 Doreen Kamori
Doreen Kamori Takamasa Ueno
Takamasa Ueno
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Microbiol. , 30 January 2017
Sec. Virology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.00080
This article is part of the Research Topic Highly mutable animal RNA viruses: adaptation and evolution View all 37 articles
Despite the effective use of antiretroviral therapy, the remainder of a latently HIV-1-infected reservoir mainly in the resting memory CD4+ T lymphocyte subset has provided a great setback toward viral eradication. While host transcriptional silencing machinery is thought to play a dominant role in HIV-1 latency, HIV-1 protein such as Tat, may affect both the establishment and the reversal of latency. Indeed, mutational studies have demonstrated that insufficient Tat transactivation activity can result in impaired transcription of viral genes and the establishment of latency in cell culture experiments. Because Tat protein is one of highly variable proteins within HIV-1 proteome, it is conceivable that naturally occurring Tat mutations may differentially modulate Tat functions, thereby influencing the establishment and/or the reversal of viral latency in vivo. In this mini review, we summarize the recent findings of Tat naturally occurring polymorphisms associating with host immune responses and we highlight the implication of Tat sequence variations in relation to HIV latency.
Viral latency is a reversible state whereby a pathogenic virus becomes dormant (latent) during the viral life cycle in individual cells. HIV-1 may either actively replicate to rapidly produce progeny virions or can enter a long-lived quiescent state (viral latency), from which it may later be subsequently reactivated. The mechanisms for establishment and maintenance of HIV-1 latency mainly operate at the transcriptional level by both viral (Yukl et al., 2009; Donahue et al., 2012; Donahue and Wainberg, 2013; Ranasinghe et al., 2013) and host (Coiras et al., 2009, 2010; Donahue and Wainberg, 2013) machineries and occur at the levels of transcription, chromatin modification, and epigenetic regulations (Coiras et al., 2009; Donahue and Wainberg, 2013; Archin et al., 2014; Cary et al., 2016).
HIV-1 latency is primarily found within resting memory CD4+ T cells (Chun et al., 1995, 1997; Dahabieh et al., 2015), microglia cells (Chakrabarti et al., 1991; Davis et al., 1992), monocytes/macrophages (Battistini and Sgarbanti, 2014; Kumar et al., 2014; Abbas et al., 2015), and others (Canki et al., 2001; MacDougall et al., 2002; Valentin et al., 2002) which intrinsically have a long half-life in vivo. Because the expression level of the viral proteins is absent or poorly expressed and also the existence of immune escape mutations (Deng et al., 2015), the latently infected cells are much less susceptible to be recognized and cleared by the host immune system, viral cytopathic effects or currently available antiretroviral drugs. Thus to date, latently infected viral reservoir is one of the fundamental limitations toward HIV cure (Marsden and Zack, 2015).
Among the viral proteins, HIV-1 Tat has attracted more attention in viral latency because it potently plays a role in viral transcription regulation. Structurally, Tat is a small nuclear protein with amino acid length ranging from 86 to 101 and the molecular weight ranging from 14 to 16 kDa (Ruben et al., 1989). Functionally, Tat is divided by six domains and plays a role in nuclear translocation (Efthymiadis et al., 1998; Rana and Jeang, 1999), binding for viral RNA (Roy et al., 1990), several host factors and co-factors (Jeang et al., 1993; Garber et al., 1998; Marzio et al., 1998), and the transactivation of 5′ long terminal repeat (LTR) (Ruben et al., 1989; Roy et al., 1990; Jeang et al., 1993; Tong-Starksen et al., 1993; Neuveut and Jeang, 1996). Despite such fundamental functions in the virus life cycle, Tat is a highly polymorphic protein comparable to other HIV-1 polymorphic proteins such as Env, Vpu, and Nef (Yusim et al., 2002; Rossenkhan et al., 2012). Recent studies indicate that a substantial part of viral polymorphisms including in Tat is caused by viral mutational escape from cellular immune responses (Allen et al., 2000; Mason et al., 2009; John et al., 2010; Carlson et al., 2012). It is conceivable that naturally occurring mutations in Tat may modulate transactivation or other Tat functions, and that consequently affect the establishment and reversal of HIV-1 latency in vivo. In this mini review, we will describe the role of HIV-1 Tat toward HIV-1 latency establishment and reactivation, and discuss the possibility that naturally occurring Tat mutations may influence viral latency. The details of host machinery in relation to HIV-1 latency have been well described in recent reviews (Ruelas and Greene, 2013; Dahabieh et al., 2015; Cary et al., 2016) and are not discussed here.
Tat ensures high levels of viral transcription during the virus life cycle (Das et al., 2011). The protein stimulates transcription from the viral 5′ LTR promoter and controls RNA polymerase II (RNAP II) elongation. This is achieved by Tat binding to the TAR hairpin in the nascent RNA transcript and the complex of positive transcription elongation factor b (P-TEFb) composed of Cyclin T1 (CycT1) and cyclin-dependent kinase 9 (CDK9) which phosphorylates the C-terminal domain of the RNAP II that consequently promote transcriptional elongation from the viral promoter (Figure 1) (Dahmus, 1996; Parada and Roeder, 1996; Das et al., 2011; Peterlin et al., 2012). Importantly, the absence or inactivation of Tat in HIV-1 infection has been observed to predominantly generate short non-polyadenylated transcripts of less than 100 nucleotides in length that forms the TAR stem-loop structure, and resulted in reduction of viral transcription and replication (Feng and Holland, 1988; Roy et al., 1990; Yedavalli et al., 2003; Pagans et al., 2005; Das et al., 2011) (Figure 1).
