 Ben Fan
Ben Fan Jochen Blom
Jochen Blom Hans-Peter Klenk
Hans-Peter Klenk Rainer Borriss
Rainer Borriss- 1Co-Innovation Center for Sustainable Forestry in Southern China, College of Forestry, Nanjing Forestry University, Nanjing, China
- 2Bioinformatics and Systems Biology, Justus-Liebig-Universität Giessen, Giessen, Germany
- 3School of Biology, Newcastle University, Newcastle upon Tyne, UK
- 4Fachgebiet Phytomedizin, Institut für Agrar- und Gartenbauwissenschaften, Humboldt Universität zu Berlin, Berlin, Germany
- 5Nord Reet UG, Greifswald, Germany
The plant growth promoting model bacterium FZB42T was proposed as the type strain of Bacillus amyloliquefaciens subsp. plantarum (Borriss et al., 2011), but has been recently recognized as being synonymous to Bacillus velezensis due to phylogenomic analysis (Dunlap C. et al., 2016). However, until now, majority of publications consider plant-associated close relatives of FZB42 still as “B. amyloliquefaciens.” Here, we reinvestigated the taxonomic status of FZB42 and related strains in its context to the free-living soil bacterium DSM7T, the type strain of B. amyloliquefaciens. We identified 66 bacterial genomes from the NCBI data bank with high similarity to DSM7T. Dendrograms based on complete rpoB nucleotide sequences and on core genome sequences, respectively, clustered into a clade consisting of three tightly linked branches: (1) B. amyloliquefaciens, (2) Bacillus siamensis, and (3) a conspecific group containing the type strains of B. velezensis, Bacillus methylotrophicus, and B. amyloliquefaciens subsp. plantarum. The three monophyletic clades shared a common mutation rate of 0.01 substitutions per nucleotide position, but were distantly related to Bacillus subtilis (0.1 substitutions per nucleotide position). The tight relatedness of the three clusters was corroborated by TETRA, dDDH, ANI, and AAI analysis of the core genomes, but dDDH and ANI values were found slightly below species level thresholds when B. amyloliquefaciens DSM7T genome sequence was used as query sequence. Due to these results, we propose that the B. amyloliquefaciens clade should be considered as a taxonomic unit above of species level, designated here as “operational group B. amyloliquefaciens” consisting of the soil borne B. amyloliquefaciens, and plant associated B. siamensis and B. velezensis, whose members are closely related and allow identifying changes on the genomic level due to developing the plant-associated life-style.
Introduction
At the time of writing, the genus Bacillus (Gordon et al., 1973), consisted of 318 species with validly published names (http://www.bacterio.net/bacillus.html) with Bacillus subtilis as the type species (Cohn, 1872; Skerman et al., 1980). The industrial important species B. subtilis, Bacillus amyloliquefaciens, Bacillus licheniformis, and Bacillus pumilus are representing a group of phylogenetically and phenetically homogeneous species called, in the vernacular, the B. subtilis species complex (Fritze, 2004). For many years, it has been recognized that these species are hardly to distinguish on the basis of traditional phenotypic methods. Moreover, phylogenetic analysis of the 16S rRNA gene also fails to differentiate species within the complex due to the highly conserved nature of the gene (Rooney et al., 2009).
All members of this species complex are placed in 16S rRNA/DNA group 1. Its separation was based mainly on the significantly low DNA relatedness values experimentally determined by DDH, and their different fatty acid profiles (Priest et al., 1987). Besides the “original members” B. subtilis, B. licheniformis, and B. pumilus, early described by Gordon et al. (1973), many novel species belonging to the B. subtilis species complex have been described in last decades: B amyloliquefaciens (Priest et al., 1987), Bacillus atrophaeus (Nakamura, 1989), Bacillus mojavensis (Roberts et al., 1994), Bacillus vallismortis (Roberts et al., 1996), Bacillus sonorensis (Palmisano et al., 2001), Bacillus velezensis (Ruiz-García et al., 2005a), Bacillus axarquiensis (Ruiz-García et al., 2005b), Bacillus tequilensis (Gatson et al., 2006), Bacillus aerius, Bacillus aerophilus, Bacillus stratosphericus, Bacillus altitudinis (Shivaji et al., 2006), Bacillus safensis (Satomi et al., 2006), Bacillus methylotrophicus (Madhaiyan et al., 2010), Bacillus siamensis (Sumpavapol et al., 2010), Bacillus xiamenensis (Lai et al., 2014), Bacillus vanillea (Chen et al., 2014), Bacillus paralicheniformis (Dunlap C. et al., 2015), Bacillus glycinifermentas (Kim et al., 2015), Bacillus oryzicola (Chung et al., 2015), Bacillus gobiensis (Liu et al., 2016), and Bacillus nakamurai (Dunlap C. A. et al., 2016). B. vanillea, B. oryzicola, and B. methylotrophicus could not be corroborated as valid species and were identified as later heterotypic synonyms of either B. siamensis (Dunlap, 2015), or B. velezensis (Dunlap C. et al., 2016). B. subtilis has been subdivided into the three subspecies: B. subtilis subsp. subtilis, B. subtilis subsp. spizizenii (Nakamura et al., 1999), and B. subtilis subsp. inaquosorum (Rooney et al., 2009). In recent time, methods based on genome sequences (complete and WGS), such as ANI (Richter and Rosselló-Móra, 2009), AAI (Konstantinidis and Tiedje, 2005), dDDH (Meier-Kolthoff et al., 2013), and TETRA (Teeling et al., 2004), were used to finally discriminate a wide spectrum of bacterial taxons including the B. subtilis species complex (Federhen, 2015).
Some representatives of B. amyloliquefaciens were found plant-root-associated and to act beneficial on plant growth (Idriss et al., 2002). Reva et al. (2004) reported that seven Bacillus isolates from plants or soil are closely related to but distinct from B. amyloliquefaciens type strain DSM7T. These strains are more proficient for rhizosphere colonization than other members of the B. subtilis group (Hossain et al., 2015). B. amyloliquefaciens strains GB03 (Choi et al., 2014), and FZB42 (Chen et al., 2007) are widely used in different commercial formulations to promote plant growth.
With the advent of comparative genomics and the availability of an increasing number of whole genome sequences, it became possible to distinguish two subspecies within B. amyloliquefaciens: B. amyloliquefaciens subsp. amyloliquefaciens (type strain DSM7T), and B. amyloliquefaciens subsp. plantarum (type strain: FZB42T). Spectroscopic DDH performed with hydroxylapatite-purified chromosomal DNA from DSM7T and FZB42T yielded DNA-DNA relatedness values ranging between 63.7 and 71.2% which apparently did not sufficiently support discrimination of both taxons on the species level (Borriss et al., 2011). According to this view the subspecies “plantarum” represented a distinct ecotype of plant-associated B. amyloliquefaciens strains (Reva et al., 2004), which is increasingly used as biofertilizer and biocontrol agents in agriculture (Borriss, 2011).
Whilst many researchers are still using this classification (e.g., Hossain et al., 2015), recent phylogenomic studies showed a high degree of similarity between the genomes of the B. methylotrophicus, B. velezensis, B. oryzicola, and B. vanillea type strains, and the genome of the B. amyloliquefaciens subsp. plantarum type strain FZB42T (= DSM 23117T = BGSC 10A6T). Due to this finding it was proposed that the taxon B. amyloliquefaciens subsp. plantarum should be considered as a later heterotypic synonym of either B. methylotrophicus (Dunlap C. A. et al., 2015) or, more correctly due to priority rule, of B. velezensis (Dunlap C. et al., 2016). In spite of this increasingly complex taxonomic situation, we conducted here an extended phylogenomic analysis based on 66 core genomes displaying a high degree of similarity with the type strain of B. amyloliquefaciens DSM7T. It ruled out that three tightly linked clades including a conspecific group consisting of FZB42T, B. methylotrophicus KACC 13103T, and B. velezensis KCTC13012T, could be distinguished. The tight relatedness of the three clades consisting of representatives of B. amyloliquefaciens, B. velezensis, and B. siamensis was validated by rpoB gene sequence homology, and, ANI, AAI, dDDH, and TETRA analysis of the core genomes. We propose to introduce the term “operational group B. amyloliquefaciens” to underline their close phylogenomic relationship.
