 Jinzhen Jiao
Jinzhen Jiao Jian Wu1,3
Jian Wu1,3 Min Wang
Min Wang- 1Key Laboratory for Agro-Ecological Processes in Subtropical Region, Hunan Research Center of Livestock and Poultry Sciences, South Central Experimental Station of Animal Nutrition and Feed Science in the Ministry of Agriculture, Institute of Subtropical Agriculture, The Chinese Academy of Sciences, Changsha, China
- 2Hunan Co-Innovation Center of Animal Production Safety, CICAPS, Changsha, China
- 3Graduate University of Chinese Academy of Sciences, Beijing, China
The establishment of gut microbiota is increasingly recognized as a crucial action in neonatal development, host health and productivity. We hypothesized that the ileal microbiome shifted as goats matured, and this colonization process would be associated with host fermentation capacity. To this end, 18 Liuyang black grazing goats were randomly slaughtered at d 0, 7, 28, 42, and 70. Ileal microbiota was profiled by Miseq sequencing of 16S rRNA gene of bacteria, and fermentation capacity [volatile fatty acid, activities of amylase, carboxymethylcellulase (CMCase) and xylanase] was determined using digesta sample. Principal coordinate analysis revealed that each age group harbored its distinct bacteria. Total bacteria copy number and most alpha diversity indexes increased (P < 0.01) from d 0 to 70. At the phylum level, abundances of Cyanobacteria (P = 0.018) and TM7 (P = 0.010) increased linearly, abundances of Bacteroidetes (P = 0.075) and Fibrobacteres (P = 0.076) tended to increase linearly, whist Proteobacteria abundance tended to decline quadratically (P = 0.052) with age. At the genus level, Enterococcus (30.9%), Lactobacillus (32.8%), and Escherichia (2.0%) dominated at d 0, while Prevotella, Butyrivibrio, Ruminococcus, SMB53, and Fibrobacter surged in abundance after day 20. The highest amylase activity was observed at day 42, while xylanase activity increased quadratically (P = 0.002) from days 28 to 70. Correlation analysis indicated that abundances of Bacteroides, Clostridium, Lactobacillus, Propionibacterium, Enterococcus, and p-75-a5 positively correlated with enzyme activity. Collectively, ileal bacteria in grazing goats assemble into distinct communities throughout development, and might participate in the improvement of host fermentation capacity.
Introduction
The mammal intestine resides 100 trillion microorganisms, with major bacterial phyla comprising of Bacteroidetes, Firmicutes, and Proteobacteria (Donaldson et al., 2016). Many intestinal commensals can promote various physiological functions in terms of nutrition, immunity and defense in normal mammals (Round and Mazmanian, 2009; Bernalier-Donadille, 2010). Using in vitro cultivation technique, our previous studies have suggested that except for the rumen and large intestine, the ileum also serves as an indispensable fermentation site in goats (Jiao et al., 2014a,b). Furthermore, Zeng et al. (2015) proposed that sheep ileum harbored a larger number of cellulolytic bacteria, particularly Clostridium cluster IV (108 copies per gram digesta). Generally, anaerobic digestion of carbohydrate depends on a wide range of microbial groups, especially fibrolytic bacteria, and leads to the formation of volatile fatty acids (VFA), carbon dioxide (CO2), hydrogen (H2), and microbial biomass (Bernalier-Donadille, 2010).
Next generation sequencing technology has advanced our knowledge about intestinal microbial diversity and function. Using pyrosequencing, Malmuthuge et al. (2014) pointed out that in pre-weaned calves, the ileal digesta bacterial community contained primarily Firmicutes (97.7%), consisting of Lactobacillus (44.5%), Clostridium (16.7%), and Sharpea (8.9%). In goat ileum, barcoded DNA pyrosequencing revealed that the proportions of genera Acetitomaculum, Enterococcus, Atopobium, unclassified Coriobacteriaceae, and unclassified Planctomycetaceae were decreased in dietary treatment containing greater proportion of corn grain (50% vs. 25%, 0%) (Mao et al., 2013). This implies that the ileal bacterial community is diet-dependant.
During animal development process, ruminants undergo a drastic change in nutrient supply from high-fat milk diets during non-rumination phase to forage based diets during rumination phase. Since ruminants depend on rumen microbes to degrade plant lignocellulosic material, investigating colonization process of the rumen microbiome has become the research interest of several scientists. Rey et al. (2014) reported that rumen digesta bacteria community undergoes a three-stage implantation process with a progressive but important shift of composition from day 1 to weaning at day 83 of age. For goat rumen epithelial bacteria, Escherichia genus (80.79%) dominated at day 0, while Prevotella, Butyrivibrio, and Campylobacter genera surged in abundance at days 42 and 70 (Jiao et al., 2015b). Compared to considerable studies focusing on rumen microbial succession, few studies have attempted to investigate bacteria colonization process in ruminant ileum, especially in grazing animals. Therefore, the present study aimed to explore age-related changes in ileal bacterial community for grazing goats and their potential roles in host fermentation capacity.
Materials and Methods
Animals
Eighteen newly born Liuyang black goat kids (average weight of 1.35 ± 0.12 kg) were separated from the nanny, placed in individual pens, and trained to suckle milk from nipple pails. From days 0 to 20, kids were provided with goat milk. From days 20 to 40, the kids were provided with goat milk and grazed on pasture. After day 40, kids received no milk and just grazed on pasture. Three kids were slaughtered at days 0 and 7, and four kids were slaughtered at days 28, 42, and 70, respectively. Detailed feeding management, ingredients of forage (mainly twitch grass, Miscanthus) have been described in our previous parallel study (Jiao et al., 2015b). All experiments were approved and performed following the guidelines of the Animal Care Committee, Institute of Subtropical Agriculture, Chinese Academy of Sciences, Changsha, China.
Sampling Procedures
The ileal digesta were collected immediately after slaughter. One gram digesta was snap-frozen in liquid nitrogen and kept frozen at -80°C until processing for DNA extraction. Three grams digesta were homogenized with 1 mL of 25% (wt/vol) metaphosphoric acid and 6 mL distilled water and then centrifuged (17,000 × g at 4°C for 10 min), and the supernatant was stored at -20°C for VFA analysis. Three grams digesta were diluted 1:3 (wt:vol) with pre-warmed (39°C), anaerobic, sterile buffer (0.1 M citrate-phosphate buffer, pH 6.6) and stored at -20°C for assay of enzyme activity.
