
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 19 August 2016
Sec. Microbiotechnology
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.01303
 Mathilde Jeanbille1*
Mathilde Jeanbille1* Jérôme Gury1
Jérôme Gury1 Robert Duran1
Robert Duran1 Jacek Tronczynski2
Jacek Tronczynski2 Jean-François Ghiglione3
Jean-François Ghiglione3 Hélène Agogué4Olfa Ben Saïd5Najwa Taïb6
Hélène Agogué4Olfa Ben Saïd5Najwa Taïb6 Didier Debroas6
Didier Debroas6 Cédric Garnier7
Cédric Garnier7 Jean-Christophe Auguet8
Jean-Christophe Auguet8Benthic microorganisms are key players in the recycling of organic matter and recalcitrant compounds such as polyaromatic hydrocarbons (PAHs) in coastal sediments. Despite their ecological importance, the response of microbial communities to chronic PAH pollution, one of the major threats to coastal ecosystems, has received very little attention. In one of the largest surveys performed so far on coastal sediments, the diversity and composition of microbial communities inhabiting both chronically contaminated and non-contaminated coastal sediments were investigated using high-throughput sequencing on the 18S and 16S rRNA genes. Prokaryotic alpha-diversity showed significant association with salinity, temperature, and organic carbon content. The effect of particle size distribution was strong on eukaryotic diversity. Similarly to alpha-diversity, beta-diversity patterns were strongly influenced by the environmental filter, while PAHs had no influence on the prokaryotic community structure and a weak impact on the eukaryotic community structure at the continental scale. However, at the regional scale, PAHs became the main driver shaping the structure of bacterial and eukaryotic communities. These patterns were not found for PICRUSt predicted prokaryotic functions, thus indicating some degree of functional redundancy. Eukaryotes presented a greater potential for their use as PAH contamination biomarkers, owing to their stronger response at both regional and continental scales.
Coastal ecosystems ecologically support numerous human activities (e.g., fishing, aquaculture, tourism, urban development, transport and refining of oil, industrial activities, etc.), which exert considerable anthropogenic pressure, potentially leading to the erosion of ecosystem health (Halpern et al., 2008; Borja et al., 2011). Hydrocarbon pollution constitutes the most significant threat, as it is estimated to represent worldwide between 1.3 and 8.8 million tons of discharge per year (National Research Council (US) Committee on Oil in the Sea, 2003). The ecological, economic and social repercussions of oil spills, such as the recent Deepwater Horizon oil spill in the Gulf of Mexico, are considerable. However, estimates of oil contamination show that oil spills are quantitatively less important than chronic pollution. For instance, they represent less than 30% of total input into the Mediterranean Sea (European Environment Agency, 2006). Additionally, coastal ecosystems receive significant and continuous inputs of pyrogenic hydrocarbons generated by combustion of fossil fuels (coal and oil) and other organic material such as wood (Page et al., 1999; Ravindra et al., 2008).
A huge diversity and density of Bacteria (Lozupone and Knight, 2007) and Archaea (Auguet et al., 2009), as well as a rich eukaryotic microfauna and meiofauna (Creer et al., 2010; Pernice et al., 2013), inhabit the marine sediment. Both prokaryotes and eukaryotes play pivotal roles in marine habitats. They support major ecosystem services like nutrient and organic matter cycling (Danovaro et al., 2008; Falkowski et al., 2008) and organic pollutant biodegradation, particularly hydrocarbons (Leahy and Colwell, 1990; Widdel and Rabus, 2001; Pérez-Pantoja et al., 2010; Coulon et al., 2012). The response of sediment microbial communities to acute PAH pollution events is very consistent across studies and usually characterized by a decrease in alpha-diversity due to the proliferation of hydrocarbonoclastic organisms (Head et al., 2006; Nogales et al., 2011; Kimes et al., 2014; King et al., 2015). However, the response to chronic pollution is largely unknown. It has recently been shown that pollution history influenced the response of coastal bacterial and nanoeukaryote communities to pollution (Sauret et al., 2012). Chronic pollution seems to lead to higher bacterial diversity, as a result of induced stability caused by long-term exposure (Nogales et al., 2007; Païssé et al., 2008; Zhang et al., 2008). However, several recent