Materials and Methods
Retrieval of rpoB Sequences
Complete rpoB gene sequences with homology to B. amyloliquefaciens DSM7T were retrieved from the respective genomes of Bacillus strains available at NCBI (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi?organism$=$microb). Sequence comparisons were obtained by NCBI BlastN (http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD$=$Web&PAGE_TYPE$=$BlastHome).
Alignment of DNA rpoB Sequences
Alignment of DNA rpoB sequences was performed by the Clustal Omega program accessible at http://www.ebi.ac.uk/Tools/msa/clustalo/. A distance matrix was calculated from this alignment by DNA distance matrix calcuation (DNADIST program), and the matrix was then transformed into a tree by the NEIGHBOR program. In order to verify the accuracy of the tree multiple data sets were generated with the SEQBOOT program using 200 bootstrap replicates. A tree was built from each replicate with the DNADIST program, and then bootstrap values were computed with the CONSENSE program. The phylogenetic tree was visualized with TreeViewX (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html). The programs used to construct the phylogenetic tree were obtained from the PHYLIP package, v.3.65 (Felsenstein, 1989), which is accessible at http://evolution.genetics.washington.edu/phylip.html.
Comparative Genome Analysis
Comparative genome analysis was performed using the EDGAR 1.3 software framework. For orthology estimation EDGAR uses a generic orthology threshold calculated from the similarity statistics of the compared genomes (Blom et al., 2016; http://edgar.computational.bio.uni-giessen.de). A private project was constructed comprising 66 genomes closely related to B. amyloliquefaciens DSM7T and selected other representatives of the B. subtilis species complex. To construct a phylogenetic tree for this project, around 2000 core genes were computed by pairwise iterative comparison of a set of genomes (Blom et al., 2016). In a following step multiple alignments of the core genes were generated using MUSCLE, non-matching parts of the alignment were masked by GBLOCKS and subsequently removed. The remaining parts of all alignments were concatenated to one large alignment. The PHYLIP package was used to generate a phylogenetic tree of this alignment, represented in newick format.
The EDGAR software framework was also used to calculate average nucleotide identity (ANI) and average amino acid identity (AAI), matrices for a selected set of genomes. The blast hits between the orthologous genes of the core of the selected genome were analyzed for their mean/median percent identity values. The recommended species cut-off was 95% for the ANI and AAI indices (Richter and Rosselló-Móra, 2009). In addition, JSpeciesWS (http://jspecies.ribohost.com/jspeciesws/) was used to determine ANIb (average nucleotide identity based on BLAST+) and ANIm (average nucleotide identity based on MUMmer) values by pairwise genome comparisons. Correlation indexes of their Tetra-nucleotide signatures (TETRA) were determined by using the JSpeciesWS software (Richter et al., 2016).
Digital DNA–DNA Hybridization (dDDH)
The genome-to-genome-distance calculator (GGDC) version 2.1 provided by DSMZ (http://ggdc.dsmz.de/) was used for genome-based species delineation (Meier-Kolthoff et al., 2013) and genome-based subspecies delineation (Meier-Kolthoff et al., 2014). Distances were calculated by (i) comparing two genomes using the chosen program to obtain HSPs/MUMs and (ii) inferring distances from the set of HSPs/MUMs using three distinct formulas. Next, the distances were transformed to values analogous to DDH. The DDH estimates were based on an empirical reference dataset comprising real DDH values and genome sequences. The DDH estimate resulted from a generalized linear model (GLM) which also provided the estimate's confidence interval (after the ± sign). Three formulas are available for the calculation: Formula: 1 (HSP length/total length), formula: 2 (identities/HSP length) and formula 3 (identities/total length). Formula 2, which is especially appropriate to analyze draft genomes, was used.
Results
Phylogenomics of the B. Subtilis Species Complex
The core genomes of 20 type strains of the B. subtilis species complex were used for phylogenomic analysis applying the EDGAR software package (Figure 1). Four main monophyletic groups were corroborated by 100% bootstrap values. Clade I (“subtilis”) is early diverged into two branches comprising B. atrophaeus, and B. subtilis and its close relatives; clade II (“amyloliquefaciens”) comprises B. amyloliquefaciens, B. siamensis, and a conspecific group containing the type strains of B. amyloliquefaciens subsp. plantarum, B. velezensis, and B. methylotrophicus; clade III (“licheniformis”) consists of B. licheniformis and B. sonorensis; and clade IV (“pumilus”) comprises B. pumilus, B. safensis, B. xiamenensis, and a conspecific group involving the type strains of B. altitudinis, B. stratosphericus, and B. aerophilus. The members of clade II appeared closely related. This is indicated by the high number of orthologous CDSs (2794) shared by the five type strains of clade II. A similar cladogram has been published recently (Dunlap C. et al., 2016) suggesting that the B. subtilis species complex can be divided into four groups above species level, which need further characterization. We have directed our further analysis to clade II (named from now on “operational group B. amyloliquefaciens”), which clearly shows the highest degree of compactness.
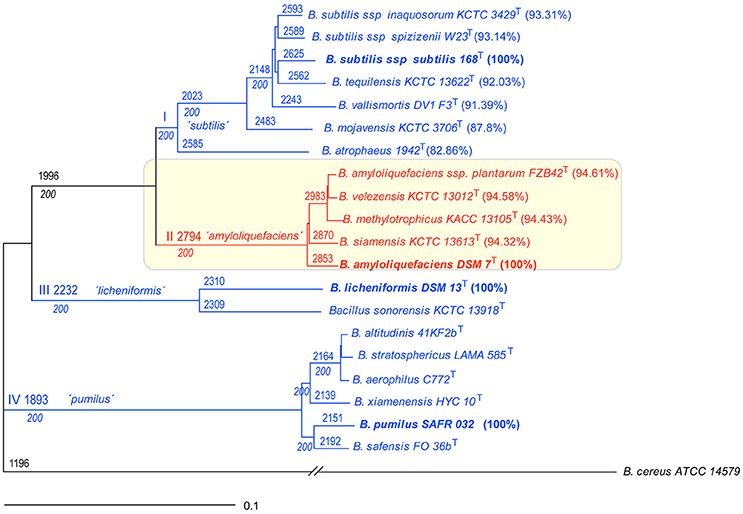
Figure 1. Phylogeny of the Bacillus subtilis species complex based on the core genomes of representative type strains. The core genome of Bacillus cereus ATCC14579 was used as outgroup. The roman letters at the branching points designate the four clades identified in this analysis. The numbers at the branching points designate the number of CDS calculated for the core genome of a given subset of genomes. Bootstrap values of 200 (100%) are indicated below the CDS numbers (see Materials and Methods). Percentage of identity according to type strains B. subtilis subsp. subtilis 168T, B. amyloliquefaciens DSM7T, B. licheniformis DSM13T, and B. pumilus SAFR032, respectively. Note that within clade II (“amyloliquefaciens”) the group with B. amyloliquefaciens subsp. plantarum FZB42T, B. velezensis KCTC 13012T, and B. methylotrophicus KACC 13105T is conspecific. The same is true for the group within clade IV (“pumilus”) consisting of B. altitudinis 41KF2bT, B. stratosphericus LAMA 585T, and B. aerophilus C772T. The scale bar corresponds to 0.1 substitutions per site.