Chemical Analysis
Volatile fatty acids were assayed from chromatograph peak areas using calibration with external standards using a gas chromatograph (7890A, Agilent, Wilmington, DE, USA). The VFA were separated with a nitroterephthalic acid modified polyethylene glycol column (30 m × 250 um × 0.25 um) with 0.8 mL/min nitrogen gas flow rate, and detected with a flame ionization detector. Amylase, carboxymethylcellulase (CMCase), and xylanase activities were assayed according to the methods detailed by Jiao et al. (2014b), using starch, carboxymethylcellulose (CMC), and xylan (all purchased from Sigma Chemicals, St. Louis, MO, USA) as substrates, respectively. Briefly, rumen digesta samples were thawed at 4°C, and submitted to ultrasonic disintegration for three cycles for 15 s with 10-s intervals at 4°C on a Vibra CellTM sonicator (Bertin technologies, Montigny le Bretonneux, France). The samples were centrifuged (20,000 × g for 15 min at 4°C) to separate cellular debris and enzymes, and only the supernatants were used to assay enzyme activities. The enzyme reactions were initiated by addition of 0.5 mL enzyme solution into a tube containing a mixture of 1 mL substrate and 0.5 mL citrate phosphate buffer, which were prior incubated at 39°C for 10 min. The reaction times for amylase, CMCase, and xylanase were 15, 30, and 15 min, respectively. One enzyme activity unit (U) was defined as the amount of enzyme required to release 1 mmol of xylose or glucose equivalents per min per g of wet ileal content.
DNA Extraction and Quantification of Total Bacteria
The genomic DNA was extracted from the ileal digesta samples using the QIAamp DNA Stool Mini Kits (Qiagen GmbH, Hilden, Germany). Absolute quantitative real-time PCR was performed to quantify copy numbers of 16S rRNA genes of total bacteria according to the methods detailed in our previous study (Jiao et al., 2015b). The copy number of total bacteria 16SrRNA gene per gram of fresh digesta was calculated as according to the formula suggested by Zhou et al. (2009). The copy numbers were converted to log10 for further statistical analysis.
PCR and High-Throughput Sequencing
The extracted DNA was subjected to PCR amplification of the V2 and V3 region of 16S rRNA gene using universal primer 104F (5′-NNNNNN GGCGVACGGGTGAGTAA-3′) and 530R (5′-CCGCNGCNGCTGGCAC-3′) using procedures as described by Jiao et al. (2015a). The forward primer, 104F, contained 6-base barcodes represented by the italicized poly (N) section of the primer. The PCR products were purified using Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, USA). The amplicons generated for each sample was quantified using QuantiFluorTM -ST system (Promega, Madison, WI, USA), and pooled in equimolar concentrations. The 16S rRNA library was prepared according to the instruction of Nextera XT Index kits (Illumina, San Diego, CA, USA). Sequencing was conducted on an Illumina MiSeq PE300 instrument, according to the procedures of Miseq reagent kits v3 (Illumina, San Diego, CA, USA).
Data Analysis
The bacterial 16S rRNA reads were analyzed using QIIME (Caporaso et al., 2010) and mothur (Schloss et al., 2009) pipeline. Pair-end reads were deconvoluted into individual samples based on their barcodes and assembled base on their overlap sequence using Connecting Overlapped Pair-End (COPE; Liu et al., 2012). After primer trimming, the assembled sequences were then assigned to operational taxonomic units (OTUs) at 97% similarity using UPASE (Edgar, 2013). Sequence alignment was performed by PyNAST and phylogenetic tree was constructed using FastTree (Price et al., 2009). Taxonomic assignments within the latest Greengenes database (Greengenes May, 2013 release) were generated using the mothur-based implementation of the RDP Bayesian classifier with a 0.80 confidence threshold (DeSantis et al., 2006).
Alpha diversity of ileal bacterial communities of grazing kids at different ages was obtained using the alpha rarefaction pipeline. Principal coordinate analysis (PCoA) was performed using unweighted Unifrac distance. Transformation-based principal components analysis (tb-PCA) was applied to ordinate the 18 samples according to OTU abundance data (Legendre and Gallagher, 2001). Only the top 10 OTUs for which variation of abundance contributed most to the clustering obtained from the tb-PCA were selected and represented.
Statistical Analysis
A power test using GPower software was performed on the major bacteria genera (Lactobacillus, Ruminococcus and SMB53), as well as on enzyme activity. The results indicated that the within-group variation is low, with four replicates reaching a power > 0.80 when comparing two ages.
The effect of age on ileal bacterial community and fermentation parameters was investigated using the MIXED procedures of SAS (SAS Inst. Inc., Cary, NC, USA). Tukey’s test was used to compare least squares means. The model included the fixed effect of age and the random effect of animal within age. Linear and quadratic effects of age were analyzed using orthogonal contrasts. Statistical significance was accepted at P < 0.05 and a trend was considered at P < 0.10. All presented data are expressed as least-squares means.
The PROC CORR procedure of SAS was used to determine the Spearman’s rank correlations between ileal bacterial community and fermentation parameters. Only bacterial genus that represented >1% of the total community in one sample at least and that were detected in >50% of the samples were included in the analysis.
Nucleotide Sequence Accession Numbers
Sequencing data in this study were deposited in the NCBI Sequence Read Archive (SRA) under accession numbers SRR3524811 to SRR3524822, SRR3524899, SRR3524924, SRR3524946, SRR3524987, SRR3524998, and SRR3525010.
Results
Fermentation Parameters
The values for VFA and enzyme activities were not presented for the reason that they were not detected due to insufficient sample amount. Meanwhile, except for acetate other VFA (i.e., propionate and butyrate etc.) concentrations were not detectable at experimental period, which were thereby not listed in Table 1. Age exerted no effects on acetate concentration (P = 0.317, linearly) and CMCase activity (P = 1.000, linearly) (Table 1). Age exhibited quadratic effect on amylase activity (P < 0.001), while exerted quadratical increasing effect (P = 0.002) on xylanase activity from days 28 to 70.
