 Bosheng Chen1†
Bosheng Chen1† Chao Sun2†Xili Liang1Xingmeng Lu1Qikang Gao2
Chao Sun2†Xili Liang1Xingmeng Lu1Qikang Gao2 Pol Alonso-Pernas3
Pol Alonso-Pernas3 Beng-Soon Teh3
Beng-Soon Teh3 Alexey L. Novoselov3
Alexey L. Novoselov3 Wilhelm Boland3
Wilhelm Boland3 Yongqi Shao1*
Yongqi Shao1*- 1Laboratory of Invertebrate Pathology, College of Animal Sciences, Zhejiang University, Hangzhou, China
- 2Analysis Center of Agrobiology and Environmental Sciences, Zhejiang University, Hangzhou, China
- 3Department of Bioorganic Chemistry, Max Planck Institute for Chemical Ecology, Jena, Germany
Introduction
Insects are the most abundant and diverse animal class on Earth, and they are associated with an amazing variety of symbiotic microorganisms, which participate in many relationships with the hosts (Douglas, 2015). For example, the fungal symbiont (Leucoagaricus gongylophorus) of leaf-cutting ants produces diverse enzymes for the degradation of plant material (Kooij et al., 2016). Similarly, Bacillus pumilus isolated from the gut of wood boring Mesomorphus sp. (Coleoptera: Tenebrionidae) exhibits significant cellulolytic and xylose isomerase activities (Balsingh et al., 2016).
The Lepidoptera, including moths and butterflies, is one of the most widespread and widely recognizable insect orders in the world. Although butterflies and moths play an important role in the natural ecosystem as pollinators and as food in the food chain, their leaf-chewing larvae are often problematic in agriculture, as their main source of food is live plants (Mithöfer and Boland, 2012). The leafworm Spodoptera littoralis (Lepidoptera: Noctuidae) is a highly polyphagous lepidopteran pest found worldwide and also an important model system used in a variety of biological research. Recent extensive surveys of its microbiome reveal that Enterococcus mundtii is one of the predominant gut microorganisms of S. littoralis and present at high frequency (Tang et al., 2012; Chen et al., 2016; Teh et al., 2016). Particularly, a stable isotope labeling-based approach suggested that this phylotype was also highly metabolically active inside the host across life history of S. littoralis, indicating the significant role played by E. mundtii in host biology (Shao et al., 2014). Therefore, the symbiotic E. mundtii probably constitutes a key factor for the success of this generalist herbivore in adapting to different environments and food sources. The aim of this study was to produce a genome sequence of the strain SL 16, which would assist in understanding of the coevolution of the microbe and the insect host. The dataset has been submitted to NCBI Whole Genome Shotgun (WGS) projects and is reported here, providing an overview of the genome sequence and relevant features of gut symbiotic E. mundtii.
Materials and Methods
Isolation of the Bacterial Strain
E. mundtii strain SL 16 was isolated from the mature 5th instar larva using standard microbiology methods. Briefly, the normal larvae were washed and sedated on ice for at least 1 h to anesthetize them. Then the whole gut sections were dissected from larvae using a fine Vannas scissor and forceps under a binocular microscope (Shao et al., 2013). The fresh gut tissues were put into phosphate buffered saline (PBS: 137 mM NaCl, 10 mM Na2HPO4, 2 mM KH2PO4, and 2.7 mM KCl) and homogenized by hand with a sterile pestle. Bacterial isolates were made by plating the homogenized gut tissues on the Enterococcus Selective Agar (45183, Fluka). After incubation for 24 h at 30°C, the growing bacterial colonies were sub-cultured twice on the same agar medium. 2, 3, 5-Triphenyltetrazolium chloride (TTC) in the medium is reduced to insoluble formazan inside the bacterial cells, which gives pink or red coloration to enterococcal colonies. These purified enterococcal colonies were tested for key phenotypic traits including carbohydrate fermentation capability, motility, and pigment production as previously described (Manero and Blanch, 1999). Furthermore, the taxonomy was validated by colony PCR and sequencing of the amplified 16S rRNA gene. The representative E. mundtii isolate, designated strain SL 16, was selected for this WGS project.
