was unable to stop t-FtsKαβγEC translocation. As a consequence, the XerCD complex is dissociated from difGGI and the excision process is inhibited.
EludIMEX-1 is a 29.1-kb IMEX found in E. ludwigii (ECAA-01) that carries the blaNMC-A gene that encodes for a serine carbapenemase. It was first characterized by Antonelli et al. (2015) when they sequenced the whole genome of a NMC-A-positive isolate of E. ludwigii. The results indicated the presence of a new 29-kb region with lower GC content when compared to the bacterial genome, indicating a possible gene transfer acquisition (Wu et al., 2012). Further analysis revealed that this region is flanked by putative XerC/XerD recombination sites with high homology at the XerC-binding site. They also determined that EludIMEX-1 insertion site in the genome was the same for two distinct species of the E. cloacae complex suggesting a possible acquisition via a XerC/XerD dependent recombination event at a specific dif-like site (Antonelli et al., 2015). Understanding of IMEX control and excision processes will provide us a better idea of how counteract the acquisition of antibiotic resistance genes in pathogenic microorganisms.
The difSL/XerS Model
The E. coli pathway of dimer resolution has been found to be highly conserved among bacteria with circular chromosomes. It was initially demonstrated by Recchia and Sherratt (1999) when they analyzed 16 eubacterial and five archaeal genomes for XerCD-CodV/RipX homologs. They showed that most eubacterial genomes possess two putative Xer recombinases whereas Archaea presented a single recombinase in three of the five genomes analyzed (Recchia and Sherratt, 1999). Subsequently, Carnoy and Roten (2009) demonstrated by doing an exhaustive computational analysis of 234 chromosomes from 156 proteobacterial species, that 87.8% of the genomes analyzed presented XerCD-like and dif-related sequences. Moreover, Kono et al. (2011) predicted by a recursive hidden Markov model method (including XerCD orthologs) that 578 out of 592 bacterial genomes with a single chromosome and 63 out of 66 genomes with multiple chromosomes presented a dif-like sequence. Additionally, they remarked how XerC and XerD are conserved in almost 60–70% of bacterial species, and 85% in proteobacterial species (Debowski et al., 2012). These results among many others led to the general view that the E. coli pathway is predominant for dimer resolution. However, dimer resolution machinery or regulation of strand exchange may differ: some processes may require or disregard accessory proteins, others may or may not require activation by translocases, some will be mediated by a XerC-first strand exchange whereas others by XerD-first strand exchange and others may need two recombinases or only one. Among these divergences and unique characteristics for each bacteria to solve dimer formation, the less studied ones are the unconventional single recombinases.
Recchia and Sherratt (1999) first mentioned the presence of single recombinases from the identification of two eubacterial genomes harboring only one Xer homolog. It was later confirmed when Le Bourgeois et al. (2007) demonstrated that some species of Lactococcus and Streptococcus use an alternative Xer recombination machinery. This new Xer complex is based on a single tyrosine recombinase called XerS (356 aa) that acts on an atypical 31 bp recombination site called difSL in the presence of dimers. Unlike E. coli, the xerS gene is found immediately adjacent to the recombination site difSL acting as a single module. The difSL site differs from most dif sites because of its large central region of 11 bp as opposed to the normally found 6–8 bp in all other dif regions (Figure 3I) (Leroux et al., 2011). Thus, difSL consists of two imperfect inverted repeat sites of different sizes separated by the central region where DNA exchange occurs. The inverted repeat region is one nucleotide longer in difSL and contains an extra nucleotide in the middle of the right inverted repeat (TTTTCTTGAAA) versus the left part of the sequence (TTTCCGAAAA). This additional spacing suggests XerS/difSL may be biased to favor binding in one-half site over the other. It was later confirmed by Leroux et al. (2011), where they also showed that XerS presented stronger interaction with the left-half site of difSL than the right-half site, and a preference for initiating the recombination reaction on the bottom strand of the difSL site. These results indicate that, although the difSL site is relatively symmetric and XerS is a single tyrosine recombinase, there is a bias for where the proteins initially bind to difSL and where they initiate the strand cleavage reaction. Thus, the left-bound monomer could activate the right-bound monomer by bending the DNA or changing the conformation of the second monomer which could explain the preferential cleavage and exchange of the bottom strand. This behavior resembles what XerC displays with weak binding but stronger strand exchange when compared to XerD (Nolivos et al., 2010; Leroux et al., 2011). This intrinsic bias alone cannot control the preference of the directionality of the strand cleavage reaction. The achievement of proper control requires the action of a SpoIIIE-like homolog translocase called FtsKSL, a protein of 758 aa in length in Streptococcus mutans or 816 aa in S. agalactiae with low similarity at the N-terminal region between them. This low similarity does not affect its binding preference to the division septum commonly found in most proteins of the FtsK-HerA superfamily (Le Bourgeois et al., 2007). The C-terminal domain of FtsKSL shows 41% similarity at the amino acid level in relation to FtsKE. coli with four of the five amino acids similar (QR-GN motif) involved in XerD interaction (Keller et al., 2016). On the other hand, FtsKSL is unable to read E. coli KOPS motifs as demonstrated by Nolivos et al. (2012), probably due to the lack of common skewed octamers sequences called Architecture Imparting Sequences (AIMS) in Firmicutes, which means that KOPS sequences in Firmicutes are not as conserved as in proteobacteria (Hendrickson and Lawrence, 2006). This would also explain the divergence between FtsKγ domains even among Firmicutes. Additionally, AIMS found in Lactococcus lactis differ in both in length and in sequence from traditional KOPS/SRS motifs, being A-rich heptamer motifs instead of the GC-rich octamer motifs (Nolivos et al., 2012). XerS also lacks critical residues found in XerD to interact with FtsK (residues RQ-QQ). Interestingly, XerS/difSL recombination occurs almost in a similar fashion to that of E. coli. Both Xer systems require FtsKN localization at the division septum and FtsKC translocation to achieved Xer dimer resolution. Additionally, XerS/difSL proved to be functional in E. coli, despite the lack of homology in their FtsK proteins. Further analyses on FtsK-Xer interactions are required since the exact mode of action is still speculative (Nolivos et al., 2010).
