
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 November 2016
Sec. Virology
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.01740
This article is part of the Research Topic Virus discovery by metagenomics: the (im)possibilities View all 20 articles
 Lavinia Gambelli1
Lavinia Gambelli1 Geert Cremers1
Geert Cremers1 Rob Mesman1
Rob Mesman1 Simon Guerrero1
Simon Guerrero1 Bas E. Dutilh2,3
Bas E. Dutilh2,3 Mike S. M. Jetten1
Mike S. M. Jetten1 Huub J. M. Op den Camp1
Huub J. M. Op den Camp1 Laura van Niftrik1*
Laura van Niftrik1*With its capacity for anaerobic methane oxidation and denitrification, the bacterium Methylomirabilis oxyfera plays an important role in natural ecosystems. Its unique physiology can be exploited for more sustainable wastewater treatment technologies. However, operational stability of full-scale bioreactors can experience setbacks due to, for example, bacteriophage blooms. By shaping microbial communities through mortality, horizontal gene transfer, and metabolic reprogramming, bacteriophages are important players in most ecosystems. Here, we analyzed an infected Methylomirabilis sp. bioreactor enrichment culture using (advanced) electron microscopy, viral metagenomics and bioinformatics. Electron micrographs revealed four different viral morphotypes, one of which was observed to infect Methylomirabilis cells. The infected cells contained densely packed ~55 nm icosahedral bacteriophage particles with a putative internal membrane. Various stages of virion assembly were observed. Moreover, during the bacteriophage replication, the host cytoplasmic membrane appeared extremely patchy, which suggests that the bacteriophages may use host bacterial lipids to build their own putative internal membrane. The viral metagenome contained 1.87 million base pairs of assembled viral sequences, from which five putative complete viral genomes were assembled and manually annotated. Using bioinformatics analyses, we could not identify which viral genome belonged to the Methylomirabilis- infecting bacteriophage, in part because the obtained viral genome sequences were novel and unique to this reactor system. Taken together these results show that new bacteriophages can be detected in anaerobic cultivation systems and that the effect of bacteriophages on the microbial community in these systems is a topic for further study.
The importance of microorganisms in biogeochemical cycles and global warming is well-known (Falkowski et al., 2008). Particularly, microorganisms that take part in the carbon and nitrogen cycles are of great interest for both the scientific community and the public at large due to the growing awareness of climate change. In fact, among greenhouse gasses that affect the climate, carbon dioxide (CO2), methane (CH4), and nitrous oxide (N2O) are the most relevant (Pachauri et al., 2014).
Aerobic and anaerobic methanotrophic microorganisms oxidize CH4 to CO2 mitigating the release of CH4 to the atmosphere. The newly discovered anaerobic denitrifying methanotroph “Candidatus Methylomirabilis oxyfera” was enriched from freshwater sediments and couples methane oxidation to nitrite reduction, thereby linking carbon and nitrogen cycles via a postulated unique intra-aerobic pathway (Ettwig et al., 2009, 2010). Next to its remarkable physiology, M. oxyfera stands out from other bacteria also with respect to its cell shape and structure. These Gram-negative, rod-shaped bacteria have a polygonal cell shape constituted by several longitudinal ridges running along the cell length and converging in a cap-like structure at the cell poles (Wu et al., 2012). Furthermore, these microorganisms are not only interesting from a fundamental scientific point of view, but could also be important players in natural ecosystems (Deutzmann et al., 2014; Hu et al., 2014) and be implemented in the removal of dissolved methane and ammonium from digester effluents in combination with anaerobic ammonium oxidizing bacteria (Luesken et al., 2011; Shi et al., 2013).
Bacteria constitute the vast majority of the biomass on Earth (about 90%), yet viruses are the most abundant “biological entities,” comprising ~94% of the nucleic-acid containing-particles (Suttle, 2007). The discovery that viruses indiscriminately occupy marine environments (Suttle, 2005) and freshwater ecosystems (Middelboe et al., 2008) opened a new research area concerning the effect of bacteriophages on microbial populations and the role of bacteriophages in elemental and nutrient cycles. By inducing bacterial lysis, bacteriophages affect abundance, diversity and functioning of microbial populations (Weinbauer and Rassoulzadegan, 2004).
Marine and freshwater viruses show a great variety of different morphologies (Wommack and Colwell, 2000; Sulcius et al., 2011; Borrel et al., 2012). Viruses have a huge heterogeneity in size and genetic structure. The size ranges from 20 to 200 nm in diameter (Brum et al., 2013), and the genetic material can be either dsDNA, ssDNA (+), dsRNA, or ssRNA (±) (Dimmock et al., 2007). Viruses infecting Bacteria and Archaea generally consist of a proteinaceous capsid that contains the genetic material. They can also have a tail, which is used to inject the genetic material into the host. Tailed viruses belong to three families: the Siphoviridae (non-contractile, long tail), the Myoviridae (contractile, medium-length tail), and the Podoviridae (non-contractile, short tail). Tailless viruses have been described with very diverse morphologies, such as polyhedral, filamentous, spindle-like, cubic-like, star-like, or pleomorphic. Among tailless viruses, bacteriophages with an internal lipid bilayer enclosing the genome have been described and assigned to the Tectiviridae and Corticoviridae families (King et al., 2012).
The striking morphological diversity of viruses is also reflected in their great genetic heterogeneity (Breitbart et al., 2002). Bacteriophages assigned to a certain family often share very little or no sequence similarity. Comparative analysis of bacteriophage genomes often reveals that genes are organized in modules characterized by a different evolutionary history. This phenomenon is known as genome mosaicism, and it is the result of a high degree of horizontal gene exchange (Hatfull, 2008).
The lack of a universal genetic marker and the fact that only a minority of bacterial hosts can be cultivated in laboratories (Edwards and Rohwer, 2005) make the assessment of such heterogeneity very challenging. Although, culture-independent, large-scale metagenomics has paved the way to a more exhaustive understanding of the viral sequence space (Dutilh, 2014), this approach is not flawless. For example, small datasets may be obtained due to the limited yield of viral DNA from environmental samples, biasing downstream analysis. Moreover, viruses show a high spatiotemporal variability (Koskella and Parr, 2015), contributing to a limited overlap between samples. The analysis of viral metagenomes is laborious, partly because of the lack of reference databases and appropriate analytical tools compared to, for example, the ones for microbial metagenomics (Mokili et al., 2012; Hurwitz and Sullivan, 2013). Consequently, a high abundance of unknown viral sequences (65–95%) are reported in new surveys when these metagenomes are mapped against databases of unknown sequences (Breitbart et al., 2007; Mokili et al., 2012). Finally, viral metagenomics studies are frequently still relatively small in scope.
Several studies investigated the complex dynamics of phage-host interactions in lab-scale bioreactor systems (for example Barr et al., 2010; Shapiro et al., 2010). However, there are limited bioreactor studies on virus-host interaction that include viral metagenomics (one example is Kunin et al., 2008). The present paper describes the bacteriophage population in a Methylomirabilis bioreactor enrichment culture using (advanced) electron microscopy, viral metagenomics, and bioinformatics analysis. Several viral genomes and morphologies were found and one lytic bacteriophage was observed to infect Methylomirabilis cells. Through bioinformatics we attempted to find which of the obtained viral contigs belonged to the bacteriophage that infects Methylomirabilis cells. Finally we speculate on the significance of the infection on bacterial growth and population dynamics.
The Methylomirabilis enrichment culture (~80% Methylomirabilis sp.) was grown in a continuous sequencing batch reactor (Applicon Biotechnology BV, Applisens, Schiedam, the Netherlands) made of stainless steel and glass with a volume of 6 L. The inoculum biomass was derived from a pre-existing enrichment, originally inoculated with sediment samples from a ditch in the Ooijpolder (Ettwig et al., 2009). The culture was kept anoxic by a continuous supply of a gas mixture composed of methane and carbon dioxide (95:5, v/v) and the medium was continuously flushed with a mixture of argon and carbon dioxide (95:5, v/v). The pH was maintained at 7.3 ± 0.1, the temperature was kept stable at 30°C and the system was constantly stirred at 100 RPM. The volume of the enrichment was kept at 4 L by a level sensor controlled pump in sequential cycles of feeding (22.5 h) and rest (30 min to settle, 60 min to pump out excess medium). The HRT (hydraulic retention time) was 4 days and the composition of the medium was (per L) 0.552–2.07 g NaNO2 (8–30 mM depending on culture performance), 288 μg MgSO4, 192 μg CaCl2, 50 μg KH2PO4, 2.5 mg FeSO4*7H2O. The trace element solution was adapted from Ettwig et al. (2010) and contained per L: 150 μg ZnSO4·7H2O, 60 μg CoCl26H2O, 400 μg CuSO4·, 100μg NiCl26H2O, 10 μg H3BO3, 100 μg MnCl24H2O, 10 μg Na2WO4·2H2O, 50 μg Na2MoO4·2H2O, 15 μg SeO2, 10 μg CeCl2.
A 20 ml sample was collected from the bioreactor enrichment culture and filtered with 0.45 and 0.2 μm syringe filters, sequentially (Puradisc Cellulose Acetate, Whatman) to remove microorganisms and keep only the viral fraction. The flow-through was concentrated using spin columns (Vivaspin, GE Healthcare, 10,000 MWCO) at 5000 × g for 5 min at 4°C, (Allegra X-15R Centrifuge, Beckman Coulter) to a final volume of about 60 μl. Approximately 3 μl sample was placed on glow-discharged and carbon-Formvar-coated 100 mesh hexagonal copper grids (Veco) and incubated at room temperature for 15 min. The excess liquid was blotted off using filter paper (Ashless, Grade 589/1 Filtration Paper, Whatman). The specimens were stained with 0.5% uranyl acetate for 1 min, blotted dry, washed in Milli-Q, blotted dry with filter paper and let air dry completely. The virioplankton was observed using a JEOL JEM-2100 transmission electron microscope operated at 200 kV.