Fluorescence in situ Hybridization (FISH) was applied to localize the dominant enterococci as previously described (Shao et al., 2014). Shortly, FISH was performed on 5 μm thin cross sections of the cold polymerizing resin (Technovit 8100, Heraeus Kulzer GmbH, Wehrheim, Germany) embedded gut tissue. The specificity of probes was tested and hybridization condition was achieved as described (Tang et al., 2012). The sample was hybridized with 1.5 mM FITC-labeled Enterococcus-specific probe in hybridization buffer containing 900 mM NaCl, 20 mM Tris-HCl (pH 8.0), 20% formamide, 1% SDS. And images were taken with an Axio Imager Z1 microscope (Carl Zeiss, Jena, Germany). For scanning electron microscopy (SEM), cells were fixed in paraformaldehyde (1%), and glutaraldehyde (0.25%), dehydrated by ascending alcohol series and dried. After coating samples with gold, scanning electron micrographs were taken with a LEO 1525 instrument (Carl Zeiss, Jena, Germany).
Genomic DNA Isolation, Library Preparation and Sequencing
The genomic DNA was extracted from the cultured bacterium according to Pospiech and Neumann (1995). DNA quality was examined by 1% agarose gel electrophoresis and quantified using a NanoDrop™ spectrophotometer. The DNA library was constructed using the TruSeq™ DNA Sample Preparation Kit (Illumina Inc., San Diego, CA), and 5 μg of pure genomic DNA was prepared for a standard Illumina shotgun library construction. Briefly, genomic DNA was first sheared to a size ranging between 400 and 500 bp using the Covaris M220 per the manufacturer's recommendations. The fragmented DNA sample was end-repaired, dA-tailed, and ligated to multiplex adapters according to the manufacturer's instructions. The ligated products were purified and further enriched using PCR. The quality of the final amplified libraries were checked by running an aliquot (1 μL) on a high-sensitivity Bioanalyzer 2100 DNA Chip (Agilent Technologies). Paired-end sequencing was performed by using an Illumina MiSeq platform (Illumina Inc., San Diego, CA) at Majorbio Bio-pharm Technology Co., Ltd (Shanghai, China) according to the manufacturer's instructions (Zhang et al., 2016).
Preprocessing and Genome Assembly
The quality of sequence reads was evaluated using the FastQC tool as previously described (Balsingh et al., 2016). Reads with >10% Ns and/or 25–35 bases of low quality (≤ Q20) were filtered out, and adapter and duplication contamination were removed as well as read ends were trimmed off. The filtered reads were assembled with Short Oligonucleotide Analysis Package (SOAP) de novo version 2.04 using a range of k-mer sizes (Li et al., 2009). Then GapCloser version 1.12 was used to close any internal gaps in the optimal scaffolded assembly. Repeats were predicted by RepeatMasker and Tandem Repeats Finder (TRF) tools (Rédou et al., 2016). Barrnap version 0.4.2 and tRNAscan-SE version 1.3.1 were employed to predict rRNAs and tRNAs respectively. The genome was annotated using Glimmer version 3.02 (Xu et al., 2014). The Clusters of Orthologous Groups of proteins (COG) categories were assigned to the SL 16 genome annotation using blastp (BLAST 2.2.28+) against the COG genes collection (Von Mering et al., 2005). The translations of the identified coding sequences (CDSs) were also used to search against the Protein family (Pfam) database with E-value cut-off of 1-e5. The metabolic pathway analysis was constructed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2014).
Interpretation of Data Set
Whole Genome Sequencing of E. mundtii SL 16
Large amounts of E. mundtii closely adhere to the mucosal layer of S. littoralis gut epithelium, where they form a biofilm-like structure (Figures 1A,B). Strain SL 16 displays characteristic phenotypes of E. mundtii. It grows well on Slanetz and Bartley medium (Slanetz and Bartley, 1957), producing smooth, circular, glistening colonies (Figure 1C). The bacterial cells are 0.5–1.0 μm in diameter, and occur in the form of pairs (Figure 1D). Strain SL 16 could utilize various carbon sources, including xylose, cellobiose, and sucrose (Table 1).
