 Alex Orlek1,2*
Alex Orlek1,2* Nicole Stoesser1
Nicole Stoesser1 Muna F. Anjum2,3
Muna F. Anjum2,3 Michel Doumith4Matthew J. Ellington2,4Tim Peto1,2Derrick Crook1,2Neil Woodford2,4A. Sarah Walker1,2
Michel Doumith4Matthew J. Ellington2,4Tim Peto1,2Derrick Crook1,2Neil Woodford2,4A. Sarah Walker1,2 Hang Phan1,2†
Hang Phan1,2† Anna E. Sheppard1,2†
Anna E. Sheppard1,2†
- 1Nuffield Department of Medicine, John Radcliffe Hospital, University of Oxford, Oxford, UK
- 2National Institute for Health Research Health Protection Research Unit in Healthcare Associated Infections and Antimicrobial Resistance, University of Oxford, Oxford, UK
- 3Department of Bacteriology, Animal and Plant Health Agency, Addlestone, UK
- 4Antimicrobial Resistance and Healthcare Associated Infections Reference Unit, Public Health England, London, UK
Plasmids are extra-chromosomal genetic elements ubiquitous in bacteria, and commonly transmissible between host cells. Their genomes include variable repertoires of ‘accessory genes,’ such as antibiotic resistance genes, as well as ‘backbone’ loci which are largely conserved within plasmid families, and often involved in key plasmid-specific functions (e.g., replication, stable inheritance, mobility). Classifying plasmids into different types according to their phylogenetic relatedness provides insight into the epidemiology of plasmid-mediated antibiotic resistance. Current typing schemes exploit backbone loci associated with replication (replicon typing), or plasmid mobility (MOB typing). Conventional PCR-based methods for plasmid typing remain widely used. With the emergence of whole-genome sequencing (WGS), large datasets can be analyzed using in silico plasmid typing methods. However, short reads from popular high-throughput sequencers can be challenging to assemble, so complete plasmid sequences may not be accurately reconstructed. Therefore, localizing resistance genes to specific plasmids may be difficult, limiting epidemiological insight. Long-read sequencing will become increasingly popular as costs decline, especially when resolving accurate plasmid structures is the primary goal. This review discusses the application of plasmid classification in WGS-based studies of antibiotic resistance epidemiology; novel in silico plasmid analysis tools are highlighted. Due to the diverse and plastic nature of plasmid genomes, current typing schemes do not classify all plasmids, and identifying conserved, phylogenetically concordant genes for subtyping and phylogenetics is challenging. Analyzing plasmids as nodes in a network that represents gene-sharing relationships between plasmids provides a complementary way to assess plasmid diversity, and allows inferences about horizontal gene transfer to be made.
Introduction
Plasmid genomes generally include a ‘backbone’ of core genetic loci, which are somewhat conserved amongst broadly related plasmids of the same family (Phan et al., 2009), and associated with key plasmid-specific functions such as replication and mobility. Accessory genes may also be present, and often confer clinically relevant traits such as virulence and antibiotic resistance (Thomas and Summers, 2008). Plasmids can act as efficient vectors of horizontal gene transfer (HGT). Notably, during conjugation, a plasmid promotes its own transfer (and/or that of a co-resident plasmid) from one bacterial cell to another (Norman et al., 2009). Accessory genes are therefore frequently spread by virtue of being located on transmissible plasmids; moreover, they are commonly associated with smaller mobile elements such as transposons, facilitating intracellular mobilization amongst plasmids, or to the chromosome (Stokes and Gillings, 2011). Due to their ability to transmit genes encoding adaptive traits across bacterial populations, plasmids can enable bacteria to evolve rapidly under environmental pressure (Heuer and Smalla, 2012). A striking example of bacterial adaptive evolution is that of antibiotic resistance, which is driven, in part, by dissemination of resistance plasmids (plasmids conferring antibiotic resistance), and now threatens modern medicine (Carattoli, 2013; World Health Organization, 2014).
Classifying plasmids according to a typing scheme provides useful insights into the epidemiology of plasmid-mediated antibiotic resistance: for example, studying the composition of plasmid types can indicate whether an antibiotic resistance epidemic is driven by diverse plasmids or one dominant plasmid type (Valverde et al., 2009). In addition, hypotheses about resistance transmission during outbreaks can be refined according to the relatedness of resistance plasmids harbored by clinical strains (Pecora et al., 2015). The principal plasmid classification schemes are replicon and MOB typing, based on backbone loci encoding plasmid replication and mobility functions, respectively (Carattoli et al., 2005; Garcillán-Barcia et al., 2009). Whilst these single-locus typing schemes have been widely and successfully applied, they provide limited resolution (Fricke et al., 2009), restricting epidemiological inference: in an outbreak context, if two patients are infected by unrelated strains harboring resistance plasmids of the same type, this raises the possibility of plasmid transmission, but plasmid transmission cannot be conclusively ruled-in using single-locus plasmid typing alone; further higher-resolution investigation would be required (Foxman et al., 2005). If resistance plasmids are unrelated, plasmid transmission can be ruled-out, though a transmission link via resistance gene transposition is possible.
Plasmid typing may provide a stepping-stone to higher resolution analyses; identifying shared (‘core’) genes amongst related plasmids can inform development of plasmid multi-locus sequence typing (pMLST) schemes (García-Fernández et al., 2011), or allow phylogenetic relationships to be reconstructed based on core gene single nucleotide polymorphisms (SNPs) (de Been et al., 2014). Unfortunately, determining high-resolution plasmid relationships is challenging: the tendency of plasmids to gain, lose and rearrange genetic content means sets of plasmids – even if of the same type – will tend to share few phylogenetically concordant core genes (Fondi et al., 2010; Tazzyman and Bonhoeffer, 2014), impeding subtyping and phylogenetic analysis (Maiden, 2006). Even backbone genes may not be well conserved across all plasmids of the same type (Lanza et al., 2014), and sometimes show mosaic phylogenetic origins (Sen et al., 2013).
Whole-genome sequencing (WGS) data can now be obtained for many bacterial isolates, at relatively low cost, within short timescales (Metzker, 2010). Whilst sequencing reads from a bacterial isolate represent plasmid(s) as well as the chromosome, WGS-based studies have often focused on the host strain chromosome as the unit of interest (Croucher and Didelot, 2015). For strain-level clinical surveillance to elucidate antibiotic resistance transmission routes, dissemination should primarily involve clonal transmission of particular antibiotic-resistant strains. However, recent analyses indicate that plasmids may transmit between strains frequently, even over short timescales (Conlan et al., 2014; Sheppard et al., 2016). Therefore, the chain of transmission no longer simply corresponds to strain transmission; resistance plasmid dissemination across strains recruits different recipient strains into the outbreak too, resulting in a ‘plasmid outbreak.’ Although insight may be limited by difficulties in determining high-resolution plasmid relationships, these dynamics mean that plasmid analysis across a variety of strains is important, including for short-term surveillance studies (Adler and Carmeli, 2011).