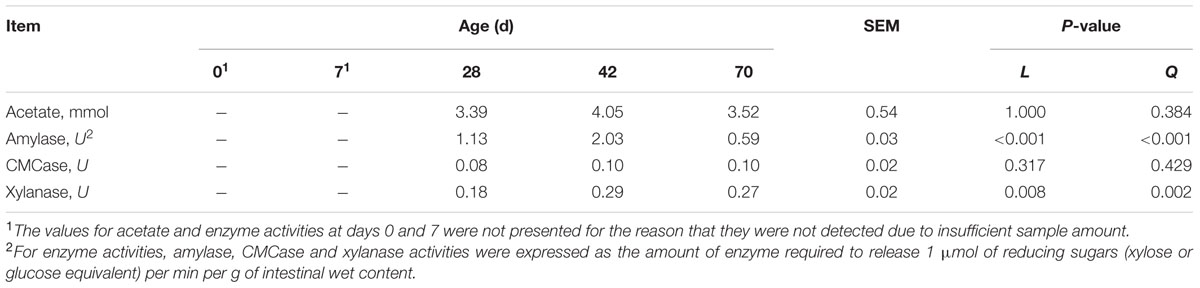
TABLE 1. Volatile fatty acid (VFA) and enzyme activities of ileal contents at different ages in grazing kids.
Total Bacteria Copy Number
Age exerted a quadratic effect (P < 0.001) on total bacteria copy number, with the greatest value observed at day 28 (Table 2). Specifically, total bacteria copy number increased sharply (almost 500 times) during the 1st week, and its value also experienced another drastic increase at day 28 (almost 100 times), followed by a decline afterward.
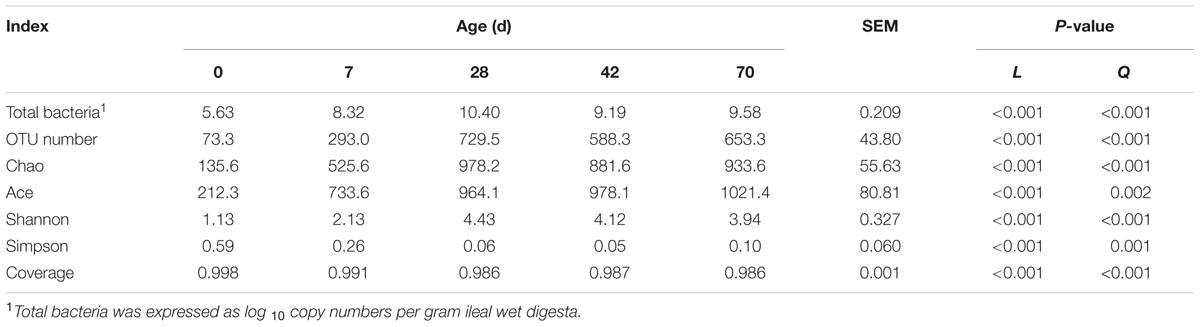
TABLE 2. Total bacteria copy number and alpha diversity measures of ileal bacterial community at different ages in grazing kids.
Sampling Depth and Alpha Diversity
Nearly 878,590 sequences were generated for ileal bacteria, with an average of 48,811 sequences for each sample (the minimum value was 16,694 and the maximum was 105,903). This yielded 3,114 representative OTUs based on 97% similarity. With subsample of 16,000 reads every sample, both the rarefaction curves (Supplementary Figure S1) and the high coverage value (from 0.986 to 0.998, Table 2) showed the sampling depth is enough. Age exhibited quadratic effect (P < 0.001) on OTU number, with the highest value detected at day 28. Chao, Ace and Shannon index increased quadratically (P < 0.001), while Simpson index and coverage declined quadratically (P < 0.001) from days 0 to 70 (Table 2).
Taxonomic Composition of Ileal Bacterial Community
Twenty-two phyla were identified, with 12 phyla abundances were <0.5%, and only a small proportion of sequences (<1%) remained unclassified at the phylum level (Table 3). Cyanobacteria (P = 0.018) and TM7 (P = 0.010) abundances increased linearly, Bacteroidetes (P = 0.075), Fibrobacteres (P = 0.076), and Tenericutes (P = 0.069) abundances tended to increase, whist Proteobacteria abundance tended to decline quadratically (P = 0.052) from days 0 to 70. Actinobacteria abundance in one sample at day 0 peaked at 9.96%, and Verrucomicrobia abundance in one sample at day 7 peaked at 11.51%, respectively. Fusobacteria was only detected at day 7 (5.47%). Firmicutes abundance fluctuated with age, whereas age had a quadratic effect on Tenericutes abundance (P = 0.007).
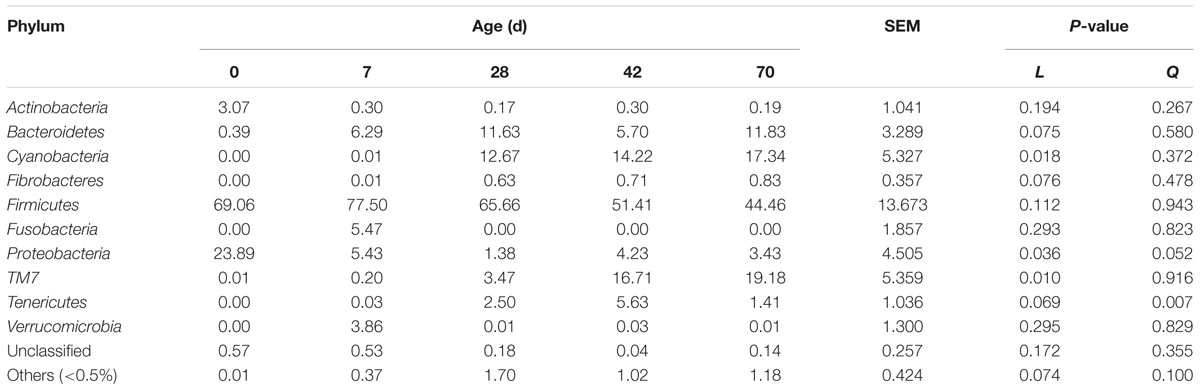
TABLE 3. Phylum level composition (% of sequences) of ileal bacterial community at different ages in grazing kids.