The Helicobacter and Campylobacter (difH/XerH) Model
Studies in Helicobacter sp and Campylobacter sp led to the discovery of another type of single recombinase called XerH that acts on a recombination site called difH in a FtsK-dependent manner. It was shown to be involved in chromosome segregation and possibly dimer resolution in Helicobacter pylori (Debowski et al., 2012). XerH (354–362 aa) differs from the traditional XerCD (298 aa) recombinases by its size and protein homology (26% of identity with respect to XerCD). It also shows more similarity to XerS (356 aa) in both the size of the protein and the high degree of homology of their recombination sites (Carnoy and Roten, 2009; Leroux et al., 2013). Another characteristic of XerH and a possible hallmark of single recombinases (XerS and XerH) is that the difH sequence is also located near the recombinase-encoding gene, indicating a possible individual genetic module for Xer expression (Le Bourgeois et al., 2007; Carnoy and Roten, 2009). Interestingly, most of the epsilon species of 𝜀-proteobacteria harbor a XerH/difH system whereas some other 𝜀-proteobacteria (Sulfurimonas denitrificans and Sulfurovum) possess a system analogous to the classical XerCD system. Additionally, unlike other tyrosine Xer recombinases, XerH activity appears to be affected by a second Xer recombinase called XerT in H. pylori (the TnPZ transposon associated recombinase) since under XerT deletion, difH recombination levels increased (Debowski et al., 2012). Recent structural studies showed that the Helicobacter difH comprises two highly conserved imperfect inverted binding sites of 11 and 10 bp (AGTTATGAAAA and AAAAGTTTGA) in the left and right sides respectively, separated by a 6–10 bp central region (Figure 3J) (Bebel et al., 2016) (Unpublished data suggest a 10 bp central region, Leroux et al., 2013). Two subunits of XerH bind cooperatively to each side with a stronger binding affinity as well as cleavage reaction efficiency in the left half site than the right half site (the outer region in dif appears to be determinant in the order of binding and cleavage reactions). The left half site preference is due to stronger interaction between XerH and difH left site ΔG = -21.3 kcal/mol compared to the right half site ΔG = -15.4 kcal/mol). The extra nucleotide thymine (T4) in the outer region of the left half site confers a specific hydrogen bond between the left arm and the lysine (K290) of XerH that favors stronger protein–DNA interaction with the other three outermost nucleotides, DNA bending and specific positioning of the nucleophilic tyrosine. Surprisingly, XerH assembly on difH does not induce strong DNA bending alone and it seems to require FtsK to generate the required conformational rearrangements to favor XerH DNA exchange (Bebel et al., 2016). Results obtained by Leroux et al. (2013) in difH of C. jenuni (difHcamp) demonstrated that XerH binding to either the left or right site of difHcamp resulted in similar affinities compared to the full difHcamp site possibly due to the similarity between the outer sequences of both arms. Additionally, XerH binding to difHcamp appears to be less efficient than XerS which suggests that it is less cooperative than XerS/difSL system. Additionally, these results contradict XerH binding affinities observed in H. pylori by Bebel et al. (2016) and in most tyrosine recombinases involved in chromosome resolution since it did not show any binding preference. On the other hand, unlike binding activity, asymmetrical cleavage reactions by XerH were found with a higher efficiency for bottom-strand substrates than top strand, in agreement with the results of Bebel et al. (2016) XerH recombination was also observed in vivo between two difHcamp sites located on the same plasmid; it is also suggested that XerH might be involved in decatenation processes because of the apparent absence of Topo IV proteins in H. pylori (Debowski et al., 2012; Leroux et al., 2013). Interestingly, despite difHcamp and difSL similarities in the recombination sites, the recombinases do not cross-react (XerH does not bind difSL and XerS does not bind difHcamp sites) (Leroux, unpublished).
The Archaea dif/XerA Model
In archaea, chromosome resolution appears to be catalyzed by a single recombinase (XerA) in a FtsK-independent manner that acts on a dif-like site located in the replication terminus region (Cortez et al., 2010; Serre et al., 2013). XerA shares a conserved C-terminal domain where the active tyrosine and the conserved catalytic residues (R-K-H-R-[H/W]-Y) reside. XerA proteins are well conserved between the archaeal species analyzed with 85% of sequence similarity. The xerA gene location is highly variable in archaea; some species exhibit separated xerA/dif sequences whereas some others harbor an individual xerA/dif module. Unlike most bacteria, the dif-like site is not normally located at 180° from oriC and conversely, it is located between 122° and 144° from oriC in the analyzed genomes, although the Methanosphaera stadtmanae genome showed a dif-like site at 180° from oriC. dif-like sequences consist of the traditional structure; two inverted repeat sequences of 11 bp separated by a central region of 6 bp. XerA catalyzes cleavage reactions without any detectable strand preference (Cortez et al., 2010; Serre et al., 2013).
Although archaea do not require a FtsK homolog to perform chromosome resolution, KOPS-like motifs have been found in Archaea. These KOPS-like motifs consist of four nucleotides (GTTG OR GTTC) called ASPS (Archaea Short Polarized Sequences) that are skewed toward dif sites, showing a similar triangle-shaped diagram observed in Bacteria of skew inversion at dif sites (Cortez et al., 2010). Serre et al. (2013) have revealed the crystal structure of XerA proteins from Pyrococcus abyssi, and Hwa Jo et al. (2016) from Thermoplasma acidophilum. Both groups reinforced the idea of cis- cleavage reaction by XerA.
Future Directions
Much information has been gained on site specific recombinases and dimer resolution. This review has highlighted the complexity of dif/Xer recombinase systems in prokaryotes and its importance for genome stability and pathogenicity factors. However, many fundamental questions remain unanswered: how do SpoIIIE and SftA from Bacillus activate SSR? Moreover, what is the selective advantage of having two chromosome DNA translocases? Additionally, 12% of the studied proteobacterial species do not possess the traditional Xer recombination machinery. Thus, it is still unknown whether these microorganisms lost the Xer recombination system, never acquired it or developed an alternative system to decatenate the chromosomes. It raises the question of how do bacterial cells handle chromosome decatenation without Xer recombinases and dif? Is there an alternative recombination system that functions as the dif/Xer system? Regarding single recombinases, have they evolved from XerC/XerD recombinases or vice versa, or did they arise from an ancestral recombinase? These and other issues already considered in this review are being gradually addressed by the use of the latest techniques in real-time imaging with super-resolution microscopy. Such as; photo-activated localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM), plus the use of other techniques as Förster (Fluorecence) resonance energy transfer (FRET), tethered fluorophore motion (TFM), single-molecule Flourescence resonance energy transfer (smFRET), among others. They are providing a powerful blueprint for investigators studying short- and long-range changes in DNA, DNA/protein, and protein/protein interactions. Researchers in SSR systems and protein–protein interactions might be the most direct beneficiaries of these techniques, especially when it is becoming urgent to further understand IMEX insertion and its subsequent influence in antibiotic resistance and bacterial virulence.