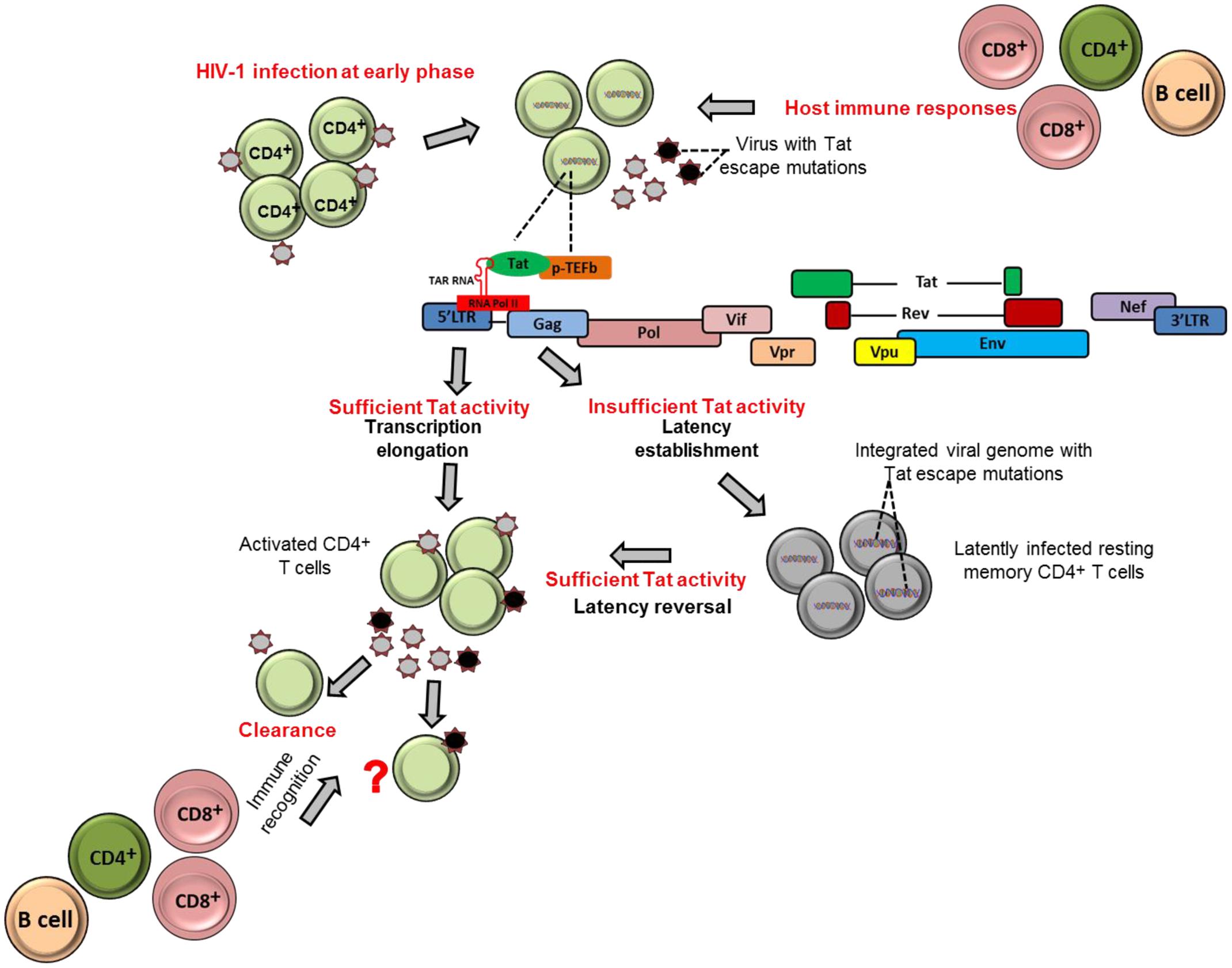
FIGURE 1. Tat role in establishment and reversion of viral latency. The schematic figures illustrate how HIV-1 Tat may contribute to establishment of viral latency and latency reversal in resting memory CD4+ T cells in HIV-1 infection.
It could be therapeutically beneficial if we could prevent or at least reduce to a large extent the size of the established latent reservoir. Evidence indicates that Tat, when present in sufficient quantities, may counteract the establishment of HIV-1 latency by promoting transcriptional initiation or elongation (Pearson et al., 2008; Donahue et al., 2012). One study demonstrated that fewer latently infected cells were established in Jurkat cells that stably express Tat compared to cells that did not express Tat (Donahue et al., 2012). These findings highlight the contribution of Tat and its abundance on prevention of establishment of viral latency. In contrast, a complete block of Tat activity may induce permanent latency as observed with use the of Tat dependent transcription inhibitors such as didehydro-cortistatin A (dCA). The agent has been shown to induce permanently the inactivation of the viral transcription in primary latently infected CD4+ T cells isolated from aviremic ART-treated subjects; and also when tested in several cell line models of latency (HeLa-CD4, promyelocytic OM-10.1 and J-Lat T-lymphocytic cell lines) (Mousseau et al., 2015). In addition, in the same study both in primary cells and latently infected cell line models, the dCA established a state of latency with an extremely impaired ability to reactivate even in the presence of conventional latency-reversing agents (such as TNF-α and prostratin). Therefore, the concomitant treatment of dCA and antiretroviral drugs may reduce the size of reactivation of latently infected cells in vivo and eventually attain a functional HIV cure. However, to date, most experiments done for dCA are limited to in vitro models of latently infected cell lines and primary CD4+ T cells. Therefore, further studies are needed to test the efficacy and safety of dCA as a viral transcription inhibitor agent in advanced experimental systems such as using humanized mice and non-human primates.