A 2 ml sample was collected from the bioreactor enrichment culture and centrifuged at 800 × g for 4 min at 30°C (Microcentrifuge 5417R, Eppendorf, Hamburg, Germany). Supernatant was decanted and the pellet was resuspended in remaining supernatant. For high-pressure freezing, a specimen sandwich was assembled in the specimen holder of a Leica EMHPF. The sandwich consisted of an aluminum spacer ring (0.2 × 3 mm) and a membrane carrier (2.6 × 0.1 mm, no. 16707898) in which 0.2 μl sample was loaded, the sample was sealed off with a gold stub (specimen carrier D3/D2, gold, no. 16770131). After closing the holder, the sample was high-pressure frozen in an EMHPF, operating at 2100 bar. Samples were stored in liquid nitrogen.
To obtain a freeze-fracture replica, the sample was placed in a detachable cold table and loaded onto the stage of a Balzers BAF400 freeze-etch machine, pre-cooled to −170°C. Specimens were fractured at −110°C and subsequently etched for 12 min at the same temperature with a vacuum below 10−7 Bar. The specimen was shadowed with 1.3 nm Pt (angle 45°) and 15 nm C (angle 90°) and a replica of the sample was obtained. The biological material underneath the replica was digested in 70% H2SO4 for 48 h. Replicas were washed twice in MilliQ water and picked up with glow discharged 700 mesh hexagonal copper grids (reference number G276OG, Agar Scientific) and investigated by a JEOL JEM-2100 transmission electron microscope operated at 200 kV.
Cells from the bioreactor enrichment culture were harvested and cryofixed by high-pressure freezing (Leica HPM 100; Leica Microsystems, Vienna, Austria). Samples were placed into a 100 μm cavity of a type A platelet (3 mm diameter; 0.1–0.2 mm depth, Leica Microsystems) and closed with the flat side of a lecithin-coated type B platelet (3 mm diameter, 0.3 mm depth). The platelets were stored in liquid nitrogen.
For epon embedding, frozen samples were freeze-substituted in acetone containing 2% OsO4, 0.2% uranyl acetate, and 1% H2O (Walther and Ziegler, 2002). The substitution followed several intervals: cells were kept at −90°C for 47 h; brought to −60°C at 2°C per hour and kept at −60°C for 8 h; brought to −30°C at 2°C per hour and kept at −30°C for 8 h in a freeze-substitution unit (AFS; Leica Microsystems, Vienna, Austria). To remove uranyl acetate, the samples were washed four times for 30 min in the AFS device at −30°C with acetone containing 2% OsO4and 1% H2O. Next, fixation was continued on ice for 1 h. OsO4 and H2O were removed by washing the sample twice in anhydrous acetone for 30 min. Samples were gradually infiltrated with epon resin and polymerized for 72 h at 60°C (Mollenhauer, 1964). Ultrathin sections of 60 nm were cut using a Leica UCT ultramicrotome (Leica Microsystems, Vienna, Austria) and collected on carbon-Formvar-coated 100 mesh hexagonal copper grids (Veco). The sections were then post-stained with 2% uranyl acetate for 20 min and lead citrate for 2 min. After each of the two steps, grids were washed in MilliQ water. Sections were investigated using a JEOL JEM1010 transmission electron microscope operated at 60 kV.
Semi-thin sections (200–300 μm) were cut using a diamond knife (Diatome, Biel, Switzerland) and an ultramicrotome (UCT, Leica microsystems) and collected on 50 mesh copper grids with a carbon coated formvar support film. After air drying, grids were post-stained with 4% uranyl acetate in MilliQ for 30 min and Reynolds lead citrate for 2 min. Ten nanometers proteinA gold (CMC, UMC Utrecht, The Netherlands) was applied to the sections to act as fiducial marker during tilt-series acquisition and reconstruction.
Virus infected cells and isolated virus particles were chosen as the region of interest. Dual axis tilt series (−60 to +60) were recorded on the JEOL JEM-2100 microscope operating at 200 kV, using SerialEM for automated image acquisition (Mastronarde, 2005).
Recorded tilt series were reconstructed using the IMOD package (Kremer et al., 1996) and tomograms were generated using both the weighted back-projection and SIRT algorithms. Reconstructed tomograms were segmented by hand using 3DMOD. Iso-surface model was generated from a sub-tomogram employing a circular mask. Summed virtual slices (36 slices) were visualized using the UCSF Chimera package (Pettersen et al., 2004), the histogram was adjusted to fit the density associated with the virus particle.
Over a period of ~3 months bioreactor material and effluent (13 L) were collected from the bioreactor enrichment culture and stored at 4°C until further analyses. Since the majority of the microbial population in the bioreactor grows in aggregates, the total sample was split in two: the bioreactor supernatant containing free bacteriophages and the bacterial biomass containing bacteriophages in the aggregates and within the cells. The two samples were obtained by centrifuging the sample at 20,000 × g for 1 h in 350 ml centrifuge tubes (Thermo Scientific, SorvallTM LYNX 4000 centrifuge). The resulting biomass pellets were resuspended in a total volume of 50 mL of the supernatant. Free-floating bacteriophages were precipitated by iron chloride flocculation, whereas bacterial biomass-associated bacteriophages were collected by PEG 8000.
To isolate free bacteriophages approximately 12.95 L of supernatant was first filtered with 0.45 and 0.2 μm filters sequentially (nuclepore track-etched polycarbonate membrane filters, Whatman) to remove microbial biomass. The sample was subsequently processed by iron chloride flocculation as described previously (John et al., 2011; Cunningham et al., 2015). Since iron is known to be an inhibitor of DNA amplification, it was removed from the sample by extensively washing the sample with MSM buffer (400 mM NaCl, 20 mM MgSO4·Ů7H2O, 50 mM Tris, pH 7.5) in spin columns (Vivaspin, GE Healthcare, 10,000 MWCO) at 2500 × g for 40 min at 4°C, using an Allegra X-15R Centrifuge (Beckman Coulter). The sample was stored at 4°C until further analyses.
About 50 ml of microbial biomass was first disrupted by pottering on ice to free the bacteriophages. To digest non-viral DNA and RNA, DNase I (Thermo Scientific, final concentration 1U/ml), and RNase A (Thermo Scientific, final concentration 5U/mL) treatments were simultaneously performed for 1 h at 37°C. The sample volume was divided in 2 ml Eppendorf tubes and PEG 8000 precipitation was performed as described previously (Guo et al., 2012). The sample was centrifuged for 30 min at 2000 × g and the obtained pellet was resuspended in MSM buffer. PEG 8000 was separated from the viral particles as described previously (Colombet and Sime-Ngando, 2012). In addition, to lower the salt concentration, the supernatant containing free phages was extensively washed with MSM buffer in spin columns (Vivaspin, GE Healthcare, 10,000 MWCO) at 2500 × g and 4°C using an Allegra X-15R Centrifuge (Beckman Coulter) to a final concentration of 20 mM KCl. The sample was stored at 4°C until further analyses.
The two viral samples obtained by iron chloride and PEG 8000 precipitation were pooled together and bacteriophages were further concentrated by ultracentrifugation (Optima XE90, Beckman-Coulter, rotor type 90 Ti, Beckman-Coulter) at 77,000 rcf for 1 h at 4°C (Szpara et al., 2011). A P1 reference bacteriophage NC_005856.1 was used as a positive control for DNA extraction. The pellet was resuspended in 1 ml of supernatant and the total DNA was extracted according to the protocol published by Thurber et al. (2009). Using the Qubit dsDNA HS assay kit (Life), the extracted DNA was quantified at 0.2 ng DNA.
To prevent amplification biases, yet obtain enough DNA for Ion Torrent sequencing, 43.2 ng of 16S ribosomal DNA of “Candidatus K. stuttgartiensis” was added to 0.1 ng of viral DNA (Cremers et al., in preparation). The total DNA was sheared for 6 cycles using the Bioruptor (Diagenode, Liege, Belgium; 1 min on, 1 min off) and prepared according to manufactures protocol (IonXpress Plus gDNA fragment library preparation Rev C.0, Life). The sample was sequenced using an Ion Torrent Personal Genome Machine (Life) on a v318 chip following manufacturer's protocol.
The sequencing reads (4,334,460) were trimmed with default quality settings (size 25–375 bp) using CLC genomics workbench (CLCbio, Aarhus, Denmark) and filtered to remove 16S ribosomal K. stuttgartiensis DNA (~87.6%) and M. oxyfera genomic DNA (~0.08%). The remaining reads were assembled using SPAdes (v.3.5.0), and ESOM (Ultsch and Mörchen, 2005; Dick et al., 2009) was used to validate the consistency of the contig tetranucleotide profiles (Supplementary File 1). This resulted in 2094 assembled contigs with a total length of 1.87 Mbp, including the five proposed genome sequences (Supplementary Files 2A–C).