Conventional PCR-based plasmid typing methods are commonly used, but in silico approaches for classifying sequenced plasmids are also available. WGS datasets from short-read sequencing projects offer exciting opportunities for large-scale plasmid analysis, while presenting the additional challenge of assembling reads to resolve individual plasmid structures. After summarizing current plasmid classification schemes (replicon and MOB typing), this review discusses the opportunities and challenges of conducting in silico plasmid typing on WGS datasets to gain insight into plasmid-mediated resistance epidemiology. We highlight novel tools for WGS-based plasmid analysis, and examine gene-sharing networks as a complementary approach for analyzing plasmid relationships. This review focuses on WGS datasets from cultured rather than metagenomic samples; for the latter, see recent reviews (Jørgensen et al., 2014; Martínez et al., 2016).
Plasmid Typing Schemes
Replicon typing schemes exploit genetic elements of the replicon region (encoding replication machinery) (Table 1). Couturier et al. (1988) typed plasmids according to Southern blot hybridization, using replicons from plasmids of different incompatibility groups as probes. However, this method is limited by probe cross-hybridization amongst closely related replicon sequences (Carattoli, 2009). PCR-based replicon typing (PBRT) – where plasmids are typed according to PCRs targeting various replicon sequences – is less laborious, and shows higher specificity in detecting replicons (Carattoli et al., 2005). For gram-negative bacteria, PBRT schemes targeting replicons found in Enterobacteriaceae and Acinetobacter baumannii plasmids are available (Carattoli et al., 2005; Bertini et al., 2010). A PBRT scheme for plasmids of gram-positive bacteria has been developed, focusing on enterococcal (Jensen et al., 2010) and staphylococcal (Lozano et al., 2012) plasmids. For common Enterobacteriaceae replicon types, pMLST schemes have been devised for subtyping (Brolund and Sandegren, 2016; Hancock et al., 2016). Availability of WGS data has motivated the development of in silico replicon typing and subtyping tools, which have been validated for Enterobacteriaceae plasmids (Carattoli et al., 2014). For plasmids from taxa not represented by existing in silico tools, ad hoc in silico methods have been derived from PBRT schemes (Shintani et al., 2015; Brodrick et al., 2016).

TABLE 1. Summary of plasmid typing and subtyping schemes.
MOB typing exploits the conserved N-terminal sequence of the relaxase proteins encoded by transmissible plasmids (Francia et al., 2004; Garcillán-Barcia et al., 2009). As with replicon typing, both PCR-based and in silico approaches are used for MOB typing (Table 1). Compared with replicon typing, MOB typing classifies plasmids at lower resolution (Garcillán-Barcia et al., 2011).
A drawback of replicon typing is that individual plasmids can contain multiple replicons, complicating classification, whereas usually just one relaxase is encoded. However, due to its finer resolution, replicon typing provides more detailed information on plasmid relatedness, particularly if a pMLST subtyping scheme is available (Garcillán-Barcia and de la Cruz, 2013). Even within relatively well-studied taxa, neither scheme classifies all plasmids, likely reflecting diversity in plasmid backbones. Shintani et al. (2015) assessed in silico typing, and found that the proportion of Enterobacteriaceae plasmids that could be replicon typed was 75%; for Acinetobacter plasmids the proportion was 67%. Only around half of plasmids from major gram-positive taxa could be replicon typed (51% Firmicutes plasmids, 49% Actinobacteria plasmids), although the proportion was higher for enterococcal and staphylococcal plasmids (83 and 85% respectively) (Supplementary Table S1 in Shintani et al., 2015). Lanza et al. (2015) also highlight gaps in replicon typing of Firmicutes plasmids. MOB typing only types transmissible plasmids (∼50% γ-Proteobacterial plasmids; ∼35% Firmicutes plasmids) (Smillie et al., 2010).
WGS Data for Plasmid Classification: Opportunities and Challenges
When analyzing whole genomic DNA, limited information can be derived from plasmid typing alone: bacterial cells may contain multiple different plasmids, and a single plasmid may contain multiple replicons, obscuring correspondence between detected replicons and the set of plasmid types within a host cell (Johnson et al., 2007). Therefore, in PCR-based studies – where genomic context of an amplicon remains unknown – plasmids are commonly isolated first, before being individually characterized by replicon and resistance typing (see Table 11 in EFSA, 2011). This is time-consuming, restricts the number of isolates that can be analyzed, and has inherent limitations: if plasmids from the same isolate are of similar size they cannot be separated by pulsed-field gel electrophoresis, and isolation by transfer to recipient cells is not always achieved (Dib et al., 2015).
Potentially, WGS enables in silico analyses in which plasmid typing and analysis of loci of interest, such as resistance genes (and their plasmid or chromosomal genetic context), are performed in a unified way. Consequently, much larger isolate collections can be analyzed – a key advantage given that plasmid studies have indicated a need for including more strains in analyses (e.g., environmental strains, or isolates exhibiting only low-level resistance) to uncover more complex transmission routes (Carrër et al., 2010; Stoesser et al., 2014, 2015a). Furthermore, a thorough mechanistic understanding of clinically important aspects of plasmid biology, such as host range and phenotypic effects, may require analysis of the wider plasmid genome rather than specific loci.
Unfortunately, there are significant obstacles to in silico analysis of WGS data. Popular high-throughput sequencing technologies (e.g., Illumina) produce short (∼100–300 bp) reads, for which assembly is inherently challenging (Nagarajan and Pop, 2013). Isolating individual plasmids prior to sequencing simplifies assembly, potentially enabling complete plasmid reconstruction (Mathers et al., 2015), but is laborious. Long-read sequencing vastly simplifies assembly, but high costs restrict its use (Koren and Phillippy, 2015). Therefore, a major challenge lies in extracting useful information from short sequencing reads derived from different sources (different co-resident plasmids, the chromosome).
Reference-Based Read Mapping versus De novo Read Assembly
Having obtained short reads from WGS of isolate DNA, reads are generally mapped to a reference and/or assembled de novo using a de Bruijn graph assembler (Zerbino and Birney, 2008; Compeau et al., 2011) (for detailed workflows see Edwards and Holt, 2013; Lynch et al., 2016). Reference-based read mapping is a fast and accurate method to characterize SNPs and detect loci of interest (Li and Durbin, 2009). Identified core genome SNPs can be used to construct strain phylogenies. In addition, rapid epidemiological surveillance of replicons and resistance genes can potentially be achieved with read mapping tools such as SRST2 (Inouye et al., 2014). However, the read mapping approach is limited when structural information is of interest: is a detected resistance gene located on chromosome or plasmid, and if the latter, which plasmid type is it associated with? One approach to overcome this is de novo read assembly, which can be less sensitive for SNP or locus detection (Inouye et al., 2014), but provides reference-free structural information, and can identify loci not represented on available references. Table 2 summarizes key in silico tools for plasmid analysis.