A great proportion of sequences (9.86 to 66.90%) remained unclassified at the genus level. Within the Bacteroidetes phylum, Bacteroides abundance declined linearly (P = 0.049), whist Prevotella abundance increased linearly (P = 0.022) from days 0 to 70 (Table 4). Within the Firmicutes phylum, abundances of Butyrivibrio, Coprococcus, Pseudobutyrivibrio, and Ruminococcus increased linearly (P < 0.05), abundances of Clostridium (P = 0.091) and Moryella (P = 0.082) tended to increase linearly, whereas Lactobacillus abundance decreased linearly (P = 0.004) from days 0 to 70. Age exerted quadratic effects on abundances of Mogibacterium (P = 0.005) and p-75-a5 (P = 0.002), and tended to have quadratic effects on abundances of Bulleidia (P = 0.095) and SMB53 (P = 0.074). Enterococcus abundance peaked at 92.09% in one sample at day 0, and the highest Oscillospira abundance were 0.83% (day 70). Within the Proteobacteria phylum, Escherichia abundance fluctuated with age. Within the Actinobacteria phylum, abundances of Bifidobacterium (0.58%) and Propionibacterium (3.11%) peaked at one sample from day 0. Moreover, age exerted quadratic effect (P = 0.020) on SHD-231 abundance, while Fibrobacter abundance tended to increase linearly (P = 0.076) from d 0 to 70. Treponema and Akkermansia abundances peaked at 0.61% (day 28) and 3.86% (day 7), respectively.
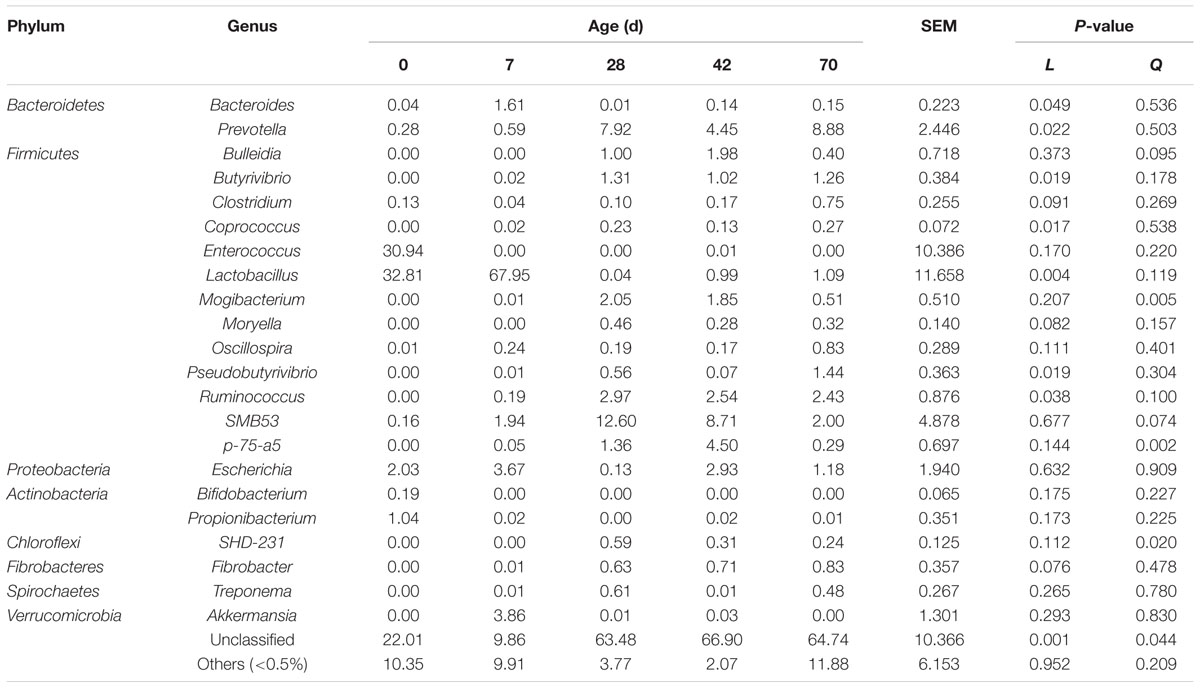
TABLE 4. Genus level composition (% of sequences) of ileal bacterial community at different ages in grazing kids.
OTU Diversity and Similarity Analysis
Principal coordinate analysis based on unweighted UniFrac distance revealed that each age group had its distinct bacteria (Figure 1). Transform-based PCA indicated that three OTUs separating day 0 from others were identified as Enterococcus genus, Halomonadaceae family and Cloacibacterium genus (Figure 2). Three OTUs discriminating day 7 from others belonged to Lactobacillus genus. Furthermore, four OTUs (Streptophyta order, F16 order and SMB53 genus) accounted for separation of days 28, 42, and 70 from others.
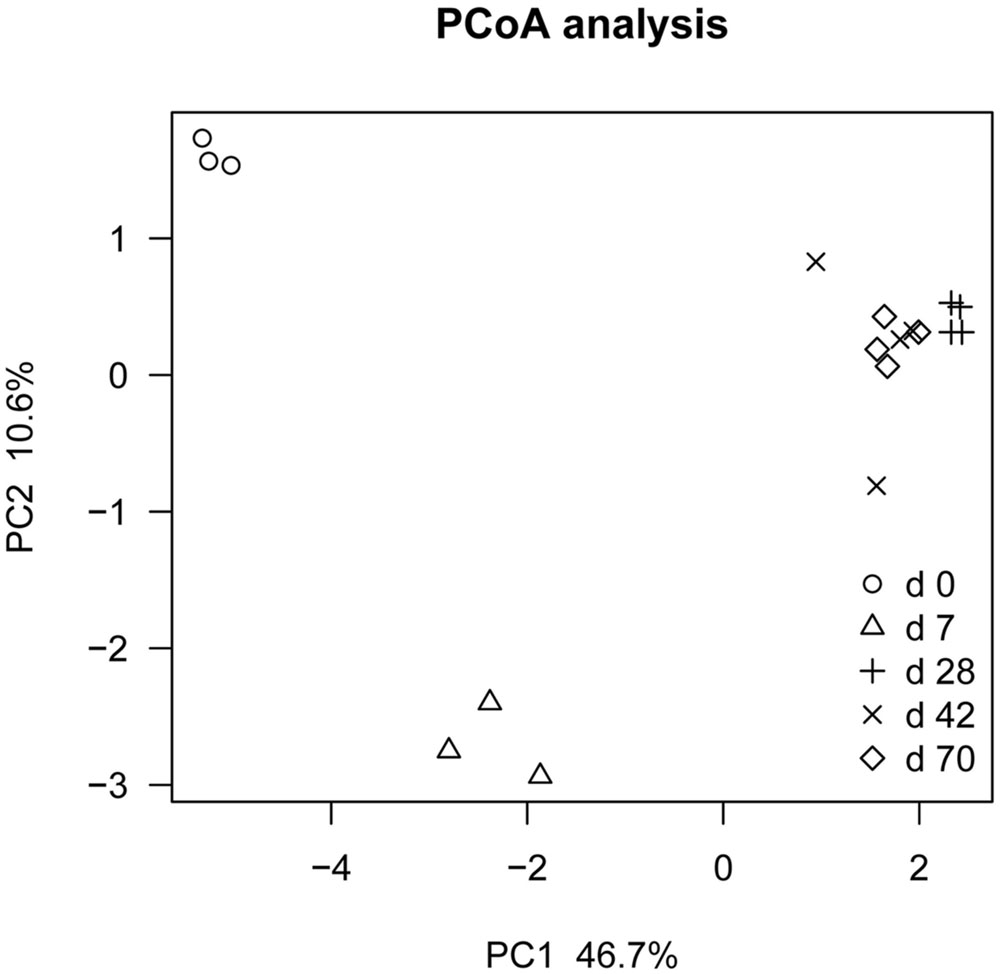
FIGURE 1. Principal co-ordinate analysis (PCoA) analysis of ileal bacterial community at different ages using Jackknifed beta diversity of 100 times resampling at a depth of 12,000 sequences in grazing kids (n = 18).
