 Paola Cremonesi1*
Paola Cremonesi1* Claudia Cortimiglia1
Claudia Cortimiglia1 Claudia Picozzi2
Claudia Picozzi2 Giulietta Minozzi3,4
Giulietta Minozzi3,4 Michela Malvisi3
Michela Malvisi3 Mario Luini5
Mario Luini5 Bianca Castiglioni1
Bianca Castiglioni1- 1Institute of Agricultural Biology and Biotechnology, National Research Council, Lodi, Italy
- 2DeFENS, University of Milan, Milan, Italy
- 3PTP Science Park, Lodi, Italy
- 4DiMeVet, University of Milan, Milan, Italy
- 5Istituto Zooprofilattico Sperimentale della Lombardia e dell'Emilia Romagna, Lodi, Italy
Dairy products can harbor various microorganisms (e.g., Campylobacter spp., Salmonella spp., Listeria monocytogenes, verocytotoxin-producing Escherichia coli) arising from animal reservoirs, and which can become important sources of foodborne illness. Therefore, early detection of food pathogens is crucial to prevent diseases. We wished to develop an accurate quantitative protocol based on a droplet digital polymerase chain reaction (ddPCR) involving eight individual TaqMan™ reactions to detect simultaneously, without selective enrichment, Listeria spp., L. monocytogenes, Salmonella spp., verocytotoxin-producing E. coli and Campylobacter spp. in cheese. ddPCR (a “third-generation PCR”) provides absolute quantification of target DNAs without requirement of a standard curve, which simplifies experimentation and data comparability. The accuracy, specificity and sensitivity of the developed ddPCR system were assessed using purified DNA from 50 reference pathogenic and non-pathogenic strains from international or Italian collections and analyzing soft cheese samples artificially contaminated with serial dilutions (from 4 × 106 to 4 × 101 CFU/g) of pure cultures from the American Type Culture Collection. Finally, the performance of our ddPCR system was compared by parallel testing with quantitative PCR: it gave higher sensitivity (102 CFU/g for the Listeria spp. assay) without the necessity of a standard curve. In conclusion, this is the first ddPCR system developed for simultaneous detection of common foodborne pathogens in cheese using a single set of amplification conditions. As such, it could become a useful strategy for high-throughput screening of microorganisms to evaluate the quality and safety of food products.
Introduction
Over the past three decades, the incidence of foodborne illnesses has increased dramatically to become a major public-health issue. The US Center of Disease Control and Prevention estimates that each year ≈48 million Americans (1 in 6) become ill, 128,000 are hospitalized, and 3000 die of foodborne diseases [Centers for Disease Control and Prevention (CDC), 2011]. In Europe in 2013, 5196 outbreaks of foodborne illnesses with 43,183 cases, 5946 hospitalizations, and 11 deaths were reported [European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC), 2015].
Foodborne illnesses are usually caused by consumption of food/drinking water contaminated with pathogenic bacteria, bacterial toxins, viruses, or parasites that invade the body via the gastrointestinal tract (where the first symptoms usually occur). Everyone is at risk, but the most severe consequences are for infants, the elderly, and people with a compromised immune system [European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC), 2015].
Among the bacteria that can contaminate food, some have an animal reservoir. Milk and dairy products can become contaminated during production and harbor various microorganisms (e.g., Campylobacter spp., Salmonella spp., Listeria spp., verocytotoxin-producing Escherichia coli, including E. coli O157) that can be important sources of foodborne diseases. Raw milk and raw-milk products are experiencing increasing market demand worldwide due to their alleged superior nutritional properties (Quigley et al., 2013). Therefore, it is necessary to: (i) establish the absence of pathogens or their toxins to ensure food safety; (ii) monitor the effectiveness of hygienic processing; and (iii) verify product quality and shelf-life stability. Hence, food safety is dependent upon rapid detection of these pathogens in foodstuffs through sensitive, fast and cost-effective technologies to prevent illnesses.
Beside conventional, laborious, and time-consuming culturing approaches, molecular methods with higher sensitivity and specificity have been developed. Such methods can be categorized into those based on nucleic acids (e.g., polymerase chain reaction (PCR), multiplex PCR, real-time PCR, nucleic acid sequence-based amplification, loop-mediated isothermal amplification, oligonucleotide DNA microarray), biosensors (electrochemical, optical, mass-sensitive) and immunologic (enzyme-linked immunosorbent assay, lateral flow immunoassay) (Mortari and Lorenzelli, 2014; Law et al., 2015).
In vitro amplification of nucleic acids via PCR remains the most widely applied method in research and clinical laboratories for the detection, identification, and enumeration of foodborne pathogens (Postollec et al., 2011). During the past decade, quantitative PCR (qPCR) has emerged as a method for rapid detection of foodborne pathogens in dairy microbiology due to its accuracy and precision (Fukushima et al., 2010). Several qPCR protocols have been applied to Campylobacter jejuni (Yang et al., 2003), E. coli O157 (Paul et al., 2013) and Salmonella spp. (Hein et al., 2006).
If the concentration of pathogens in complex biologic food matrices is very low, the quantification step of qPCR can affect the accuracy of template quantification considerably (Ramakers et al., 2003). To circumvent this problem, droplet digital PCR (ddPCR) has been considered. This approach partitions the sample into hundreds of millions of water-in-oil droplets before thermal cycling (McDermott et al., 2013). These droplets are monitored for positive amplification after endpoint PCR amplification using fluorescent target-specific hydrolysis probes (Floren et al., 2015). Until now, this method has been adopted for: routine analyses of genetically modified organisms in food and animal feed (Morisset et al., 2013; Gerdes et al., 2016); detection and quantification of pathogenic bacteria such as Salmonella spp., Campylobacter jejuni and Listeria monocytogenes in environmental water (Rothrock et al., 2013); exact quantification of different species in meat and processed meat products (Floren et al., 2015); monitoring the dynamics of microbial populations in soils with different population levels (Kim et al., 2014).