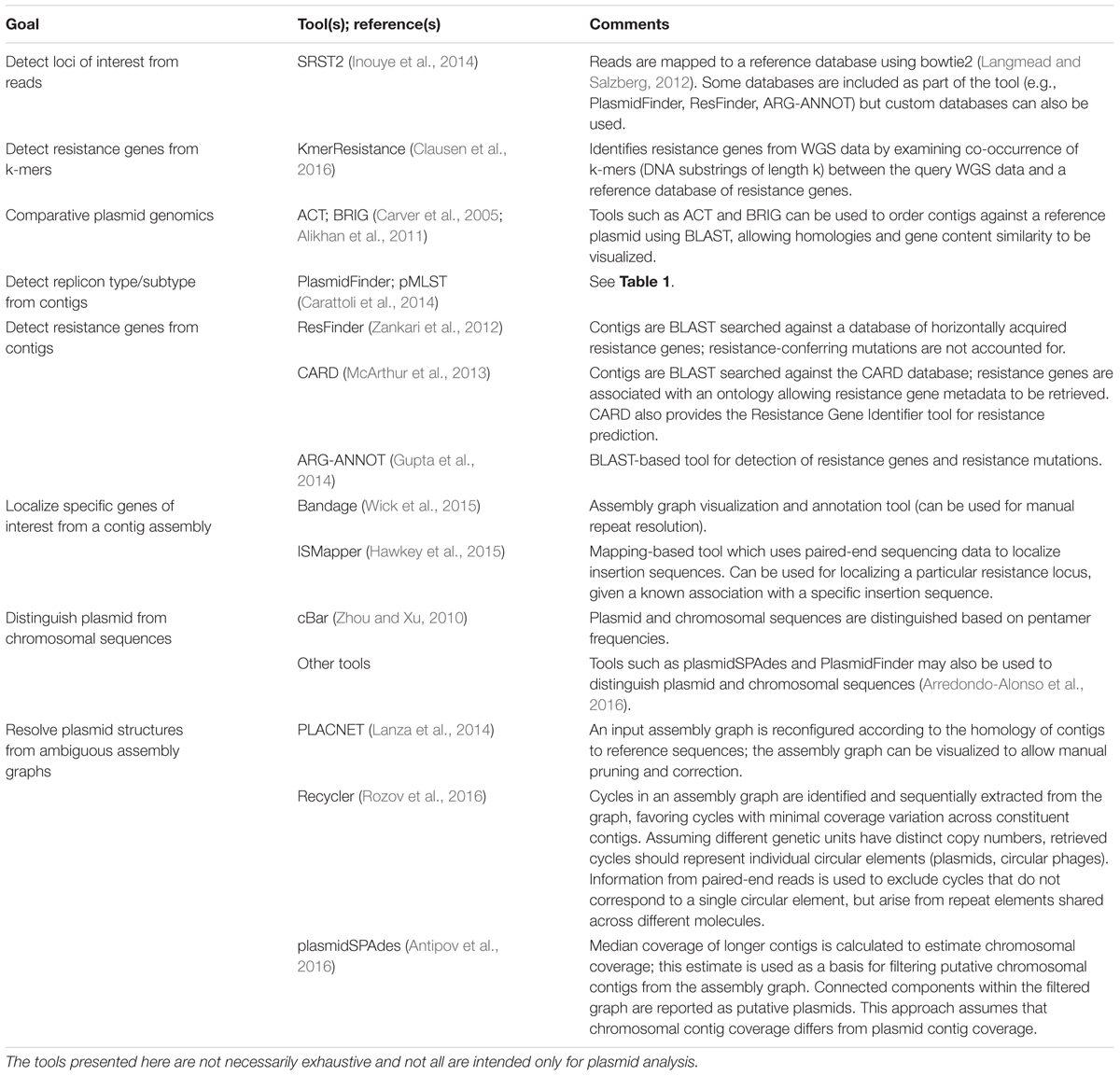
TABLE 2. Summary of common in silico tools used for plasmid analysis.
Determining the Genetic Context of Resistance Genes from De novo Assemblies
Sometimes, short reads can be assembled into complete plasmid structures, and plasmid-localized resistance genes can be identified using a combination of in silico plasmid typing and resistance gene typing methods (e.g., PlasmidFinder and ResFinder). However, complete plasmid assembly is frequently not possible (Arredondo-Alonso et al., 2016). Notably, the presence of multiple copies of the same repeat structure – a common situation in plasmid genomes – introduces assembly ambiguity, which can fragment assemblies (Pevzner et al., 2001; Treangen and Salzberg, 2011). Paired-end sequencing data can resolve repeat location, but only if the paired reads span the length of the repeat. If contigs contain sufficient informative sequence, plasmid and chromosomal contigs can be distinguished by BLAST searching against the Genbank nucleotide database (Seni et al., 2016).
Resistance genes are frequently flanked by repetitive mobile elements, and are therefore prone to poor assembly, obscuring their genetic context. There are several tools which can help resolve the location of specific loci of interest (Holt, 2015). Bandage allows visualization and annotation of the assembly graph; for example, users can zoom to unresolved repeat regions and BLAST search connecting contigs (Wick et al., 2015). If all connecting contigs match either plasmid or chromosomal references, then an ambiguously linked region can be assigned accordingly. For example, Bandage helped to reveal diverse plasmid contexts for the mcr-1 colistin resistance gene in UK clinical isolates (Doumith et al., 2016). However, this manual approach is unfeasible when analyzing large datasets. In some cases, the genetic context of resistance genes may be inferred for large datasets using the ISMapper tool, as demonstrated by a study of 1832 isolates belonging to the successful H58 Salmonella Typhi lineage (Wong et al., 2015).
Reference-Based Mapping to Track Plasmid Transmission during Short-term Outbreaks
As well as detecting loci of interest, reference-based mapping has been used to track plasmids during short-term outbreaks. Specifically, ‘index’ plasmids of an outbreak are fully assembled, often through long-read sequencing. Short reads or contigs from subsequent isolates are then mapped to the index plasmid, which is deemed present if homology is demonstrated across a given length of the reference sequence (Mathers et al., 2015; Pecora et al., 2015; Stoesser et al., 2015b). This approach implicitly assumes that the index plasmid is important throughout the study period, and that plasmid structures are relatively conserved in the short-term. In some cases, these assumptions may hold (Stoesser et al., 2014), but other studies show major structural changes can occur, including recombination of large segments (Conlan et al., 2016) as well as mobilization of resistance genes (Sheppard et al., 2016). Crucially, Sheppard et al. (2016) demonstrated that reference-based mapping can be misleading if plasmid plasticity is high. Specifically, a reference blaKPC resistance plasmid from the index isolate was detected across diverse strains by a contig-alignment approach, leading to the initial interpretation that resistance had spread via the original blaKPC plasmid. Instead, long-read sequencing showed that often the blaKPC gene was actually present on a co-resident plasmid, suggesting that mobilization of blaKPC had recruited diverse plasmids to the outbreak. It was usually not possible to determine the genetic context of blaKPC without long reads due to long repetitive flanking sequences (Sheppard et al., 2016).