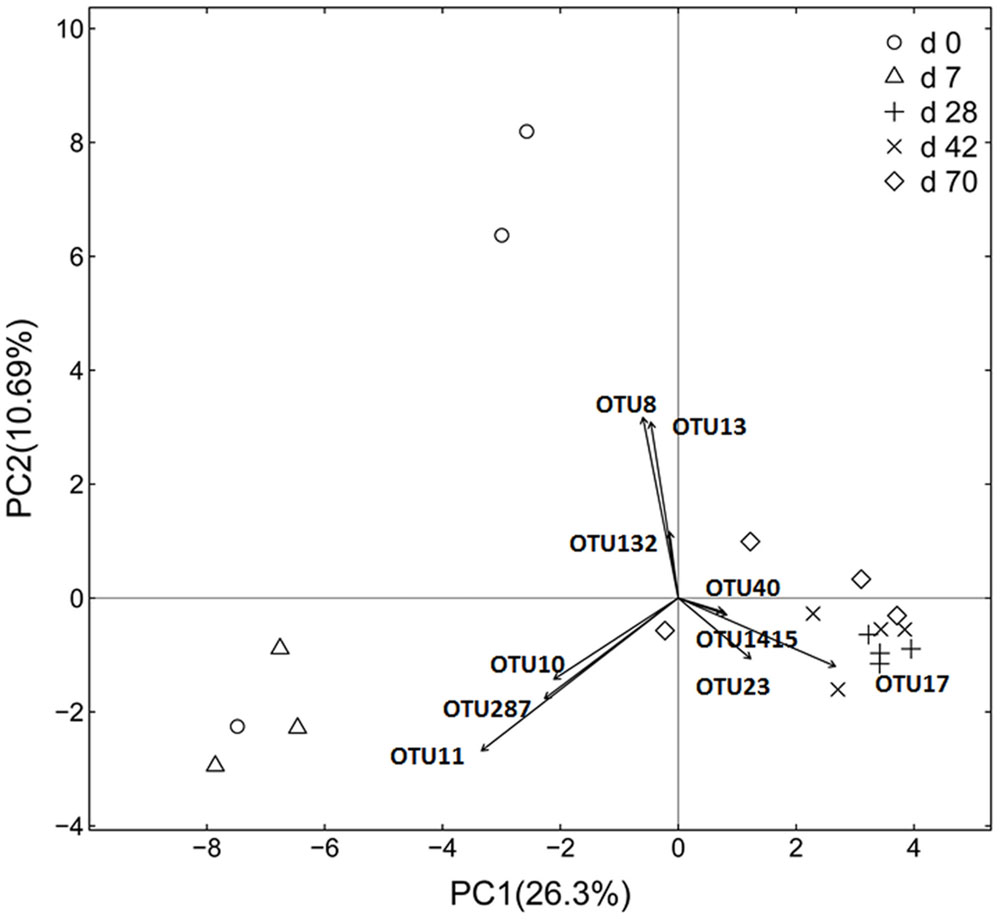
FIGURE 2. Transform-based PCA analysis of ileal bacterial community at different ages using the Hellinger distance in grazing kids (n = 18). The top 10 operational taxonomic units (OTUs) contributing to the separation of samples are displayed on the figure (OTU number). OTU11, Lactobacillus genus; OTU10, Lactobacillus genus; OTU287, Lactobacillus genus; OTU8, Enterococcus genus; OUT13, Halomonadaceae family; OTU132, Cloacibacterium family; OTU17, Streptophyta order; OTU23, SMB53 genus; OTU40, F16 family; OTU1415, F16 family.
Correlation between Bacterial Community and Fermentation Parameters
To further investigate the potential role of specific bacteria genera in the consummation of host fermentation capacity, Spearman’s rank correlations were conducted between genus proportion and fermentation parameters. As indicated in Table 5, amylase activity negatively correlated with abundances of Escherichia (R = -0.50, P < 0.05), Pseudobutyrivibrio (R = -0.55, P < 0.05) and Treponema (R = -0.61, P < 0.05), while positively correlated with p-75-a5 abundance (R = 0.79, P < 0.01). CMCase activity was positively associated with Enterococcus abundance (R = 0.50, P < 0.05), and xylanase activity was positively associated with abundances of Bacteroides (R = 0.56, P < 0.05), Clostridium (R = 0.50, P < 0.05), Lactobacillus (R = 0.50, P < 0.05), and Propionibacterium (R = 0.63, P < 0.05).