Tat can also contribute to reactivation of latently infected cells. For example, previous studies demonstrated that Tat is responsible for directly activating viral transcription in the patient-derived latently infected resting memory CD4+ T cells without requiring cellular activation (Lin et al., 2003; Lassen et al., 2006). This is also supported by the Jurkat model of latency showing that the introduction of exogenous Tat was sufficient to reactivate most of the latently infected population (Donahue et al., 2012). Similarly, HIV-1 latently infected cells, at least in Jurkat cells, can be reactivated by cellular superinfection in a Tat-dependent manner (Donahue et al., 2013). Moreover, both experimental and computational methods have revealed that Tat is more effective than cellular activation approaches in reactivation of full-length transcription of latent HIV. In a recent study, Razooky et al. (2015) showed that removal of cell activation stimuli in HIV-infected primary CD4+ T cells resulted in a drastic decline in cellular activation, but viral transcription activity as measured by GFP expression of productively infected cells remained relatively unchanged. Furthermore, the same study revealed by a computational method of HIV transcriptional modulation that Tat in abundance alone is sufficient for reactivation of the latently infected cells (Razooky et al., 2015). In addition, the depletion of some host factors or molecules that inhibit Tat transactivation activities, such as the long non-coding RNAs (NRON) that degrades Tat protein, in combination with a histone deacetylase (HDAC) inhibitor, has also been shown to significantly reactivate HIV-1 latency in CD4+ T lymphocytes (Li et al., 2016). Furthermore, in a recent mutational study, a Tat mutant, Tat-R5M4 that comprises of V36A, Q66A, V67A, S66A, and S77A mutations, exhibited a potent ability to reactivate latently infected CD4+ T lymphocytes (Geng et al., 2016). Taken together, these findings provide a potential alternative approach toward reactivation of the latently infected cells with Tat protein.
Sequence analysis of plasma viral RNA isolated from cross-sectional and longitudinal collection of HIV-infected individuals exhibited that HIV-1 Tat is a highly variable protein even among the rapidly mutating HIV-1 proteins such as Env, Vpu, and Nef (Yusim et al., 2002; Li et al., 2015). The high genetic variability of HIV-1 Tat is observed across the subtypes, such as subtypes B and C, in the major HIV-1 group M, and also across HIV-1 groups O and N as well as HIV-2 (Yusim et al., 2002; Rossenkhan et al., 2012; Li et al., 2015; Roy et al., 2015b). Interestingly, Bayesian evolutionary analysis model demonstrated that subtype B Tat has evolved relatively faster than other subtypes (Roy et al., 2015a). The extent of amino acid variability in Tat as estimated by the Shannon entropy score in subtype B sequences published in Los Alamos sequence database is illustrated in Figure 2.
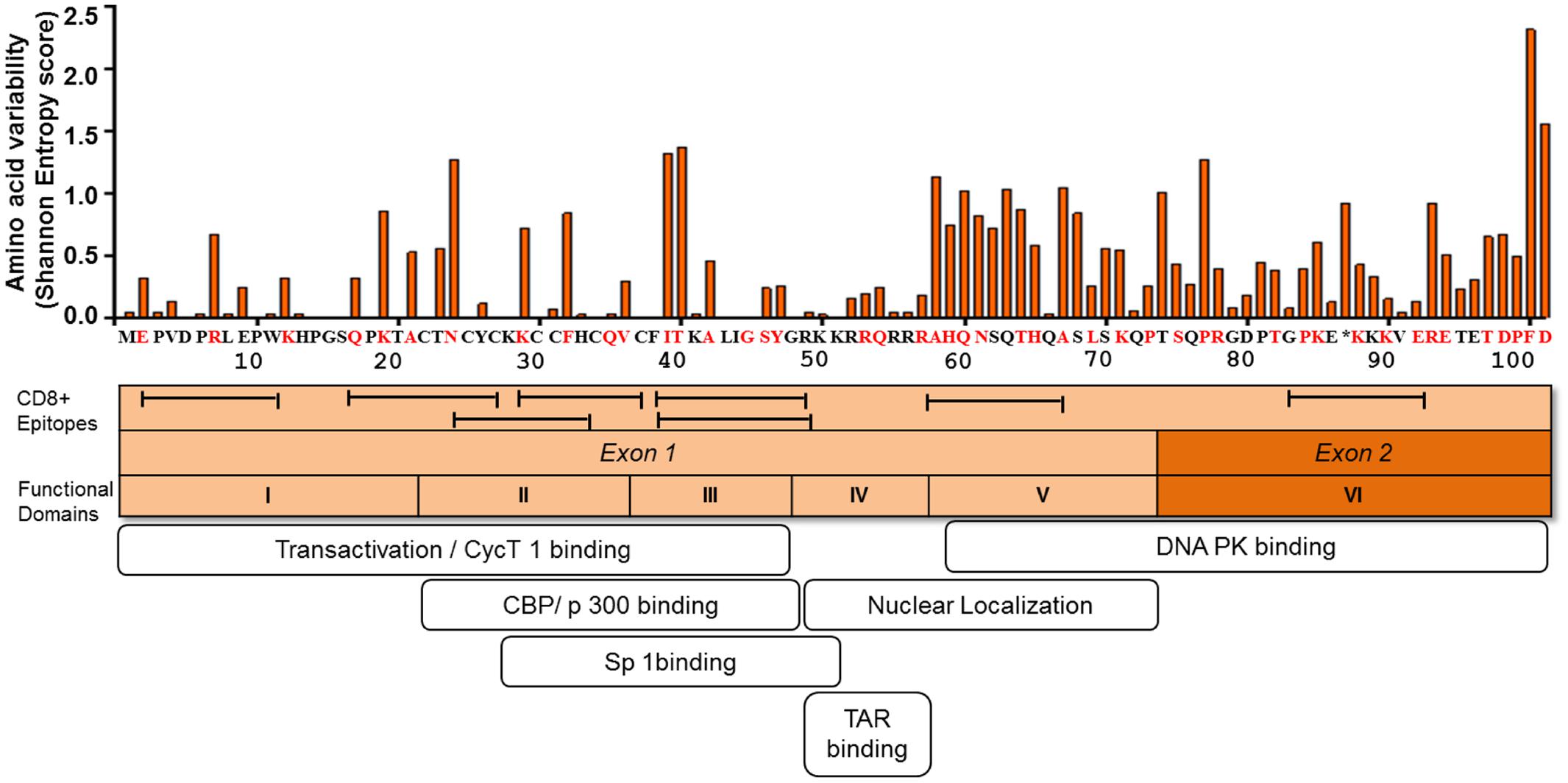
FIGURE 2. Amino acid variability, immunogenic sites for CTL, and functionally important sites for transactivation activity of HIV-1 Tat. The figure depicts the amino acid variability of Tat as measured by the Shannon entropy score for subtype B Tat sequences (n = 378) obtained from a public database (Los Alamos Sequence Database). The immune reactive sites are shown with location of CTL epitopes and the amino acid residues that are associated with CTL escape as determined by statistical association with host HLA class I allele (red). TatHXB2 is used as reference. The domains associated with the transactivation activity are also indicated.