Phylogenetic Analysis of Clade II Based on Complete rpoB Nucleotide Sequence
It is obvious, that 16S rRNA sequences are not sufficient to discriminate representatives of the B. subtilis species complex. For example, comparison of the complete 16S rRNA sequences of B. amyloliquefaciens DSM7T and B. subtilis 168T revealed 99.48% identity (Table 1), which is well above of the recommended threshold of >98.65% for species delineation (Kim et al., 2014). In order to elucidate more precisely the phylogenetic and taxonomic relationship of the members of the B. subtilis species complex belonging to the “operational group B. amyloliquefaciens,” we used two methods. (i) Tetra correlation search (TCS, Richter et al., 2016) was performed with the complete genome of DSM7T and (ii) the complete RNA polymerase beta-subunit (rpoB) gene of DSM7T was used for BLASTN comparison with the corresponding sequences extracted from complete genomes or genome assemblies. Fifty-Two genomes, which were in range with the intraspecific Tetra-nucleotide signature correlation index (>0.99) were detected in the JSpecies data bank. The TCS value determined for B. subtilis was only 0.954, suggesting that using this alignment-free parameter allows discriminating of B. subtilis and B. amyloliquefaciens (Table 1). Complete rpoB gene sequencing has been proposed as phylogenetic marker (Klenk et al., 1994) and as a supplement to DDH (Adékambi et al., 2008). The power and potential of complete rpoB gene sequence in taxonomic, phylogenetic and evolutionary studies has been previously reported (Sharma and Patil, 2011). Our BLASTN search revealed that at least 66 genomes present in the NCBI data bank contain rpoB gene sequences with more than 98% identity to the rpoB gene from DSM7T, the type strain of B. amyloliquefaciens (Priest et al., 1987). For comparison, the rpoB gene from B. subtilis subsp. subtilis 168T displayed only 90.3% identity to B. amyloliquefaciens. The rpoB gene identities among strains assigned as being B. amyloliquefaciens, B. siamensis, B. amyloliquefaciens subsp. plantarum, B. methylotrophicus, B. velezensis, and B. vanillea are listed in Table 1. The list of strains containing rpoB genes with high similarity to B. amyloliquefaciens DSM7T includes also strains obviously not correctly assigned, such as B. subtilis, Bacillus sp., or Paenibacillus polymyxa. It is interesting to note that majority of the strains representing the conspecific B. velezensis/B.methylotrophicus/B.amyloliquefaciens subsp. plantarum group were isolated from plant sources, whilst B. amyloliquefaciens sensu stricto seems to be soil-borne. The main source of the salt tolerant B. siamensis/B.vanillea group was fermented plant food (Table 1).
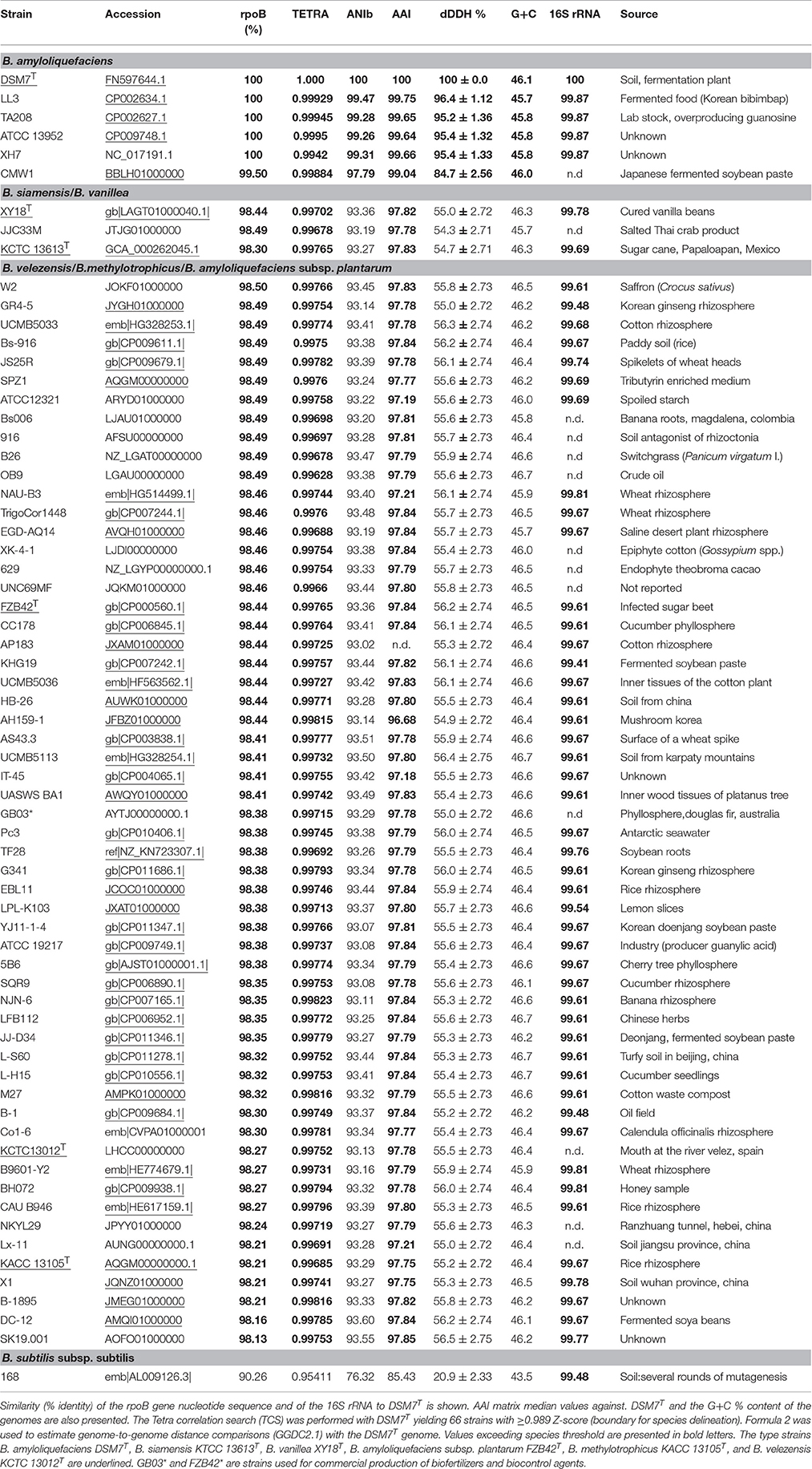
Table 1. Genomes containing rpoB sequences displaying ≥98% similarity to B. amyloliquefaciens DSM7T.
The phylogenetic tree based on complete rpoB gene sequence suggests existence of three tightly connected monophyletic groups: (i) B. amyloliquefaciens containing six strains including type strain DSM7T; (ii) B. siamensis cluster consists of three strains: the type strain KCTC 13613T, strain XY18, originally assigned as type strain for B. vanillea (Chen et al., 2014) but recently reclassified as being B. siamensis (Dunlap, 2015), and a strain assigned as being B. amyloliquefaciens JJC33M; (3) the conspecific complex comprising B. velezensis, B. methylotrophicus, and B. amyloliquefaciens subsp. Plantarum contained 57 strains. The tree is robust displaying high bootstrap values for all three groupings, although the three clusters are closely related and separated by only 0.01–0.02 substitutions per nucleotide position. By contrast, taxonomic distance to B. subtilis is around tenfold larger (Figure 2).