Author Contributions
All the authors contributed equally to the work by providing: Substantial contributions to the conception or design of the work; the acquisition, analysis, and interpretation of data for the work; Drafting the work or revising it critically for important intellectual concepts; Final approval of the version to be published; and Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
The authors acknowledge funding from Discovery Grant 106085-2013 from the Natural Sciences and Engineering Research Council of Canada. FC received salary support from the Département de Microbiologie, Infectiologie et Immunologie, and AB received salary support from the Tunisian Ministry of Higher Education.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Antonelli, A., D’Andrea, M. M., Di Pilato, V., Viaggi, B., Torricelli, F., and Rossolini, G. M. (2015). Characterization of a novel putative xer-dependent integrative mobile element carrying the blaNMC-A carbapenemase gene, inserted into the chromosome of members of the Enterobacter cloacae complex. Antimicrob. Agents Chemother. 59, 6620–6624. doi: 10.1128/AAC.01452-15
Aussel, L., Barre, F. X., Aroyo, M., Stasiak, A., Stasiak, A. Z., and Sherratt, D. (2002). FtsK is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 108, 195–205. doi: 10.1016/S0092-8674(02)00624-4
Austin, S., Ziese, M., and Sternberg, N. (1981). A novel role for site-specific recombination in maintenance of bacterial replicons. Cell 25, 729–736. doi: 10.1016/0092-8674(81)90180-X
Ayora, S., Carrasco, B., Cárdenas, P. P., César, C. E., Cañas, C., Yadav, T., et al. (2011). Double-strand break repair in bacteria: a view from Bacillus subtilis. FEMS Microbiol. Rev. 35, 1055–1081. doi: 10.1111/j.1574-6976.2011.00272.x
Azeroglu, B., Mawer, J. S. P., Cockram, C. A., White, M. A., Hasan, A. M. M., Filatenkova, M., et al. (2016). RecG directs DNA synthesis during double-strand break repair. PLoS Genet. 12:e1005799. doi: 10.1371/journal.pgen.1005799
Barre, F. X. (2007). FtsK and SpoIIIE: the tale of the conserved tails. Mol. Microbiol. 66, 1051–1055. doi: 10.1111/j.1365-2958.2007.05981.x
Barre, F. X., Søballe, B., Michel, B., Aroyo, M., Robertson, M., and Sherratt, D. (2001). Circles: the replication-recombination-chromosome segregation connection. Proc. Natl. Acad. Sci. U.S.A. 98, 8189–8195. doi: 10.1073/pnas.111008998
Bebel, A., Karaca, E., Kumar, B., and Stark, W. M. (2016). Structural snapshots of Xer recombination reveal activation by synaptic complex remodeling and DNA bending. Elife 5:e19706. doi: 10.7554/eLife.19706
Berezuk, A. M., Goodyear, M., and Khursigara, C. M. (2014). Site-directed fluorescence labeling reveals a revised N-terminal membrane topology and functional periplasmic residues in the Escherichia coli cell division protein FtsK. J. Biol. Chem. 289, 23287–23301. doi: 10.1074/jbc.M114.569624
Berza, I., Dishlers, A., Petrovskis, I., Tars, K., and Kazaks, A. (2013). Plasmid dimerization increases the production of hepatitis B core particles in E. coli. Biotechnol. Bioprocess Eng. 18, 850–857. doi: 10.1007/s12257-013-0188-5
Besprozvannaya, M., and Burton, B. M. (2014). Do the same traffic rules apply? Directional chromosome segregation by SpoIIIE and FtsK. Mol. Microbiol. 93, 599–608. doi: 10.1111/mmi.12708
Besprozvannaya, M., Pivorunas, V. L., Feldman, Z., and Burton, B. M. (2013). SpoIIIE protein achieves directional DNA translocation through allosteric regulation of ATPase activity by an accessory domain. J. Biol. Chem. 288, 28962–28974. doi: 10.1074/jbc.M113.484055
Bigot, S., Corre, J., Louarn, J. M., Cornet, F., and Barre, F. X. (2004). FtsK activities in Xer recombination, DNA mobilization and cell division involve overlapping and separate domains of the protein. Mol. Microbiol. 54, 876–886. doi: 10.1111/j.1365-2958.2004.04335.x
Bigot, S., Sivanathan, V., Possoz, C., Barre, F. X., and Cornet, F. (2007). FtsK, a literate chromosome segregation machine. Mol. Microbiol. 64, 1434–1441. doi: 10.1111/j.1365-2958.2007.05755.x
Biller, S. J., and Burkholder, W. F. (2009). The Bacillus subtilis SftA (YtpS) and SpoIIIe DNA translocases play distinct roles in growing cells to ensure faithful chromosome partitioning. Mol. Microbiol. 74, 790–809. doi: 10.1111/j.1365-2958.2009.06893.x
Bischerour, J., Spangenberg, C., and Barre, F.-X. (2012). Holliday junction affinity of the base excision repair factor Endo III contributes to cholera toxin phage integration. EMBO J. 31, 3757–3767. doi: 10.1038/emboj.2012.219
Bisicchia, P., Steel, B., and Debela, M. H. M. (2013). The N-terminal membrane-spanning domain of the Escherichia coli DNA translocase FtsK hexamerizes at midcell. MBio 4:e00800-13. doi: 10.1128/mBio.00800-13
Biswas, T., Aihara, H., Radman-Livaja, M., Filman, D., Landy, A., and Ellenberger, T. (2005). A structural basis for allosteric control of DNA recombination by lambda integrase. Nature 435, 1059–1066. doi: 10.1038/nature03657
Blaby, I. K., and Summers, D. K. (2009). The role of FIS in the Rcd checkpoint and stable maintenance of plasmid ColE1. Microbiology 155, 2676–2682. doi: 10.1099/mic.0.029777-0
Blakely, G., May, G., Mcculloch, R., Arciszewska, L. K., Burke, M., Lovett, S. T., et al. (1993). Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 75, 351–361. doi: 10.1016/0092-8674(93)80076-q
Bose, B., Reed, S. E., Besprozvannaya, M., and Burton, B. M. (2016). Missense mutations allow a sequence-blind mutant of spoIIIE to successfully translocate chromosomes during sporulation. PLoS ONE 11:e0148365. doi: 10.1371/journal.pone.0148365
Brown, W. R. A., Lee, N. C. O., Xu, Z., and Smith, M. C. M. (2011). Serine recombinases as tools for genome engineering. Methods 53, 372–379. doi: 10.1016/j.ymeth.2010.12.031
Bruning, J. G., Howard, J. L., and McGlynn, P. (2015). Accessory replicative helicases and the replication of protein-bound DNA. J. Mol. Biol. 426, 3917–3928. doi: 10.1016/j.jmb.2014.10.001
Burton, B. M., Marquis, K. A., Sullivan, N. L., Rapoport, T. A., and Rudner, D. Z. (2010). The ATPase SpoIIIE transports DNA across fused septal membranes during sporulation in Bacillus subtilis. Cell 131, 1301–1312. doi: 10.1016/j.cell.2007.11.009
Carnoy, C., and Roten, C.-A. (2009). The dif/Xer recombination systems in proteobacteria. PLoS ONE 4:e6531. doi: 10.1371/journal.pone.0006531
Carrasco, B., Cozar, M. C., Lurz, R., Alonso, J. C., and Ayora, S. (2004). Genetic recombination in Bacillus subtilis 168: contribution of holliday junction processing functions in chromosome segregation. J. Bacteriol. 186, 5557–5566. doi: 10.1128/JB.186.17.5557
Cattoni, D. I., Thakur, S., Godefroy, C., Le Gall, A., Lai-Kee-Him, J., Milhiet, P. E., et al. (2014). Structure and DNA-binding properties of the Bacillus subtilis SpoIIIE DNA translocase revealed by single-molecule and electron microscopies. Nucleic Acids Res. 42, 2624–2636. doi: 10.1093/nar/gkt1231
Chen, Y., Narendra, U., Iype, L. E., Cox, M. M., and Rice, P. A. (2000). Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol. Cell 6, 885–897. doi: 10.1016/S1097-2765(05)00088-2
Chen, Z., Yang, H., and Pavletich, N. P. (2008). Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453, 489–494. doi: 10.1038/nature06971
Christie, P. J., Whitaker, N., and González-Rivera, C. (2014). Mechanism and structure of the bacterial type IV secretion systems. Biochim. Biophys. Acta 1843, 1578–1591. doi: 10.1016/j.bbamcr.2013.12.019
Colloms, S. D. (2013). The topology of plasmid-monomerizing Xer site-specific recombination. Biochem. Soc. Trans. 41, 589–594. doi: 10.1042/BST20120340
Colloms, S. D., McCulloch, R., Grant, K., Neilson, L., and Sherratt, D. J. (1996). Xer-mediated site-specific recombination in vitro. EMBO J. 15, 1172–1181.