Putative ORFs (open reading frames) were predicted and automatically annotated on all contigs (Supplementary File 3) by using Prokka (Seemann, 2014). The five longest and putative complete viral genomes were selected for manual annotation, and visualized by ARTEMIS (Rutherford et al., 2000). The automatic annotation of each putative ORF was manually curated using BLASTp (Johnson et al., 2008). An e < 10−3 was chosen as a cut-off for annotation of genes and domains. The amino acid sequences were also analyzed for the presence of conserved domains consulting: InterProScan (Jones et al., 2014), PROSITE (De Castro et al, 2006), Motif Scan (Pagni et al., 2007), Pfam (Finn et al., 2016), and SMART (Letunic et al., 2014). For each putative ORF, percentage identity and overlap to the first annotated/validated hit obtained through BLASTp were calculated using Pairwise Alignment (Wu et al., 2003). Classification of annotated genes was performed according to the following criteria. Hypothetical protein; >20% identity to hypothetical protein, conserved hypothetical protein; >30% identity to hypothetical protein (similar length), similar to; >20% identity to known protein (similar length), strongly similar to; >40% identity to known protein (similar length). ORFs that contained conserved domains (but no annotated Blast hits) or (strong) similarity to a known protein but different length were described as putative [protein name].
To assess the presence of known viruses and bacteria in the viral metagenome, all contigs obtained with SPAdes and their predicted ORFs were compared against the nr databases from NCBI (using BLASTn and BLASTp, respectively) and the Earth's virome database (Paez-Espino et al., 2016; Supplementary File 4).
The viral genomes are available from GenBank under accession numbers, KX853510, KX853511, KX853512, KX853513, KX853514 for Genomes 1–5, respectively. The remaining contigs are submitted under number MKFH00000000.
Computational phage-host prediction signals (Edwards et al., 2015) were calculated to identify which of the 2094 assembled contigs belonged to the bacteriophage infecting Methylomirabilis. First, genetic homology between the Methylomirabilis genomes (Methylomirabilis oxyfera; Ettwig et al., 2010 and a new Methylomirabilis species; Guerrero et al., in preparation) and the viral contigs was determined by using blastn and tblastx 2.2.30+ with default settings and E ≤ 10−5 (Camacho et al., 2009), and the total bitscore for each viral contig was recorded. Second, both Methylomirabilis genomes were checked for CRISPR sequences using CRISPRfinder (http://crispr.u-psud.fr/Server/) and CRASS (Skennerton et al., 2013). The resulting spacer sequences were subsequently cross-referenced with the viral genomes and contigs from the SPAdes assembly using CRISPRtarget (Biswas et al., 2013). Third, similarity in oligonucleotide usage was calculated as 1—E, where E is the Euclidean distance between the k-mer profiles of each viral contig and the Methylomirabilis genomes (k = 2, 4, 6).
We investigated the bacteriophage population in a bioreactor containing an enrichment culture of the anaerobic methanotroph Methylomirabilis sp. (Table 1). Several free bacteriophages with different morphologies were observed using negative staining. Among these, four morphotypes were identified. All bacteriophages had putative icosahedral capsid symmetry and three of them possessed a tail. Capsids and tails varied in diameter and length, thereby allowing a clear distinction between the morphotypes (Figure 1).

Table 1. Properties of the 4 viral morphotypes observed in the Methylomirabilis enrichment culture.
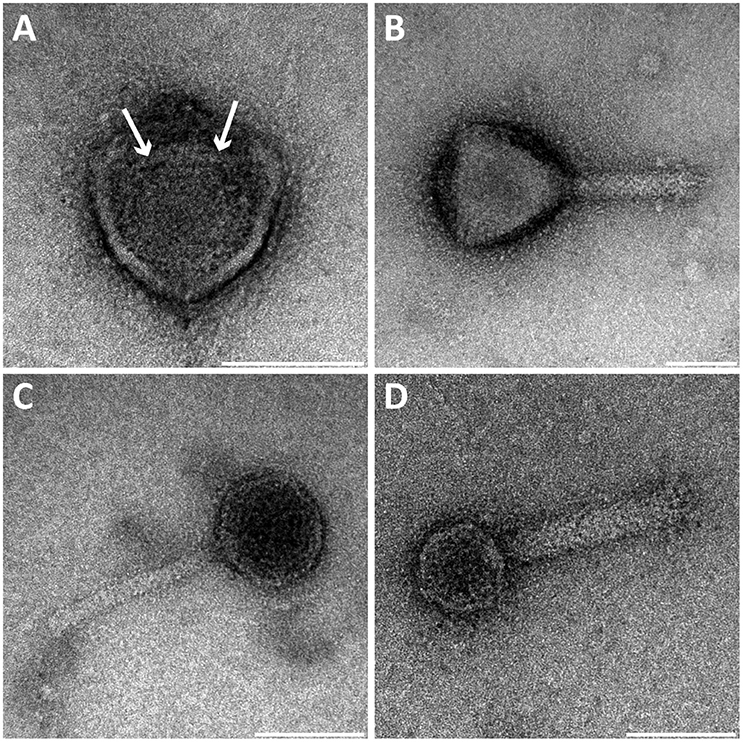
Figure 1. Transmission electron micrographs of negative-stained bacteriophages present in the Methylomirabilis bioreactor enrichment culture. Four viral morphotypes were observed. (A) Viral morphotype with putative icosahedral symmetry of the head (~71 nm diameter), without a tail or other appendages but with a central round internal core (~25 nm diameter, see arrows). (B–D) Three additional viral morphotypes all with a tail and a putative icosahedral symmetry of the head (~87, ~50, and ~41 nm diameter respectively). Scale bars; 50 nm.
In addition to free bacteriophages, the microbial population was investigated for phage infection using thin sections, freeze-etching, and electron tomography. Thin sections of high-pressure frozen, freeze-substituted, and epon embedded biomass samples showed a lytic phage infection in Methylomirabilis cells (Figure 2). The infection rate (amount of infected Methylomirabilis cells in the total Methylomirabilis population) was calculated to be 2.3%. The infection did not affect bioreactor performance with respect to activity (methane and nitrite consumption). The intracellular bacteriophages had a hexagonal capsid (ca. 55 nm wide, vertex-to-vertex), indicating an icosahedral 3D symmetry. The capsid contained an electron dense and round central core (ca. 20 nm diameter), probably enclosing the genetic material. The bacteriophages were present both outside and inside the Methylomirabilis cells. Infected Methylomirabilis cells displayed different stages of bacteriophage assembly. In addition to completely assembled bacteriophages, entities were observed inside the cells that contained only the central core, i.e., the capsid was not yet assembled. As more and more bacteriophages were assembled, they were organized in a highly packed formation within the cell (Figures 2C,D). This caused the infected cell to swell approximately 1.9x in size compared to a not infected cell [area measured on TEM sections based on 10 infected (0.44 ± 0.2 μm2) and 10 not infected cells (0.23 ± 0.06 μm2)]. Afterwards the infected cell lysed thereby releasing the bacteriophages (Figures 2E,F). Comparing the intracellular Methylomirabilis bacteriophage to the free bacteriophages observed in the bioreactor enrichment culture using negative staining, its morphology resembled that of the bacteriophage depicted in Figure 1A based on size and symmetry. Freeze-etching of infected Methylomirabilis cells revealed the surface of the bacteriophage capsid (Figure 3). The capsomeres, the building subunits of the capsid, were arranged in triangular faces that constitute the icosahedral symmetry of the proteinaceous capsid.
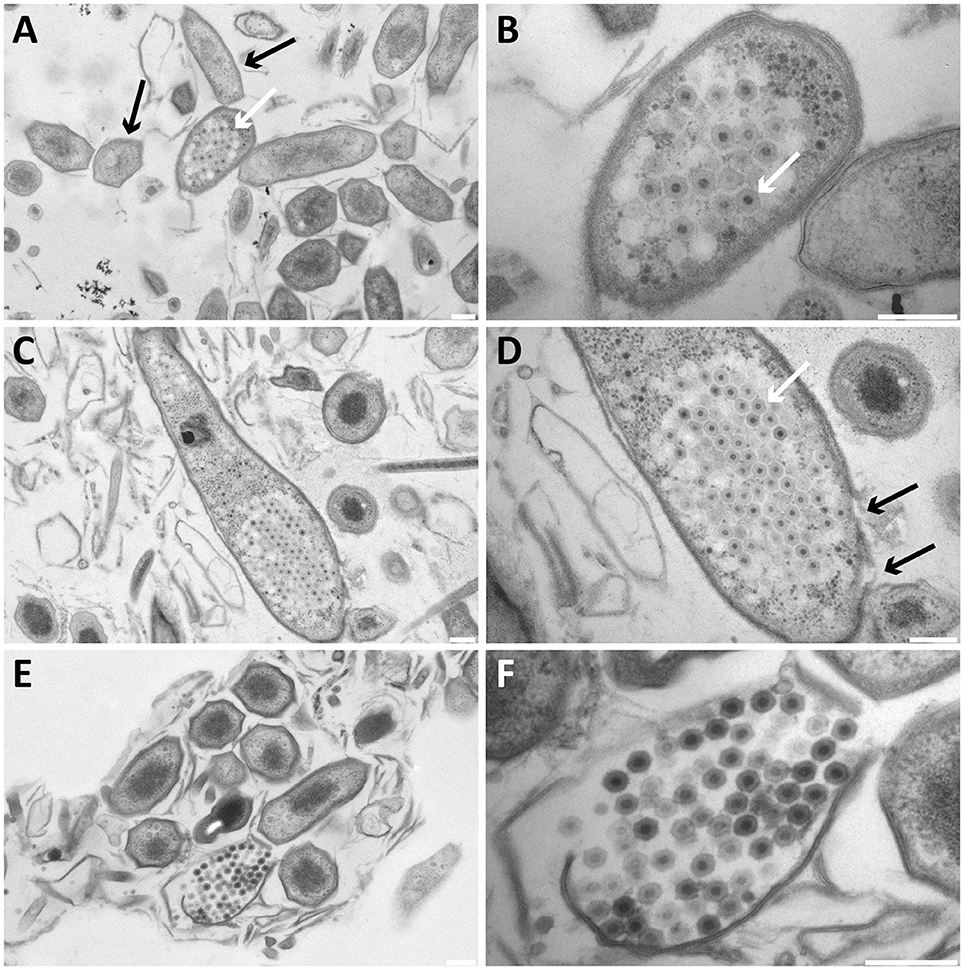
Figure 2. Transmission electron micrographs of high-pressure frozen, freeze-substituted, resin-embedded, and thin-sectioned Methylomirabilis cells taken from a bioreactor enrichment culture. (A,B) Infected Methylomirabilis cells (white arrow in A) are in clusters among non-infected cells (black arrows in A). The bacteriophage (white arrow in B) has a hexagonal shape and an internal electron dense core. (C,D) The bacteriophages are organized in a highly packed formation (white arrow in D). The replication and assembly of bacteriophages causes the Methylomirabilis cell to swell and eventually the cell wall breaks (black arrows in D). (E,F) Lysed Methylomirabilis cell releasing the viral progeny. (B,D,F) are enlargements of (A,C,E), respectively. Scale bars; 200 nm.