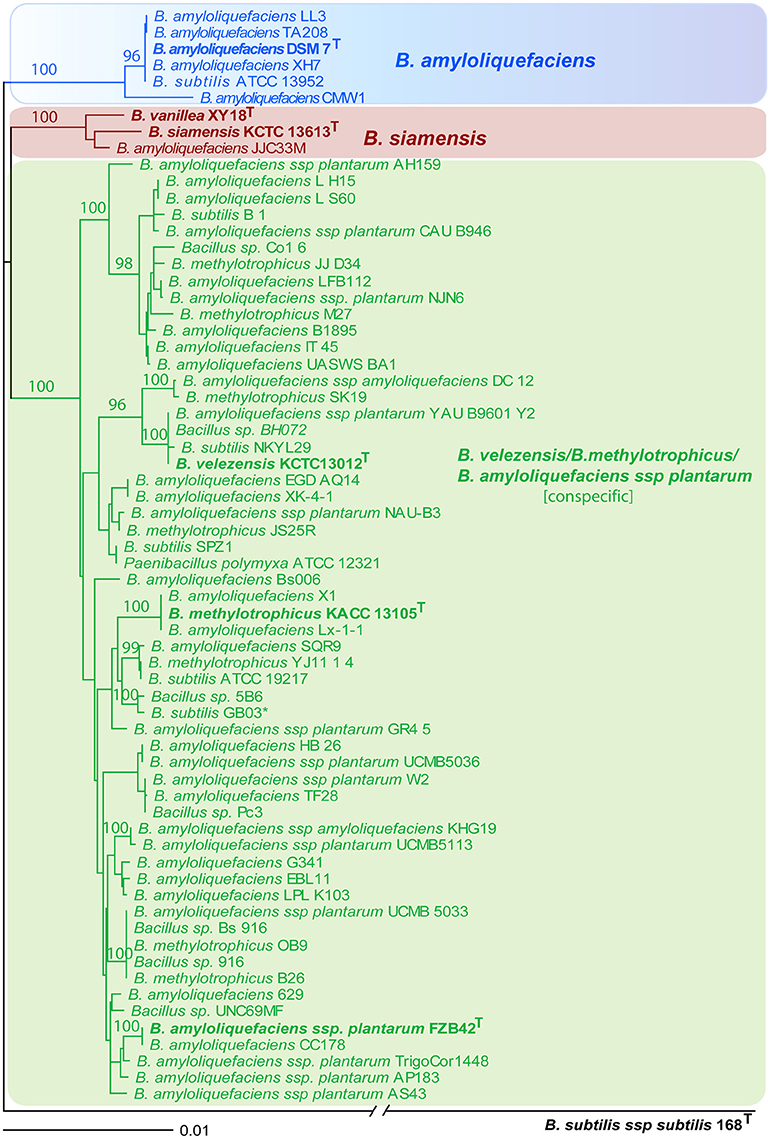
Figure 2. NJ phylogenetic tree, extracted from 66 complete rpoB nucleotide sequences with high similarity to B. amyloliquefaciens DSM7T (>98% identity). B. subtilis subsp. Subtilis 168T was used as outgroup. The consensus tree was reconstructed from 1000 trees according to the extended majority rule (SEQBOOT program). Bootstrap values >90%, based on 1000 repetitions, are indicated at branch points. Strain and accession numbers are indicated. Type strains for B. amyloliquefaciens (DSM7T), B. siamensis (KCTC13613T) and B. vanillea (XY18T), and the conspecific group containing FZB42T as the type strain for B. amyloliquefaciens subsp. Plantarum, B. velezensis KCTC13012T, and B. methylotrophicus KACC13105T are in bold. Bar, 0.01 substitutions per nucleotide position. For further characterization of strains and genomes see Table 1.
Phylogenomic Analysis of Clade II (Operational Group B. amyloliquefaciens)
In order to confirm the phylogenetic analysis based on rpoB sequences we calculated the core genomes using the EDGAR 1.3 program package. A total of 1998 CDSs were shared by the 66 core genome sequences extracted in that analysis. It ruled out that the phylogenomic tree based on complete core genome sequences (Figure 3) did reflect the phylogenomic distances similar as the phylogenetic tree based on rpoB nucleotide sequences (Figure 2). The same robust monophyletic groups as in Figure 2 were obtained. The B. siamensis cluster consisting of three representatives shared a core genome of 3097 CDSs; the B. amyloliquefaciens cluster consisting of six representatives shared a core genome of 3139 CDSs; and the conspecific group containing 57 plant-growth promoting Bacilli including FZB42T shared a relatively small core genome consisting of only 2295 CDSs, which is mainly due to the high number of genomes included in this analysis. Subgroups of this cluster shared core genomes ranging from 2659 to 3137 CDSs (Figure 3). Again, the NJ tree suggested that B. amyloliquefaciens subsp. Plantarum, B. methylotrophicus, and B. velezensis formed a monophyletic group corroborating recent findings (Dunlap C. A. et al., 2015; Wu et al., 2015; Dunlap C. et al., 2016).

Figure 3. NJ phylogenomic tree, constructed from the 66 core genomes with the highest similarity to DSM7T (Table 1). The B. subtilis genome was used as outgroup. The number of core genome CDSs is indicated at the nodes. They were calculated for the respective subsets of genomes. Bootstrap values obtained from 200 repetitions are also indicated at the nodes. Type strains (T) are indicated by bold letters. Bar, 0.02 substitutions per nucleotide position.
At next we tried to elucidate the taxonomic status of these closely related genomes. Different phylogenetic and phylogenomic methods were used to analyze relationship of all 65 genomes with that of B. amyloliquefaciens DSM7T. As shown above, rpoB sequence similarity, exceeding threshold of species delineation, and the intraspecific Tetra-nucleotide signature correlation index (>0.99) suggested that all strains analyzed belong to the species B. amyloliquefaciens. TETRA analysis (Jspecies) demonstrated that the six type strains of clade II were closely related and yielded pairwise Tetra results (tetranucleotide signature correlation index) in species range (≥0.989, Figure 4 lower part). Deviations of the mean G+C content calculated for the whole genomes were less than one percent which does not contradict species definition (Table 2). Grouping of all strains into a single species, B. amyloliquefaciens, was further supported by the AAI values (Table 1). The mean AAI values of the 66 core genomes selected by their rpoB similarity to DSM7T were ≥96.5%, exceeding the proposed cut-off of 96% for species delineation. However, parameters, considered recently as being most important for genome-based species delineation, such as ANI and dDDH (Federhen et al., 2016), did not support this conclusion (Table 1).
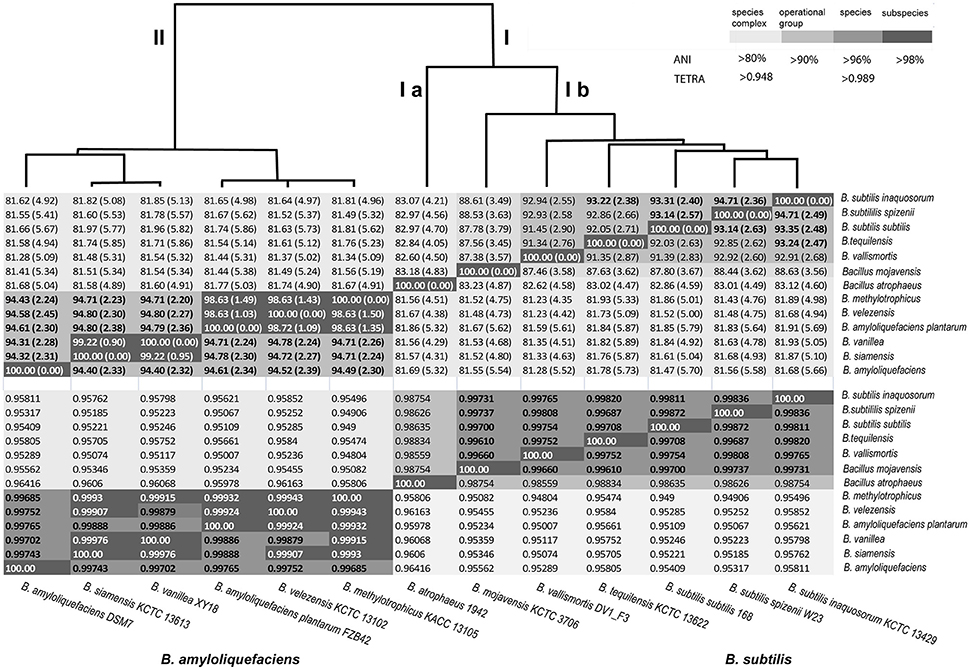
Figure 4. Dendrogram of the type strains of clade I (operational group “B.subtilis”) and II (operational group “B. amyloliquefaciens”) based on their median ANIb values (upper part of the Table) and Tetra-nucleotide correlation signatures (lower part of the Table). The median nucleotide percent identity values between the orthologous genes of the core of the selected genomes after pairwise BLASTN comparison are indicated. Standard deviation values are given in parentheses.