Algorithms to Improve Plasmid Reconstruction from Fragmented Assemblies
Approaches described so far aim to address specific questions, such as the location of a resistance gene, or the short-term transmission of particular plasmids. It is yet more challenging to generate complete structures of diverse plasmids from fragmented assemblies, although several algorithms attempt to do this. The Plasmid Constellation Network (PLACNET) method (Lanza et al., 2014) takes the assembly graph and adds reference genomes as nodes; references are linked to homologous contigs, and the assembly graph is reconfigured according to the additional links. Finally, manual reconfiguration is required to retrieve disjoint connected components that should represent distinct genetic units (chromosome/plasmid), with plasmids identified by presence of replication or relaxase proteins. However, poor assembly of repetitive sequences remains a challenge for resistance gene localization, and reliance on reference sequences to order the network can lead to large-scale errors in structure. For example, de Been et al. (2014) used long-read sequencing to validate PLACNET reconstructions; in one case, plasmid contigs had been incorrectly reconstructed as two distinct plasmids rather than one, probably because the plasmid was a fusion of two previously-observed reference plasmids.
Alternative algorithms such as Recycler (Rozov et al., 2016) and plasmidSPAdes (Antipov et al., 2016) are entirely automated, and independent of reference sequences. Instead, read coverage is used to reconstruct plasmids, with the assumption that contigs from the same genetic unit should share similar coverage. However, read coverage will not distinguish different plasmids if they maintain similar copy numbers within the same host cell, nor distinguish chromosomal from plasmid contigs if a plasmid is maintained at a copy number of one across sampled cells. A recent assessment shows that Recycler and plasmidSPAdes fail to accurately reconstruct all plasmid structures from short-read WGS datasets, though the goal of identifying plasmid-derived sequence (regardless of structural accuracy) is more attainable (Arredondo-Alonso et al., 2016).
Long-Read Sequencing
Long-read sequencing technologies, notably single molecule real-time sequencing (SMRT, Pacific Biosciences) and nanopore sequencing (Oxford Nanopore) promise to revolutionize plasmid analysis (Chin et al., 2013; Loman et al., 2015). The accurate plasmid structures generated (Ashton et al., 2014) allow for a detailed picture of plasmid epidemiology and evolution (Conlan et al., 2014; Johnson et al., 2016). However, current cost considerations have so far restricted analyses to small isolate collections. Conlan et al. (2016) showed how analysis of short-read data can guide economical use of SMRT sequencing: to examine plasmids during an outbreak, short reads were mapped to a reference, whilst unmapped reference regions, PCR amplification of marker genes, and excess reads were used as indicators of structural change, justifying investigation with long-read sequencing. However, minor structural variation (which may include resistance gene mobilization) is unlikely to be detected by this approach. Hybrid (short/long-read) assembly, using reduced long-read coverage, may help to partially mitigate current costs (Koren and Phillippy, 2015).
Networks for Plasmid Analysis
Current plasmid typing schemes exploit a relatively small number of loci thought to best reflect the vertical (tree-like) component of plasmid evolution. However, plasmid genomes tend not to conform to tree-like evolution: co-integration events and genetic exchanges amongst plasmids mean that different parts of a plasmid genome may have different evolutionary origins (Bapteste et al., 2009). A complementary way to assess plasmid diversity involves ordering plasmids into dendrograms (de Been et al., 2014) or networks according to their gene content similarity, irrespective of whether gene-sharing stems from vertical inheritance or horizontal acquisition. In a gene-sharing network, plasmids are nodes that are linked to other plasmids if they share genes at a given sequence identity threshold (Brilli et al., 2008; Corel et al., 2015). The network topology can identify plasmids with interesting gene-sharing patterns. For example, some plasmids may act as ‘bridges’ in the gene-sharing network, straddling different groups that respectively share few genes (in graph theory terms, these plasmids have high ‘betweenness centrality’). Such plasmids may represent co-integrate plasmids, or may be important in shuttling genes across the network (Halary et al., 2010).
Making well-supported inferences from gene-sharing networks is challenging since gene-sharing might result from vertical inheritance, horizontal acquisition, or acquisition from a source not represented within the network. However, there are various ways to infer HGT events (Fondi and Fani, 2010; Tamminen et al., 2012; Fondi et al., 2016). For example, finding genes with very high sequence identity from plasmids that otherwise share few genes has been used to identify putative recent HGT events (Yamashita et al., 2014). Overall, networks are a powerful complementary tool for visualizing relationships across diverse plasmids, and generating hypotheses about the horizontal component of plasmid evolution. Gene-sharing networks only require assembly of genes, not complete plasmids, so are suited to analysis of fragmented assemblies. Since network topology is determined by constituent nodes, large unbiased plasmid datasets will produce the most informative network analyses.
Future Prospects: WGS and Beyond
In future, optical mapping of intact plasmids could complement sequencing-based analysis. Specifically, AT-rich plasmid sequence can be fluorescently labeled, after which plasmids are elongated within nanofluidic channels and visualized with fluorescence microscopy. This allows a course-grained optical ‘barcode’ to be obtained. Optical plasmid barcoding could be used for classifying plasmids, and the course-grained structural information from the barcodes could guide plasmid assembly (Nyberg et al., 2016).
Meanwhile, current algorithms fail to accurately reconstruct all plasmids from short-read WGS datasets. Long-read sequencing will be increasingly used to determine accurate plasmid structures. Use of replicon and MOB typing for plasmid taxonomy will probably continue, but future developments could include methods to incorporate plasmid structural variation into a phylogenetic framework. Networks are a powerful tool for assessing plasmid relationships, from a functional rather than a phylogenetic perspective. Perhaps future plasmid databases could be structured as networks; when novel plasmids are added, information about their potential importance (e.g., centrality within the network) could be determined, and putative HGT events could be continually inferred. Future advances in plasmid metagenomics will enhance our knowledge of plasmids across a range of environments, and improve understanding of resistance gene reservoirs.
Author Contributions
AO undertook literature searching, and wrote a draft manuscript. AO, HP, AS, NS, AW, MA, ME, MD, and NW suggested/implemented revisions. HP, AS, NS, AW, MA, and MD helped in planning the manuscript. AO, NS, MA, MD, ME, TP, DC, NW, AW, HP, and AS read and approved the manuscript.