Colloms, S. D., Sykora, P., Szatmari, G., and Sherratt, D. J. (1990). Recombination at ColEl cer requires the Escherichia coli xerC gene product, a member of the lambda integrase family of site-specific recombinases. J. Bacteriol. 172, 6973–6980. doi: 10.1128/jb.172.12.6973-6980.1990
Cornet, F., Hallet, B., and Sherratt, D. J. (1997). Xer recombination in Escherichia coli. J. Biol. Chem. 272, 21927–21931. doi: 10.1074/jbc.272.35.21927
Cornet, F., Mortier, I., Patte, J., and Louarn, J. M. (1994). Plasmid pSC101 harbors a recombination site, psi, which is able to resolve plasmid multimers and to substitute for the analogous chromosomal Escherichia coli site dif. J. Bacteriol. 176, 3188–3195. doi: 10.1128/jb.176.11.3188-3195.1994
Cortez, D., Quevillon-Cheruel, S., Gribaldo, S., Desnoues, N., Sezonov, G., Forterre, P., et al. (2010). Evidence for a Xer/dif system for chromosome resolution in archaea. PLoS Genet. 6:e1001166. doi: 10.1371/journal.pgen.1001166
Costes, A., and Lambert, S. (2012). Homologous recombination as a replication fork escort: fork-protection and recovery. Biomolecules 3, 39–71. doi: 10.3390/biom3010039
Cox, M. M., Goodman, M. F., Kreuzer, K. N., Sherratt, D. J., Sandler, S. J., and Marians, K. J. (2000). The importance of repairing stalled replication forks. Nature 404, 37–41. doi: 10.1038/35003501
Cromie, G. A., and Leach, D. R. (2000). Control of crossing over. Mol. Cell 6, 815–826. doi: 10.1016/S1097-2765(05)00095-X
Crozat, E., Fournes, F., Cornet, F., Hallet, B., and Rousseau, P. (2014). Resolution of multimeric forms of circular plasmids and chromosomes. Microbiol. Spectr. 2, 1–16. doi: 10.1128/microbiolspec.PLAS-0025-2014.f1
Crozat, E., and Grainge, I. (2010). FtsK DNA translocase: the fast motor that knows where it’s going. ChemBioChem 11, 2232–2243. doi: 10.1002/cbic.201000347
Darmon, E., Eykelenboom, J. K., Lopez-Vernaza, M. A., White, M. A., and Leach, D. R. F. (2014). Repair on the go: E. Coli maintains a high proliferation rate while repairing a chronic DNA double-strand break. PLoS ONE 9:e110784. doi: 10.1371/journal.pone.0110784
Das, B. (2014). Mechanistic insights into filamentous phage integration in Vibrio cholerae. Front. Microbiol. 5:650. doi: 10.3389/fmicb.2014.00650
Das, B., Martínez, E., Midonet, C., and Barre, F.-X. (2013). Integrative mobile elements exploiting Xer recombination. Trends Microbiol. 21, 23–30. doi: 10.1016/j.tim.2012.10.003
Debowski, A. W., Carnoy, C., Verbrugghe, P., Nilsson, H.-O., Gauntlett, J. C., Fulurija, A., et al. (2012). Xer recombinase and genome integrity in Helicobacter pylori, a pathogen without topoisomerase IV. PLoS ONE 7:e33310. doi: 10.1371/journal.pone.0033310
Demarre, G., Galli, E., and Barre, F.-X. (2013). “The FtsK family of DNA pumps,” in DNA Helicases and DNA Motor Proteins: Advances in Experimental, ed. M. Spies (New York, NY: Springer), 245–262. doi: 10.1007/978-1-4614-5037-5
Demarre, G., Galli, E., Muresan, L., Paly, E., David, A., Possoz, C., et al. (2014). Differential management of the replication terminus regions of the two Vibrio cholerae chromosomes during cell division. PLoS Genet. 10:e1004557. doi: 10.1371/journal.pgen.1004557
Ðermić, D. (2015). Double-strand break repair mechanisms in Escherichia coli: recent insights. Adv. Genomics Genet. 5, 35–42. doi: 10.2147/AGG.S51699
Diagne, C. T., Salhi, M., Crozat, E., Salomé, L., Cornet, F., Rousseau, P., et al. (2014). TPM analyses reveal that FtsK contributes both to the assembly and the activation of the XerCD-dif recombination synapse. Nucleic Acids Res. 42, 1721–1732. doi: 10.1093/nar/gkt1024
Domínguez, N. M., Hackett, K. T., and Dillard, J. P. (2011). XerCD-mediated site-specific recombination leads to loss of the 57-kilobase gonococcal genetic Island. J. Bacteriol. 193, 377–388. doi: 10.1128/JB.00948-10
Dubarry, N., Possoz, C., and Barre, F. X. (2010). Multiple regions along the Escherichia coli FtsK protein are implicated in cell division. Mol. Microbiol. 78, 1088–1100. doi: 10.1111/j.1365-2958.2010.07412.x
Elshenawy, M. M., Jergic, S., Xu, Z.-Q., Sobhy, M. A., Takahashi, M., Oakley, A. J., et al. (2015). Replisome speed determines the efficiency of the Tus-Ter replication termination barrier. Nature 525, 394–398. doi: 10.1038/nature14866
Faruque, S. M., and Mekalanos, J. J. (2012). Phage-bacterial interactions in the evolution of toxigenic Vibrio cholerae. Virulence 3, 556–565. doi: 10.4161/viru.22351
Fiche, J. B., Cattoni, D. I., Diekmann, N., Langerak, J. M., Clerte, C., Royer, C. A., et al. (2013). Recruitment, assembly, and molecular architecture of the SpoIIIE DNA pump revealed by superresolution microscopy. PLoS Biol. 11:e1001557. doi: 10.1371/journal.pbio.1001557
Field, C. M., and Summers, D. K. (2011). Multicopy plasmid stability: revisiting the dimer catastrophe. J. Theor. Biol. 291, 119–127. doi: 10.1016/j.jtbi.2011.09.006
Fijalkowska, I. J., Schaaper, R. M., and Jonczyk, P. (2012). DNA replication fidelity in Escherichia coli: a multi-DNA polymerase affair. FEMS Microbiol. Rev. 36, 1105–1121. doi: 10.1111/j.1574-6976.2012.00338.x
Fournes, F., Crozat, E., Pages, C., Tardin, C., Salomé, L., Cornet, F., et al. (2016). FtsK translocation permits discrimination between an endogenous and an imported Xer/dif recombination complex. Proc. Natl. Acad. Sci. U.S.A. 113, 7882–7887. doi: 10.1073/pnas.1523178113
Gopaul, D. N., and Van Duyne, G. D. (1999). Structure and mechanism in site-specific recombination. Curr. Opin. Struct. Biol. 9, 14–20. doi: 10.1016/S0959-440X(99)80003-7