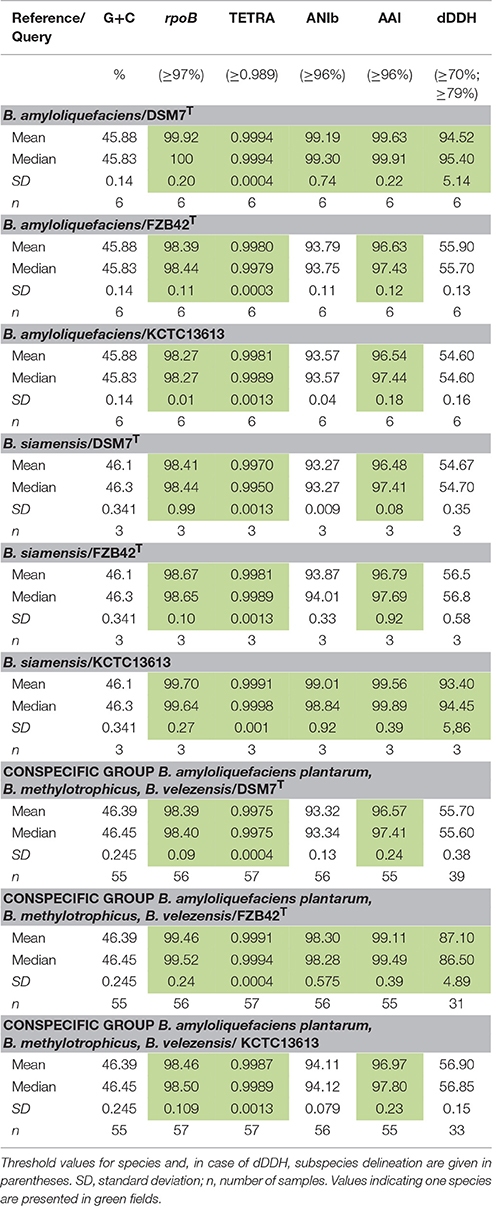
Table 2. Summary of phylogenetic (rpoB) and phylogenomic parameters calculated for B. amyloliquefaciens, B. siamensis and conspecific group consisting of B. amyloliquefaciens plantarum, B. methylotrophicus and B. velezensis against corresponding type strains.
ANI analysis performed with the EDGAR program package discriminated clearly two clusters corresponding to clades I and II of the B. subtilis species complex (Figure 4). Clade II representing the B. amyloliquefaciens group was divided into three groups consisting of B. amyloliquefaciens DSM7T (i), B. siamensis and B. vanillea (ii), and the conspecific complex formed by the type strains B. methylotrophicus KACC13105T, B. velezensis KCTC 13102T and B. amyloliquefaciens subsp. plantarum FZB42T (iii). The latter group displayed ANI values of >98% exceeding the cut-off for species delineation when compared with each other suggesting that the members of the conspecific complex belong to a single species. B. methylotrophicus KACC 13105T, B. velezensis KCTC 13102T, and B. amyloliquefaciens subsp. plantarum FZB42T displayed similar median ANI values ranging between 94.3 and 94.8% when compared with B. amyloliquefaciens DSM7T (1) and B. siamensis KCTC-13613T (2), respectively. Given a calculated deviation of ±2.2–2.3% the ANI matrix values suggests a high degree of relatedness to B. amyloliquefaciens, B. siamensis and the conspecific group formed by B. amyloliquefaciens subsp. plantarum, B. methylotrophicus, and B. velezensis, but did not sufficiently support species delineation (Figure 4, upper part). According to more recent findings the recommended cut-off point for species delineation corresponds to ~96% ANI (Colston et al., 2014). Similar results were obtained when ANIb and ANIm values were determined by using the JSpecies program package for all the 66 genomes included in this analysis. Threshold values sufficient for species delineation were only obtained, when representatives of B. amyloliquefaciens (6 genomes), B. siamensis (3 genomes), and of the conspecific group (57 genomes) were compared with their respective type strains. However, comparison of the 57 strains of the conspecific group (e.g., FZB42, B. velezensis) with either B. amyloliquefaciens DSM7T or B. siamensis KCTC 13613T yielded ANI values slightly below the cut-off for species delineation. The same was true when the three members of the B. siamensis group were compared with either FZB42T or DSM7T (Table 2) suggesting that according to ANI analysis the members of clade II represent three discrete, although closely related, species.
In order to finally decide, whether all strains of clade II belong to one species or not, electronic DNA-DNA hybridization (dDDH) was applied in a quantitative analysis involving all 66 genomes. As shown previously, dDDH is useful to mimic the wet-lab DDH and can be used for genome-based species delineation and genome-based subspecies delineation (Meier-Kolthoff et al., 2013, 2014). For calculating dDDH three different formulas can be applied (see Materials and Methods), but only results obtained with the recommended formula 2 were used in our analysis (Table 2). When comparing members of the “siamensis group 2” and the “conspecific B. velezensis group” with B. amyloliquefaciens DSM7T, dDDH values of <70%, the defined threshold for species delineation, were obtained. All in all, dDDH supports our previous finding about a close relationship within clade II, but did not support their classification into one single species. The results are summarized in Table 2 and Supplementary Table 1.
Gene Clusters Involved in Nonribosomal Synthesis of Secondary Metabolites
Compared to other members of the B. subtilis species complex, the plant-associated B. amyloliquefaciens possess an enormous potential to synthesize bioactive secondary metabolites. Besides five gene clusters, known from B. subtilis to mediate nonribosomal synthesis of secondary metabolites, four giant gene clusters absent in B. subtilis 168 were identified in FZB42 (Chen et al., 2007). The nine gene clusters that direct the synthesis of bioactive peptides and polyketides by modularly organized mega-enzymes define both nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKS). Three (bmyD, dfn, and mln) are not present in B. subtilis 168, but occur in all members of the “operational group B. amyloliquefaciens.” Except for the gene cluster encoding bacilysin synthesis, the functional activities of the remaining gene clusters depend on Sfp, an enzyme that transfers 4′-phosphopantetheine from coenzyme A to the carrier proteins of nascent peptide or polyketide chains. A direct comparison revealed that the nine gene cluster responsible for nonribosomal synthesis of bioactive secondary metabolites including macrolactin are only present in FZB42 and in the other members of the conspecific B. velezensis group, whilst the gene cluster involved in macrolactin synthesis was not detected in B. siamensis and B. amyloliquefaciens (Table 3). Noteworthy, the gene cluster responsible for synthesis of the polyketide difficidin was present in B. siamensis, but not in any other member of the B. subtilis species complex suggesting a stepwise loss of the ability to synthesize secondary metabolites in the order B. velezensis (including FZB42)  B. siamensis B. amyloliquefaciens.
B. siamensis B. amyloliquefaciens.
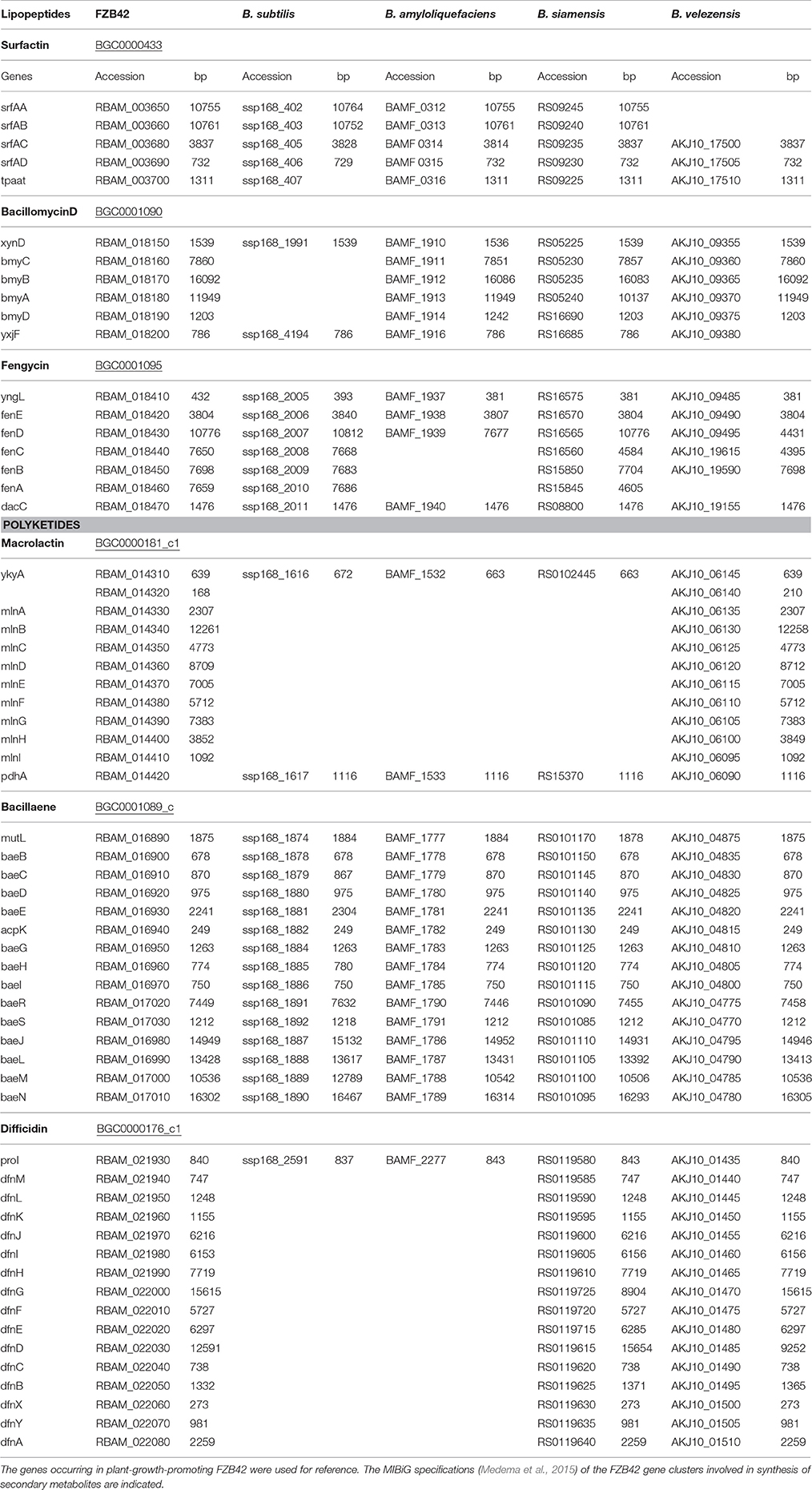
Table 3. Gene clusters encoding nonribosomal synthesis of lipopeptides and polyketides in type strains of B. subtilis, B. amyloliquefaciens, B. siamensis, and B. velezensis.