We wished to develop an accurate quantitative protocol based on ddPCR involving eight individual TaqMan™ reactions to detect simultaneously, without selective enrichment, Listeria spp., L. monocytogenes, Salmonella spp., verocytotoxin-producing E. coli, and Campylobacter spp. in cheese.
Materials and Methods
Bacterial Strains and Growth Conditions
Strains and culture conditions (culture media, temperature, incubation time) are listed in Table 1. Most of the bacteria tested originated from international (American Type Colture Collection; Deutsche Sammlung von Mikroorganismen und Zellkulturen; Collection of Institute Pasteur; Salmonella Genetic Stock Centre; Culture Collection, University of Göteborg, Sweden) and Italian collections.
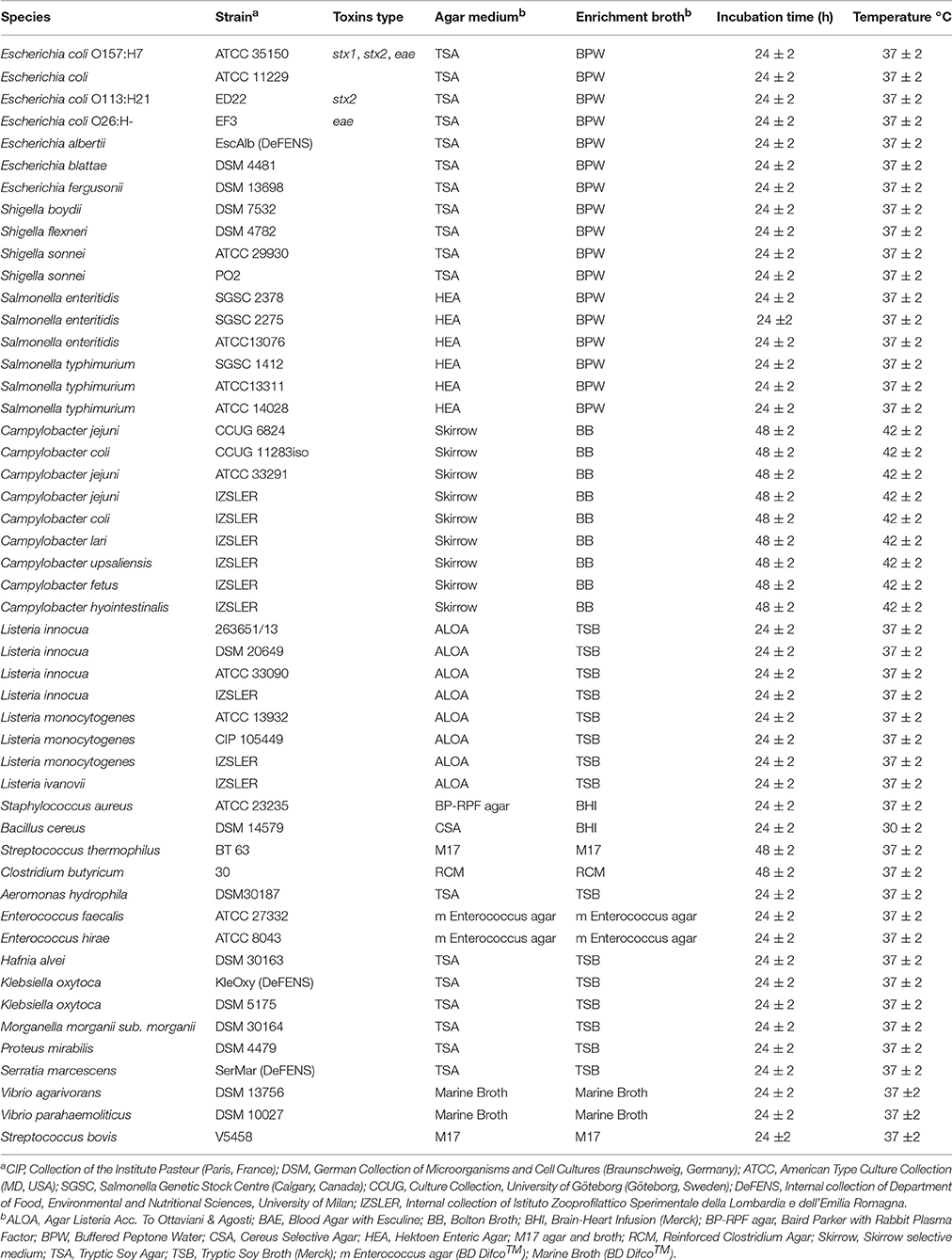
Table 1. List of target and non-target species with growth conditions.
E. coli ED226 and EF3 strains were provided by Istituto Superiore di Sanità (Rome, Italy); Shigella sonnei PO2 is part of the Centro Enteropatogeni Italia Settentrionale (Milan, Italy) collection; L. innocua 263651/13 was isolated from an environmental sample from Istituto Zooprofilattico Sperimentale della Lombardia e dell'Emilia Romagna (Brescia, Italy), which also supplied L. innocua, L. ivanovii, C. jejuni, C. coli, C. lari, C. upsaliensis, C. fetus, and C. hyointestinalis. Streptococcus thermophilus BT63, St. bovis V5458 and Clostridium butyricum 30 were supplied by ISPA-CNR (Milan, Italy). E. albertii (isolated from lake water), Klebsiella oxytoca (isolated from fresh cheese) and Serratia marcescens (isolated from fresh cheese) were provided by the Department of Food, Environmental and Nutritional Sciences of the University of Milan.
All strains were cultivated aerobically except for Campylobacter spp., the isolates of which were grown under microaerophilic conditions. Stock cultures were thawed on selective agar plates; then single colonies were inoculated into appropriate enrichment broth for 24–48 h (Table 1). Five hundred microliters of each culture were used for DNA extraction.