Funding
The research was funded by the National Institute for Health Research Health Protection Research Unit (NIHR HPRU) in Healthcare Associated Infections and Antimicrobial Resistance at Oxford University in partnership with Public Health England (PHE) [grant HPRU-2012-10041]. The report presents independent research funded by the National Institute for Health Research. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research, the Department of Health or Public Health England. NS is currently funded through an NIHR/University of Oxford Academic Clinical Lectureship. TP is an NIHR Senior Investigator.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adler, A., and Carmeli, Y. (2011). Dissemination of the Klebsiella pneumoniae carbapenemase in the health care settings: tracking the trails of an elusive offender. mBio 2:e00280-11. doi: 10.1128/mBio.00280-11
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Alvarado, A., Garcillán-Barcia, M. P., and de la Cruz, F. (2012). A degenerate primer MOB typing (DPMT) method to classify gamma-proteobacterial plasmids in clinical and environmental settings. PLoS ONE 7:e40438. doi: 10.1371/journal.pone.0040438
Antipov, D., Hartwick, N., Shen, M., Raiko, M., Lapidus, A., and Pevzner, P. A. (2016). Genome analysis plasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics 33, 3380–3387.
Arredondo-Alonso, S., van Schaik, W., Willems, R. J., and Schurch, A. C. (2016). On the (im)possibility to reconstruct plasmids from whole genome short-read sequencing data. bioRxiv 1–18. doi: 10.1101/086744
Ashton, P. M., Nair, S., Dallman, T., Rubino, S., Rabsch, W., Mwaigwisya, S., et al. (2014). MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat. Biotechnol. 33, 296–300. doi: 10.1038/nbt.3103
Bapteste, E., O’Malley, M. A., Beiko, R. G., Ereshefsky, M., Gogarten, J. P., Franklin-Hall, L., et al. (2009). Prokaryotic evolution and the tree of life are two different things. Biol. Direct 4:34. doi: 10.1186/1745-6150-4-34
Bertini, A., Poirel, L., Mugnier, P. D., Villa, L., Nordmann, P., and Carattoli, A. (2010). Characterization and PCR-based replicon typing of resistance plasmids in Acinetobacter baumannii. Antimicrob Agents Chemother. 54, 4168–4177. doi: 10.1128/AAC.00542-10
Bousquet, A., Henquet, S., Compain, F., Genel, N., Arlet, G., and Decré, D. (2015). Partition locus-based classification of selected plasmids in Klebsiella pneumoniae, Escherichia coli and Salmonella enterica spp.: an additional tool. J. Microbiol. Methods 110, 85–91. doi: 10.1016/j.mimet.2015.01.019
Brilli, M., Mengoni, A., Fondi, M., Bazzicalupo, M., Liò, P., and Fani, R. (2008). Analysis of plasmid genes by phylogenetic profiling and visualization of homology relationships using Blast2Network. BMC Bioinformatics 9:551. doi: 10.1186/1471-2105-9-551
Brodrick, H. J., Raven, K. E., Harrison, E. M., Blane, B., Reuter, S., Török, M. E., et al. (2016). Whole-genome sequencing reveals transmission of vancomycin-resistant Enterococcus faecium in a healthcare network. Genome Med. 8:4. doi: 10.1186/s13073-015-0259-7
Brolund, A., and Sandegren, L. (2016). Characterization of ESBL disseminating plasmids. Infect. Dis. 48, 18–25. doi: 10.3109/23744235.2015.1062536
Carattoli, A. (2009). Resistance plasmid families in Enterobacteriaceae. Antimicrob Agents Chemother. 53, 2227–2238. doi: 10.1128/AAC.01707-08
Carattoli, A. (2013). Plasmids and the spread of resistance. Int. J. Med. Microbiol. 303, 298–304. doi: 10.1016/j.ijmm.2013.02.001
Carattoli, A., Bertini, A., Villa, L., Falbo, V., Hopkins, K. L., and Threlfall, E. J. (2005). Identification of plasmids by PCR-based replicon typing. J. Microbiol. Methods 63, 219–228. doi: 10.1016/j.mimet.2005.03.018
Carattoli, A., Zankari, E., Garcia-Fernández, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Carrër, A., Poirel, L., Yilmaz, M., Akan,ÖA., Feriha, C., Cuzon, G., et al. (2010). Spread of OXA-48-encoding plasmid in Turkey and beyond. Antimicrob. Agents Chemother 54, 1369–1373. doi: 10.1128/AAC.01312-09
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M. A., Barrell, B. G., and Parkhill, J. (2005). ACT: the artemis comparison tool. Bioinformatics 21, 3422–3423. doi: 10.1093/bioinformatics/bti553
Center for Genomic Epidemiology (2016). PlasmidFinder / pMLST. Available at: http://www.genomicepidemiology.org/ [accessed August 5, 2016].
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Clausen, P., Zankari, E., Aarestrup, F. M., and Lund, O. (2016). Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J. Antimicrob. Chemother. 71, 2484–2488. doi: 10.1093/jac/dkw184
Compain, F., Poisson, A., Le Hello, S., Branger, C., Weill, F. X., Arlet, G., et al. (2014). Targeting relaxase genes for classification of the predominant plasmids in Enterobacteriaceae. Int. J. Med. Microbiol. 304, 236–242. doi: 10.1016/j.ijmm.2013.09.009
Compeau, P. E. C., Pevzner, P. A., and Tesler, G. (2011). How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 29, 987–991. doi: 10.1038/nbt.2023
Conlan, S., Park, M., Deming, C., and Thomas, P. J. (2016). Plasmid dynamics in KPC-positive Klebsiella pneumoniae during long-term patient colonization. MBio 7:e00742-16. doi: 10.1128/mBio.00742-16
Conlan, S., Thomas, P. J., Deming, C., Park, M., Lau, A. F., Dekker, J. P., et al. (2014). Single-molecule sequencing to track plasmid diversity of hospital-associated carbapenemase-producing Enterobacteriaceae. Sci. Transl. Med. 6, 254ra126. doi: 10.1126/scitranslmed.3009845
Corel, E., Lopez, P., Méheust, R., and Bapteste, E. (2015). Network-thinking: graphs to analyze microbial complexity and evolution. Trends Microbiol. 24, 224–237. doi: 10.1016/j.tim.2015.12.003
Couturier, M., Bex, F., Bergquist, P. L., and Maas, W. K. (1988). Identification and classification of bacterial plasmids. Microbiol. Rev. 52, 375–395. doi: 10.1016/0042-6822(62)90018-1
Croucher, N. J., and Didelot, X. (2015). The application of genomics to tracing bacterial pathogen transmission. Curr. Opin. Microbiol. 23, 62–67. doi: 10.1016/j.mib.2014.11.004
de Been, M., Lanza, V. F., de Toro, M., Scharringa, J., Dohmen, W., Du, Y., et al. (2014). Dissemination of cephalosporin resistance genes between Escherichia coli strains from farm animals and humans by specific plasmid lineages. PLoS Genet. 10:e1004776. doi: 10.1371/journal.pgen.1004776
Dealtry, S., Ding, G. C., Weichelt, V., Dunon, V., Schlüter, A., Martini, M. C., et al. (2014a). Cultivation-independent screening revealed hot spots of IncP-1, IncP-7 and IncP-9 plasmid occurrence in different environmental habitats. PLoS ONE 9:e89922. doi: 10.1371/journal.pone.0089922
Dealtry, S., Holmsgaard, P. N., Dunon, V., Jechalke, S., Ding, G. C., Krögerrecklenfort, E., et al. (2014b). Shifts in abundance and diversity of mobile genetic elements after the introduction of diverse pesticides into an on-farm biopurification system over the course of a year. Appl. Environ. Microbiol. 80, 4012–4020. doi: 10.1128/AEM.04016-13
Diatheva (2016). PBRT KIT- PCR-Based Replicon Typing. Available at: http://www.diatheva.com/molecular-biology/pcr-based-replicon-typing/ pbrt-kit-pcr-based-replicon-typing-details [accessed June 22, 2016].