Discussion
The B. subtilis species complex consists of a steadily increasing number of validly described species (see Introduction), which display an extremely high degree of similarity. They are very difficult to distinguish by using classical taxonomy parameters: morphological and physiological characteristics, cell wall compositions, 16S rRNA sequence, G+C content, and FAME. Also, experimental determination of DNA-DNA relatedness (DDH), gold-standard of bacterial taxonomy for 50 years, yields often erroneous and variable results (Auch et al., 2010). Therefore, the taxonomic status of these species constantly brings confusion to researchers, especially for non-professional taxonomy researchers. Our analysis using the available core genomes of 23 type strains suggests that within the B. subtilis species complex four clades can be distinguished: clade I consisting of B. subtilis including its three subspecies subtilis, spizenii, and inaquosorum, B. tequilensis, B. vallismortis, B. mojavensis, and B. atrophaeus, clade II consisting of B. siamensis, B. amyloliquefaciens, and a conspecific complex consisting of B. methylotrophicus, B. velezensis, and B. amyloliquefaciens subsp. plantarum, clade III consisting of B. licheniformis and related species, and clade IV consisting of B. pumilus and related species (Figure 1).
We have chosen here clade II comprising B. amyloliquefaciens and related species for a deeper analysis. Due to the high number of available genomic sequences, we applied a quantitative phylogenomic approach including 66 genomes with a high degree of similarity to DSM7T, the type strain of B. amyloliquefaciens. In accordance to Dunlap C. A. et al. (2015) we could demonstrate existence of three distinct monophyletic groups within this clade. Six core genomes represented the species B. amyloliquefaciens and three strains were assigned as being B. siamensis. The results of our extensive phylogenomic analysis (Table 2) corroborates the monophyletic nature of the conspecific group consisting of B. amyloliquefaciens subsp. plantarum, B. methylotrophicus, and B. velezensis, suggesting that this unique taxon is closely related to B. amyloliquefaciens (Borriss et al., 2011). B. velezensis is a heterotypic synonym of B. methylotrophicus, B. amyloliquefaciens subsp. plantarum, and Bacillus oryzicola, and is used to control plant fungal diseases. This idea is further supported by a recent phylogenetic and phylogenomic analysis in which B. amyloliquefaciens, B. siamensis, and B. amyloliquefaciens subsp. plantarum were established as closely related monophyletic groups harboring a common ancestor based on their gyrB and core genome (729,383 bp) sequences (Hossain et al., 2015). The conspecific group consisting of B. amyloliquefaciens subsp. plantarum, B. methylotrophicus, and B. velezensis was recently classified as being B. velezensis (Dunlap C. et al., 2016), because the valid publication of B. velezensis (Ruiz-García et al., 2005a) predates the publication of B. methylotrophicus (Madhaiyan et al., 2010) and B. amyloliquefaciens subsp. plantarum (Borriss et al., 2011). The tight relatedness of B. siamensis and B. velezensis with B. amyloliquefaciens is indicated by:
(i) highly conserved rpoB nucleotide sequence with more than 98% identity to DSM7T;
(ii) Mean G+C % content is only 0.5% different ranging between 45.9% (subsp. amyloliquefaciens), 46.1% (subsp. siamensis), and 46.4% (subsp. plantarum);
(iii) Tetranucleotide signatures, TETRA, were determined as above the cut-off for species delineation (>0.989);
(iv) AAI values are well above 96%, representing the intraspecific threshold.
On the other hand, ANIb and ANIm were calculated as around 93 to 94% identity to B. amyloliquefaciens on the nucleotide level, which is slightly lower than the threshold proposed for species delineation (95–96% ANI, Kim et al., 2014). Moreover, electronic DDH calculation using formula 2 yielded only 56% identity, which is clearly below the cut-off for species delineation. In spite of these contradictory results, we have to conclude that three discrete species exist within clade II, given that results obtained by ANI and dDDH are more important in modern taxonomy (Auch et al., 2010; Meier-Kolthoff et al., 2013) and outcompete the other results favoring a B. amyloliquefaciens subspecies concept. However, due to the close relationship of all three species comprised in clade II we propose an “operational group B. amyloliquefaciens” comprising B. amyloliquefaciens, B. siamensis, and B. velezensis. The introduction of this “operational group” above species level should improve hierarchical classification within the B. subtilis species complex. The members of the “operational group B. amyloliquefaciens are distinguished from B. subtilis and its closest relatives by their ability to synthesize nonribosomally antifungal acting lipopopeptides of the iturin group, mostly bacillomycin D or iturin A. The ecotype of plant-associated B. amyloliquefaciens is well introduced since many years (Reva et al., 2004) and includes the most important biocontrol- and plant-growth-promoting Bacilli, which are successfully used as environmental-friendly means in agriculture (Borriss, 2011). In addition, numerous studies have been published in recent years in order to identify and to understand the specific features of the group of B. amyloliquefaciens strains able to colonize plant organs and to withstand strong plant response reactions. As in B. subtilis it is now widely recognized that a relevant part of metabolism of plant-associated B. amyloliquefaciens is devoted to metabolic interactions with plants (Belda et al., 2013). Metabolites produced by the plant-associated B. amyloliquefaciens FZB42 and other members of the conspecific B. velezensis group represent a substantial part of the diversity of nonribosomal secondary metabolites from the genus Bacillus. For example, they produce three types of polyene polyketides (difficidin, macrolactin, and bacillaene) with strong antibiotic action (Chen et al., 2007). By contrast, B. siamensis does only produce two (difficidin and bacillaene) and soil-borne B. amyloliquefaciens does only produce one polyketide (bacillaene). It is highly desirable to apply a correct taxonomic designation to distinguish the plant-associated (= B. velezensis) and the soil-borne B. amyloliquefaciens” (= B. amyloliquefaciens) strains, but also to take into consideration their high degree of relatedness. This should be reflected by their grouping into the “operational B. amyloliquefaciens group,” as a novel taxonomic unit above species level but below the “B. subtilis species complex.” Introduction of the novel taxonomic unit seems also be recommended in spite of a permanent misuse in describing taxonomy important Bacillus biocontrol strains such as GB03 (Choi et al., 2014) and QST713 (Kinsella et al., 2009), which are often designated as B. subtilis although they are true representatives of B. velezensis and simultaneously members of the “operational group B. amyloliquefaciens.”
In summary, due to their differences in ANI and dDDH values, which are slightly below species level thresholds, we propose that B. amyloliquefaciens, B. velezensis, and B. siamensis should keep their status as species of its own, as proposed by Dunlap (Dunlap C. et al., 2016). The close relatedness of the three species is well reflected by the novel taxonomic unit “operational group B. amyloliquefaciens.” Introducing of this novel taxonomic unit should improve also understanding of previous and recent scientific investigations performed with “plant-associated B. amyloliquefaciens” strains which often have not been designated correctly.