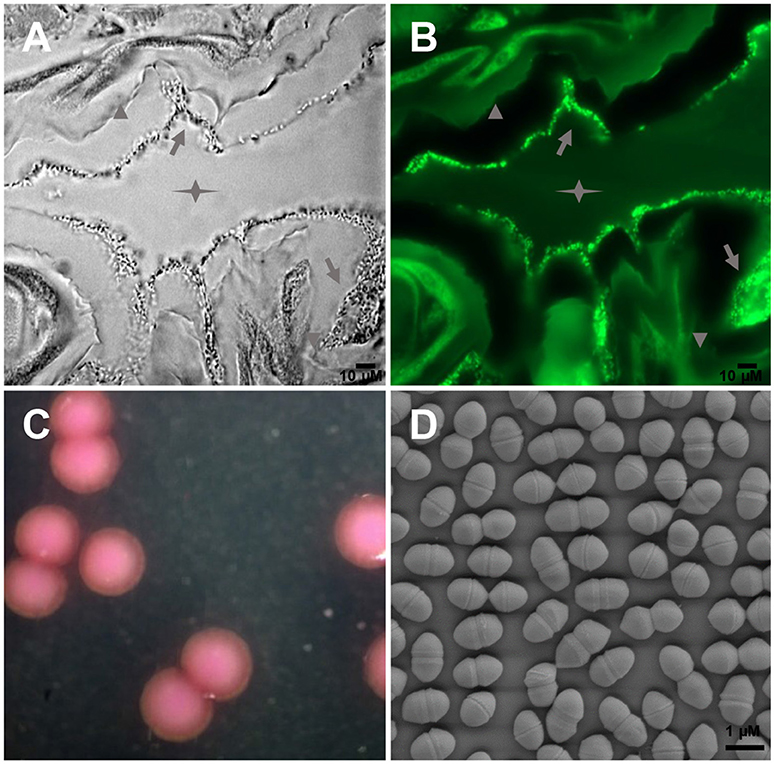
Figure 1. Images of E. mundtii from S. littoralis reveal bacterial gut localization and phenotypic characteristics. (A) Phase-contrast micrograph and (B) FISH with a FITC-labeled Enterococcus-specific probe (green) show a high density of bacterial cells adhere on the mucus layer lining the gut epithelium. Star indicates the gut lumen, arrowhead indicates the gut epithelium tissue, and arrow indicates bacteria. (C) Photomicrograph of source organism on Enterococcus selective agar. 2, 3, 5-Triphenyltetrazolium chloride (TTC) in the agar is reduced to insoluble formazan inside the bacterial cells, which gives pink or red coloration to colonies. (D) Scanning electron micrograph of E. mundtii SL 16, showing cell division.
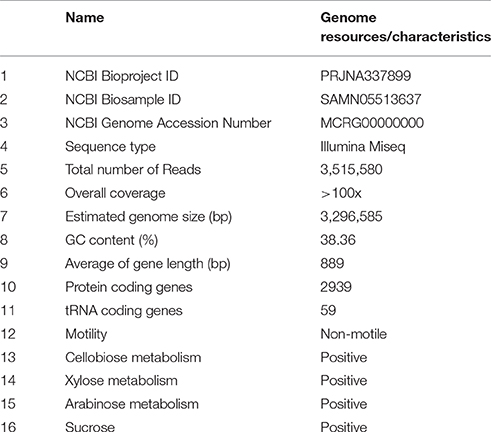
Table 1. E. mundtii SL 16 genome resources and characteristics.