Spiking of Food Samples
L. innocua 263651/13, S. typhimurium ATCC 14028 and E. coli ATCC 35150 strains were used to contaminate soft cheese samples artificially to evaluate the performance of qPCR and ddPCR. Pure cultures of each bacteria type were grown for 24–48 h (as described above) and the concentration was determined by inoculation of the tenfold dilution series onto appropriate agar plates. Serial dilutions (108–101 CFU/mL for L. innocua; 109–101 CFU/mL for S. typhimurium and E. coli) in 0.9% NaCl (Sigma–Aldrich, St Louis, MO, USA) were prepared: 1 mL of each dilution was used to artificially contaminate 25 g of soft cheese. The latter was weighed in a 50-mL sterile Falcon tube (Orange Scientific, Belgium), then 1 mL of bacteria suspension added. The Falcon tube was vortexed for 10 s. Then, 5 g of contaminated samples was mixed with 45 mL of 2% (w/v) K2HPO4 buffer solution (Sigma–Aldrich) and homogenized in a Stomacher® paddle blender (PBI, Milan, Italy) for 60 s. A negative control (sample of uncontaminated cheese in sterile buffer) was included. After homogenization, 500 μL were subjected to DNA extraction.
DNA Extraction from Pure Cultures and from Samples of Spiked Soft Cheese
DNA was extracted from 500 μL of pure cultures and from the samples of spiked soft cheese according to our previous protocol (Cremonesi et al., 2006) starting from step 2. For artificially spiked samples, few modifications were applied to the protocol. Briefly, 300 μL of binding solution and 400 μL of lysis solution, washing solution, and ethanol solution were used. All centrifugations were carried out at 500 × g, with a final centrifugation of 550 × g. DNA was eluted in 100 μL of elution buffer. Quality and quantity of DNA were evaluated by spectrophotometric (NanoDrop Technologies, Wilmington, DE, USA) means at an absorbance of 260 and 280 nm, respectively. DNA was stored at −20°C.
Probe Design for PCR Target Genes
Candidate assay targets for the eight bacteria of interest were chosen on the basis of published data. The yccT gene (which codes for a conserved protein of unknown function) was chosen to identify E. coli and the closely related Shigella spp. (Clifford et al., 2012). For Shiga toxin-producing E. coli (STEC), two probes for shigatoxin1 (stx1) and shigatoxin 2 (stx2) were designed by considering the conserved region screened in the National Centre for Biotechnology Information. The eae (intimin) probe has been described by our research team (Cremonesi et al., 2014). The assay for Campylobacter spp. was designed on a specific region of the 16S rRNA gene to identify all the bacteria belonging to this species. For Listeria spp. and Salmonella spp., phosphoribosylpyrophosphate synthetase (prs) and invasion protein A (invA) were chosen because of their specificity for these species, respectively. The L. monocytogenes assay was designed on the inlA gene (which codes for a virulence protein that mediates adhesion and internalization into host cells).
After selection of target genes, specific target probes were designed using Primer Express® v3.0 (Applied Biosystems, Foster City, CA, USA) by setting the annealing temperature of primers and probes at 60 and 70°C, respectively. The nucleotide BLAST tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to confirm the specificity of oligonucleotides in silico. Primers and TaqMan probes were synthesized by Applied Biosystems (Life Technologies Inc, Italy). Primers, 5′6-fluorescein-labeled (FAM) TaqMan probes, target genes, and reference sequences are listed in Table 2.
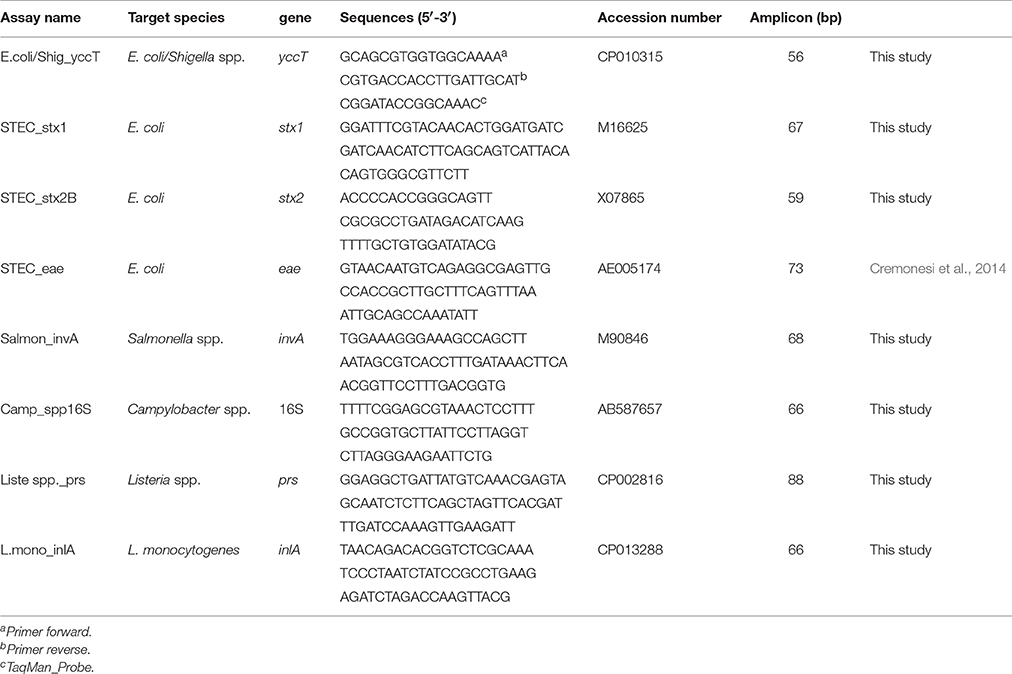
Table 2. TaqMan™ assays used for qPCR and ddPCR.