Another less surprising finding from our analysis was that many of the publically available Bacillus genomes that we analyzed are inconsistently assigned. Fortunately, a recent initiative has been started to correct such mistakes in Genbank entries (Federhen et al., 2016).
Author Contributions
BF, JB, HK, and RB performed phylogenomic analyses. All authors were involved in preparing the manuscript. The final version of the manuscript was prepared by RB.
Funding
The financial support for BF by the National Natural Science Foundation of China (No. 31100081), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, and Natural Science Foundation of Jiangsu Province (No. BK20151514) is gratefully acknowledged.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00022/full#supplementary-material
Abbreviations
AAI, average amino acid identity; ANI, average nucleotide identity; CDS, coding sequence; DDH, DNA–DNA hybridization; dDDH, digital DNA–DNA hybridization; GGDC, Genome-to-Genome Distance Calculator; TETRA, tetranucleotide frequency distribution.
References
Adékambi, T., Shinnick, T. M., Raoult, D., and Drancourt, M. (2008). Complete rpoB gene sequencing as a suitable supplement to DNA-DNA hybridization for bacterial species and genus delineation. Int. J. Syst. Evol. Microbiol. 58, 1807–1814. doi: 10.1099/ijs.0.65440-0
Auch, A. F., Jan, M., von Klenk, H. P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Belda, E., Sekowska, A., Le Fevre, F., Morgat, A., Mornico, D., Ouzunis, C., et al. (2013). An updated metabolic view of the Bacillus subtilis 168 genome. Microbiology 159, 757–770. doi: 10.1099/mic.0.064691-0
Blom, J., Kreis, J., Spänig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Borriss, R. (2011). “Use of plant-associated Bacillus strains as biofertilizers and biocontrol agents,” in Bacteria in Agrobiology: Plant Growth Response, ed D. K. Maheshwari (Heidelberg: Springer), 41–76.
Borriss, R., Chen, X. H., Rueckert, C., Blom, J., Becker, A., Baumgarth, B., et al. (2011). Relationship of Bacillus amyloliquefaciens clades associated with strains DSM 7T and FZB42T: a proposal for Bacillus amyloliquefaciens subsp. amyloliquefaciens subsp. nov. and Bacillus amyloliquefaciens subsp. plantarum subsp. nov. based on complete genome sequence comparisons. Int. J. Syst. Evol. Microbiol. 61, 1786–1801. doi: 10.1099/ijs.0.023267-0
Chen, X. H., Koumoutsi, A., Scholz, R., Eisenreich, A., Schneider, K., Heinemeyer, I., et al. (2007). Comparative analysis of the complete genome sequence of the plant growth promoting bacterium Bacillus amyloliquefaciens FZB42. Nat. Biotechnol. 25, 1007–1014. doi: 10.1038/nbt1325
Chen, Y., Gu, F., Li, J., Xu, F., He, S., and Fang, Y. (2014). Bacillus vanillea sp. nov., isolated from the cured vanilla bean. Curr. Microbiol. 70, 235–239. doi: 10.1007/s00284-014-0707-4
Choi, S. K., Jeong, H., Kloepper, J. W., and Ryu, C. M. (2014). Genome Sequence of Bacillus amyloliquefaciens GB03, an active ingredient of the first commercial biological control product. Genome Announc. 2:e01092-14. doi: 10.1128/genomeA.01092-14
Chung, E. J., Hossain, M. T., Khan, A., Kim, K. H., Jeon, C. O., and Chung, Y. R. (2015). Bacillus oryzicola sp. nov., an endophytic bacterium isolated from the roots of rice with antimicrobial, plant growth promoting, and systemic resistance inducing activities in rice. Plant Pathol. J. 31, 152–164. doi: 10.5423/PPJ.OA.12.2014.0136
Colston, S. M., Fullmer, M., Beka, L., Lamy, B., Gogarten, J. P., and Graf, J. (2014). Bioinformatic genome comparisons for taxonomic and phylogenetic assignments using Aeromonas as a test case. mBio 5, e02136–e02114. doi: 10.1128/mBio.02136-14
Dunlap, C. A. (2015). Phylogenomic analysis shows that ‘Bacillus vanillea’ is a later heterotypic synonym of Bacillus siamensis. Int. J. Syst. Evol. Microbiol. 65, 3507–3510. doi: 10.1099/ijsem.0.000444
Dunlap, C. A., Kim, S. J., Kwon, S. W., and Rooney, A. P. (2015). Phylogenomic analysis shows that Bacillus amyloliquefaciens subsp. plantarum is a later heterotypic synonym of Bacillus methylotrophicus. Int. J. Syst. Evol. Microbiol. 65, 2104–2109. doi: 10.1099/ijs.0.000226
Dunlap, C. A., Saunders, L. P., Schisler, D. A., Leathers, T. D., Naeem, N., Cohan, F. M., et al. (2016). Bacillus nakamurai sp. nov., a black pigment producing strain. Int. J. Syst. Evol. Microbiol. 66, 2987–2991. doi: 10.1099/ijsem.0.001135
Dunlap, C., Kim, S. J., Kwon, S. W., and Rooney, A. (2016). Bacillus velezensis is not a later heterotypic synonym of Bacillus amyloliquefaciens, Bacillus methylotrophicus, Bacillus amyloliquefaciens subsp. plantarum and ‘Bacillus oryzicola’ are later heterotypic synonyms of of Bacillus velezensis based on phylogenomics. Int. J. Syst. Evol. Microbiol. 66, 1212–1217. doi: 10.1099/ijsem.0.000858
Dunlap, C., Kwon, S. W., Rooney, A., and Kim, S. J. (2015). Bacillus paralicheniformis sp. nov., isolated from fermented soybean paste. Int. J. Syst. Evol. Microbiol. 65, 3487–3492. doi: 10.1099/ijsem.0.000441
Federhen, S. (2015). Type material in the NCBI taxonomy database. Nucleic Acids Res. 43, D1086–D1098. doi: 10.1093/nar/gku1127
Federhen, S., Rossello-Mora, R., Klenk, H. P., et al. (2016). Meeting report: Genbank microbial genomic taxonomy workshop (12–13 May, 2015). Stand. Genomic Sci. 11:15. doi: 10.1186/s40793-016-0134-1
Fritze, D. (2004). Taxonomy of the genus Bacillus and related genera: the aerobic endospore-forming bacteria. Phytopathology 94, 1245–1248. doi: 10.1094/PHYTO.2004.94.11.1245
Gatson, J. W., Benz, B. F., Chandrasekaran, C., Satomi, M., Venkateswaran, K., and Hart, M. E. (2006). Bacillus tequilensis sp. nov., isolated from 2000-year-old Mexican shaft-tomb, is closely related to Bacillus subtilis. Int. J. Syst. Evol. Microbiol. 56, 1475–1484. doi: 10.1099/ijs.0.63946-0
Gordon, R. E., Haynes, W. C., and Pang, C. H.-N. (1973). The Genus Bacillus. Washington, DC: United States Department of Agriculture.