Mutational studies of HIV-1 Tat revealed that Tat is divided into six functional domains (Kuppuswamy et al., 1989) (Figure 2). The first three domains are responsible for Tat transactivation activity and binding with the transcription cofactors (Feng and Holland, 1988; Feinberg et al., 1991; Garber et al., 1998; Wei et al., 1998; Rusnati et al., 1999); while the fourth domain is a TAR binding domain (Dingwall et al., 1989; Roy et al., 1990; Weeks and Crothers, 1991). The fourth and fifth domains are important for Tat nuclear localization (Ruben et al., 1989), the sixth domain binds to DNA PK and also contribute to viral infectivity (Smith et al., 2003). Importantly in regard to viral latency the functional domains II and III, spanning amino acid positions 22 to 48, are shown to be responsible for transactivation activity (Figure 2). The several mutations at positions 22 to 40 amino acid residues (including highly conserved cysteine residues) have been shown to be deleterious with respect to Tat transactivation activity; whereas those at positions 1 to 21 amino acid residues are relatively functionally tolerated (Kuppuswamy et al., 1989; Ruben et al., 1989). Tat plays active role in productive viral replication mainly through enhancement of transcription at viral LTR promoter. Mutational studies have shown there is a strong correlation between Tat transactivation activity and viral replication capacity, whereby the functionally defective Tat has ability to severely inhibit viral replication in vitro (Verhoef et al., 1997; Das et al., 2011). This suggests that provirus with functionally defective Tat influences the viral replication and size of the latent reservoir in vivo. In respect to the naturally occurring mutations from HIV-1-infected individuals, the Cys-22 to Ser mutation (C22S) in HIV-1 Oyi strain resulted in loss of transactivation activity and was enriched in long-term non-progressive patients (Huet et al., 1989; Peloponese et al., 1999; Watkins et al., 2006). Moreover, several naturally occurring polymorphisms, including P10S, W11R, K19R, A42V, and Y47H, that were observed in 5 HIV-infected subjects at acute or early infection stage, demonstrated impaired transactivation activity and were statistically significantly enriched in the latently infected CD4+ T cells (Yukl et al., 2009). These findings suggest that certain naturally occurring mutations can influence Tat transactivation activity and the establishment of viral latency or reactivation of latent reservoirs during the course of HIV-1 infection in vivo. Therefore, this issue warrants for more comprehensive study using a large number of HIV-infected subjects.
It is becoming evident that mutational escape from CD8+ cytotoxic T lymphocyte (CTL) responses represents a potent ongoing driver of global HIV-1 diversification (Price et al., 1997; Goulder et al., 2001; Brumme et al., 2009; Carlson et al., 2012). Tat has also been shown to be frequently targeted by the host HLA-restricted CTL responses (Addo et al., 2001, 2002; Westrop et al., 2009). A number of CTL epitopes have been identified, including PW9 (3PVDPRLEPW11) and EW10 (2EPVDPNLEPW11) restricted by the protective HLA-I alleles, HLA-B∗57 and HLA-B∗5801, respectively (Schellens et al., 2008; Zhai et al., 2008; Chopera et al., 2011). Additional epitopes are well summarized at the web site, http://www.hiv.lanl.gov/content/immunology/maps/ctl/Tat.html. CTL epitopes are distributed in both highly conserved and polymorphic regions in Tat; however, more number of CTL epitopes are reported at the relatively conserved regions to date (Figure 2). A number of Tat mutations in both conserved and variable regions have been reported to be associated with host cellular immune responses in various viral subtypes and host populations (Figure 2) (Allen et al., 2000; Guillon et al., 2006; Mason et al., 2009; John et al., 2010; Carlson et al., 2012). Importantly, some of the CTL escape mutations in Tat such as F32L and V36S observed in a frequently recognized (or immunodominant) Tat epitope, CC8 (30CCFHCQVC37) restricted by HLA-C∗12:03 (Cao et al., 2003; Liu et al., 2007, 2011), are located at sites that are important for transactivation and co-factor binding (Figure 2). Some other CTL escape mutations are located at functionally important regions; N24K, N24T, K29R, and K29S in NF9 (24NCYCKRCCF32) epitope restricted by HLA-A∗29:02 (Jones et al., 2004), K40T in FY10 (38FQKKGLGISY47) restricted by HLA-B∗15:03 (Liu et al., 2013), and R7S, R7K, and E9D in PW9 (3PVDPRLEPW11) restricted by HLA-A∗25:01 (Liu et al., 2007). These data suggest that CTL escape mutations in Tat, especially those located at functionally important conserved regions, have a potential to differentially influence Tat activity. However, it remains elusive as to what extent CTL responses to Tat or CTL escape mutations in Tat may influence viral latency kinetics both at establishment and reversal stages. Also, it is intriguing to ask whether Tat mutations may influence immune recognition of latently infected cells after reactivation. It is also worth to mention that despite the predominant effect of CTL selection pressure on Tat sequence polymorphism, other host immune responses such as those mediated by CD4+ T cells (Lichterfeld et al., 2012; Ranasinghe et al., 2013) and B cells (Goldstein et al., 2001; Moreau et al., 2004) also target Tat; and may therefore potentially impose selection pressure leading to escape mutations which may differentially affect Tat activity.