Gowrishankar, J. (2015). End of the beginning: elongation and termination features of alternative modes of chromosomal replication initiation in bacteria. PLoS Genet. 11:e1004909. doi: 10.1371/journal.pgen.1004909
Graham, J. E., Sherratt, D. J., and Szczelkun, M. D. (2010). Sequence-specific assembly of FtsK hexamers establishes directional translocation on DNA. Proc. Natl. Acad. Sci. U.S.A. 107, 20263–20268. doi: 10.1073/pnas.1007518107
Graham, J. E., Sivanathan, V., Sherratt, D. J., and Arciszewska, L. K. (2009). FtsK translocation on DNA stops at XerCD-dif. Nucleic Acids Res. 38, 72–81. doi: 10.1093/nar/gkp843
Grainge, I., Bregu, M., Vazquez, M., Sivanathan, V., Ip, S. C. Y., and Sherratt, D. J. (2007). Unlinking chromosome catenanes in vivo by site-specific recombination. EMBO J. 26, 4228–4238. doi: 10.1038/sj.emboj.7601849
Grainge, I., Lesterlin, C., and Sherratt, D. J. (2011). Activation of XerCD-dif recombination by the FtsK DNA translocase. Nucleic Acids Res. 39, 5140–5148. doi: 10.1093/nar/gkr078
Grindley, N. D. F., Whiteson, K. L., and Rice, P. A. (2006). Mechanisms of site-specific recombination. Annu. Rev. Biochem. 75, 567–605. doi: 10.1146/annurev.biochem.73.011303.073908
Groothuizen, F. S., and Sixma, T. K. (2015). The conserved molecular machinery in DNA mismatch repair enzyme structures. DNA Repair 38, 14–23. doi: 10.1016/j.dnarep.2015.11.012
Hallet, B., Arciszewska, L. K., and Sherratt, D. J. (1999). Reciprocal control of catalysis by the tyrosine recombinases XerC and XerD: an enzymatic switch in site-specific recombination. Mol. Cell 4, 949–959. doi: 10.1016/S1097-2765(00)80224-5
Handa, N., Morimatsu, K., Lovett, S. T., and Kowalczykowski, S. C. (2009). Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev. 23, 1234–1245. doi: 10.1101/gad.1780709
Harrison, P. W., Lower, R. P. J., Kim, N. K. D., and Young, J. P. W. (2010). Introducing the bacterial “chromid”: not a chromosome, not a plasmid. Trends Microbiol. 18, 141–148. doi: 10.1016/j.tim.2009.12.010
Hassan, F., Kamruzzaman, M., Mekalanos, J. J., and Faruque, S. M. (2010). Satellite phage TLCφ enables toxigenic conversion by CTX phage through dif site alteration. Nature 467, 982–985. doi: 10.1038/nature09469
Heller, R. C., and Marians, K. J. (2006). Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 7, 932–943. doi: 10.1038/nrm2058
Hendrickson, H., and Lawrence, J. G. (2006). Selection for chromosome architecture in bacteria. J. Mol. Evol. 62, 615–629. doi: 10.1007/s00239-005-0192-2
Hickman, A. B., Waninger, S., Scocca, J. J., and Dyda, F. (1997). Molecular organization in site-specific recombination: the catalytic domain of bacteriophage HP1 integrase at 2.7 A resolution. Cell 89, 227–237. doi: 10.1016/S0092-8674(00)80202-0
Hirano, N., Muroi, T., Takahashi, H., and Haruki, M. (2011). Site-specific recombinases as tools for heterologous gene integration. Appl. Microbiol. Biotechnol. 92, 227–239. doi: 10.1007/s00253-011-3519-5
Huber, K. E., and Waldor, M. K. (2002). Filamentous phage integration requires the host recombinases XerC and XerD. Nature 417, 656–659. doi: 10.1038/nature00782
Hwa Jo, C., Kim, J., Han, A., Yong Park, S., Yeon Hwang, K., Hyun Nam, K., et al. (2016). Crystal structure of Thermoplasma acidophilum XerA recombinase shows large C-shape clamp conformation and cis-cleavage mode for nucleophilic tyrosine. FEBS Lett. 590, 848–856. doi: 10.1002/1873-3468.12109
Jha, J. K., Baek, J. H., Venkova-Canova, T., and Chattoraj, D. K. (2012). Chromosome dynamics in multichromosome bacteria. Biochim. Biophys. Acta 1819, 826–829. doi: 10.1016/j.bbagrm.2012.01.012
Jha, J. K., Ramachandran, R., and Chattoraj, D. K. (2016). Opening the strands of replication origins—still an open question. Front. Mol. Biosci. 3:62. doi: 10.3389/fmolb.2016.00062
Johnston, C., Mortier-Barrière, I., Granadel, C., Polard, P., Martin, B., and Claverys, J. P. (2015). RecFOR is not required for pneumococcal transformation but together with XerS for resolution of chromosome dimers frequently formed in the process. PLoS Genet. 11:e1004934. doi: 10.1371/journal.pgen.1004934
Kaimer, C., González-Pastor, J. E., and Graumann, P. L. (2009). SpoIIIE and a novel type of DNA translocase, SftA, couple chromosome segregation with cell division in Bacillus subtilis. Mol. Microbiol. 74, 810–825. doi: 10.1111/j.1365-2958.2009.06894.x
Kaimer, C., and Graumann, P. L. (2011). Players between the worlds: multifunctional DNA translocases. Curr. Opin. Microbiol. 14, 719–725. doi: 10.1016/j.mib.2011.10.004
Kaimer, C., Schenk, K., and Graumann, P. L. (2011). Two DNA translocases synergistically affect chromosome dimer resolution in Bacillus subtilis. J. Bacteriol. 193, 1334–1340. doi: 10.1128/JB.00918-10
Keller, A. N., Xin, Y., Boer, S., Reinhardt, J., Baker, R., Arciszewska, L. K., et al. (2016). Activation of Xer-recombination at dif: structural basis of the FtsKγ–XerD interaction. Sci. Rep. 6, 1–12. doi: 10.1038/srep33357
Kirkup, B. C., Chang, L., Chang, S., Gevers, D., and Polz, M. F. (2010). Vibrio chromosomes share common history. BMC Microbiol. 10:137. doi: 10.1186/1471-2180-10-137
Kisker, C., Kuper, J., and Van Houten, B. (2013). Prokaryotic nucleotide excision repair. Cold Spring Harb. Perspect. Biol. 5, 1–18. doi: 10.1101/cshperspect.a012591
Kono, N., Arakawa, K., and Tomita, M. (2011). Comprehensive prediction of chromosome dimer resolution sites in bacterial genomes. BMC Genomics 12:19. doi: 10.1186/1471-2164-12-19
Kuempel, P., Henson, J., Dircks, L., Tecklenburg, M., and Lim, D. (1991). dif, a recA-independent recombination site in the terminus region of the chromosome of Escherichia coli. New Biol. 3, 799–811.