Hossain, M. J., Ran, C., Liu, K., Ryu, C. M., Rasmussen-Ivey, C. R., Williams, M. A., et al. (2015). Deciphering the conserved genetic loci implicated in plant disease control through comparative genomics of Bacillus amyloliquefaciens subsp. plantarum strains. Front. Plant Sci. 6:631. doi: 10.3389/fpls.2015.00631
Idriss, E. E., Makarewicz, O., Farouk, A., Rosner, K., Greiner, R., Bochow, H., et al. (2002). Extracellular phytase activity of Bacillus amyloliquefaciens FZB45 contributes to its plant-growth-promoting effect. Microbiology 148, 2097–2109. doi: 10.1099/00221287-148-7-2097
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Kim, S. J., Dunlap, C. A., Kwon, S. W., and Rooney, A. P. (2015). Bacillus glycinifermentans sp. nov., isolated from fermented soybean paste. Int. J. Syst. Evol. Microbiol. 65, 3586–9350. doi: 10.1099/ijsem.0.000462
Kinsella, K., Schulthess, C. P., Morris, T. F., and Stuart, J. D. (2009). Rapid quantification of Bacillus subtilis antibiotics in the rhizosphere. Soil Biol. Biochem. 41, 374–379. doi: 10.1016/j.soilbio.2008.11.019
Klenk, H. P., Palm, P., and Zillig, W. (1994). DNA dependent RNA polymerases as phylogenetic marker molecules. Syst. Appl. Microbiol. 16, 638–647. doi: 10.1016/S0723-2020(11)80335-8
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 2005, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Lai, Q., Liu, Y., and Shao, Z. (2014). Bacillus xiamenensis sp. nov., isolated from intestinal tract contents of a flathead mullet (Mugil cephalus). Antonie Van Leeuwenhoek 105, 99–107. doi: 10.1007/s10482-013-0057-4
Liu, B., Liu, G. H., Cetin, S., Schumann, P., Pan, Z. Z., and Chen, Q. Q. (2016). Bacillus gobiensis sp. nov., isolated from a soil sample collected from Xinjiang of China. Int. J. Syst. Evol. Microbiol. 66, 379–384. doi: 10.1099/ijsem.0.000729
Madhaiyan, M., Poonguzhali, S., Kwon, S.-W., and Sa, T.-M. (2010). Bacillus methylotrophicus sp. nov., a methanol utilizing, plant-growth-promoting bacterium isolated from rice rhizosphere soil. Int. J. Syst. Evol. Microbiol. 60, 2490–2495. doi: 10.1099/ijs.0.015487-0
Medema, M. H., Kottmann, R., Yilmaz, P., Cummings, M., Biggins, J. B., Blin, K., et al. (2015). The minimum information about a biosynthetic gene cluster (MIBiG) specification. Nat. Chem. Biol. 11, 625–631. doi: 10.1038/nchembio.1890
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Hahnke, R. L., Petersen, J., Scheuner, C., Michael, V., Fiebig, A., et al. (2014). Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic Sci. 10:2. doi: 10.1186/1944-3277-9-2
Nakamura, L. K. (1989). Taxonomic relationship of black-pigmented Bacillus subtilis strains and a proposal for Bacillus atrophaeus sp. nov. Int. J. Syst. Bacteriol. 39, 295–300. doi: 10.1099/00207713-39-3-295
Nakamura, L. K., Roberts, M. S., and Cohan, F. M. (1999).Relationship of Bacillus subtilis clades associated with strains 168 and W23: a proposal for Bacillus subtilis subsp. subtilis subsp. nov. and Bacillus subtilis subsp. spizizenii subsp. nov. Int. J. Syst. Bact. 49, 1211–1215. doi: 10.1099/00207713-49-3-1211
Palmisano, M. M., Nakamura, L. K., Duncan, K. E., Istock, C. A., and Cohan, F. M. (2001). Bacillus sonorensis sp. nov., a close relative of B. licheniformis, isolated from soil in the Sonoran Desert, Arizona. Int. J. Syst. Evol. Microbiol. 51, 1671–1679. doi: 10.1099/00207713-51-5-1671
Priest, F. G., Goodfellow, M., Shute, L. A., and Berkeley, W. (1987). Bacillus amyloliquefaciens sp. nov., nom. rev. Int. J. Syst. Bact. 37, 69–71. doi: 10.1099/00207713-37-1-69
Reva, O. N., Dixelius, C., Meijer, J., and Priest, F. G. (2004). Taxonomic characterization and plant colonizing abilities of some bacteria related to Bacillus amyloliquefaciens and Bacillus subtilis. FEMS Microbiol. Ecol. 48, 249–259. doi: 10.1016/j.femsec.2004.02.003
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Richter, M., Rosselló-Móra, R., Glöckner, F. O., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Roberts, M. S., Nakamura, L. K., and Cohan, F. M. (1994). Bacillus mojavensis sp. nov., distinguishable from Bacillus subtilis by sexual isolation, divergence in DNA sequence, and differences in fatty acid composition. Int. J. Syst. Bacteriol. 44, 256–264. doi: 10.1099/00207713-44-2-256
Roberts, M. S., Nakamura, L. K., and Cohan, F. M. (1996). Bacillus vallismortis sp. nov., a close relative of Bacillus subtilis, isolated from soil in death valley, California. Int. J. Syst. Bacteriol. 46, 470–475. doi: 10.1099/00207713-46-2-470
Rooney, A. P., Price, N. P., Ehrhardt, C., Swezey, J. L., and Bannan, J. D. (2009). Phylogeny and molecular taxonomy of the Bacillus subtilis species complex and description of Bacillus subtilis subsp. inaquosorum subsp. nov. Int. J. Syst. Evol. Microbiol. 59, 2420–2436. doi: 10.1099/ijs.0.009126-0
Ruiz-García, C., Béjar, V., Martinez-Checa, F., Llamas, I., and Quesada, E. (2005a). Bacillus velezensis sp. nov., a surfactant-producing bacterium isolated from the river Velez in Malaga, southern Spain. Int. J. Syst. Evol. Microbiol. 55, 191–195. doi: 10.1099/ijs.0.63310-0
Ruiz-García, C., Quesada, E., Martínez-Checa, F., Llamas, I., Urdaci, M. C., and Béjar, V. (2005b). Bacillus axarquiensis sp. nov. and Bacillus malacitensis sp. nov., isolated from river-mouth sediments in southern Spain. Int. J. Syst. Evol. Microbiol. 55, 1279–1285. doi: 10.1099/ijs.0.63567-0
Satomi, M., La Duc, M. T., and Venkateswaran, K. (2006). Bacillus safensis sp. nov., isolated from spacecraft and assembly-facility surfaces. Int. J. Syst. Evol. Microbiol. 56, 1735–1740. doi: 10.1099/ijs.0.64189-0
Sharma, V., and Patil, P. B. (2011). Resolving the phylogenetic and taxonomic relationship of Xanthomonas and Stenotrophomonas strains using complete rpoB gene sequence. PLoS Curr. 3:RRN1239. doi: 10.1371/currents.RRN1239
Shivaji, S., Chaturvedi, P., Suresh, K., Reddy, G. S., Dutt, C. B., Wainwright, M., et al. (2006). Bacillus aerius sp. nov., Bacillus aerophilus sp. nov., Bacillus stratosphericus sp. nov. and Bacillus altitudinis sp. nov., isolated from cryogenic tubes used for collecting air samples from high altitudes. Int. J. Syst. Evol. Microbiol. 56, 1465–1473. doi: 10.1099/ijs.0.64029-0
Skerman, V. B. D., McGowan, V., and Sneath, P. H. A. (eds.). (1980). Approved lists of bacterial names. Int. J. Syst. Bacteriol. 30, 225–420. doi: 10.1099/00207713-30-1-225
Sumpavapol, P., Tongyonk, L., Tanasupawat, S., Chokesajjawatee, N., Luxananil, P., and Visessanguan, W. (2010). Bacillus siamensis sp. nov., isolated from salted crab (poo-khem) in Thailand. Int. J. Syst. Evol. Microbiol. 60, 2364–2370. doi: 10.1099/ijs.0.018879-0
Teeling, H., Meyerdierks, A., Baurer, M., Amman, R., and Glockner, F. O. (2004). Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ. Microbiol. 2004, 938–947. doi: 10.1111/j.1462-2920.2004.00624.x
Keywords: phylogenomics, Bacillus subtilis group, Bacillus amyloliquefaciens, Bacillus taxonomy, digital DNA–DNA hybridization, average nucleotide identity (ANI), average amino acid identity (AAI)
Citation: Fan B, Blom J, Klenk H-P and Borriss R (2017) Bacillus amyloliquefaciens, Bacillus velezensis, and Bacillus siamensis Form an “Operational Group B. amyloliquefaciens” within the B. subtilis Species Complex. Front. Microbiol. 8:22. doi: 10.3389/fmicb.2017.00022
Received: 19 October 2016; Accepted: 04 January 2017;
Published: 20 January 2017.
Edited by:
Rakesh Sharma, Institute of Genomics and Integrative Biology (CSIR), IndiaReviewed by:
Prabhu B. Patil, Institute of Microbial Technology (CSIR), IndiaBo Liu, Fujian Academy of Agaricultural Sciences, China
Copyright © 2017 Fan, Blom, Klenk and Borriss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rainer Borriss, cmFpbmVyLmJvcnJpc3NAcnouaHUtYmVybGluLmRl