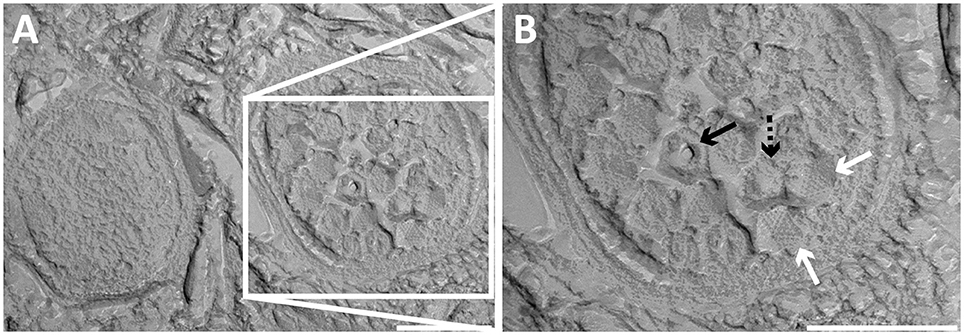
Figure 3. Transmission electron micrographs of high-pressure frozen and freeze-etched Methylomirabilis cells taken from a bioreactor enrichment culture. (A) Cross-section of a non-infected (left) and infected (right) Methylomirabilis cell. (B) The bacteriophages (white arrows) contain a proteinaceous capsid. The capsid is made of triangular faces built by capsomeres. Concave (dashed black arrow) and convex (black arrow) printing of the internal core is visible in two of the viral particles. Scale bars; 200 nm.
Electron tomography was used to study the 3D structure of the bacteriophage and the infected Methylomirabilis cells (Figure 4). 3D reconstruction and modeling showed that the cytoplasmic membrane of infected cells was broken at many places while the cell wall was often still intact (Figures 4A,B and Supplementary Movie 1). Inside infected cells, both completely assembled bacteriophages were present as well as incompletely assembled bacteriophages (with only the electron dense core visible—no capsid yet). 3D reconstruction and modeling of free bacteriophages suggested the presence of a putative internal membrane surrounding the electron dense core (Figures 4C–F). In addition, no tail was observed on the free bacteriophages.
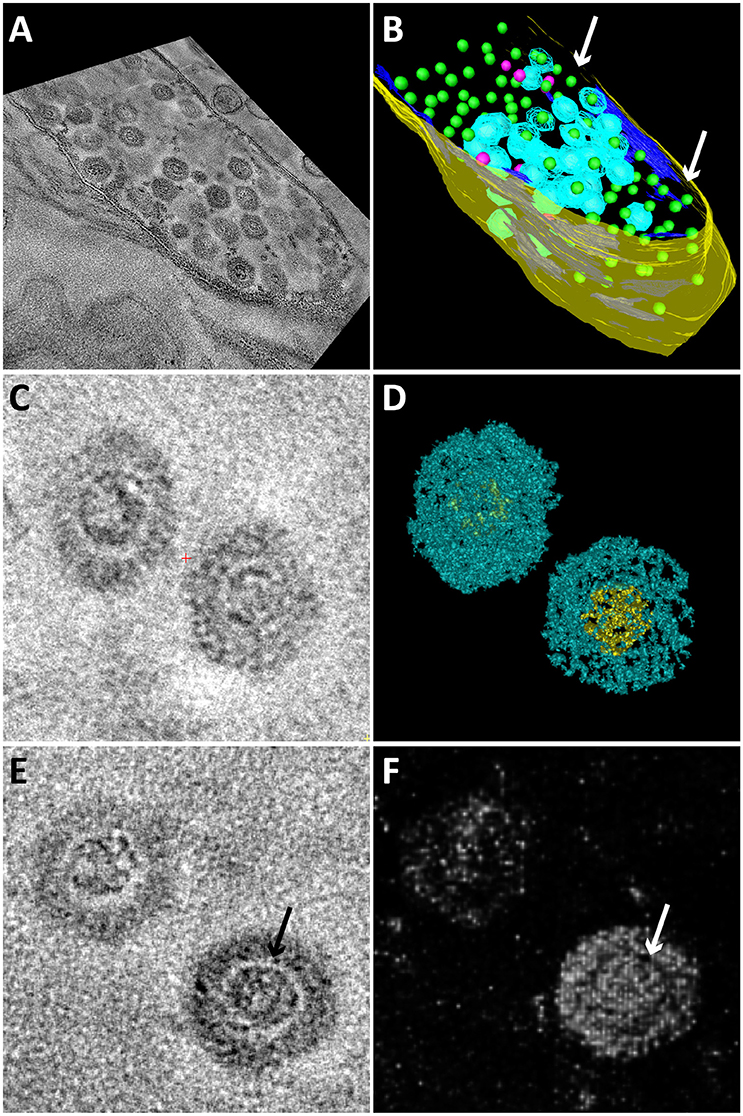
Figure 4. Snapshots of electron tomograms and models of free and intracellular bacteriophages infecting Methylomirabilis cells. Tomogram (A) and model (B) of an infected Methylomirabilis cell. Most bacteriophages have the capsid (blue) assembled around the electron dense core (green). Some bacteriophages are still in the process of assembly and only consist of the electron dense core (pink). The cell is swollen and the cytoplasmic membrane (dark blue) is broken at many places (arrows). The cell wall (yellow) is still intact. All green electron dense cores were surrounded by a capsid, but not all capsids were modeled for reasons of clarity. Tomogram (C) and isosurface density model (D) of two free bacteriophages showing the icosahedral capsid (blue) and electron dense core (yellow). Tomogram (E) and Chimera model (F) showing two free bacteriophages. The electron dense core is enclosed by a putative membrane (arrows).
To characterize the bacteriophage population present in the Methylomirabilis bioreactor enrichment culture, the total viral DNA was extracted from the bioreactor and the effluent and sequenced. After assembly, 2094 contigs were obtained (Supplementary Files 2A,B). The five longest and putative complete viral genomes (Table 2) were selected for manual annotation (Figure 5 and Supplementary Files 5A–E). Four of these contigs (197, 85, 71, 16 kb) contained identical sequences at both ends, indicating a full genome and a circular genomic arrangement. The 41 kb contig did not have overlapping end sequences, possibly representing a linear genome.
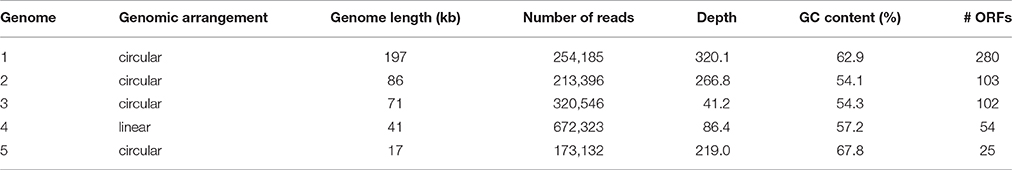
Table 2. Genomic characteristics of the five putative complete viral genomes retrieved from the viral metagenome of the bacteriophage population in the Methylomirabilis bioreactor enrichment culture.