qPCR
DNAs extracted from all pure cultures and from soft cheese contaminated artificially by several dilutions of L. innocua, E. coli and S. typhimurium were tested by qPCR. Reactions were carried out in 96-well plates sealed with adhesive optical covers (Applied Biosystems) and run on a QuantStudio™ 3 Real-Time PCR system (Applied Biosystems) at 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C. An identical thermal cycle was used for each target. All PCRs were done in duplicate. Each 20 μL of amplification reaction mix contained 1 μL of DNA (or water for negative controls), 10 μL of TaqMan Environmental Master Mix 2.0 (2 ×), 1 μL of TaqMan assay 20 × (18 μM for each primer, 5 μM for probe), TaqMan Exogenous Internal Positive Control (IPC) Reagents VIC™-labeled (2 μL of the ExoIPC Mix, Applied Biosystems), 0.4 μL of the Exo IPC DNA (target DNA) and 5.6 μL of molecular-grade water.
ddPCR
DNA was detected and quantified using an QX100™ Droplet Digital™ PCR system (Bio-Rad Laboratories, Hercules, CA, USA). Reaction mixtures were set-up in a specific manner. Briefly, 10 μL of 2 × ddPCR Master Mix (Bio-Rad Laboratories) and 1 μL of TaqMan assay 20 × (18 μM for each primer, 5 μM for probe) were mixed with 1 μL of DNA from pure cultures, and nuclease- and protease-free water to complete a reaction volume of 20 μL. For samples of spiked food, a different amount of DNA template (2 μL for E. coli and 4 μL for Salmonella spp or L. innocua DNA) was used in the reaction mixture.
To generate the droplets, 20 μL of ddPCR and 70 μL of Droplet Generation oil for Probes (Bio-Rad Laboratories) were inserted in an eight-well cartridge using a QX100 droplet generator (Bio-Rad Laboratories) according to manufacturer instructions. Then, 40 μL of the generated droplet emulsion was transferred to a new 96-well PCR plate (Eppendorf, Hamburg, Germany) and amplified in a T100™ thermal cycler (Bio-Rad Laboratories). Amplification conditions started with 10 min of activation of DNA polymerase at 95°C, followed by 40 cycles of a two-step thermal profile of 15 s at 95°C for denaturation, and 1 min at 60°C for annealing and extension. A final hold of 10 min at 98°C was used for droplet stabilization followed by cooling to 4°C. No optimization of ddPCR was necessary with respect to qPCR annealing or probe concentration.
After thermal cycling, plates were transferred to a droplet reader (Bio-Rad Laboratories). The software provided with the ddPCR system (QuantaSoft 1.3.2.0; Bio-Rad Laboratories) was used for data acquisition to calculate the concentration of target DNA in copies/mL from the fraction of positive reactions using Poisson distribution analyses (McDermott et al., 2013) (Supplementary Table 2).
Specificity and Sensitivity
The specificity of each TaqMan assay was assessed using qPCR with purified genomic DNA from the reference strains described in Table 1. For each target assay, the DNA of other non-target bacteria was used as the negative control.
The limit of detection (LoD) for each qPCR and ddPCR assay was determined with pure culture, starting from 50 ng/μL of the DNA template, using a 100-fold dilution up to 5 fg/μL. The LoD for qPCR and ddPCR was also evaluated using soft cheese samples contaminated artificially by tenfold dilution from 4 × 106 CFU/g up to 4 × 101 CFU/g. Linearity over the dynamic range was determined by the coefficient of correlation (R2) calculated on the mean value of target copy numbers measured in the replicated dilution series for qPCR and ddPCR.
Intra- and Inter-Assay Repeatability
Repeatability was determined on a sub-sample of the TaqMan assay (STEC_eae, Salmon_invA, Liste spp_prs) using: (i) the DNA of three reference strains (50 pg/μL of L. innocua 263651/13, S. typhimurium ATCC 14028 and E. coli ATCC 35150); (ii) DNA samples extracted from artificially contaminated soft cheese (4 × 105 CFU/g for each of the three types of bacteria); (iii) three DNAs extracted from artificially contaminated soft cheese (4 × 106 CFU/g). Then, these sub-samples were mixed to form a pooled sample. For these tests, the same DNA was used as the technical replicate.
Intra-assay repeatability was assessed by calculation of the coefficient of variation (CV) of measured percentages from quadruplicate ddPCR measurements conducted in 1 day on a single sample run. The inter-assay test was evaluated by calculation of the CV of each sample, processed in duplicate for 5 consecutive days.
Results
Probe Design
Each TaqMan assay, tested initially in silico through the BLAST tool, did not reveal identical sequences other than those targeted (100% of query cover and max identity). For verocytotoxin-producing E. coli, two assays (Table 2) were designed to detect virulence-specific genes such as stx1 and stx2. The assay for detection of the intimin gene (eae) was taken from our previous data (Cremonesi et al., 2014).
Assay Specificity
The specificity of the eight TaqMan assays was assessed first by qPCR with 50 pathogenic target and non-target strains (Table 1). All trials identified the target strains correctly without generating false-positive or false-negative results, thereby confirming assay specificity. All TaqMan assays amplified their targets under identical qPCR conditions, and optimization was not done with ddPCR for annealing temperature or probe concentration. An identical protocol was used for qPCR and ddPCR, so the specificity test was not repeated for ddPCR.
Assay Sensitivity
Reference Strains
For qPCR, the analytical sensitivity of all TaqMan assays tested in triplicate was ≈0.5 pg/μL of total DNA, with mean cycle threshold (CT) values from 28.9 ± 0.03 for Campylobacter spp. to 38.4 ± 0.91 for Listeria spp. (Table 3A). TaqMan assays for E. coli/Shig_yccT and Campylobacter spp. showed good sensitivity at 0.05 (33.8 ± 0.45) and 0.005 (35.2 ± 0.28) pg/μL, respectively.