Kuzminov, A. (1999). Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 63, 751–813.
Kwon, H. J., Tirumalai, R., Landy, A., and Ellenberger, T. (1997). Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science 276, 126–131. doi: 10.1126/science.276.5309.126
Labib, K., and Hodgson, B. (2007). Replication fork barriers: pausing for a break or stalling for time? EMBO Rep. 8, 346–353. doi: 10.1038/sj.embor.7400940
Le Bourgeois, P., Bugarel, M., Campo, N., Daveran-Mingot, M.-L., Labonté, J., Lanfranchi, D., et al. (2007). The unconventional Xer recombination machinery of Streptococci/Lactococci. PLoS Genet. 3:e117. doi: 10.1371/journal.pgen.0030117
Lee, J. Y., Finkelstein, I. J., Arciszewska, L. K., Sherratt, D. J., and Greene, E. C. (2014). Single-molecule imaging of FtsK translocation reveals mechanistic features of protein-protein collisions on DNA. Mol. Cell 54, 832–843. doi: 10.1016/j.molcel.2014.03.033
Lee, J. Y., Finkelstein, I. J., Crozat, E., Sherratt, D. J., and Greene, E. C. (2012). Single-molecule imaging of DNA curtains reveals mechanisms of KOPS sequence targeting by the DNA translocase FtsK. Proc. Natl. Acad. Sci. U.S.A. 109, 6531–6536. doi: 10.1073/pnas.1201613109
Lenhart, J. S., Schroeder, J. W., Walsh, B. W., and Simmons, L. A. (2012). DNA repair and genome maintenance in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 76, 530–564. doi: 10.1128/MMBR.05020-11
Leroux, M., Jia, F., and Szatmari, G. (2011). Characterization of the Streptococcus suis XerS recombinase and its unconventional cleavage of the difSL site. FEMS Microbiol. Lett. 324, 135–141. doi: 10.1111/j.1574-6968.2011.02398.x
Leroux, M., Rezoug, Z., and Szatmari, G. (2013). The Xer/dif site-specific recombination system of Campylobacter jejuni. Mol. Genet. Genomics 288, 495–502. doi: 10.1007/s00438-013-0765-5
Lilley, D. M. J., and White, M. F. (2001). The junction-resolving enzymes. Nat. Rev. Mol. cell Biol. 2, 433–443. doi: 10.1038/35073057
Löwe, J., Ellonen, A., Allen, M. D., Atkinson, C., Sherratt, D. J., and Grainge, I. (2008). Molecular mechanism of sequence-directed DNA loading and translocation by FtsK. Mol. Cell 31, 498–509. doi: 10.1016/j.molcel.2008.05.027
Martin, S. S., Pulido, E., Chu, V. C., Lechner, T. S., and Baldwin, E. P. (2002). The order of strand exchanges in Cre-LoxP recombination and its basis suggested by the crystal structure of a Cre-LoxP holliday junction complex. J. Mol. Biol. 319, 107–127. doi: 10.1016/S0022-2836(02)00246-2
Martini, E., Diaz, R. L., Hunter, N., and Keeney, S. (2006). Crossover homeostasis in yeast meiosis. Cell 126, 285–295. doi: 10.1016/j.cell.2006.05.044
Massey, T. H., Aussel, L., Barre, F.-X., and Sherratt, D. J. (2004). Asymmetric activation of Xer site-specific recombination by FtsK. EMBO Rep. 5, 399–404. doi: 10.1038/sj.embor.7400116
Massey, T. H., Mercogliano, C. P., Yates, J., Sherratt, D. J., and Löwe, J. (2006). Double-stranded DNA translocation: structure and mechanism of hexameric FtsK. Mol. Cell 23, 457–469. doi: 10.1016/j.molcel.2006.06.019
May, P. F. J., Zawadzki, P., Sherratt, D. J., Kapanidis, A. N., and Arciszewska, L. K. (2015). Assembly, translocation, and activation of XerCD-dif recombination by FtsK translocase analyzed in real-time by FRET and two-color tethered fluorophore motion. Proc. Natl. Acad. Sci. U. S. A. 112, 5133–5141. doi: 10.1073/pnas.1510814112
Mazin, A. V., Timchenko, T. V., Saparbaev, M. K., and Mazina, O. M. (1996). Dimerization of plasmid DNA accelerates selection for antibiotic resistance. Mol. Microbiol. 20, 101–108. doi: 10.1007/978-3-642-28951-4
McLeod, S. M., and Waldor, M. K. (2004). Characterization of XerC- and XerD-dependent CTX phage integration in Vibrio cholerae. Mol. Microbiol. 54, 935–947. doi: 10.1111/j.1365-2958.2004.04309.x
Meinke, G., Bohm, A., Hauber, J., Pisabarro, M. T., and Buchholz, F. (2016). Cre recombinase and other tyrosine recombinases. Chem. Rev. 116, 12785–12820. doi: 10.1021/acs.chemrev.6b00077
Merrikh, H., Zhang, Y., Grossman, A. D., and Wang, J. D. (2012). Replication–transcription conflicts in bacteria. Nat. Rev. Microbiol. 10, 449–458. doi: 10.1038/nrmicro2800
Michel, B., Recchia, G. D., Penel-Colin, M., Ehrlich, S. D., and Sherratt, D. J. (2000). Resolution of Holliday junctions by RuvABC prevents dimer formation in rep mutants and UV-irradiated cells. Mol. Microbiol. 37, 180–191. doi: 10.1046/j.1365-2958.2000.01989.x
Midonet, C., and Barre, F.-X. (2016). How Xer-exploiting mobile elements overcome cellular control. Proc. Natl. Acad. Sci. U.S.A. 113, 8343–8345. doi: 10.1073/pnas.1608539113
Midonet, C., Das, B., Paly, E., and Barre, F.-X. (2014). XerD-mediated FtsK-independent integration of TLCupphi into the Vibrio cholerae genome. Proc. Natl. Acad. Sci. U.S.A. 111, 16848–16853. doi: 10.1073/pnas.1404047111
Million-Weaver, S., and Camps, M. (2014). Mechanisms of plasmid segregation: Have multicopy plasmids been overlooked? Plasmid 75, 27–36. doi: 10.1017/CBO9781107415324.004
Nolivos, S., Pages, C., Rousseau, P., Le Bourgeois, P., and Cornet, F. (2010). Are two better than one? Analysis of an FtsK/Xer recombination system that uses a single recombinase. Nucleic Acids Res. 38, 6477–6489. doi: 10.1093/nar/gkq507
Nolivos, S., Touzain, F., Pages, C., Coddeville, M., Rousseau, P., El Karoui, M., et al. (2012). Co-evolution of segregation guide DNA motifs and the FtsK translocase in bacteria: identification of the atypical Lactococcus lactis KOPS motif. Nucleic Acids Res. 40, 5535–5545. doi: 10.1093/nar/gks171