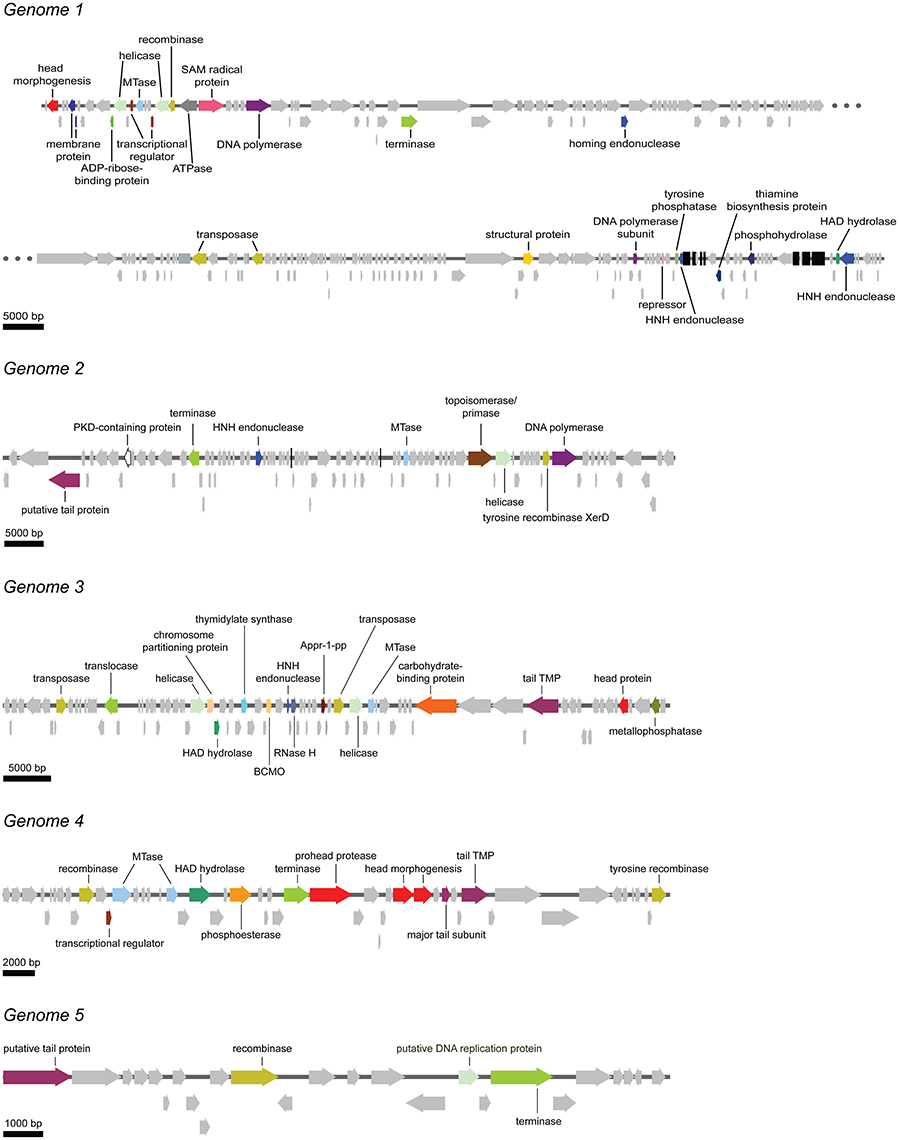
Figure 5. Schematic overview of the five putative complete viral genomes retrieved from the viral metagenome of the bacteriophage population in the Methylomirabilis bioreactor enrichment culture. Arrows with the same color represent genes encoding for the same putative functional category. Light gray arrows indicate hypothetical proteins. Black bars in genome 1 and genome 2 represent tRNAs. Appr-1-pp, ADP-ribose 1-phosphate phosphatase; BCMO, β-carotene 15, 15′-monooxygenase; HAD hydrolase, haloacid dehalogenase; MTase, methyltransferase; PKD-containing protein, polycystic kidney disease-containing protein; RNase H, ribonuclease H; tail TMP, tail tape measure protein.
In this genome, one head morphogenesis protein was found, involved in initial stages of head assembly (Hsiao and Black, 1978; Leiman et al., 2003). DNA replication enzymes are represented by one DNA polymerase and one DNA polymerase subunit. In addition, the two helicases may also take part in the replication process. The DNA packaging machinery in genome 1 includes three terminases. Compared to the other five annotated genomes, genome 1 was the only annotated genome that does not contain a clear hit to a tail protein. In addition, genome 1 encodes for one putative ATPase, maybe involved in DNA packaging (Kondabagil et al., 2006), and two ORFs with a blast hit against a membrane protein. The genome encodes no less than 41 tRNAs. The acquisition of these tRNAs from the host might function in providing the phage with a similar GC content as the host, thereby facilitating the recruitment of the host DNA replication enzymes (Bailly-Bechet et al., 2007; Enav et al., 2012).
Genome 2 encodes for one putative tail protein. No capsid-related genes could be annotated. The DNA replication machinery in genome 2 consists of one DNA polymerase, one helicase and one primase. Interestingly, a PKD (polycystic kidney disease) domain-containing protein was also annotated. These proteins may have a function in mediating phage-host contact (Fraser et al., 2006, 2007; Sathaliyawala et al., 2010) and BAM (bacteriophage adherence to mucus) mechanisms (Barr et al., 2013).
Genome 3 contains one tail TMP (tape measure protein) and one head protein. Two helicases and one RNase (ribonuclease) H are the only three annotated genes that could be part of the DNA replication machinery. One chromosome partitioning protein similar to ParB was found in this genome. A ParB protein is also encoded by the E. coli phage P1, which can be present in the cell as a plasmid or integrated in the bacterial DNA (Abeles et al., 1985). The presence of a ParB-like protein in genome 3 may indicate that this bacteriophage might also be capable of integrating in the host genome or forming a plasmid. A putative phage-host interaction protein was also identified. This is a carbohydrate-binding protein with an Ig-like fold. Carbohydrate-binding domains have been found on tail spike proteins (Andres et al., 2010) and head spikes (Westbye et al., 2016), in both cases mediating the recognition of the target polysaccharides in the initial steps of infection.
Genome 4 has three proteins involved in head assembly, being a prohead protease and two head morphogenesis proteins. Next to them in the genome two tail proteins are located (one tail TMP and one major tail subunit).
Genome 5 encodes for a putative tail protein but no head proteins could be annotated. Most of the ORFs in this small genome are hypothetical proteins.
All annotated genomes show the putative ability to establish lysogeny. One transposase was found in genomes 2 and 5, two transposases in genomes 3 and 4 and three transposases in genome 1. These enzymes mediate unidirectional and site-specific recombination between two DNA sequences, one on the viral genome and one on the host genome (Groth and Calos, 2004). All genomes encode for one or two terminases (or translocase). These enzymes are responsible for packaging the genome inside the capsid (Catalano, 2000; Duffy and Feiss, 2002). HNH endonucleases found in double copy in genome 1 and in single copy in genomes 2 and 3 may also be involved in cutting the genome while DNA packaging is taking place (Moodley et al., 2012; Kala et al., 2014). Methyltransferases were also frequently found in single copy (genomes 1, 2, and 3) and double copy (genome 4). In bacteriophages methyltransferases are often thought to protect the viral genome from degradation by host-encoded edonucleases. However, few studies hypothesize a role in DNA replication and regulation both in lysogenic and lytic cycles (Murphy et al., 2013).
A main focus of the viral metagenomics in this study was to genomically identify the bacteriophage infecting Methylomirabilis cells (Figures 2–4). Based on the five viral genomes and over two thousand contig sequences obtained above, it is not directly clear which cellular hosts the recovered bacteriophages infect. Thus, to address this question we applied several computational approaches to predict this phage-host relationship from sequence signals (Edwards et al., 2015) that linked the assembled viral contigs to both the original genome and that of the new Methylomirabilis species. As described in Edwards et al. (2015), we calculated three main categories of phage-host prediction signals: genetic homology, similarity in CRISPR spacers, and similarity in oligonucleotide usage (Supplementary File 6). Genetic homology is expected to occur between the viral and bacterial genome sequences due to processes including transduction. This was identified at the nucleotide level (blastn, E ≤ 10−5) and at the protein level (tblastx, E ≤ 10−5), the former detecting only five short fragments, the latter detecting many more contigs including the long genomes 1–4. However, in all cases the matches between the bacteriophages and the Methylomirabilis genomes were based on relatively short, scattered regions of similarity with low sequence identity. As it was also showed by Edwards et al. (2015), the similarities in oligonucleotide usage profiles (k = 2, 4, 6) are not strong as phage-host predictors.
In the previously described M. oxyfera genome (Ettwig et al., 2010) one CRISPR array was present, which contained 23 spacers (Supplementary File 7). In the genome of a new Methylomirabilis species (Guerrero et al., in preparation) derived from the reactor from which the virome was sequenced, two CRISPR arrays were observed; one of which contained 26 spacers and the other one six spacers (Supplementary File 7). No significant nucleotide similarity was detected between these spacers and any of the 2094 possible viral contig sequences with fewer than 11/37 mismatches, which means these hits are indistinguishable from random noise (Edwards et al., 2015).
We also looked for signs of lysogeny in the M. oxyfera metagenome (Ettwig et al., 2010) and the new Methylomirabilis sp. metagenome (Guerrero et al., in preparation). The viral contigs were compared using BLASTn searching for contigs that were partly viral and partly bacterial. However, no such contigs were present in neither of the two metagenomes.
We analyzed the bacteriophage community in a bioreactor enrichment culture of the anaerobic methane oxidizing bacterium Methylomirabilis sp. Four different bacteriophage morphologies were observed inside the bioreactor system, one of which was observed to infect Methylomirabilis cells. From the viral metagenome data at least five putative complete viral genomes were assembled. Based on bioinformatics including CRISPR analyses, we could not identify the viral genome of the Methylomirabilis infecting bacteriophage.
Viral metagenome analysis of bioreactor enrichment cultures is challenging. In this case, the viral abundance was low and it required 13 L bioreactor material to obtain 0.2 ng viral DNA. We used a newly developed method for obtaining the (dsDNA) viral metagenome from the low amounts of viral DNA (Cremers et al., in preparation). In the end, 2094 contigs were assembled. Among these we identified five putative complete viral genomes. The annotation of these five genomes resulted in many hypothetical proteins. Metagenomics reads are often short and will not lead to improved annotation if viruses are not isolated and further experiments performed. Therefore, the viral sequences in the databases represent only a small fraction of the entire viral sequence space (Mokili et al., 2012; Dutilh, 2014). Nevertheless, we could in most cases identify putative genes involved in DNA processing and tail and capsid formation.