Sequencing the genome of E. mundtii SL 16 produced a raw data set of 1,764,821,160 total bases. During the quality control, Illumina PCR adapter reads and low-quality reads were removed, and a total of 3,469,570 mate-pair reads (total bases 1,698,525,052 bp) were retained. The cleaned sequence reads were assembled with a k-mer setting of 125, which was determined by the optimal assembly result. The resulting genome sequence has an estimated size of 3,296,585 bp and a G+C content of 38.36%. 43,977 bp were repeats as predicted by RepeatMasker and TRF tools, which constituted 1.33% of the entire assembled genome.
A total of 3125 genes with sequence length of 2,780,928 bp were predicted, which account for 84.4% of the genome, and 59 tRNA genes were identified by tRNAscan-SE. CDSs were searched against the NR, GO, string, Swiss-Prot, COG, and KEGG databases to analyze gene functions and metabolic pathways. In all, 1493 CDSs were assigned to COG families and 1411 CDSs were included in 154 pathways. Several physiological traits that may explain the successful adaptation of this bacterium to the environment of the gut have been found. In particular, a large amount of the coding capacity encountered in the genome of SL 16 (almost 12%) is dedicated to genes assigned to functions related to carbohydrate transport and metabolism, which matches well with the observed physiological characteristics of this strain (Table 1). This feature is shared with other colonic inhabitants, such as Bacteroides fragilis (Flint et al., 2008), and reflects the ecological niche of the organism presented inside a herbivore gut. The genome encodes several ABC-type sugar transporters, sugar-binding proteins, and a rich suite of glycosyl hydrolases, such as β-N-acetylhexosaminidase, α-galactosidase, β-glucosidase, β-galactosidase, and α-glucosidase. Moreover, the pyruvate dissipation pathways predicted for SL 16 include the capacity to produce L-lactate and several other fermentation metabolites, like short-chain fatty acids formate and acetate. This metabolic flexibility is expected to aid in efficient digestion and conversion of plant saccharides, thus promoting host development.
In conclusion, here we report a 3.30 Mbp draft genome sequence of E. mundtii strain SL 16, isolated from the generalist herbivore S. littoralis. The final de novo assembly is based on 1765 Mbp of Illumina data which provides an average coverage of 535 ×. Analysis of the genome shows high correlation between the genotypes and the phenotypes.
Direct Link to Deposited Data and Information to Users
The dataset submitted to NCBI include the assembled consensus sequence of E. mundtii SL 16 in Fasta format. The genome sequence can be accessed at DDBJ/EMBL/GenBank under the accession no. MCRG00000000. This paper describes the first version of the genome (https://www.ncbi.nlm.nih.gov/nuccore/MCRG00000000).
Author Contributions
Work was planned by YS and WB, and executed jointly by BC and CS. XLi and BT were associated with isolation of the bacterium. AN and PA performed bioinformatics analyses. QG and XLu contributed to the DNA sequencing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 31601906 to YS), the “Hundred Talents Program to YS” from Zhejiang University, the Modern Agricultural Technology System (No. CARS-22-ZJ0202) and the National Natural Science Foundation of China (Grant No. 31302033 to XLu).
References
Balsingh, J., Radhakrishna, S., and Ulaganathan, K. (2016). Draft genome sequence of Bacillus pumilus ku-bf1 isolated from the gut contents of wood boring Mesomorphus sp. Front. Microbiol. 7:1037. doi: 10.3389/fmicb.2016.01037
Chen, B., Teh, B. S., Sun, C., Hu, S., Lu, X., Boland, W., et al. (2016). Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 6:29505. doi: 10.1038/srep29505
Douglas, A. E. (2015). Multiorganismal insects: diversity and function of resident microorganisms. Annu. Rev. Entomol. 60, 17–34. doi: 10.1146/annurev-ento-010814-020822
Flint, H. J., Bayer, E. A., Rincon, M. T., Lamed, R., and White, B. A. (2008). Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 6, 121–131. doi: 10.1038/nrmicro1817
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Kooij, P. W., Pullens, J. W., Boomsma, J. J., and Schiøtt, M. (2016). Ant mediated redistribution of a xyloglucanase enzyme in fungus gardens of Acromyrmex echinatior. BMC Microbiol. 16:81. doi: 10.1186/s12866-016-0697-4
Li, R., Yu, C., Li, Y., Lam, T. W., Yiu, S. M., Kristiansen, K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Manero, A., and Blanch, A. R. (1999). Identification of Enterococcus spp. with a biochemical key. Appl. Environ. Microbiol. 65, 4425–4430.