To date, the highly genetic viral variability and the existence of latently infected resting CD4+ T lymphocytes and other cells in vivo are among the setbacks toward achievement of complete HIV control and eradication. It is generally thought that virus can acquire mutations and evade host immune responses while maintain their fitness effects as minimal as possible. However, similar to the cases in the other HIV-1 proteins such as Gag (Goulder et al., 2001; Troyer et al., 2009) and Nef (Mwimanzi et al., 2013; Kuang et al., 2014), certain naturally occurring immune-associated mutations in Tat may impose fitness cost to the virus. However, it remains poorly described how immune-mediated Tat polymorphisms affect either establishment of viral latency or reactivation of the latently infected cells and also the consequence of such viral polymorphisms on immune recognition. These points could open a new venue to modulate HIV latency and reversal of latency in vivo for future therapeutic application toward cure.
DK and TU conceived, designed, compiled the data, and wrote the manuscript.
This work was supported in part by a grant from JSPS KAKENHI Grant number JP16K15284 and JP16H05822, AIDS International Collaborative Research Grant from the Ministry of Education, Science, Sports, and Culture (MEXT) of Japan, and Japan Agency for Medical Research and Development, AMED (Research Program on HIV/AIDS). DK is supported by the scholarship for The International Priority Graduate Programs; Advanced Graduate Courses for International Students (Doctoral Course), MEXT, Japan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors also wish to thank M. Mahiti and other lab members for helpful discussion.
Abbas, W., Tariq, M., Iqbal, M., Kumar, A., and Herbein, G. (2015). Eradication of HIV-1 from the macrophage reservoir: an uncertain goal? Viruses 7, 1578–1598. doi: 10.3390/v7041578
Addo, M. M., Altfeld, M., Rosenberg, E. S., Eldridge, R. L., Philips, M. N., Habeeb, K., et al. (2001). The HIV-1 regulatory proteins Tat and Rev are frequently targeted by cytotoxic T lymphocytes derived from HIV-1-infected individuals. Proc. Natl. Acad. Sci. U.S.A. 98, 1781–1786. doi: 10.1073/pnas.98.4.1781
Addo, M. M., Yu, X. G., Rosenberg, E. S., Walker, B. D., and Altfeld, M. (2002). Cytotoxic T-lymphocyte (CTL) responses directed against regulatory and accessory proteins in HIV-1 infection. DNA Cell Biol. 21, 671–678. doi: 10.1089/104454902760330219
Allen, T. M., O’Connor, D. H., Jing, P., Dzuris, J. L., Mothe, B. R., Vogel, T. U., et al. (2000). Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407, 386–390. doi: 10.1038/35030124
Archin, N. M., Sung, J. M., Garrido, C., Soriano-Sarabia, N., and Margolis, D. M. (2014). Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 12, 750–764. doi: 10.1038/nrmicro3352
Battistini, A., and Sgarbanti, M. (2014). HIV-1 latency: an update of molecular mechanisms and therapeutic strategies. Viruses 6, 1715–1758. doi: 10.3390/v6041715
Brumme, Z. L., John, M., Carlson, J. M., Brumme, C. J., Chan, D., Brockman, M. A., et al. (2009). HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS ONE 4:e6687. doi: 10.1371/journal.pone.0006687
Canki, M., Thai, J. N., Chao, W., Ghorpade, A., Potash, M. J., and Volsky, D. J. (2001). Highly productive infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) indicates no intracellular restrictions to HIV-1 replication in primary human astrocytes. J. Virol. 75, 7925–7933. doi: 10.1128/JVI.75.17.7925-7933.2001
Cao, J., McNevin, J., Malhotra, U., and McElrath, M. J. (2003). Evolution of CD8+ T cell immunity and viral escape following acute HIV-1 infection. J. Immunol. 171, 3837–3846. doi: 10.4049/jimmunol.171.7.3837
Carlson, J. M., Brumme, C. J., Martin, E., Listgarten, J., Brockman, M. A., Le, A. Q., et al. (2012). Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J. Virol. 86, 13202–13216. doi: 10.1128/JVI.01998-12
Cary, D. C., Fujinaga, K., and Peterlin, B. M. (2016). Molecular mechanisms of HIV latency. J. Clin. Invest. 126, 448–454. doi: 10.1172/JCI80565
Chakrabarti, L., Hurtrel, M., Maire, M. A., Vazeux, R., Dormont, D., Montagnier, L., et al. (1991). Early viral replication in the brain of SIV-infected rhesus monkeys. Am. J. Pathol. 139, 1273–1280.
Chopera, D. R., Mlotshwa, M., Woodman, Z., Mlisana, K., de Assis Rosa, D., Martin, D. P., et al. (2011). Virological and immunological factors associated with HIV-1 differential disease progression in HLA-B 58:01-positive individuals. J. Virol. 85, 7070–7080. doi: 10.1128/JVI.02543-10
Chun, T. W., Carruth, L., Finzi, D., Shen, X., DiGiuseppe, J. A., Taylor, H., et al. (1997). Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387, 183–188. doi: 10.1038/387183a0
Chun, T. W., Finzi, D., Margolick, J., Chadwick, K., Schwartz, D., and Siliciano, R. F. (1995). In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1, 1284–1290. doi: 10.1038/nm1295-1284
Coiras, M., Lopez-Huertas, M. R., Perez-Olmeda, M., and Alcami, J. (2009). Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 7, 798–812. doi: 10.1038/nrmicro2223
Coiras, M., Lopez-Huertas, M. R., Sanchez del Cojo, M., Mateos, E., and Alcami, J. (2010). Dual role of host cell factors in HIV-1 replication: restriction and enhancement of the viral cycle. AIDS Rev. 12, 103–112.