Electron microscopy analyses showed a bacteriophage infecting Methylomirabilis cells at a relatively low infection rate of 2.3%. The bacteriophage performed a lytic cycle as bacteriophages were replicated inside the host cell, causing the host to swell. In the end, the host cell lysed and released the viral particles. The bacteriophage had a putative icosahedral capsid and no tail. Inside the bacteriophage, a round electron dense core was visible, which most probably represented the highly packed genome. Electron tomography suggested the presence of a putative internal membrane, which appeared as an electron-light area surrounding the genome. Such an electron-light structure around a viral genome has not been observed for the other described morphotypes, therefore this could be an indication that this genome was enclosed by a structure, most likely a lipid membrane, as it was also shown for bacteriophages with similar morphology (Harrison et al., 1971; Mindich et al., 1982). Inside the infected Methylomirabilis cells incompletely assembled bacteriophages were observed constituted by only the electron dense core. The assembly of this bacteriophage might thus proceed by the following steps: membrane formation, packaging of the genome, and assembly of the capsid.
The presence of an internal membrane surrounding the viral genome has so far only been described for two bacteriophage families: Tectiviridae (four phages described and PRD1 as type species) and Corticoviridae (one phage described; PM2; King et al., 2012). These bacteriophages infect Gram-negative bacteria, have a relatively small dsDNA genome [10 kb circular, Corticoviridae (Männistö et al., 1999); 15 kb linear, Tectiviridae (Bamford et al., 1991)] and have an icosahedral capsid [66 nm from facet to facet for Tectiviridae (Abrescia et al., 2004) and 55 nm from facet to facet for Corticoviridae (Kivelä et al., 2002)] with no tail. It was described that these bacteriophages obtain their internal membrane from the host cytoplasmic membrane using so called membrane proteins (Espejo and Canelo, 1968; Mindich et al., 1982). Also in the present study, the cytoplasmic membrane of infected Methylomirabilis cells was not intact anymore as was apparent from 3D electron tomography models. The similar ultrastructure of the Methylomirabilis bacteriophage to the Corticoviridae and Tectiviridae might indicate that it belongs to one of these families.
By using a bioinformatics approach, we tried to predict which viral contigs (2094) assembled from the viral metagenome data belonged to the Methylomirabilis- infecting bacteriophage. However, we were not able to obtain any convincing result. Homology searches yielded only short spurious hits, while the oligonucleotide usage profiles were too broad and did not provide a specific signal to any one of the longer contigs. In addition, the CRISPR arrays that were identified in the Methylomirabilis genomes did not have a clear hit to any of the assembled bacteriophage contigs. There are several possible explanations for the latter. First, since bacteriophage genomes with high similarity to one or more spacers in a CRISPR array are precluded from infection, CRISPR spacers are a focal point of positive selection in bacteriophage genome sequences, because the only bacteriophages that survive are the ones with mutations in the protospacer (Sun et al., 2013; Paez-Espino et al., 2015). Indeed, the closest match between CRISPR spacer and viral contigs already showed 11 mismatches. Second, the spacer composition of CRISPR arrays in a bacterial community can be very volatile due to ecological selection for bacterial strains with spacers acquired from the current bacteriophages in the environment (Rho et al., 2012; Koskella and Brockhurst, 2014). Thus, even though the Methylomirabilis genomes that were mined for CRISPR spacers were obtained from the same bioreactor (Ettwig et al., 2010; Guerrero et al., in preparation), not a single one of the CRISPR spacers was found in common. Taken together, computational phage-host signals did not result in a strong candidate for the bacteriophage infecting Methylomirabilis in this bioreactor.
We also investigated signs of lysogeny by in depth analyses of the M. oxyfera metagenome (Ettwig et al., 2010) and the new Methylomirabilis sp. metagenome (Guerrero et al., in preparation), looking for contigs that were partly bacterial and partly viral. However, no such sequences were detected. This could indicate that the bacteriophage does not perform a lysogenic cycle but only a lytic one. In the end, we can only speculate about which viral genome might belong to the bacteriophage observed to infect the Methylomirabilis cells. Unfortunately none of the five putative complete viral genomes matched all (indirect) criteria. The bacteriophage morphology (icosahedral capsid, no tail, genome surrounded by putative internal membrane) indicated a relation to the Tectiviridae and Corticoviridae families that have a relatively small genome size, although the bacteriophage could also belong to a yet-undiscovered family. The only small (complete) genome (genome 5–17 kb) contained a putative tail protein indicating that most likely it was not our tailless bacteriophage. There was one genome without an annotated tail gene but it was fairly large (genome 1–197 kb) which did not fit to the described genome size in these families. Other bacteriophages with an internal membrane and a genome size bigger than 15 kb have been described (Aalto et al., 2012; Rissanen et al., 2013) but they are not yet assigned to a viral family.
In our study we focused on DNA-containing viruses. One explanation why the applied bioinformatic approaches could not find any phage-host interaction signals could be because the Methylomirabilis-infecting bacteriophage might have a ssDNA or an RNA genome. However, ssDNA viruses and especially RNA viruses comprise only a small minority among prokaryotic viruses, dominated instead by dsDNA viruses (Koonin et al., 2015). Also, especially considering the low yield of viral DNA, we might not have retrieved the entire bacteriophage population from the bioreactor. Not until methods as phageFISH (Allers et al., 2013) are optimized to be used for non-model systems it remains difficult to link a phage to a host through non bioinformatic methods.
The Methylomirabilis infection rate was observed to be relatively low (2.3% of all infected Methylomirabilis cells) at different time points throughout the period of ~1 year and did not affect bioreactor performance with respect to activity and growth. Indeed, the bacteriophage did not kill the Methylomirabilis population. Metagenomics analysis of the enrichment culture indicated the presence of a new Methylomirabilis species (Guerrero et al., in preparation), which was not present in the bioreactor when the genome of the original M. oxyfera species was published (Ettwig et al., 2010). An interesting speculation could be that the bacteriophage was specific for only the original M. oxyfera species (Ettwig et al., 2010) and in the end instigated the shift from the original to the new Methylomirabilis species. Indeed currently, the infection has almost disappeared from the enrichment culture, the published M. oxyfera 16S rRNA gene (NCBI, FP565575) cannot be detected anymore and the enrichment culture is dominated by the new Methylomirabilis species.
Bacteriophages can have profound effects on bacterial populations also in bioreactor enrichment systems. This has to be taken into account in the application of microorganisms in, for example, wastewater treatment systems or other industrial applications. In general, how bacteriophages can shape environmental or industrial microbial communities is still largely unexplored and is an exciting topic for further study.
LV and MJ designed the project. LG and LV designed the experiments. SG maintained and enriched the Methylomirabilis culture. LG and RM performed all TEM related experiments. LG performed the concentration of viral particles and DNA extraction. GC performed the library preparation and sequencing. GC, BD, and HO designed, performed and analyzed the bioinformatics research. LG manually annotated the complete viral genomes. LV supervised the research. LG and LV performed data analysis, data interpretation and wrote the manuscript with input from GC, RM, SG, BD, MJ, and HO.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Jeremy J. Barr for critical reading of the manuscript and for the bacteriophage DNA isolation protocol; Christina Ferousi, Dave Allen, Felipe H. Coutinho, Diego D. Cambuy, and Bruno G. Andrade for practical assistance; Theo van Alen, and Daan Speth for advice on (all) and assistance with (TvA) running the ion torrent sequencer and analyzing the data; Boran Kartal for input for manuscript preparation; Geert-Jan Janssen and the General Instruments department for maintenance of TEM equipment. BD was supported by the Netherlands Organization for Scientific Research (NWO) Vidi grant 864.14.004. RM was supported by NWO Spinozapremie 2012 of MJ. MJ and LG are supported by ERC 339880, and MJ and GC also by OCW/NWO Gravitation grant (SIAM 024002002). HO is supported by ERC-AG 669371.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01740/full#supplementary-material
Aalto, A. P., Bitto, D., Ravantti, J. J., Bamford, D. H., Huiskonen, J. T., and Oksanen, H. M. (2012). Snapshot of virus evolution in hypersaline environments from the characterization of a membrane-containing Salisaeta icosahedral phage 1. Proc. Natl. Acad. Sci. U.S.A. 109, 7079–7084. doi: 10.1073/pnas.1120174109
Abeles, A. L., Friedman, S. A., and Austin, S. J. (1985). Partitioning of unit-copy miniplasmids to daughter cells. III. The DNA sequence and functional organization of the P1 partition region. J. Mol. Biol. 185, 261–272. doi: 10.1016/0022-2836(85)90402-4
Abrescia, N. G. A., Cockburn, J. J. B., Grimes, J. M., Sutton, G. C., Diprose, J. M., Butcher, S. J., et al. (2004). Insight into assembly from structural analysis of bacteriophage PRD1. Nature 432, 68–74. doi: 10.1038/nature03056
Allers, E., Moraru, C., Duhaime, M. B., Beneze, E., Solonenko, N., Barrero-Canosa, J., et al. (2013). Single-cell and population level viral infection dynamics revealed by phageFISH, a method to visualize intracellular and free viruses. Environ. Microbiol. 15, 2306–2318. doi: 10.1111/1462-2920.12100
Andres, D., Baxa, U., Hanke, C., Seckler, R., and Barbirz, S. (2010). Carbohydrate binding of Salmonella phage P22 tailspike protein and its role during host cell infection. Biochem. Soc. Trans. 38, 1386–1389. doi: 10.1042/BST0381386
Bailly-Bechet, M., Vergassola, M., and Rocha, E. (2007). Causes for the intriguing presence of tRNAs in phages. Genome Res. 17, 1486–1495. doi: 10.1101/gr.6649807
Bamford, J. K. H., Hänninen, A. L., Pakula, T. M., Ojala, P. M., Kalkkinen, N., Frilander, M., et al. (1991). Genome organization of membrane-containing bacteriophage PRD1. Virology 183, 658–676. doi: 10.1016/0042-6822(91)90995-N
Barr, J. J., Auro, R., Furlan, M., Whiteson, K. L., Erb, M. L., Pogliano, J., Stotland, A., Wolkowicz, R., et al. (2013). Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 10771–10776. doi: 10.1073/pnas.1305923110
Barr, J. J., Slater, F. R., Fukushima, T., and Bond, P. L. (2010). Evidence for bacteriophage activity causing community and performance changes in a phosphorus-removal activated sludge. FEMS Microbiol. Ecol. 74, 631–642. doi: 10.1111/j.1574-6941.2010.00967.x
Biswas, A., Gagnon, J. N., Brouns, S. J., Fineran, P. C., and Brown, C. M. (2013). CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 10, 817–827. doi: 10.4161/rna.24046
Borrel, G., Colombet, J., Robin, A., Lehours, A. C., Prangishvili, D., and Sime-Ngando, T. (2012). Unexpected and noved putative viruses in the sediments of a deep-dark permanently anoxic freshwater habitat. ISME J. 6, 2119–2127. doi: 10.1038/ismej.2012.49
Breitbart, M., Salamon, P., Andersen, B., Mahaffy, J. M., Segall, A. M., Mead, D., et al. (2002). Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. U.S.A. 99, 14250–14255. doi: 10.1073/pnas.202488399
Breitbart, M., Thompson, L. R., Suttle, C. A., and Sullivan, B. (2007). Exploring the vast diversity of marine viruses. Oceanography 20, 135–139. doi: 10.5670/oceanog.2007.58
Brum, J. R., Schenck, R. O., and Sullivan, M. B. (2013). Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 7, 1738–1751. doi: 10.1038/ismej.2013.67
Camacho, C., Coulouris, G., Avagyan, V., MA, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421–430. doi: 10.1186/1471-2105-10-421
Catalano, C. E. (2000). The terminase enzyme from bacteriophage lambda: a DNA-packaging machine. Cell. Mol. Life Sci. 57, 128–148. doi: 10.1007/s000180050503
Colombet, J., and Sime-Ngando, T. (2012). “Use of PEG, Polyethylene glycol, to characterize the diversity of environmental viruses,” in Current Microscopy Contributions to Advances in Science and Technology, ed A. Méndez-Vilas (Badajoz: Formatex Research Center), 316–322.