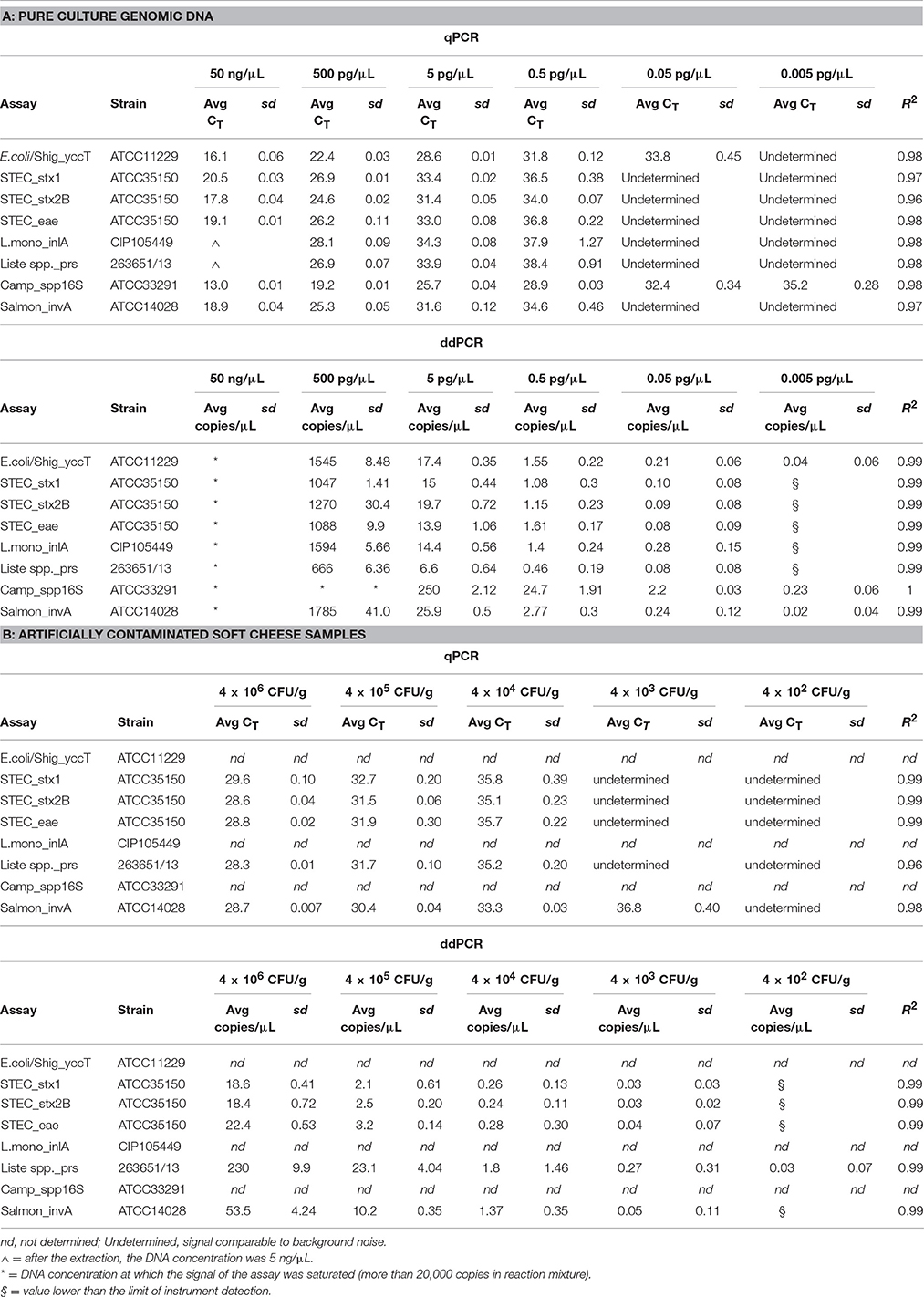
Table 3. Sensitivity and efficiency of the TaqMan™ assays obtained by series of 100-fold dilutions of the pure culture genomic DNA (from 50 ng/μL up to 0.005 pg/μL; A) and with artificially contaminated soft cheese sample using tenfold dilution of 3 bacterial pure cultures (from 4 × 106 CFU/g up to 4 × 104 CFU/g; B) by qPCR and ddPCR.
To identify the lowest LoD in ddPCR, eight replicates were run with the two lowest concentrations of the DNA samples used to construct the standard curve. Good linearity was reached for all TaqMan assays revealing, with 0.05 pg/μL of total DNA, a mean of 0.08 ± 0.08 copies/μL for E. coli eae and Liste spp_prs assays and ≤ 2.2 ± 0.03 copies/μL for Campylobacter spp. Moreover, TaqMan assays for E. coli/Shig_yccT, Campylobacter spp. and Salmonella spp. showed good sensitivity for ≤5 fg of total DNA (0.04 ± 0.06; 0.23 ± 0.06; 0.02 ± 0.04 copies/μL, respectively) (Table 3B). TaqMan assays with qPCR and ddPCR showed good linearity in the range of quantification, with R2 of 0.96% and 1%, respectively. And more, with Campylobacter spp. assay, reaction saturation was reached at a concentration of 500 ng/μL (more than 20,000 positive droplets) and therefore it was impossible to quantify this concentration. The negative control for qPCR and ddPCR did not show amplification (data not shown). Examples of the results obtained are represented in Figure 1A and in Supplementary Figure 1.