Dahabieh, M. S., Battivelli, E., and Verdin, E. (2015). Understanding HIV latency: the road to an HIV cure. Annu. Rev. Med. 66, 407–421. doi: 10.1146/annurev-med-092112-152941
Dahmus, M. E. (1996). Phosphorylation of mammalian RNA polymerase II. Methods Enzymol. 273, 185–193. doi: 10.1016/S0076-6879(96)73019-7
Das, A. T., Harwig, A., and Berkhout, B. (2011). The HIV-1 Tat protein has a versatile role in activating viral transcription. J. Virol. 85, 9506–9516. doi: 10.1128/JVI.00650-11
Davis, L. E., Hjelle, B. L., Miller, V. E., Palmer, D. L., Llewellyn, A. L., Merlin, T. L., et al. (1992). Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 42, 1736–1739. doi: 10.1212/WNL.42.9.1736
Deng, K., Pertea, M., Rongvaux, A., Wang, L., Durand, C. M., Ghiaur, G., et al. (2015). Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 517, 381–385. doi: 10.1038/nature14053
Dingwall, C., Ernberg, I., Gait, M. J., Green, S. M., Heaphy, S., Karn, J., et al. (1989). Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc. Natl. Acad. Sci. U.S.A. 86, 6925–6929. doi: 10.1073/pnas.86.18.6925
Donahue, D. A., Bastarache, S. M., Sloan, R. D., and Wainberg, M. A. (2013). Latent HIV-1 can be reactivated by cellular superinfection in a Tat-dependent manner, which can lead to the emergence of multidrug-resistant recombinant viruses. J. Virol. 87, 9620–9632. doi: 10.1128/JVI.01165-13
Donahue, D. A., Kuhl, B. D., Sloan, R. D., and Wainberg, M. A. (2012). The viral protein Tat can inhibit the establishment of HIV-1 latency. J. Virol. 86, 3253–3263. doi: 10.1128/JVI.06648-11
Donahue, D. A., and Wainberg, M. A. (2013). Cellular and molecular mechanisms involved in the establishment of HIV-1 latency. Retrovirology 10:11. doi: 10.1186/1742-4690-10-11
Efthymiadis, A., Briggs, L. J., and Jans, D. A. (1998). The HIV-1 Tat nuclear localization sequence confers novel nuclear import properties. J. Biol. Chem. 273, 1623–1628. doi: 10.1074/jbc.273.3.1623
Feinberg, M. B., Baltimore, D., and Frankel, A. D. (1991). The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. U.S.A. 88, 4045–4049. doi: 10.1073/pnas.88.9.4045
Feng, S., and Holland, E. C. (1988). HIV-1 tat trans-activation requires the loop sequence within tar. Nature 334, 165–167. doi: 10.1038/334165a0
Garber, M. E., Wei, P., KewalRamani, V. N., Mayall, T. P., Herrmann, C. H., Rice, A. P., et al. (1998). The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 12, 3512–3527. doi: 10.1101/gad.12.22.3512
Geng, G., Liu, B., Chen, C., Wu, K., Liu, J., Zhang, Y., et al. (2016). Development of an attenuated tat protein as a highly-effective agent to specifically activate HIV-1 latency. Mol. Ther. 24, 1528–1537. doi: 10.1038/mt.2016.117
Goldstein, G., Tribbick, G., and Manson, K. (2001). Two B cell epitopes of HIV-1 Tat protein have limited antigenic polymorphism in geographically diverse HIV-1 strains. Vaccine 19, 1738–1746. doi: 10.1016/S0264-410X(00)00393-5
Goulder, P. J., Brander, C., Tang, Y., Tremblay, C., Colbert, R. A., Addo, M. M., et al. (2001). Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412, 334–338. doi: 10.1038/35085576
Guillon, C., Stankovic, K., Ataman-Onal, Y., Biron, F., and Verrier, B. (2006). Evidence for CTL-mediated selection of Tat and Rev mutants after the onset of the asymptomatic period during HIV type 1 infection. AIDS Res. Hum. Retroviruses 22, 1283–1292. doi: 10.1089/aid.2006.22.1283
Huet, T., Dazza, M. C., Brun-Vezinet, F., Roelants, G. E., and Wain-Hobson, S. (1989). A highly defective HIV-1 strain isolated from a healthy Gabonese individual presenting an atypical western blot. AIDS 3, 707–715. doi: 10.1097/00002030-198911000-00004
Jeang, K. T., Chun, R., Lin, N. H., Gatignol, A., Glabe, C. G., and Fan, H. (1993). In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J. Virol. 67, 6224–6233.
John, M., Heckerman, D., James, I., Park, L. P., Carlson, J. M., Chopra, A., et al. (2010). Adaptive interactions between HLA and HIV-1: highly divergent selection imposed by HLA class I molecules with common supertype motifs. J. Immunol. 184, 4368–4377. doi: 10.4049/jimmunol.0903745
Jones, N. A., Wei, X., Flower, D. R., Wong, M., Michor, F., Saag, M. S., et al. (2004). Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J. Exp. Med. 200, 1243–1256. doi: 10.1084/jem.20040511
Kuang, X. T., Li, X., Anmole, G., Mwimanzi, P., Shahid, A., Le, A. Q., et al. (2014). Impaired Nef function is associated with early control of HIV-1 viremia. J. Virol. 88, 10200–10213. doi: 10.1128/JVI.01334-14
Kumar, A., Abbas, W., and Herbein, G. (2014). HIV-1 latency in monocytes/macrophages. Viruses 6, 1837–1860. doi: 10.3390/v6041837
Kuppuswamy, M., Subramanian, T., Srinivasan, A., and Chinnadurai, G. (1989). Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis. Nucleic Acids Res. 17, 3551–3561. doi: 10.1093/nar/17.9.3551
Lassen, K. G., Ramyar, K. X., Bailey, J. R., Zhou, Y., and Siliciano, R. F. (2006). Nuclear retention of multiply spliced HIV-1 RNA in resting CD4+ T cells. PLoS Pathog. 2:e68. doi: 10.1371/journal.ppat.0020068
Li, G., Piampongsant, S., Faria, N. R., Voet, A., Pineda-Pena, A. C., Khouri, R., et al. (2015). An integrated map of HIV genome-wide variation from a population perspective. Retrovirology 12:18. doi: 10.1186/s12977-015-0148-6
Li, J., Chen, C., Ma, X., Geng, G., Liu, B., Zhang, Y., et al. (2016). Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 7:11730. doi: 10.1038/ncomms11730
Lichterfeld, M., Gandhi, R. T., Simmons, R. P., Flynn, T., Sbrolla, A., Yu, X. G., et al. (2012). Induction of strong HIV-1-specific CD4+ T-cell responses using an HIV-1 gp120/NefTat vaccine adjuvanted with AS02A in antiretroviral-treated HIV-1-infected individuals. J. Acquir. Immune Defic. Syndr. 59, 1–9. doi: 10.1097/QAI.0b013e3182373b77