Dib, J. R., Wagenknecht, M., Farías, M. E., and Meinhardt, F. (2015). Strategies and approaches in plasmidome studies-uncovering plasmid diversity disregarding of linear elements? Front. Microbiol. 6:463. doi: 10.3389/fmicb.2015.00463
Doumith, M., Godbole, G., Ashton, P., Larkin, L., Dallman, T., Day, M., et al. (2016). Detection of the plasmid-mediated mcr-1 gene conferring colistin resistance in human and food isolates of Salmonella enterica and Escherichia coli in England and Wales. J. Antimicrob. Chemother. 71, 2300–2305. doi: 10.1093/jac/dkw093
Edwards, D. J., and Holt, K. E. (2013). Beginner’s guide to comparative bacterial genome analysis using next-generation sequence data. Microb. Inform. Exp. 3:2. doi: 10.1186/2042-5783-3-2
EFSA (2011). Scientific opinion on the public health risks of bacterial strains producing extended-spectrum β-lactamases and/or AmpC β-lactamases in food and food-producing animals. EFSA J. 9, 1–95. doi: 10.2903/j.efsa.2011.2322
Fondi, M., Bacci, G., Brilli, M., Papaleo, M. C., Mengoni, A., Vaneechoutte, M., et al. (2010). Exploring the evolutionary dynamics of plasmids: the Acinetobacter pan-plasmidome. BMC Evol. Biol. 10:59. doi: 10.1186/1471-2148-10-59
Fondi, M., and Fani, R. (2010). The horizontal flow of the plasmid resistome: clues from inter-generic similarity networks. Environ. Microbiol. 12, 3228–3242. doi: 10.1111/j.1462-2920.2010.02295.x
Fondi, M., Karkman, A., Tamminen, M., Bosi, E., Virta, M., Fani, R., et al. (2016). Every gene is everywhere but the environment selects: global geo-localization of gene sharing in environmental samples through network analysis. Genome Biol. Evol. 8, 1388–1400. doi: 10.1093/gbe/evw077
Foxman, B., Zhang, L., Koopman, J. S., Manning, S. D., and Marrs, C. F. (2005). Choosing an appropriate bacterial typing technique for epidemiologic studies. Epidemiol. Perspect. Innov. 2:10. doi: 10.1186/1742-5573-2-10
Francia, M. V., Varsaki, A., Garcillán-Barcia, M. P., Latorre, A., Drainas, C., and De La Cruz, F. (2004). A classification scheme for mobilization regions of bacterial plasmids. FEMS Microbiol. Rev. 28, 79–100. doi: 10.1016/j.femsre.2003.09.001
Freitas, A. R., Novais, C., Tedim, A. P., Francia, M. V., Baquero, F., Peixe, L., et al. (2013). Microevolutionary events involving narrow host plasmids influences local fixation of vancomycin-resistance in Enterococcus populations. PLoS ONE 8:e60589. doi: 10.1371/journal.pone.0060589
Freitas, A. R., Tedim, A. P., Francia, M. V., Jensen, L. B., Novais, C., Peixe, L., et al. (2016). Multilevel population genetic analysis of vanA and vanB Enterococcus faecium causing nosocomial outbreaks in 27 countries (1986-2012). J. Antimicrob. Chemother. doi: 10.1093/jac/dkw312 [Epub ahead of print].
Fricke, W. F., Welch, T. J., McDermott, P. F., Mammel, M. K., LeClerc, J. E., White, D. G., et al. (2009). Comparative genomics of the IncA/C multidrug resistance plasmid family. J. Bacteriol. 191, 4750–4757. doi: 10.1128/JB.00189-09
García-Fernández, A., Villa, L., Moodley, A., Hasman, H., Miriagou, V., Guardabassi, L., et al. (2011). Multilocus sequence typing of IncN plasmids. J. Antimicrob. Chemother. 66, 1987–1991. doi: 10.1093/jac/dkr225
Garcillán-Barcia, M. P., Alvarado, A., and De la Cruz, F. (2011). Identification of bacterial plasmids based on mobility and plasmid population biology. FEMS Microbiol. Rev. 35, 936–956. doi: 10.1111/j.1574-6976.2011.00291.x
Garcillán-Barcia, M. P., and de la Cruz, F. (2013). Ordering the bestiary of genetic elements transmissible by conjugation. Mob. Genet. Elements 3, e24263. doi: 10.4161/mge.24263
Garcillán-Barcia, M. P., Francia, M. V., and De La Cruz, F. (2009). The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 33, 657–687. doi: 10.1111/j.1574-6976.2009.00168.x
Goicoechea, P., Romo, M., and Coque, T. (2008). “Identification of enterococcal plasmids by multiplex-PCR-based relaxase typing,” in Proceedings of the 18th European Congress of Clinical Microbiology and Infectious Diseases, Barcelona.
Guglielmini, J., Quintais, L., Garcillán-Barcia, M. P., de la Cruz, F., and Rocha, E. P. C. (2011). The repertoire of ice in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet. 7:e1002222. doi: 10.1371/journal.pgen.1002222
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-annot, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/AAC.01310-13
Halary, S., Leigh, J. W., Cheaib, B., Lopez, P., and Bapteste, E. (2010). Network analyses structure genetic diversity in independent genetic worlds. Proc. Natl. Acad. Sci. U.S.A. 107, 127–132. doi: 10.1073/pnas.0908978107
Hancock, S. J., Phan, M.-D., Peters, K. M., Forde, B. M., Chong, T. M., Yin, W.-F., et al. (2016). Identification of IncA/C plasmid replication and maintenance genes and development of a plasmid multi-locus sequence-typing scheme. Antimicrob. Agents Chemother. 61, e01740-16. doi: 10.1128/AAC.01740-16
Hawkey, J., Hamidian, M., Wick, R. R., Edwards, D. J., Billman-Jacobe, H., Hall, R. M., et al. (2015). ISMapper: identifying transposase insertion sites in bacterial genomes from short read sequence data. BMC Genomics 16:667. doi: 10.1186/s12864-015-1860-2
Hedges, R. W., and Datta, N. (1973). Plasmids determining I Pili constitute a compatibility complex. J. Gen. Microbiol. 77, 19–25. doi: 10.1099/00221287-77-1-19
Heuer, H., and Smalla, K. (2012). Plasmids foster diversification and adaptation of bacterial populations in soil. FEMS Microbiol. Rev. 36, 1083–1104. doi: 10.1111/j.1574-6976.2012.00337.x
Holt, K. E. (2015). Locating drug Resistance Regions in Short Reads, Using Bandage and ISMapper. Available at: https://holtlab.net/2015/07/20/locating-drug-resistance-regions-in-short-reads-using-bandage-and-ismapper/ [Accessed July 11, 2016].