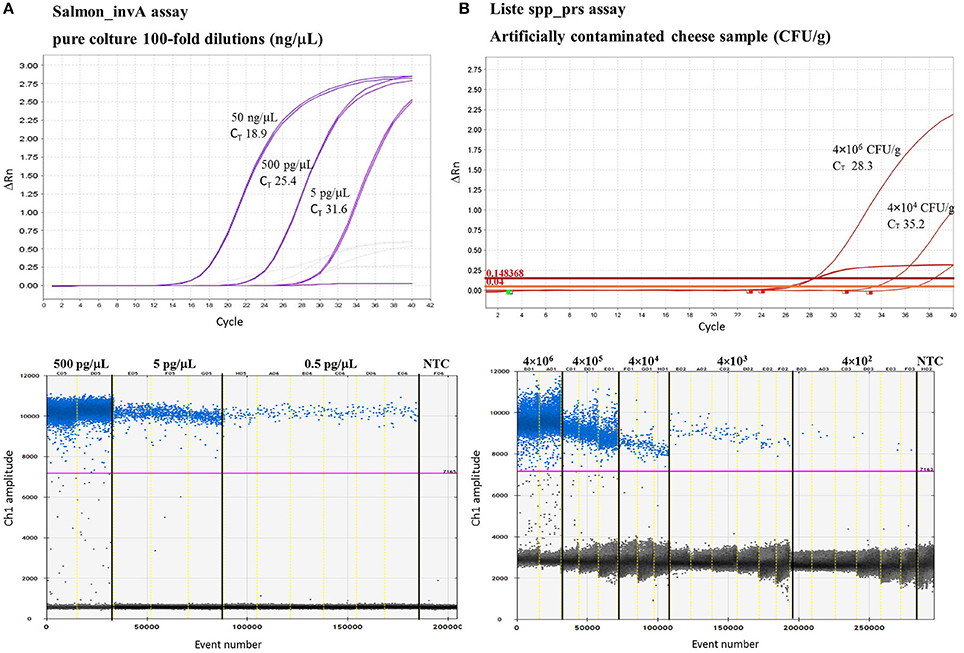
Figure 1. Example of TaqMan™ assays analytical sensitivity by qPCR and ddPCR by using 100-fold dilution of DNA S. enteritidis SGSC2378 pure culture (A) and tenfold dilution of L. innocua 263651/13 in artificially contaminated soft cheese samples (B). For the qPCR (above) two replicates are shown while for ddPCR (1D Droplet Plots) up to six replicates were run with the lowest concentrations. NTC = negative template control.
Artificially Contaminated Soft Cheese
To evaluate the performance of qPCR and ddPCR, soft cheese samples were contaminated artificially with a tenfold dilution series of three cultures of pure bacteria. Cheese samples spiked with verocytotoxin-producing E. coli, L. innocua and S. typhimurium showed good linearity within the range of quantification, giving R2 between 0.96 and 1 for qPCR and ddPCR, respectively. With qPCR and ddPCR, a sensitivity of 104 CFU/g and 103 CFU/g was reached for all the TaqMan assays tested. Moreover, the assays for Salmonella spp. and Listeria spp. showed good linearity at ≤103 CFU/g and 102 CFU/g, respectively. Examples of the results obtained are represented in Figure 1B.
Inter- and Intra-Assay Repeatability
For intra-assay experiments with the (i) DNA of three strains, (ii) three DNA samples extracted from artificially contaminated soft cheese (4 × 105 CFU/g), (iii) DNA extracted from artificially contaminated soft cheese (4 × 106 CFU/g) and then mixed in a pooled sample, the CV was 3.63, 10.41, and 10.62% for STEC_eae, 5.66, 7.73, and 3.44% for the Salmon_invA, and 10.8, 1.74, and 4.5% for Liste spp_prs, respectively. Inter-assay experiments showed a CV <12.99% and <15.91% for Salmon_invA and STEC_eae, respectively, and from 3.05 to 24.68% for Liste spp_prs (Supplementary Table 1).
Discussion
Early detection of food pathogens is crucial to prevent foodborne illnesses. In the present study, eight individual TaqMan reactions were developed to detect Listeria spp., L. monocytogenes, Salmonella spp., verocytotoxin-producing E. coli and Campylobacter spp. directly and simultaneously in cheese. In a second step, a soft cheese was contaminated with three out of the five microorganisms under study.
After DNA extraction from cheese, an assay using a ddPCR instrument (a “third-generation PCR”) was developed to provide absolute quantification of target DNAs without the requirement of a standard curve. This procedure represents an important advantage in comparison with an assay based on qPCR because construction of a standard curve requires accurate quantification of the template DNA, which might be difficult to obtain (especially if working with food samples) (Kim et al., 2014). qPCR remains the most popular choice for the detection and quantification of a wide variety of microorganisms in food samples due to quantification of real samples, the shorter time required to obtain results, and lower costs (Hudecova, 2015). However, the presence of inhibiting substances decreases the efficiency of qPCR.
Given its advantages, the ddPCR system developed in the present study represents a new strategy to quantify pathogens directly in food samples, as described also by Floren et al. (2015) and Verhaegen et al. (2016). First, the ddPCR system optimized in the present study has increased the tolerance to inhibitors arising from cheese samples (e.g., fats, proteins, high concentration of Ca2+) to improve the LoD compared with qPCR. As reported by Rački et al. (2014) and Yang et al. (2014), this effect is probably due to partitioning of the PCR that reduces interference by PCR inhibitors (Huggett et al., 2013). Second, our approach was very effective when used for detection of DNA traces without the need for a pre-amplification step, and showed higher precision, sensitivity, and reproducibility over qPCR.
For the design of quantitative assays optimized in the present study, target genes described previously were used, such as the highly conserved region 16S rRNA for detection of Campylobacter spp., or bacterial virulence genes such as stx1, stx2 and eae for verocytotoxin-producing E. coli (Verhaegen et al., 2016), invA for Salmonella spp., and inlA for L. monocytogenes (Rothrock et al., 2013). Using this strategy, good specificity and sensitivity were achieved.