Pagès, V. (2016). Single-strand gap repair involves both RecF and RecBCD pathways. Curr. Genet. 62, 519–521. doi: 10.1007/s00294-016-0575-5
Pandey, M., Elshenawy, M. M., Jergic, S., Takahashi, M., Dixon, N. E., Hamdan, S. M., et al. (2015). Two mechanisms coordinate replication termination by the Escherichia coli Tus– Ter complex. Nucleic Acids Res. 43, 5924–5935. doi: 10.1093/nar/gkv527
Pérals, K., Cornet, F., Merlet, Y., Delon, I., and Louarn, J. M. (2000). Functional polarization of the Escherichia coli chromosome terminus: the dif site acts in chromosome dimer resolution only when located between long stretches of opposite polarity. Mol. Microbiol. 36, 33–43. doi: 10.1046/j.1365-2958.2000.01847.x
Pomerantz, R. T., and O’Donnell, M. (2010). What happens when replication and transcription complexes collide? Cell Cycle 9, 2537–2543. doi: 10.4161/cc.9.13.12122
Rajeev, L., Malanowska, K., and Gardner, J. F. (2009). Challenging a paradigm: the role of DNA homology in tyrosine recombinase reactions. Microbiol. Mol. Biol. Rev. 73, 300–309. doi: 10.1128/MMBR.00038-08
Recchia, G. D., Aroyo, M., Wolf, D., Blakely, G., and Sherratt, D. J. (1999). FtsK-dependent and -independent pathways of Xer site-specific recombination. EMBO J. 18, 5724–5734. doi: 10.1093/emboj/18.20.5724
Recchia, G. D., and Sherratt, D. J. (1999). Conservation of xer site specific recombinaion genes in bacteria. Mol. Microbiol. 34, 1146–1148. doi: 10.1046/j.1365-2958.1999.01668.x
Reijns, M., Lu, Y., Leach, S., and Colloms, S. D. (2005). Mutagenesis of PepA suggests a new model for the Xer/cer synaptic complex. Mol. Microbiol. 57, 927–941. doi: 10.1111/j.1365-2958.2005.04716.x
Reyes-Lamothe, R., Nicolas, E., and Sherratt, D. J. (2012). Chromosome replication and segregation in bacteria. Annu. Rev. Genet. 46, 121–143. doi: 10.1146/annurev-genet-110711-155421
Rocha, E. P. C., Cornet, E., and Michel, B. (2005). Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet. 1:e15. doi: 10.1371/journal.pgen.0010015
Roghanian, M., Zenkin, N., and Yuzenkova, Y. (2015). Bacterial global regulators DksA/ppGpp increase fidelity of transcription. Nucleic Acids Res. 43, 1529–1536. doi: 10.1093/nar/gkv003
Saleh, O. A., Bigot, S., Barre, F.-X., and Allemand, J.-F. (2005). Analysis of DNA supercoil induction by FtsK indicates translocation without groove-tracking. Nat. Struct. Mol. Biol. 12, 436–440. doi: 10.1038/nsmb926
Sciochetti, S. A., Piggot, P. J., and Garry, W. (2001). Identification and Characterization of the dif Site from Bacillus subtilis. J. Bacteriol. 183, 1058–1068. doi: 10.1128/JB.183.3.1058
Sciochetti, S. A., Piggot, P. J., Sherratt, D. J., and Blakely, G. (1999). The ripX locus of Bacillus subtilis encodes a site-specific recombinase involved in proper chromosome partitioning. J. Bacteriol. 181, 6053–6062.
Sénéchal, H., Delesques, J., and Szatmari, G. (2010). Escherichia coli ArgR mutants defective in cer/Xer recombination, but not in DNA binding. FEMS Microbiol. Lett. 305, 162–169. doi: 10.1111/j.1574-6968.2010.01921.x
Sengupta, M., and Austin, S. (2011). Prevalence and significance of plasmid maintenance functions in the virulence plasmids of pathogenic bacteria. Infect. Immun. 79, 2502–2509. doi: 10.1128/IAI.00127-11
Serre, M. C., El Arnaout, T., Brooks, M. A., Durand, D., Lisboa, J., Lazar, N., et al. (2013). The carboxy-terminal αN helix of the archaeal XerA tyrosine recombinase is a molecular switch to control site-specific recombination. PLoS ONE 8:e63010. doi: 10.1371/journal.pone.0063010
Sherratt, D. J., Arciszewska, L. K., Blakely, G., Colloms, S., Grant, K., Leslie, N., et al. (1995). Site-specific recombination and circular chromosome segregation. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 347, 37–42. doi: 10.1098/rstb.1995.0006
Sherratt, D. J., Søballe, B., Barre, F.-X., Filipe, S., Lau, I., Massey, T., et al. (2004). Recombination and chromosome segregation. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 359, 61–69. doi: 10.1098/rstb.2003.1365
Sivanathan, V., Allen, M. D., de Bekker, C., Baker, R., Arciszewska, L. K., Freund, S. M., et al. (2006). The FtsK γ domain directs oriented DNA translocation by interacting with KOPS. Nat. Struct. Mol. Biol. 13, 965–972. doi: 10.1038/nsmb1158
Spies, M., and Kowalczykowski, S. C. (2005). “RecBCD and RecF Pathways,” in The Bacterial Chromosome, ed. N. Patrick Higgins (Washington, DC: ASM press), 389–403. doi: 10.1128/9781555817640.ch21
Steiner, W., Liu, G., Donachie, W. D., and Kuempel, P. (1999). The cytoplasmic domain of FtsK protein is required for resolution of chromosome dimers. Mol. Microbiol. 31, 579–583. doi: 10.1046/j.1365-2958.1999.01198.x
Steiner, W. W., and Kuempel, P. L. (1998). Sister chromatid exchange frequencies in Escherichia coli analyzed by recombination at the dif resolvase site. J. Bacteriol. 180, 6269–6275.
Stirling, C. J., Colloms, S. D., Collins, J. F., Szatmari, G., and Sherratt, D. J. (1989). xerB, an Escherichia coli gene required for plasmid ColE1 site-specific recombination, is identical to pepA, encoding aminopeptidase A, a protein with substantial similarity to bovine lens leucine aminopeptidase. EMBO J. 8, 1623–1627.
Stirling, C. J., Stewart, G., and Sherratt, D. J. (1988a). Multicopy plasmid stability in Escherichia coli. Mol. Gen. Genet. 214, 80–84. doi: 10.1007/BF00340183
Stirling, C. J., Szatmari, G., Stewart, G., Smith, M. C. M., and Sherratt, D. J. (1988b). The arginine repressor is essential for plasmid-stabilizing site-specific recombination at the ColE1 cer locus. EMBO J. 7, 4389–4395.