Lin, X., Irwin, D., Kanazawa, S., Huang, L., Romeo, J., Yen, T. S., et al. (2003). Transcriptional profiles of latent human immunodeficiency virus in infected individuals: effects of Tat on the host and reservoir. J. Virol. 77, 8227–8236. doi: 10.1128/JVI.77.15.8227-8236.2003
Liu, M. K., Hawkins, N., Ritchie, A. J., Ganusov, V. V., Whale, V., Brackenridge, S., et al. (2013). Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J. Clin. Invest. 123, 380–393. doi: 10.1172/JCI65330
Liu, Y., McNevin, J., Zhao, H., Tebit, D. M., Troyer, R. M., McSweyn, M., et al. (2007). Evolution of human immunodeficiency virus type 1 cytotoxic T-lymphocyte epitopes: fitness-balanced escape. J. Virol. 81, 12179–12188. doi: 10.1128/JVI.01277-07
Liu, Y., McNevin, J. P., Holte, S., McElrath, M. J., and Mullins, J. I. (2011). Dynamics of viral evolution and CTL responses in HIV-1 infection. PLoS ONE 6:e15639. doi: 10.1371/journal.pone.0015639
MacDougall, T. H., Shattock, R. J., Madsen, C., Chain, B. M., and Katz, D. R. (2002). Regulation of primary HIV-1 isolate replication in dendritic cells. Clin. Exp. Immunol. 127, 66–71. doi: 10.1046/j.1365-2249.2002.01715.x
Marsden, M. D., and Zack, J. A. (2015). Double trouble: HIV latency and CTL escape. Cell Host Microbe 17, 141–142. doi: 10.1016/j.chom.2015.01.008
Marzio, G., Tyagi, M., Gutierrez, M. I., and Giacca, M. (1998). HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. U.S.A. 95, 13519–13524. doi: 10.1073/pnas.95.23.13519
Mason, R. D., De Rose, R., and Kent, S. J. (2009). Differential patterns of immune escape at Tat-specific cytotoxic T cell epitopes in pigtail macaques. Virology 388, 315–323. doi: 10.1016/j.virol.2009.03.020
Moreau, E., Belliard, G., Partidos, C. D., Pradezinsky, F., Le Buanec, H., Muller, S., et al. (2004). Important B-cell epitopes for neutralization of human immunodeficiency virus type 1 Tat in serum samples of humans and different animal species immunized with Tat protein or peptides. J. Gen. Virol. 85(Pt 10), 2893–2901. doi: 10.1099/vir.0.80365-0
Mousseau, G., Kessing, C. F., Fromentin, R., Trautmann, L., Chomont, N., and Valente, S. T. (2015). The tat inhibitor Didehydro-Cortistatin A prevents HIV-1 reactivation from latency. MBio 6:e00465-15. doi: 10.1128/mBio.00465-15
Mwimanzi, P., Markle, T. J., Martin, E., Ogata, Y., Kuang, X. T., Tokunaga, M., et al. (2013). Attenuation of multiple Nef functions in HIV-1 elite controllers. Retrovirology 10:1. doi: 10.1186/1742-4690-10-1
Neuveut, C., and Jeang, K. T. (1996). Recombinant human immunodeficiency virus type 1 genomes with tat unconstrained by overlapping reading frames reveal residues in Tat important for replication in tissue culture. J. Virol. 70, 5572–5581.
Pagans, S., Pedal, A., North, B. J., Kaehlcke, K., Marshall, B. L., Dorr, A., et al. (2005). SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 3:e41. doi: 10.1371/journal.pbio.0030041
Parada, C. A., and Roeder, R. G. (1996). Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 384, 375–378. doi: 10.1038/384375a0
Pearson, R., Kim, Y. K., Hokello, J., Lassen, K., Friedman, J., Tyagi, M., et al. (2008). Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 82, 12291–12303. doi: 10.1128/JVI.01383-08
Peloponese, J. M. Jr., Collette, Y., Gregoire, C., Bailly, C., Campese, D., Meurs, E. F., et al. (1999). Full peptide synthesis, purification, and characterization of six Tat variants. Differences observed between HIV-1 isolates from Africa and other continents. J. Biol. Chem. 274, 11473–11478.
Peterlin, B. M., Brogie, J. E., and Price, D. H. (2012). 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip. Rev. RNA 3, 92–103. doi: 10.1002/wrna.106
Price, D. A., Goulder, P. J., Klenerman, P., Sewell, A. K., Easterbrook, P. J., Troop, M., et al. (1997). Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. U.S.A. 94, 1890–1895. doi: 10.1073/pnas.94.5.1890
Rana, T. M., and Jeang, K. T. (1999). Biochemical and functional interactions between HIV-1 Tat protein and TAR RNA. Arch. Biochem. Biophys. 365, 175–185. doi: 10.1006/abbi.1999.1206
Ranasinghe, S., Cutler, S., Davis, I., Lu, R., Soghoian, D. Z., Qi, Y., et al. (2013). Association of HLA-DRB1-restricted CD4(+) T cell responses with HIV immune control. Nat. Med. 19, 930–933. doi: 10.1038/nm.3229
Razooky, B. S., Pai, A., Aull, K., Rouzine, I. M., and Weinberger, L. S. (2015). A hardwired HIV latency program. Cell 160, 990–1001. doi: 10.1016/j.cell.2015.02.009
Rossenkhan, R., Novitsky, V., Sebunya, T. K., Musonda, R., Gashe, B. A., and Essex, M. (2012). Viral diversity and diversification of major non-structural genes vif, vpr, vpu, tat exon 1 and rev exon 1 during primary HIV-1 subtype C infection. PLoS ONE 7:e35491. doi: 10.1371/journal.pone.0035491
Roy, C. N., Khandaker, I., Furuse, Y., and Oshitani, H. (2015a). Molecular characterization of full-length Tat in HIV-1 subtypes B and C. Bioinformation 11, 151–160. doi: 10.6026/97320630011151
Roy, C. N., Khandaker, I., and Oshitani, H. (2015b). Intersubtype genetic variation of HIV-1 Tat Exon 1. AIDS Res. Hum. Retroviruses 31, 641–648. doi: 10.1089/AID.2014.0346
Roy, S., Delling, U., Chen, C. H., Rosen, C. A., and Sonenberg, N. (1990). A bulge structure in HIV-1 TAR RNA is required for Tat binding and Tat-mediated trans-activation. Genes Dev. 4, 1365–1373. doi: 10.1101/gad.4.8.1365
Ruben, S., Perkins, A., Purcell, R., Joung, K., Sia, R., Burghoff, R., et al. (1989). Structural and functional characterization of human immunodeficiency virus tat protein. J. Virol. 63, 1–8.