For a quantitative protocol based on ddPCR developed in the present study, the dynamic range was comparable with qPCR. qPCR and ddPCR exhibited excellent linearity and efficiency, but ddPCR was more sensitive, improving the LoD in spiked cheese by one order of magnitude with respect to qPCR according to previous studies (Yang et al., 2014; Porcellato et al., 2016).
ddPCR was found to exhibit a saturation limit lower than that of qPCR, by which DNA samples must be diluted to a value <20,000 copies in the reaction mixture to quantify bacteria populated densely in a reference sample. As suggested by Yang et al. (2014), to determine the optimal dilution factor for ddPCR, the first step is the set-up TaqMan assays on qPCR using reference material. This statement was confirmed in our study by Campylobacter spp. assay that, because of its high efficiency, gave saturation signal at 500 pg/μl. When the artificially contaminated food samples were analyzed with ddPCR, no saturation was observed. This was probably due to the matrix effect on the efficiency of bacterial DNA extraction.
With this protocol sensitivity level, of 103 CFU/g was reached for all the TaqMan assays (102 CFU/g for Listeria spp.) in food matrices. These results could be improved or by a short selective enrichment of cheese sample or by the use of a higher efficiency DNA extraction method. Further studies should be necessary to evaluate new approaches.
Finally, although ddPCR is considered to be more expensive and time-consuming than qPCR (Verhaegen et al., 2016), its use to investigate simultaneously a sample for different pathogens, without standard curves, could reduce the difference in cost.
Conclusions
Our results show the applicability of ddPCR to target the main foodborne pathogens in cheese. This technology is more sensitive for detection of low quantities of target DNA than qPCR, and reveals higher tolerance to inhibitors arising from food matrices. This is the first ddPCR system developed for simultaneous detection in cheese of common foodborne pathogens using a single set of amplification conditions. Hence, the good performance of this approach could be the starting point for becoming a useful approach for a high-throughput foodborne pathogens screening to evaluate quality and safety of the products. To be employed in routine testing, this ddPCR method shall be properly validated through intra-laboratories trials in order to demonstrate its efficiency.
Author Contributions
CP and ML provided, cultivated and characterized the strains and prepared the artificial contamination of cheese samples. PC and CC made the probes design and verify the “in silico” probe specificity; extracted the DNA from pure culture and artificially contaminated cheese samples. PC and CC performed part of the qPCR experiments and all the ddPCR experiments. GM and MM performed part of the qPCR experiments with the DNA extracted from pure culture strains. BC collaborated in ddPCR experiments and supervised the experimental study. PC, CC, CP, and BC drafted the manuscript. All the authors read, correct and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by MIUR with SAFE&SMART project (CTN01_00230_248064)—Nuove tecnologie abilitanti per la food safety e l'integrità delle filiere agro-alimentari in uno scenario globale (Progetto tematica 2 Sicurezza del Cluster Nazionale CL.AN). The authors want to thank Dr. Alessandro Martino (Biorad) for his technical support in the experiment set up.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01725/full#supplementary-material
Supplementary Table 1. Results obtained during the intra- and inter-assay repeatability.
Supplementary Table 2. dMIQE checklist for authors, reviewers and editors.
Supplementary Figure 1. Concentration plots show data for individual replicate wells across a dilution series. Results of Listeria spp. assay, L. monocytogenes assay (A) Campylobacter spp. assay and Salmonella spp. assay (B) are reported. The error bars represent Poisson 95% confidence intervals.
References
Centers for Disease Control Prevention (CDC) (2011). CDC Estimates of Foodborne Illness in the United States. Available online at: www.cdc.gov/foodnet/reports/index.html (Accessed 18 May, 2016).
Clifford, R. J., Milillo, M., Prestwood, J., Quintero, R., Zurawski, D. V., Kwak, Y. I., et al. (2012). Detection of bacterial 16S rRNA and identification of four clinically important bacteria by real-time PCR. PLoS ONE 7:e48558. doi: 10.1371/journal.pone.0048558
Cremonesi, P., Castiglioni, B., Malferrari, G., Biunno, I., Vimercati, C., Moroni, P., et al. (2006). Technical note: improved method for rapid DNA extraction of mastitis pathogens directly from milk. J. Dairy Sci. 89, 163–169. doi: 10.3168/jds.S0022-0302(06)72080-X
Cremonesi, P., Pisani, L. F., Lecchi, C., Ceciliani, F., Martino, P., Bonastre, A. S., et al. (2014). Development of 23 individual TaqMan® real-time PCR assays for identifying common foodborne pathogens using a single set of amplification conditions. Food Microbiol. 43, 35–40. doi: 10.1016/j.fm.2014.04.007
European Food Safety Authority European Centre for Disease Prevention Control (EFSA ECDC) (2015). The European Union summary Report on trends and sources of zoonoses, zoonotic agents and food-borne Outbreaks in 2013. EFSA J. 13:165. doi: 10.2903/j.efsa.2015.3991