Mithöfer, A., and Boland, W. (2012). Plant defense against herbivores: chemical aspects. Annu. Rev. Plant Biol. 63, 431–450. doi: 10.1146/annurev-arplant-042110-103854
Pospiech, A., and Neumann, B. (1995). A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends Genet. 11, 217–218. doi: 10.1016/S0168-9525(00)89052-6
Rédou, V., Kumar, A., Hainaut, M., Henrissat, B., Record, E., Barbier, G., et al. (2016). Draft genome sequence of the deep-sea basidiomycetous yeast Cryptococcus sp. strain Mo29 reveals its biotechnological potential. Genome Announc. 4:e00461-16. doi: 10.1128/genomeA.00461-16
Shao, Y., Arias-Cordero, E., Guo, H., Bartram, S., and Boland, W. (2014). In vivo Pyro-SIP assessing active gut microbiota of the cotton leafworm, Spodoptera littoralis. PLoS ONE 9:e85948. doi: 10.1371/journal.pone.0085948
Shao, Y., Arias-Cordero, E. M., and Boland, W. (2013). Identification of metabolically active bacteria in the gut of the generalist Spodoptera littoralis via DNA stable isotope probing using 13C-glucose. J. Vis. Exp. e50734. doi: 10.3791/50734
Slanetz, L. W., and Bartley, C. H. (1957). Numbers of enterococci in water, sewage, and feces determined by the membrane filter technique with an improved medium. J. Bacteriol. 74, 591–595.
Tang, X., Freitak, D., Vogel, H., Ping, L., Shao, Y., Cordero, E. A., et al. (2012). Complexity and variability of gut commensal microbiota in polyphagous lepidopteran larvae. PLoS ONE 7:e36978. doi: 10.1371/journal.pone.0036978
Teh, B. S., Apel, J., Shao, Y., and Boland, W. (2016). Colonization of the intestinal tract of the polyphagous pest Spodoptera littoralis with the GFP-Tagged indigenous gut bacterium Enterococcus mundtii. Front. Microbiol. 7:928. doi: 10.3389/fmicb.2016.00928
Von Mering, C., Jensen, L. J., Snel, B., Hooper, S. D., Krupp, M., Foglierini, M., et al. (2005). STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, D433–D437. doi: 10.1093/nar/gki005
Xu, G., Hu, J., Fang, X., Zhang, X., Wang, J., Guo, Y., et al. (2014). Genome sequence of Pseudomonas aeruginosa strain LCT-PA220, which was selected after space flight by using biolog's powerful carbon source utilization technology. Genome Announc. 2:e00169–14. doi: 10.1128/genomeA.00169-14
Keywords: Enterococcus mundtii, genome sequencing, symbiosis, Spodoptera littoralis, intestinal tract
Citation: Chen B, Sun C, Liang X, Lu X, Gao Q, Alonso-Pernas P, Teh B-S, Novoselov AL, Boland W and Shao Y (2016) Draft Genome Sequence of Enterococcus mundtii SL 16, an Indigenous Gut Bacterium of the Polyphagous Pest Spodoptera littoralis. Front. Microbiol. 7:1676. doi: 10.3389/fmicb.2016.01676
Received: 26 August 2016; Accepted: 06 October 2016;
Published: 25 October 2016.
Edited by:
Malka Halpern, University of Haifa, IsraelReviewed by:
John Everett Parkinson, Oregon State University, USAPeng Bao, Institute of Urban Environment (CAS), China
Copyright © 2016 Chen, Sun, Liang, Lu, Gao, Alonso-Pernas, Teh, Novoselov, Boland and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongqi Shao, eXNoYW9Aemp1LmVkdS5jbg==
†These authors have contributed equally to this work.