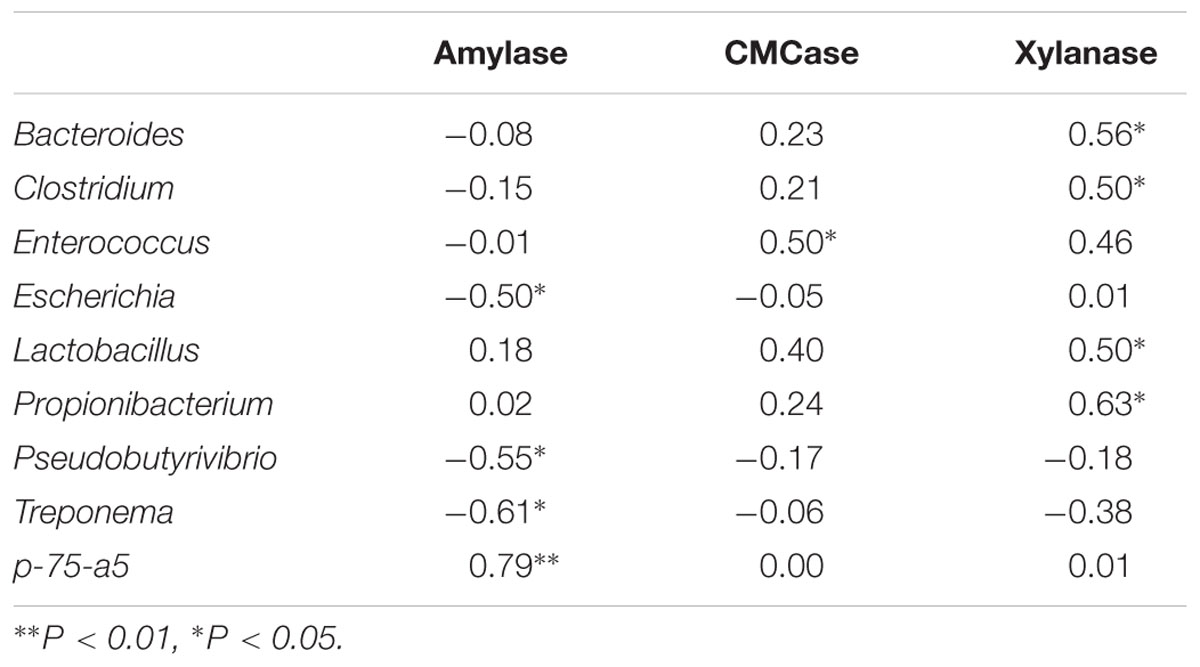
TABLE 5. Spearman’s rank correlation coefficiences between ileal bacterial community and fermentation parameters (n = 12).
Discussion
The microbial community of animal gut has been described as an additional host ‘organ.’ The synergic crosstalk between animals and microbes profoundly influence the maturation and homeostasis of adult life (Hooper and Gordon, 2001). Currently, the colonization process of ileal bacteria in grazing ruminants is poorly understood. In this study, we for the first time characterized the colonization of the ileal bacteria at five developing time points (starting from birth) of grazing goats.
Our results highlighted the notion that ileal bacteria compositions undergo stage-specific changes over development. In mammals, the diversity and quantity of gastrointestinal microbiota increase during development (Combes et al., 2011; Jami et al., 2013; Jiao et al., 2015a; Zhao et al., 2015). In goat ileum, our results demonstrated that total bacteria copy number increased with age, reaching relatively stable values after 1 month, similar to previous observation in goat rumen (Jiao et al., 2015b). PCoA revealed that each age group had its distinct microbiota. Likewise, in human (Jost et al., 2012), piglet (Niu et al., 2015), and cattle (Rey et al., 2014), previous researches indicated that diet, especially intake of breast milk and weaning could exert crucial effect on the microbial community structure. Other potentially important factors that affect the microbial colonization process include the environmental microbial exposure and host physiology (Thompson et al., 2008).
In mammal small intestine, dominant phyla are Bacteroidetes, Firmicutes, and Proteobacteria (Malmuthuge et al., 2014), but the proportion of each phylum is usually fluctuant and affected by multiple factors such as animal species, diet, age and genotype. For new born goats, total bacteria copy number was relatively low in the current study, being consistent with previous observation in calf small intestine (Malmuthuge et al., 2015). For bacteria composition, Proteobacteria phylum accounted for a great proportion (23.9%), with Escherichia genus constituted 2.0% of total sequences. This was in accordance with previous observations in pig small intestine (Zhao et al., 2015) and bovine rumen (Jami et al., 2013). Enterococcus genus also reached high proportion (31.0%) within the first day. Most members of Escherichia and Enterococcus genera are facultative anaerobic bacteria and can create a reduced environment (Franz et al., 2011; Kalita et al., 2014), allowing the succession establishment of obligate anaerobes to dominant population levels. Enterococcus comprises important lactic acid bacteria and produce moderate amounts of lactic acid, which could stimulate growth of lactic acid utilisers and stabilize gastrointestinal pH, and can be used as probiotics to improve animal health (Franz et al., 2011). Moreover, other lactic acid bacteria, Lactobacillus also accounted for a great proportion (32.8%), and these lactic acid bacteria could degrade lactose and other oligosaccharides in the milk into acetate and lactate (Walter, 2008), similar to previous trend in the rumen when goat were fed exclusively milk (Jiao et al., 2015b). Another genus needs to be taken into consideration was Bifidobacterium, which constituted 0.19% of total sequences in the ileum at day 0. Malmuthuge et al. (2015) has reported the relative higher levels of Bifidobacterium (10.4%) within 30 min after birth in neonatal calves.
At day 7, Proteobacteria declined drastically from 23.9 to 5.4%. Similarly, in dairy calf rumen, this phylum abundance decreased from 70.4% at day 2 to 16.9% between days 3 and 12 (Rey et al., 2014). Enterococcus genus declined significantly to minimal values, consistent with previous observations that Enterococcus number showed a continuous decrease from days 3 to 60 of age in the feces of young Creole goats (Draksler et al., 2002). Strikingly, Lactobacillus abundance almost doubled at day 7 vs. day 0, accounting for 68.0% of total sequences. Some cultural Lactobacillus species found in the gastrointestinal tracts (GIT) have received tremendous attention due to their health-promoting properties, especially in modulating host defenses (Walter, 2008). Moreover, the increase in abundances of two other genera, Bacteroides and Akkermansia was also observed in this study, and some members of these two were capable of producing mucin-degrading enzymes (Derrien et al., 2004; Troy and Kasper, 2010). These two genera could also be found in a relatively higher proportion in calf rumen fed exclusively milk (Jami et al., 2013).