Cunningham, B. R., Brum, J. R., Schwenck, S. M., Sullivan, M. B., and John, S. G. (2015). An inexpensive, accurate and precise wet-mount method for enumerating aquatic viruses. Appl. Environ. Microbiol. 81, 2995–3000. doi: 10.1128/AEM.03642-14
De Castro, E., Sigrist, C. J. A., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and Pro-Rule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, 362–365. doi: 10.1093/nar/gkl124
Deutzmann, J. S., Stief, P., Brandes, J., and Schink, B. (2014). Anaerobic methane oxidation coupled to denitrification is the dominant methane sink in a deep lake. Proc. Natl. Acad. Sci. U.S.A. 111, 18273–18278. doi: 10.1073/pnas.1411617111
Dick, G. J., Andersson, A. F., Baker, B. J., Simmons, S. L., Thomas, B. C., Yelton, A. P., et al. (2009). Community-wide analysis of the microbial genome sequence signatures. Genome Biol. 10:R85. doi: 10.1186/gb-2009-10-8-r85
Dimmock, N. I., Easton, A. J., and Leppard, K. N. (2007). Introduction to Modern Virology. Coventry: Department of Biological Sciences, University of Warwick.
Duffy, C., and Feiss, M. (2002). The large subunit of bacteriophage λ's terminase plays a role in DNA translocation and packaging termination. J. Mol. Biol. 316, 547–561. doi: 10.1006/jmbi.2001.5368
Dutilh, B. E. (2014). Metagenomic ventures into outer sequence space. Bacteriophage 4:e979664. doi: 10.4161/21597081.2014.979664
Edwards, R. A., McNair, K., Faust, K., Raes, J., and Dutilh, B. E. (2015). Computational approaches to predict phage-host relationships. FEMS Microbial. Rev. 40, 258–272. doi: 10.1093/femsre/fuv048
Edwards, R. A., and Rohwer, F. (2005). Viral metagenomics. Nat. Rev. Microbiol. 3, 504–510. doi: 10.1038/nrmicro1163
Enav, H., Béjà, O., and Mandel-Gutfreund, Y. (2012). Cyanophage tRNAs may have a role in cross-infectivity of oceanic Prochlorococcus and Synechococcus hosts. ISME J. 6, 619–628. doi: 10.1038/ismej.2011.146
Espejo, R. T., and Canelo, E. S. (1968). Origin of phospholipid in bacteriophage PM2. J. Virol. 2, 1235–1240.
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M. M., et al. (2010). Nitrate-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–550. doi: 10.1038/nature08883
Ettwig, K. F., van Alen, T., van de Pas-Schoonen, K. T., Jetten, M. S. M., and Strous, M. (2009). Enrichment and molecular detection of denitrifying methanotrophic bacteria of the NC10 phylum. Appl. Environ. Microbiol. 75, 3656–3662. doi: 10.1128/AEM.00067-09
Falkowski, P. G., Fenchel, T., and Delong, E. F. (2008). The microbial engines that drive earth's biogeochemical cycles. Science 320, 1034–1039. doi: 10.1126/science.1153213
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, 279–285. doi: 10.1093/nar/gkv1344
Fraser, J. S., Maxwell, K. L., and Davidson, A. R. (2007). Immunoglobulin-like domains on bacteriophage: weapons of modest damage? Curr. Opin. Microbiol. 10, 382–387. doi: 10.1016/j.mib.2007.05.018
Fraser, J. S., Yu, Z., Maxwell, K. L., and Davidson, A. R. (2006). Ig-like domains on bacteriophages: a tale of promiscuity and deceit. J. Mol. Biol. 359, 496–507. doi: 10.1016/j.jmb.2006.03.043
Groth, A. C., and Calos, M. P. (2004). Phage integrases: biology and applications. J. Mol. Biol. 335, 667–678 doi: 10.1016/j.jmb.2003.09.082
Guo, P., El-Gohary, Y., Prasadan, K., Shiota, C., Xiao, X., Wiersch, J., et al. (2012). Rapid and simplified purification of recombinant adeno-associated virus. J. Virol. Methods 183, 139–146. doi: 10.1016/j.jviromet.2012.04.004
Harrison, S. C., Caspar, D. L. D., Camerini-Otero, R. D., and Franklin, R. M. (1971). Lipid and protein arrangement in bacteriophage PM2. Nature New Biol. 229, 197–201. doi: 10.1038/newbio229197a0
Hatfull, G. F. (2008). Bacteriophage genomics. Curr. Opin. Microbiol. 11, 447–453. doi: 10.1016/j.mib.2008.09.004
Hsiao, C. L., and Black, L. W. (1978). Head morphogenesis of bacteriophage T4. II. The role of gene 40 in initiating prehead assembly. Virology 91, 15–25. doi: 10.1016/0042-6822(78)90351-3
Hu, B. L., Shen, L. D., Lian, X., Zhu, Q., Liu, S., Huang, Q., et al. (2014). Evidence for nitrite-dependent anaerobic methane oxidation as a previously overlooked microbial methane sink in wetlands. Proc. Natl. Acad. Sci. U.S.A. 111, 4495–4500. doi: 10.1073/pnas.1318393111
Hurwitz, B. L., and Sullivan, M. B. (2013). The Pacific Ocean Virome (POV): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS ONE 8:e57355. doi: 10.1371/journal.pone.0057355
John, S. G., Mendez, C. B., Deng, L., Poulos, B., Kauffman, A. K. M., Kern, S., et al. (2011). A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 3, 195–202. doi: 10.1111/j.1758-2229.2010.00208.x
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9. doi: 10.1093/nar/gkn201
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics. 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kala, S., Cumby, N., Sadowski, P. D., Zafar Hyder, B., Kanelis, V., Davidson, A. R., et al. (2014). HNH proteins are a widespread component of phage DNA packaging machines. Proc. Natl. Acad. Sci. U.S.A. 111, 6022–6027. doi: 10.1073/pnas.1320952111
King, A. M. Q., Adams, M. J., Carstens, E. B., and Lefkowitz, E. J. (2012). Virus Taxonomy. Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego, CA: Elsevier Academic Press.