Subramanya, H. S., Arciszewska, L. K., Baker, R. A., Bird, L. E., Sherratt, D. J., and Wigley, D. B. (1997). Crystal structure of the site-specific recombinase, XerD. EMBO J. 16, 5178–5187. doi: 10.1093/emboj/16.17.5178
Summers, D. K., Beton, C. W., and Withers, H. L. (1993). Multicopy plasmid instability: the dimer catastrophe hypothesis. Mol. Microbiol. 8, 1031–1038. doi: 10.1111/j.1365-2958.1993.tb01648.x
Summers, D. K., and Sherratt, D. J. (1984). Multimerization of high copy number plasmids causes instabilityϕ: ColEI encodes a determinant essential for plasmid monomerization and stability. Cell 36, 1097–1103. doi: 10.1016/0092-8674(84)90060-6
Swalla, B. M., Gumport, R. I., and Gardner, J. F. (2003). Conservation of structure and function among tyrosine recombinases: homology-based modeling of the lambda integrase core-binding domain. Nucleic Acids Res. 31, 805–818. doi: 10.1093/nar/gkg142
Tehranchi, A. K., Blankschien, M. D., Zhang, Y., Halliday, J., Peng, J., Herman, C., et al. (2010). The transcription factor DksA prevents disruption of DNA replication upon nutritional stress. Cell 141, 595–605. doi: 10.1016/j.cell.2010.03.036
Val, M. E., Bouvier, M., Campos, J., Sherratt, D., Cornet, F., Mazel, D., et al. (2005). The single-stranded genome of phage CTX is the form used for integration into the genome of Vibrio cholerae. Mol. Cell 19, 559–566. doi: 10.1016/j.molcel.2005.07.002
Val, M.-E., Kennedy, S. P., El Karoui, M., Bonné, L., Chevalier, F., and Barre, F.-X. (2008). FtsK-dependent dimer resolution on multiple chromosomes in the pathogen Vibrio cholerae. PLoS Genet. 4:e1000201. doi: 10.1371/journal.pgen.1000201
Val, M.-E., Marbouty, M., de Lemos Martins, F., Kennedy, S. P., Kemble, H., Bland, M. J., et al. (2016). A checkpoint control orchestrates the replication of the two chromosomes of Vibrio cholerae. Sci. Adv. 2, 1–14. doi: 10.1126/sciadv.1501914
van der Veen, S., and Tang, C. M. (2015). The BER necessities: the repair of DNA damage in human-adapted bacterial pathogens. Nat. Rev. Microbiol. 13, 83–94. doi: 10.1038/nrmicro3391
Van Gool, A. J., Hajibagheri, N. M. A., Stasiak, A., and West, S. C. (1999). Assembly of the Escherichia coli RuvABC resolvasome directs the orientation of Holliday junction resolution. Genes Dev. 13, 1861–1870. doi: 10.1101/gad.13.14.1861
Vanhooff, V., Normand, C., Galloy, C., Segall, A. M., and Hallet, B. (2009). Control of directionality in the DNA strand-exchange reaction catalysed by the tyrosine recombinase TnpI. Nucleic Acids Res. 38, 2044–2056. doi: 10.1093/nar/gkp1187
Veiga, H., and Pinho, M. (2016). Staphylococcus aureus requires at least one FtsK/SpoIIIE protein for correct chromosome segregation. Mol. Microbiol. 103, 504–517. doi: 10.1111/mmi.13572
Vos, M., and Didelot, X. (2009). A comparison of homologous recombination rates in bacteria and archaea. ISME J. 3, 199–208. doi: 10.1038/ismej.2008.93
Wang, X., Llopis, P. M., and Rudner, D. Z. (2013). Organization and segregation of bacterial chromosomes. Nat. Rev. Genet. 14, 1–22. doi: 10.1038/nrg3375
Warren, D., Laxmikanthan, G., and Landy, A. (2013). Integrase family of site-specific recombinases. Brenners Encycl. Genet. 4, 100–105. doi: 10.1016/B978-0-12-374984-0.01520-5
Wu, H., Zhang, Z., Hu, S., and Yu, J. (2012). On the molecular mechanism of GC content variation among eubacterial genomes. Biol. Direct 7:2. doi: 10.1186/1745-6150-7-2
Wu, L. J. (2009). It takes two DNA translocases to untangle chromosomes from the division septum. Mol. Microbiol. 74, 773–776. doi: 10.1111/j.1365-2958.2009.06892.x
Xu, Q., Dziejman, M., and Mekalanos, J. J. (2003). Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 1286–1291. doi: 10.1073/pnas.0337479100
Yates, J., Aroyo, M., Sherratt, D. J., and Barre, F.-X. (2003). Species specificity in the activation of Xer recombination at dif by FtsK. Mol. Microbiol. 49, 241–249. doi: 10.1046/j.1365-2958.2003.03574.x
Yeeles, J. T. P., and Dillingham, M. S. (2010). The processing of double-stranded DNA breaks for recombinational repair by helicase-nuclease complexes. DNA Repair 9, 276–285. doi: 10.1016/j.dnarep.2009.12.016
Yeeles, J. T. P., Poli, J., Marians, K. J., and Pasero, P. (2013). Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 5, 1–16. doi: 10.1101/cshperspect.a012815
Yen Shin, J., Lopez-Garrido, J., Lee, S.-H., Diaz-Celis, C., Fleming, T., Bustamante, C., et al. (2015). Visualization and functional dissection of coaxial paired SpoIIIE channels across the sporulation septum. Elife 4, 1–24. doi: 10.7554/eLife.06474
Yu, X. C., Weihe, E. K., and Margolin, W. (1998). Role of the C terminus of FtsK in Escherichia coli chromosome segregation. J. Bacteriol. 180, 6424–6428.
Zawadzki, P., May, P. F. J., Baker, R. A., Pinkney, J. N. M., Kapanidis, A. N., and Sherratt, D. J. (2013). Conformational transitions during FtsK translocase activation of individual XerCD–dif recombination complexes. PNAS 110, 17302–17307. doi: 10.1073/pnas.1311065110
Keywords: site-specific recombination, tyrosine recombinases, single recombinases, XerS, XerH, XerA, IMEX
Citation: Castillo F, Benmohamed A and Szatmari G (2017) Xer Site Specific Recombination: Double and Single Recombinase Systems. Front. Microbiol. 8:453. doi: 10.3389/fmicb.2017.00453
Received: 01 December 2016; Accepted: 03 March 2017;
Published: 20 March 2017.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Ian Grainge, University of Newcastle, AustraliaAlan Leonard, Florida Institute of Technology, USA
Gregory Marczynski, McGill University, Canada
Copyright © 2017 Castillo, Benmohamed and Szatmari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Castillo, ZmFiaW8uYW5kcmVzLmNhc3RpbGxvLm1hcnRpbmV6QHVtb250cmVhbC5jYQ== George Szatmari, Z2VvcmdlLnN6YXRtYXJpQHVtb250cmVhbC5jYQ==