Bacteria density increased almost one hundred times, and bacterial composition changed significantly after the introduction of forage-based diet, highlighting the role of forage diet on bacterial colonization (in both density and diversity). Prevotella genus surged in abundance while Bacteroides genus declined drastically in abundance at day 28. A similar compositional change in these two genera has been reported in the gut microbiota of children from rural Africa (De Filippo et al., 2010), which was composed of high abundance of Prevotella genus (53%) while low abundance of Bacteroides (<3%). African diet is composed mainly of plant fibre, similar to the goat diet (forage) in the present study after day 20. Another noticeable surge in abundance was observed for Ruminococcus and Fibrobacter, most of which were fibrolytic bacteria, with their abundance increased more than 10 times compared to their values at day 7. This would indicate a promotion in fiber degradation capacity (Jiao et al., 2014a). Other noticeable change lay in the drastic increase in SMB53 genus (belonging to Clostridiaceae family). Most members of this family are capable of consuming mucus- and plant-derived saccharides such as glucose in the gut (Wust et al., 2011), and the pasture forage in the present study after day 20 can provide considerable substrates for its growth.
Investigation on enzyme activities provides insights on carbohydrate digestion capacity of ileal bacteria. Xylanase activity was detected at relatively low values at day 28 and increased thereafter, highlighting the maturation of fiber digestion capacity after forage was offered. Considering cellulolytic bacteria remained high proportion at day 28, there was a lag time between enrichment of cellulolytic bacteria and improvement of fiber degradation capacity. This could be explainable that microbial attachment to fibrous substrates is an important prerequisite of fiber degradation before cellulolytic microbes start to produce enzymes (Saro et al., 2012). Furthermore, age exerted quadratic effect on amylase activity, reaching its peak at day 42. As suggested, on average, 5–20% of starch consumed is digested post-ruminally, with most of that digestion in the small intestine (Huntington, 1997). Since total bacteria number in goat ileum was relatively low, most of the starch entering the ileum should be submitted to the activity of pancreatic a-amylase (Noziere et al., 2010).
Most Bacteroidetes phylum members is specialized in the breakdown of complex plant polysaccharides (Xu et al., 2003), and some members of Clostridiaceae family are also capable of consuming plant-derived saccharides (Wust et al., 2011). Thus, it is not surprising to uncover positive associations between Bacteroides and Clostridium abundances and xylanase activity. Furthermore, positive associative relationships have also been observed between some lactic acid bacteria, such as Enterococcus, Lactobacillus (Walter, 2008), and fiber degradation enzyme activity. Together these results support the notion that a diet high in forage promotes a bacterial structure and metabolite production, which in turn, facilitates fiber digestion capacity of the host.
Inter-individual variation in microbiota is a remarkable characteristic of digestive tracts across animals’ development. In the current study, for instance, Enterococcus genus accounted for 92.1% in one sample at day 0, while its abundance in other two samples at day 0 were minor (<1%). Similar inter-individual variation has also been observed in goat and calf rumen (Jami et al., 2013; Jiao et al., 2015a), as well as zebrafish and pig intestine (Niu et al., 2015; Zac Stephens et al., 2016).
Conclusion
Both the density and diversity of the ileum bacteria in grazing goats increased with increasing age. Bacterial composition changed as goats matured, with Enterococcus, Lactobacillus, and Escherichia being dominant genera at day 0, while Prevotella, Butyrivibrio, Ruminococcus, SMB53, and Fibrobacter surged in abundance after day 20. These changes were associated with elevated xylanase activity and decreased amylase activity from days 28 to 70. To our knowledge, this is the first study in which bacteria colonization and fermentation capacity in the ileum of grazing goats was related. These findings may help in the development of strategies to guide the formation of health-promoting microbiotas that could then be maintained throughout the life of the host.
Author Contributions
JJ and ZT designed the research. JJ, JW, CZ, and MW conducted research. JJ and ST analyzed data. JJ and ZT wrote the paper. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author appreciated the joint financial support from the Open Foundation of Key Laboratory of Agro-ecological Processes in Subtropical Region, Institute of Subtropical Agriculture, Chinese Academy of Sciences (Grant No. ISA2016301), and National Natural Science Foundation of China (Grants No. 31601967, 31320103917, and 31561143009).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01364
FIGURE S1 | Rarefaction curves (number of OTUs) of ileal bacterial community at different ages in grazing kids (n = 18).
References
Bernalier-Donadille, A. (2010). Fermentative metabolism by the human gut microbiota. Gastroenterol. Clin. Biol. 34, S16–S22. doi: 10.1016/S0399-8320(10)70016-6
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Combes, S., Michelland, R. J., Monteils, V., Cauquil, L., Soulie, V., Tran, N. U., et al. (2011). Postnatal development of the rabbit caecal microbiota composition and activity. FEMS Microbiol. Ecol. 77, 680–689. doi: 10.1111/j.1574-6941.2011.01148.x
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
Derrien, M., Vaughan, E. E., Plugge, C. M., and de Vos, W. M. (2004). Akkermansia muciniphila gen. nov., sp nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 54, 1469–1476. doi: 10.1099/ijs.0.2873-0
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Donaldson, G. P., Lee, S. M., and Mazmanian, S. K. (2016). Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 14, 20–32. doi: 10.1038/nrmicro3552
Draksler, D., Locascio, M., Gonzalez, S., and Oliver, G. (2002). The development of faecal flora in young Creole goats. Small Rumin. Res. 46, 67–70. doi: 10.1016/S0921-4488(02)00162-1
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Franz, C. M., Huch, M., Abriouel, H., Holzapfel, W., and Galvez, A. (2011). Enterococci as probiotics and their implications in food safety. Int. J. Food Microbiol. 151, 125–140. doi: 10.1016/j.ijfoodmicro.2011.08.014