Ruelas, D. S., and Greene, W. C. (2013). An integrated overview of HIV-1 latency. Cell 155, 519–529. doi: 10.1016/j.cell.2013.09.044
Rusnati, M., Tulipano, G., Spillmann, D., Tanghetti, E., Oreste, P., Zoppetti, G., et al. (1999). Multiple interactions of HIV-I Tat protein with size-defined heparin oligosaccharides. J. Biol. Chem. 274, 28198–28205. doi: 10.1074/jbc.274.40.28198
Schellens, I. M., Kesmir, C., Miedema, F., van Baarle, D., and Borghans, J. A. (2008). An unanticipated lack of consensus cytotoxic T lymphocyte epitopes in HIV-1 databases: the contribution of prediction programs. AIDS 22, 33–37. doi: 10.1097/QAD.0b013e3282f15622
Smith, S. M., Pentlicky, S., Klase, Z., Singh, M., Neuveut, C., Lu, C. Y., et al. (2003). An in vivo replication-important function in the second coding exon of Tat is constrained against mutation despite cytotoxic T lymphocyte selection. J. Biol. Chem. 278, 44816–44825. doi: 10.1074/jbc.M307546200
Tong-Starksen, S. E., Baur, A., Lu, X. B., Peck, E., and Peterlin, B. M. (1993). Second exon of Tat of HIV-2 is required for optimal trans-activation of HIV-1 and HIV-2 LTRs. Virology 195, 826–830. doi: 10.1006/viro.1993.1438
Troyer, R. M., McNevin, J., Liu, Y., Zhang, S. C., Krizan, R. W., Abraha, A., et al. (2009). Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 5:e1000365. doi: 10.1371/journal.ppat.1000365
Valentin, A., Rosati, M., Patenaude, D. J., Hatzakis, A., Kostrikis, L. G., Lazanas, M., et al. (2002). Persistent HIV-1 infection of natural killer cells in patients receiving highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U.S.A. 99, 7015–7020. doi: 10.1073/pnas.102672999
Verhoef, K., Koper, M., and Berkhout, B. (1997). Determination of the minimal amount of Tat activity required for human immunodeficiency virus type 1 replication. Virology 237, 228–236. doi: 10.1006/viro.1997.8786
Watkins, J. D., Lancelot, S., Campbell, G. R., Esquieu, D., de Mareuil, J., Opi, S., et al. (2006). Reservoir cells no longer detectable after a heterologous SHIV challenge with the synthetic HIV-1 Tat Oyi vaccine. Retrovirology 3:8. doi: 10.1186/1742-4690-3-8
Weeks, K. M., and Crothers, D. M. (1991). RNA recognition by Tat-derived peptides: interaction in the major groove? Cell 66, 577–588. doi: 10.1016/0092-8674(81)90020-9
Wei, P., Garber, M. E., Fang, S. M., Fischer, W. H., and Jones, K. A. (1998). A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462. doi: 10.1016/S0092-8674(00)80939-3
Westrop, S. J., Qazi, N. A., Pido-Lopez, J., Nelson, M. R., Gazzard, B., Gotch, F. M., et al. (2009). Transient nature of long-term nonprogression and broad virus-specific proliferative T-cell responses with sustained thymic output in HIV-1 controllers. PLoS ONE 4:e5474. doi: 10.1371/journal.pone.0005474
Yedavalli, V. S., Benkirane, M., and Jeang, K. T. (2003). Tat and trans-activation-responsive (TAR) RNA-independent induction of HIV-1 long terminal repeat by human and murine cyclin T1 requires Sp1. J. Biol. Chem. 278, 6404–6410. doi: 10.1074/jbc.M209162200
Yukl, S., Pillai, S., Li, P., Chang, K., Pasutti, W., Ahlgren, C., et al. (2009). Latently-infected CD4+ T cells are enriched for HIV-1 Tat variants with impaired transactivation activity. Virology 387, 98–108. doi: 10.1016/j.virol.2009.01.013
Yusim, K., Kesmir, C., Gaschen, B., Addo, M. M., Altfeld, M., Brunak, S., et al. (2002). Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. J. Virol. 76, 8757–8768. doi: 10.1128/JVI.76.17.8757-8768.2002
Keywords: HIV-1, Tat, latency, transactivation, variability, reactivation
Citation: Kamori D and Ueno T (2017) HIV-1 Tat and Viral Latency: What We Can Learn from Naturally Occurring Sequence Variations. Front. Microbiol. 8:80. doi: 10.3389/fmicb.2017.00080
Received: 06 December 2016; Accepted: 11 January 2017;
Published: 30 January 2017.
Edited by:
Hirofumi Akari, Kyoto University, JapanReviewed by:
Kazuhisa Yoshimura, National Institute of Infectious Diseases, JapanCopyright © 2017 Kamori and Ueno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takamasa Ueno, dWVub3Rha2FAa3VtYW1vdG8tdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.