Floren, C., Wiedemann, I., Brenig, B., Schütz, E., and Beck, J. (2015). Species identification and quantification in meat and meat products using droplet digital PCR (ddPCR). Food Chem. 173, 1054–1058. doi: 10.1016/j.foodchem.2014.10.138
Fukushima, H., Kawase, J., Etoh, Y., Sugama, K., Yashiro, S., Iida, N., et al. (2010). Simultaneous screening of 24 target genes of foodborne pathogens in 35 foodborne outbreaks using multiplex real-time SYBR green PCR analysis. Int. J. Microbiol. 2010:864817. doi: 10.1155/2010/864817
Gerdes, L., Iwobi, A., Busch, U., and Pecoraro, S. (2016). Optimization of digital droplet polymerase chain reaction for quantification of genetically modified organisms. Biomol. Detect. Quantif. 7, 9–20. doi: 10.1016/j.bdq.2015.12.003
Hein, I., Flekna, G., Krassnig, M., and Wagner, M. (2006). Real-time PCR for the detection of Salmonella spp. in food: an alternative approach to a conventional PCR system suggested by the FOOD-PCR project. J. Microbiol. Methods 66, 538–547. doi: 10.1016/j.mimet.2006.02.008
Hudecova, I. (2015). Digital PCR analysis of circulating nucleic acids. Clin. Biochem. 48, 948–956. doi: 10.1016/j.clinbiochem.2015.03.015
Huggett, J. F., Foy, C. A., Benes, V., Emslie, K., Garson, J. A., Haynes, R., et al. (2013). The digital MIQE guidelines: minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 59, 892–902. doi: 10.1373/clinchem.2013.206375
Kim, T. G., Jeong, S. Y., and Cho, K. S. (2014). Comparison of droplet digital PCR and quantitative real-time PCR for examining population dynamics of bacteria in soil. Appl. Microbiol. Biotechnol. 98, 6105–6113. doi: 10.1007/s00253-014-5794-4
Law, J. W., Ab Mutalib, N. S., Chan, K. G., and Lee, L. H. (2015). Rapid methods for the detection of foodborne bacterial pathogens: principles, applications, advantages and limitations. Front. Microbiol. 5:770. doi: 10.3389/fmicb.2014.00770
McDermott, G. P., Do, D., Litterst, C. M., Maar, D., Hindson, C. M., Steenblock, E. R., et al. (2013). Multiplexed target detection using DNA-binding dye chemistry in droplet digital PCR. Anal. Chem. 85, 11619–11627. doi: 10.1021/ac403061n
Morisset, D., Štebih, D., Milavec, M., Gruden, K., and Žel, J. (2013). Quantitative analysis of food and feed samples with droplet digital PCR. PLoS ONE 8:e62583. doi: 10.1371/journal.pone.0062583
Mortari, A., and Lorenzelli, L. (2014). Recent sensing technologies for pathogen detection in milk: a review. Biosens. Bioelectron. 60, 8–21. doi: 10.1016/j.bios.2014.03.063
Paul, M., Van Hekken, D. L., and Brewsterb, J. D. (2013). Detection and quantitation of Escherichia coli O157 in raw milk by direct qPCR. Int. Dairy J. 32, 53–60. doi: 10.1016/j.idairyj.2013.04.007
Porcellato, D., Narvhus, J., and Skeie, S. B. (2016). Detection and quantification of Bacillus cereus group in milk by droplet digital PCR. J. Microbiol. Methods 127, 1–6. doi: 10.1016/j.mimet.2016.05.012
Postollec, F., Falentin, H., Pavan, S., Combrisson, J., and Sohier, D. (2011). Recent advances in quantitative PCR (qPCR) applications in food microbiology. Food Microbiol. 28, 848–861. doi: 10.1016/j.fm.2011.02.008
Quigley, L., O'Sullivan, O., Stanton, C., Beresford, T. P., Ross, R. P., Fitzgerald, G. F., et al. (2013). The complex microbiota of raw milk. FEMS Microbiol. Rev. 37, 664–698. doi: 10.1111/1574-6976.12030
Rački, N., Dreo, T., Gutierrez-Aguirre, I., Blejec, A., and Ravnikar, M. (2014). Reverse transcriptase droplet digital PCR shows high resilience to PCR inhibitors from plant, soil and water samples. Plant Methods 10:42. doi: 10.1186/s13007-014-0042-6
Ramakers, C., Ruijter, J. M., Deprez, R. H., and Moorman, A. F. (2003). Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339, 62–66. doi: 10.1016/S0304-3940(02)01423-4
Rothrock, M. J., Hiett, K. L., Kiepper, B. H., Ingram, K., and Hinton, A. (2013). Quantification of zoonotic bacterial pathogens within commercial poultry processing water samples using droplet digital PCR. Adv. Microbiol. 3, 403–411. doi: 10.4236/aim.2013.35055
Verhaegen, B., De Reu, K., De Zutter, L., Verstraete, K., Heyndrickx, M., and Van Coillie, E. (2016). Comparison of Droplet Digital PCR and qPCR for the Quantification of Shiga Toxin-Producing Escherichia coli in Bovine Feces. Toxins (Basel). 8:157. doi: 10.3390/toxins8050157
Yang, C., Jiang, Y., Huang, K., Zhu, C., and Yin, Y. (2003). Application of real-time PCR for quantitative detection of Campylobacter jejuni in poultry, milk and environmental water. FEMS Immunol. Med. Microbiol. 38, 265–271. doi: 10.1016/S0928-8244(03)00168-8
Keywords: cheese, ddPCR, detection, foodborne pathogens, qPCR
Citation: Cremonesi P, Cortimiglia C, Picozzi C, Minozzi G, Malvisi M, Luini M and Castiglioni B (2016) Development of a Droplet Digital Polymerase Chain Reaction for Rapid and Simultaneous Identification of Common Foodborne Pathogens in Soft Cheese. Front. Microbiol. 7:1725. doi: 10.3389/fmicb.2016.01725
Received: 22 July 2016; Accepted: 14 October 2016;
Published: 28 October 2016.
Edited by:
Andrea Gomez-Zavaglia, Center for Research and Development in Food Cryotechnology (CIDCA, CONICET), ArgentinaReviewed by:
Analía Inés Etcheverría, National University of Central Buenos Aires, ArgentinaChristophe Nguyen, French National Institute for Agricultural Research, France
Avelino Alvarez-Ordóñez, Teagasc Food Research Centre, Ireland
Copyright © 2016 Cremonesi, Cortimiglia, Picozzi, Minozzi, Malvisi, Luini and Castiglioni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Cremonesi, Y3JlbW9uZXNpQGliYmEuY25yLml0