Hooper, L. V., and Gordon, J. I. (2001). Commensal host-bacterial relationships in the gut. Science 292, 1115–1118. doi: 10.1126/science.1058709
Huntington, G. B. (1997). Starch utilization by ruminants: from basics to the bunk. J. Anim. Sci. 75, 852–867. doi: 10.2527/1997.753852x
Jami, E., Israel, A., Kotser, A., and Mizrahi, I. (2013). Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 7, 1069–1079. doi: 10.1038/ismej.2013.2
Jiao, J., Huang, J., Zhou, C., and Tan, Z. (2015a). Taxonomic identification of ruminal epithelial bacterial diversity during rumen development in goats. Appl. Environ. Microbiol. 81, 3502–3509. doi: 10.1128/AEM.00203-15
Jiao, J., Li, X. P., Beauchemin, K. A., Tan, Z. L., Tang, S. X., and Zhou, C. S. (2015b). Rumen development process in goats as affected by supplemental feeding v. grazing: age-related anatomic development, functional achievement and microbial colonisation. Br. J. Nutr. 113, 888–900. doi: 10.1017/S0007114514004413
Jiao, J., Lu, Q., Tan, Z., Guan, L., Zhou, C., Tang, S., et al. (2014a). In vitro evaluation of effects of gut region and fiber structure on the intestinal dominant bacterial diversity and functional bacterial species. Anaerobe 28, 168–177. doi: 10.1016/j.anaerobe.2014.06.008
Jiao, J., Wang, P., He, Z., Tang, S., Zhou, C., Han, X., et al. (2014b). In vitro evaluation on neutral detergent fiber and cellulose digestion by post-ruminal microorganisms in goats. J. Sci. Food Agric. 94, 1745–1752. doi: 10.1002/jsfa.6485
Jost, T., Lacroix, C., Braegger, C. P., and Chassard, C. (2012). New insights in gut microbiota establishment in healthy breast fed neonates. PLoS ONE 7:e44595. doi: 10.1371/journal.pone.0044595
Kalita, A., Hu, J., and Torres, A. G. (2014). Recent advances in adherence and invasion of pathogenic Escherichia coli. Curr. Opin. Infect. Dis. 27, 459–464. doi: 10.1097/qco.0000000000000092
Legendre, P., and Gallagher, E. (2001). Ecologically meaningful transformations for ordination of species data. Oecologia 129, 271–280. doi: 10.1007/s004420100716
Liu, B., Yuan, J., Yiu, S. M., Li, Z., Xie, Y., Chen, Y., et al. (2012). COPE: an accurate k-mer-based pair-end reads connection tool to facilitate genome assembly. Bioinformatics 28, 2870–2874. doi: 10.1093/bioinformatics/bts563
Malmuthuge, N., Chen, Y., Liang, G., Goonewardene, L. A., and Guan le, L. (2015). Heat-treated colostrum feeding promotes beneficial bacteria colonization in the small intestine of neonatal calves. J. Dairy Sci. 98, 8044–8053. doi: 10.3168/jds.2015-9607
Malmuthuge, N., Griebel, P. J., and Guan le, L. (2014). Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl. Environ. Microbiol. 80, 2021–2028. doi: 10.1128/AEM.03864-13
Mao, S., Huo, W., and Zhu, W. (2013). Use of pyrosequencing to characterize the microbiota in the ileum of goats fed with increasing proportion of dietary grain. Curr. Microbiol. 67, 341–350. doi: 10.1007/s00284-013-0371-0
Niu, Q., Li, P., Hao, S., Zhang, Y., Kim, S. W., Li, H., et al. (2015). Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci. Rep. 5:9938. doi: 10.1038/srep09938
Noziere, P., Ortigues-Marty, I., Loncke, C., and Sauvant, D. (2010). Carbohydrate quantitative digestion and absorption in ruminants: from feed starch and fibre to nutrients available for tissues. Animal 4, 1057–1074. doi: 10.1017/S1751731110000844
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Rey, M., Enjalbert, F., Combes, S., Cauquil, L., Bouchez, O., and Monteils, V. (2014). Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 116, 245–257. doi: 10.1111/jam.12405
Round, J. L., and Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323. doi: 10.1038/nri2515
Saro, C., Ranilla, M. J., and Carro, M. D. (2012). Postprandial changes of fiber-degrading microbes in the rumen of sheep fed diets varying in type of forage as monitored by real-time PCR and automated ribosomal intergenic spacer analysis. J. Anim. Sci. 90, 4487–4494. doi: 10.2527/jas.2012-5265
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Thompson, C. L., Wang, B., and Holmes, A. J. (2008). The immediate environment during postnatal development has long-term impact on gut community structure in pigs. ISME J. 2, 739–748. doi: 10.1038/ismej.2008.29
Troy, E. B., and Kasper, D. L. (2010). Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Front. Biosci. 15:25–34. doi: 10.2741/3603
Walter, J. (2008). Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl. Environ. Microbiol. 74, 4985–4996. doi: 10.1128/AEM.00753-08
Wust, P. K., Horn, M. A., and Drake, H. L. (2011). Clostridiaceae and Enterobacteriaceae as active fermenters in earthworm gut content. ISME J. 5, 92–106. doi: 10.1038/ismej.2010.99
Xu, J., Bjursell, M. K., Himrod, J., Deng, S., Carmichael, L. K., Chiang, H. C., et al. (2003). A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299, 2074–2076. doi: 10.1126/science.1080029
Zac Stephens, W., Burns, A. R., Stagaman, K., Wong, S., Rawls, J. F., Guillemin, K., et al. (2016). The composition of the zebrafish intestinal microbial community varies across development. ISME J. 10, 644–654. doi: 10.1038/ismej.2015.140
Zeng, Y., Zeng, D., Zhang, Y., Ni, X. Q., Tang, Y. R., Zhu, H., et al. (2015). Characterization of the cellulolytic bacteria communities along the gastrointestinal tract of Chinese Mongolian sheep by using PCR-DGGE and real-time PCR analysis. World J. Microbiol. Biotechnol. 31, 1103–1113. doi: 10.1007/s11274-015-1860-z
Zhao, W., Wang, Y., Liu, S., Huang, J., Zhai, Z., He, C., et al. (2015). The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 10:e0117441. doi: 10.1371/journal.pone.0117441
Keywords: bacterial colonization, fermentation capacity, ileum, age, goats
Citation: Jiao J, Wu J, Zhou C, Tang S, Wang M and Tan Z (2016) Composition of Ileal Bacterial Community in Grazing Goats Varies across Non-rumination, Transition and Rumination Stages of Life. Front. Microbiol. 7:1364. doi: 10.3389/fmicb.2016.01364
Received: 22 June 2016; Accepted: 17 August 2016;
Published: 05 September 2016.
Edited by:
Dimitrios Georgios Karpouzas, University of Thessaly, GreeceReviewed by:
Timothy Hackmann, University of Florida, USACharles James Newbold, Aberystwyth University, UK
Copyright © 2016 Jiao, Wu, Zhou, Tang, Wang and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiliang Tan, zltan@isa.ac.cn