Inouye, M., Dashnow, H., Raven, L.-A., Schultz, M. B., Pope, B. J., Tomita, T., et al. (2014). SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6:90. doi: 10.1186/s13073-014-0090-6
Jensen, L. B., Garcia-Migura, L., Valenzuela, A. J. S., Løhr, M., Hasman, H., and Aarestrup, F. M. (2010). A classification system for plasmids from enterococci and other Gram-positive bacteria. J. Microbiol. Methods 80, 25–43. doi: 10.1016/j.mimet.2009.10.012
Johnson, T. J., Danzeisen, J. L., and Youmans, B. (2016). Separate F-Type plasmids have shaped the evolution of the H30 subclone of Escherichia coli sequence type 131. mSphere 1, 1–15. doi: 10.1128/mSphere.00121-16
Johnson, T. J., Wannemuehler, Y. M., Johnson, S. J., Logue, C. M., White, D. G., Doetkott, C., et al. (2007). Plasmid replicon typing of commensal and pathogenic Escherichia coli isolates. Appl. Environ. Microbiol. 73, 1976–1983. doi: 10.1128/AEM.02171-06
Jørgensen, T. S., Kiil, A. S., Hansen, M. A., Sørensen, S. J., and Hansen, L. H. (2014). Current strategies for mobilome research. Front. Microbiol. 5:750. doi: 10.3389/fmicb.2014.00750
Koren, S., and Phillippy, A. M. (2015). One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 23, 110–120. doi: 10.1016/j.mib.2014.11.014
Laguerre, G., Mazurier, S. I., and Amarger, N. (1992). Plasmid profiles and restriction fragment length polymorphism of Rhizobium leguminosarum bv. viciae in field populations. FEMS Microbiol. Lett. 101, 17–26. doi: 10.1016/0378-1097(92)90693-i
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lanza, V. F., de Toro, M., Garcillán-Barcia, M. P., Mora, A., Blanco, J., Coque, T. M., et al. (2014). Plasmid Flux in Escherichia coli ST131 sublineages, analyzed by plasmid constellation network (PLACNET), a new method for plasmid reconstruction from whole genome sequences. PLoS Genet. 10:e1004766. doi: 10.1371/journal.pgen.1004766
Lanza, V. F., Tedim, A. P., Martínez, J. L., Baquero, F., Coque, T. M., Lanza, V. F., et al. (2015). The plasmidome of firmicutes: impact on the emergence and the spread of resistance to antimicrobials. Microbiol. Spectr. 3:PLAS–0039–2014. doi: 10.1128/microbiolspec.PLAS-0039-2014
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
Lozano, C., García-Migura, L., Aspiroz, C., Zarazaga, M., Torres, C., and Aarestrup, F. M. (2012). Expansion of a plasmid classification system for gram-positive bacteria and determination of the diversity of plasmids in Staphylococcus aureus strains of human, animal, and food origins. Appl. Environ. Microbiol. 78, 5948–5955. doi: 10.1128/AEM.00870-12
Lynch, T., Petkau, A., Knox, N., Graham, M., and Van Domselaar, G. (2016). A primer on infectious disease bacterial genomics. Clin. Microbiol. Rev. 29, 881–913. doi: 10.1128/CMR.00001-16
Maiden, M. C. (2006). Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 60, 561–588. doi: 10.1146/annurev.micro.59.030804.121325
Martínez, J. L., Coque, T. M., Lanza, V. F., de la Cruz, F., and Baquero, F. (2016). Genomic and metagenomic technologies to explore the antibiotic resistance mobilome. Ann. N. Y. Acad. Sci. doi: 10.1111/nyas.13282 [Epub ahead of print].
Mathers, A. J., Stoesser, N., Sheppard, A. E., Pankhurst, L., Giess, A., Yeh, A. J., et al. (2015). Klebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae at a single institution: Insights into endemicity from Whole-genome sequencing. Antimicrob. Agents Chemother. 59, 1656–1663. doi: 10.1128/AAC.04292-14
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Metzker, M. L. (2010). Sequencing technologies – the next generation. Nat. Rev. Genet. 11, 31–46. doi: 10.1038/nrg2626
Nagarajan, N., and Pop, M. (2013). Sequence assembly demystified. Nat. Rev. Genet. 14, 157–167. doi: 10.1038/nrg3367
Norman, A., Hansen, L. H., and Sørensen, S. J. (2009). Conjugative plasmids: vessels of the communal gene pool. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 2275–2289. doi: 10.1098/rstb.2009.0037
Nyberg, L. K., Quaderi, S., Emilsson, G., Karami, N., Lagerstedt, E., Müller, V., et al. (2016). Rapid identification of intact bacterial resistance plasmids via optical mapping of single DNA molecules. Sci. Rep. 6, 1–10. doi: 10.1038/srep30410
Pecora, N. D., Li, N., Allard, M., Li, C., Albano, E., Delaney, M., et al. (2015). Genomically informed surveillance for carbapenem-resistant Enterobacteriaceae in a health care system. mBio 6, e01030-15. doi: 10.1128/mBio.01030-15
Pevzner, P. A., Tang, H., and Waterman, M. S. (2001). An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. U.S.A. 98, 9748–9753. doi: 10.1073/pnas.171285098
Phan, M. D., Kidgell, C., Nair, S., Holt, K. E., Turner, A. K., Hinds, J., et al. (2009). Variation in Salmonella enterica serovar typhi IncHI1 plasmids during the global spread of resistant typhoid fever. Antimicrob. Agents Chemother. 53, 716–727. doi: 10.1128/AAC.00645-08
Rose, T. M., Schultz, E. R., Henikoff, J. G., Pietrokovski, S., McCallum, C. M., and Henikoff, S. (1998). Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res. 26, 1628–1635. doi: 10.1093/nar/26.7.1628
Rosvoll, T. C. S., Lindstad, B. L., Lunde, T. M., Hegstad, K., Aasnæs, B., Hammerum, A. M., et al. (2012). Increased high-level gentamicin resistance in invasive Enterococcus faecium is associated with aac(6′)Ie-aph(2′′)Ia-encoding transferable megaplasmids hosted by major hospital-adapted lineages. FEMS Immunol. Med. Microbiol. 66, 166–176. doi: 10.1111/j.1574-695X.2012.00997.x
Rosvoll, T. C. S., Pedersen, T., Sletvold, H., Johnsen, P. J., Sollid, J. E., Simonsen, G. S., et al. (2010). PCR-based plasmid typing in Enterococcus faecium strains reveals widely distributed pRE25-, pRUM-, pIP501- and pHTβ-related replicons associated with glycopeptide resistance and stabilizing toxin-antitoxin systems. FEMS Immunol. Med. Microbiol. 58, 254–268. doi: 10.1111/j.1574-695X.2009.00633.x