Kivelä, H. M., Kalkkinen, N., and Bamford, D. H. (2002). Bacteriophage PM2 has a protein capsid surrounding a spherical proteinaceous lipid core. J. Virol. 76, 8169–8178. doi: 10.1128/JVI.76.16.8169-8178.2002
Kondabagil, K. R., Zhang, Z., and Rao, V. B. (2006). The DNA translocating ATPase of bacteriophage T4 packaging motor. J. Mol. Biol. 363, 786–799. doi: 10.1016/j.jmb.2006.08.054
Koonin, E. V., Dolja, V. V., and Krupovic, M. (2015). Origins and evolution of viruses of eukaryotes: the ultimate modularity. Virology 479–480, 2–25. doi: 10.1016/j.virol.2015.02.039
Koskella, B., and Brockhurst, M. A. (2014). Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931. doi: 10.1111/1574-6976.12072
Koskella, B., and Parr, N. (2015). The evolution of bacterial resistance against bacteriophages in the horse chestnut phyllosphere is general across both space and time. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370:20140297. doi: 10.1098/rstb.2014.0297
Kremer, J. R., Mastronarde, D. N., and McIntosh, J. R. (1996). Computer visualization of three-dimentional image data using IMOD. J. Struct. Biol. 116, 71–76. doi: 10.1006/jsbi.1996.0013
Kunin, V., He, S., Warnecke, F., Peterson, S. B., Garcia Martin, H., Haynes, M., et al. (2008). A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res. 18, 293–297. doi: 10.1101/gr.6835308
Leiman, P. G., Kanamaru, S., Mesyanzhinov, V. V., Arisaka, F., and Rossmann, M. G. (2003). Structure and morphogenesis of bacteriophage T4. Cell. Mol. Life Sci. 60, 2356–2370. doi: 10.1007/s00018-003-3072-1
Letunic, I., Doerks, T., and Bork, P. (2014). SMART: recent updates, new developments and status 2015. Nucleic Acids Res. 43, 257–260. doi: 10.1093/nar/gku949
Luesken, F. A., van Alen, T. A., van der Biezen, E., Frijters, C., Toonen, G., Kampman, C., et al. (2011). Diversity and enrichment of nitrite-dependent anaerobic methane oxidizing bacteria from wastewater sludge. Environ. Biotechnol. 92, 845–854. doi: 10.1007/s00253-011-3361-9
Männistö, R. H., Kivelä, H. M., Paulin, L., Bamford, D. H., and Bamford, J. K. H. (1999). The complete genome sequence of PM2, the first lipid-containing bacterial virus to be isolated. Virology 262, 355–363. doi: 10.1006/viro.1999.9837
Mastronarde, D. N. (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51. doi: 10.1016/j.jsb.2005.07.007
Middelboe, M., Jacquet, S., and Weinbauer, M. (2008). Viruses in freshwater ecosystems: an introduction to the exploration of viruses in new aquatic habitats. Freshwater Biol. 53, 1069–1075. doi: 10.1111/j.1365-2427.2008.02014.x
Mindich, L., Bamford, D., McGraw, T., and Mackenzie, G. (1982). Assembly of bacteriophage PRD1: particle formation with wild-type and mutant viruses. J. Virol. 44, 1021–1030.
Mokili, J. L., Rohwel, F., and Dutilh, B. E. (2012). Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2, 63–77. doi: 10.1016/j.coviro.2011.12.004
Mollenhauer, H. H. (1964). Plastic embedding mixtures for use in electron microscopy. Stain Technol. 39, 111–114.
Moodley, S., Maxwell, K. L., and Kanelis, V. (2012). The protein gp74 from the bacteriophage HK97 functions as a HNH endonuclease. Protein Sci. 21, 809–818. doi: 10.1002/pro.2064
Murphy, J., Mahony, J., Ainsworth, S., Nauta, A., and van Sinderen, D. (2013). Bacteriophage orphan DNA methyltransferases: insight from their bacterial origin, function and occurrence. Appl. Environ. Microbiol. 79, 7547–7555. doi: 10.1128/AEM.02229-13
Pachauri, R. K., Allen, M. R., Barros, V. R., Broome, J., Cramer, W., Christ, R., et al. (2014). IPCC, 2014: Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change.
Paez-Espino, D., Eloe-Fadrosh, E. A., Pavlopoulos, G. A., Thomas, A. D., Huntemann, M., Mikhailova, N., et al. (2016). Uncovering Earth's virome. Nature 536, 425–430. doi: 10.1038/nature19094
Paez-Espino, D., Sharon, I., Morovic, W., Stahl, B., Thomas, B. C., Barrangou, R., et al. (2015). CRISPR immunity drives rapid phage genome evolution in Streptococcus thermophilus. mBio 6:e00262-15. doi: 10.1128/mBio.00262-15
Pagni, M., Ioannidis, V., Cerutti, L., Zahn-Zabal, M., Jongeneel, C. V., Hau, J., et al. (2007). MyHits: improvements to an interactive resource for analyzing protein sequences. Nucleic Acids Res. 35, 433–437. doi: 10.1093/nar/gkm352
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSFChimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Rho, M., Wu, Y. W., Tang, H., Doak, T. G., and Ye, Y. (2012). Diverse CRISPRs evolving in human microbiomes. PLoS Genet. 8:e1002441. doi: 10.1371/journal.pgen.1002441
Rissanen, I., Grimes, J. M., Pawlowski, A., Mäntynen, S., Harlos, K., Bamford, J. K. H., et al. (2013). Bacteriophage P23-77 capsid protein structures reveal the archetype of an ancient branch from a major virus lineage. Structure 21, 718–726. doi: 10.1016/j.str.2013.02.026
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M. A., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Sathaliyawala, T., Islam, M. Z., Li, Q., Fokine, A., Rossmann, M. G., and Rao, V. B. (2010). Functional analysis of the highly antigenic outer capsid protein, Hoc, a virus decoration protein from T4-like bacteriophages. Mol. Microbiol. 77, 444–455. doi: 10.1111/j.1365-2958.2010.07219.x
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shapiro, O. H., Kushmaro, A., and Brenner, A. (2010). Bacteriophage predation regulates microbial abundance and diversity in a full-scale bioreactor treating industrial wastewater. ISME J. 4, 327–336. doi: 10.1038/ismej.2009.118
Shi, Y., Hu, S., Lou, J., Lu, P., Keller, J., and Yuan, Z. (2013). Nitrogen removal from wasterater by coupling anammox and methane-dependent denitrification in a membrane biofilm reactor. Environ. Sci. Technol. 47, 11577–11583. doi: 10.1021/es402775z
Skennerton, C. T., Imelfort, M., and Tyson, G. W. (2013). Crass: identification and reconstruction of CRISPS from unassembled metagenomic data. Nucleic Acids Res. 41:e105. doi: 10.1093/nar/gkt183
Sulcius, S., Staniulis, J., and Paškauskas, R. (2011). Morphology and distribution of phage-like particles in a eutrophic boreal lagoon. Oceanologia 53, 587–603. doi: 10.5697/oc.53-2.587
Sun, C. L., Barrangou, R., Thomas, B. C., Horvath, P., Fremaux, C., and Banfield, J. F. (2013). Phage mutations in response to CRISPR diversification in a bacterial population. Environ. Microbiol. 15, 463–470. doi: 10.1111/j.1462-2920.2012.02879.x
Suttle, C. A. (2007). Marine viruses – major players in the global ecosystem. Nat. Rev. 5, 801–812. doi: 10.1038/nrmicro1750
Szpara, M. L., Tafuri, Y. R., and Enquist, L. W. (2011). Preparation of viral DNA from nucleocapsids. J. Vis. Exp. 54, 1–6. doi: 10.3791/3151
Thurber, R. V., Haynes, M., Breitbart, M., Wegley, L., and Rohwer, F. (2009). Laboratory procedures to generate viral metagenomes. Nat. Protoc. 4, 470–483. doi: 10.1038/nprot.2009.10
Ultsch, A., and Mörchen, F. (2005). ESOM-Maps: Tool for Clustering, Visualization, and Classification with Emergent SOM. Technical Report Department of Mathematics and Computer Science, University of Marburg, Germany.
Walther, P., and Ziegler, A. (2002). Freeze substitution of high-pressure frozen samples: the visibility of biological membranes is improved when the substitution medium contains water. J. Microsc. 208, 3–10. doi: 10.1046/j.1365-2818.2002.01064.x
Weinbauer, M. G., and Rassoulzadegan, F. (2004). Are viruses driving microbial diversification and diversity? Environ. Microbiol. 6, 1–11. doi: 10.1046/j.1462-2920.2003.00539.x
Westbye, A. B., Kuchinski, K., Yip, C. K., and Beatty, J. T. (2016). The gene transfer agent RcGTA contains head spikes needed for binding to the Rhodobacter capsulatus polysaccharide cell capsule. J. Mol. Biol. 428, 477–491. doi: 10.1016/j.jmb.2015.12.010
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/MMBR.64.1.69-114.2000
Wu, C. H., Yeh, L. S. L., Huang, H., Arminski, L., Castro-Alvear, J., Chen, Y., Hu, Z., et al. (2003). The protein information resource. Nucleic Acids Res. 31, 345–347. doi: 10.1093/nar/gkg040
Keywords: Methylomirabilis, bacteriophage, viral metagenome, ultrastructure, bioreactor
Citation: Gambelli L, Cremers G, Mesman R, Guerrero S, Dutilh BE, Jetten MSM, Op den Camp HJM and van Niftrik L (2016) Ultrastructure and Viral Metagenome of Bacteriophages from an Anaerobic Methane Oxidizing Methylomirabilis Bioreactor Enrichment Culture. Front. Microbiol. 7:1740. doi: 10.3389/fmicb.2016.01740
Received: 07 July 2016; Accepted: 18 October 2016;
Published: 08 November 2016.
Edited by:
William Michael McShan, University of Oklahoma Health Sciences Center, USAReviewed by:
Bernard A. P. Lafont, National Institute of Allergy and Infectious Diseases, USACopyright © 2016 Gambelli, Cremers, Mesman, Guerrero, Dutilh, Jetten, Op den Camp and van Niftrik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura van Niftrik, bC52YW5uaWZ0cmlrQHNjaWVuY2UucnUubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.