Rozov, R., Brown Kav, A., Bogumil, D., Halperin, E., Mizrahi, I., and Shamir, R. (2016). Recycler: an algorithm for detecting plasmids from de novo assembly graphs. Bioinformatics 33, doi: 10.1093/bioinformatics/btw651
Sen, D., Brown, C. J., Top, E. M., and Sullivan, J. (2013). Inferring the evolutionary history of INCP-1 plasmids despite incongruence among backbone gene trees. Mol. Biol. Evol. 30, 154–166. doi: 10.1093/molbev/mss210
Seni, J., Falgenhauer, L., Simeo, N., Mirambo, M. M., Imirzalioglu, C., Matee, M., et al. (2016). Multiple ESBL-producing Escherichia coli sequence types carrying quinolone and aminoglycoside resistance genes circulating in companion and domestic farm animals in Mwanza, Tanzania, harbor commonly occurring plasmids. Front. Microbiol. 7:142. doi: 10.3389/fmicb.2016.00142
Shearer, J. E. S., Wireman, J., Hostetler, J., Forberger, H., Borman, J., Gill, J., et al. (2011). Major families of multiresistant plasmids from geographically and epidemiologically diverse staphylococci. G3 (Bethesda) 1, 581–591. doi: 10.1534/g3.111.000760
Sheppard, A. E., Stoesser, N., Wilson, D. J., Sebra, R., Kasarskis, A., Anson, L. W., et al. (2016). Nested Russian doll-like genetic mobility drives rapid dissemination of the carbapenem resistance gene blaKPC. Antimicrob. Agents Chemother. 60, 3767–3778. doi: 10.1128/AAC.00464-16
Shintani, M., Sanchez, Z. K., and Kimbara, K. (2015). Genomics of microbial plasmids: classification and identification based on replication and transfer systems and host taxonomy. Front. Microbiol. 6:242. doi: 10.3389/fmicb.2015.00242
Smillie, C., Garcillan-Barcia, M. P., Francia, M. V., Rocha, E. P., and de la Cruz, F. (2010). Mobility of plasmids. Microbiol. Mol. Biol. Rev. 74, 434–452. doi: 10.1128/MMBR.00020-10
Snyder, L., Peters, J. E., Henkin, T. M., and Champness, W. (2013). “Chapter 4 Plasmids,” in Molecular Genetics of Bacteria (Washington, DC: ASM Press), 183–218.
Stoesser, N., Giess, A., Batty, E. M., Sheppard, A. E., Walker, A. S., Wilson, D. J., et al. (2014). Genome sequencing of an extended series of NDM-producing Klebsiella pneumoniae isolates from neonatal infections in a Nepali hospital characterizes the extent of community- Versus hospital- associated transmission in an endemic setting. Antimicrob. Agents Chemother. 58, 7347–7357. doi: 10.1128/AAC.03900-14
Stoesser, N., Sheppard, A. E., Moore, C. E., Golubchik, T., Parry, C. M., Nget, P., et al. (2015a). Extensive within-host diversity in fecally carried extended-spectrum-beta-lactamase-producing Escherichia coli Isolates: Implications for transmission analyses. J. Clin. Microbiol. 53, 2122–2131. doi: 10.1128/JCM.00378-15
Stoesser, N., Sheppard, A. E., Shakya, M., Sthapit, B., Thorson, S., Giess, A., et al. (2015b). Dynamics of MDR Enterobacter cloacae outbreaks in a neonatal unit in Nepal: insights using wider sampling frames and next-generation sequencing. J. Antimicrob. Chemother. 70, 1008–1015. doi: 10.1093/jac/dku521
Stokes, H. W., and Gillings, M. R. (2011). Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 35, 790–819. doi: 10.1111/j.1574-6976.2011.00273.x
Tamminen, M., Virta, M., Fani, R., and Fondi, M. (2012). Large-scale analysis of plasmid relationships through gene-sharing networks. Mol. Biol. Evol. 29, 1225–1240. doi: 10.1093/molbev/msr292
Taylor, D., Gibreel, A., Lawley, T., and Tracz, D. (2004). “Chapter 23: Antibiotic resistance plasmids,” in Plasmid Biology, eds B. Funnell and G. Phillips (Washington, DC: ASM Press), 473–485.
Tazzyman, S. J., and Bonhoeffer, S. (2014). Why there are no essential genes on plasmids. Mol. Biol. Evol. 32, 3079–3088.
Thomas, C., and Summers, D. (2008). “Bacterial Plasmids,” in eLS (Chichester: John Wiley & Sons, Ltd).
Treangen, T. J., and Salzberg, S. L. (2011). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46. doi: 10.1038/nrg3117
Valverde, A., Cantón, R., Garcillán-Barcia, M. P., Novais,Â, Galán, J. C., Alvarado, A., et al. (2009). Spread of blaCTX-M-14 is driven mainly by IncK plasmids disseminated among Escherichia coli phylogroups A, B1, and D in Spain. Antimicrob. Agents Chemother. 53, 5204–5212. doi: 10.1128/AAC.01706-08
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wong, V. K., Baker, S., Pickard, D. J., Parkhill, J., Page, A. J., Feasey, N. A., et al. (2015). Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter- and intracontinental transmission events. Nat. Genet. 47, 632–639. doi: 10.1038/ng.3281
World Health Organization (2014). Antimicrobial Resistance: Global Report on Surveillance. Geneva: WHO Press.
Yamashita, A., Sekizuka, T., and Kuroda, M. (2014). Characterization of antimicrobial resistance dissemination across plasmid communities classified by network analysis. Pathogens 3, 356–376. doi: 10.3390/pathogens3020356
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Keywords: plasmid typing, whole-genome sequencing, antibiotic resistance, genomic epidemiology, network analysis
Citation: Orlek A, Stoesser N, Anjum MF, Doumith M, Ellington MJ, Peto T, Crook D, Woodford N, Walker AS, Phan H and Sheppard AE (2017) Plasmid Classification in an Era of Whole-Genome Sequencing: Application in Studies of Antibiotic Resistance Epidemiology. Front. Microbiol. 8:182. doi: 10.3389/fmicb.2017.00182
Received: 21 October 2016; Accepted: 25 January 2017;
Published: 09 February 2017.
Edited by:
Teresa M. Coque, Instituto Ramón y Cajal de Investigación Sanitaria, SpainReviewed by:
Christopher Morton Thomas, University of Birmingham, UKAlessandra Carattoli, Istituto Superiore di Sanità, Italy
Copyright © 2017 Orlek, Stoesser, Anjum, Doumith, Ellington, Peto, Crook, Woodford, Walker, Phan and Sheppard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alex Orlek, YWxleC5vcmxla0B3b2xmc29uLm94LmFjLnVr
†These authors have contributed equally to this work.