studies have shown a reduction of bacterial alpha-diversity in relation to elevated PAH concentrations (Rosano-Hernández et al., 2012; Sun et al., 2012, 2013; Korlević et al., 2015; Quero et al., 2015). Deltaproteobacteria and Gammaproteobacteria, which include several hydrocarbon degraders, are found to be dominant and recurrent in polluted and non-polluted sediments from coastal areas (Païssé et al., 2008; Zhang et al., 2008; Said et al., 2010; Sun et al., 2013; Quero et al., 2015). Compared to Bacteria, the response of Archaea to hydrocarbon contamination has received little attention (Röling et al., 2004; Dias et al., 2011; Stauffert et al., 2014; Sanni et al., 2015). Similarly, few studies have focused on the response of eukaryotic communities to sediment contamination by PAHs. Both fungi and algae play an important role in hydrocarbon degradation (Head et al., 2006; Prince, 2010; Coulon et al., 2012). For instance, fungi dominated the eukaryotic community in response to the Deepwater Horizon plume (Bik et al., 2012), indicating that benthic eukaryotic diversity and richness may be sensitive to hydrocarbon pollution (Johnston and Roberts, 2009). However, Chariton et al. (2010) showed that chronic pollution by hydrocarbons affected beta-diversity rather than the richness of both microfauna and meiofauna in estuarine sediments.
Understanding the behavior of highly diverse sediment communities in a complex habitat, where ecological, historical and anthropogenic processes operate at multiple spatial scales, is a complex task. To address this problem, we adopted a macro-ecological point of view and used a holistic approach in order to investigate the continental and regional patterns of communities from the three domains of life, in relation to hydrocarbon contaminations in coastal sediments from the North East Atlantic and Mediterranean regions. Based on in silico functional diversity inference, we additionally examined the relationship between prokaryotic functions and sediment contamination by PAHs.
Sediments were sampled at 46 sites across different geographical areas in order to be representative of the heterogeneity of the environment along the Atlantic and the Mediterranean coasts (Figure 1). Within each area, samples were collected from a priori PAH-contaminated and a priori uncontaminated sites according to site observations and the vicinities of potential hydrocarbon input. Among the polluted sampling sites, most samples were collected near to urbanized areas chronically impacted by shipping and industrial activities: Le Havre (samples MA1 and MA2), Lorient (GC1 and GC2), La Rochelle (LR1), Port-Vendres (PV1–PV3), Thau lagoon (M5 and M6), Toulon (M3, M4, T12, and T15), Beirut (L1 and L2), Bizerte (BI1), and Ajaccio (M1 and M2) (Figure 1). Certain a priori contaminated samples were also subjected to chronic pollution due to recreational shipping activities (BA1–BA4, Banyuls-sur-mer and GC1, Quiberon). Finally, sample GC6 was collected after the Erika oil spill (July 2000), which occurred in December 1999, while sample GC7 was collected before the Erika oil spill in August 1999 at the same sampling location. Coastal sediment samples were all taken from the surface sediment layer (1–2 cm), using a common sampling box corer. Toulon Bay samples (T12, T15, T23, and T52) have been described in detail by Tessier et al. (2011). The samples were frozen at –80°C within 1 day after sampling. Sediment particle size distribution was determined using laser diffraction, according to Tronczynski (2005) and Tessier et al. (2011). Total organic carbon (TOC) analyses were performed according to Tessier et al. (2011) and Azoury et al. (2013). TOC percentages (%TOC) and particle size distribution (% >63 μm; PSD) were available together for 32 of the 46 samples (Supplementary Table 1). Polyaromatic hydrocarbon (PAH) quantifications were made using gas chromatography coupled to mass spectrometry (GCMS), as described by Azoury et al. (2013) and Stauffert et al. (2013). In order to take the PAH partitioning and consequently the PAH bioavailability, into account in the sediment matrices, PAH concentrations were normalized to 1% of TOC (Burgess et al., 2000; Wenning, 2005). PAH isomeric ratios (or diagnostic ratios), Phenanthrene/Anthracene (P/A) and Fluoranthene/Pyrene (F/P) were calculated in order to determine the bulk origin of the PAH contamination in sediment samples. Anthracene and fluoranthene are produced during pyrosynthesis, and are less thermodynamically stable. Furthermore, the isomeres (i.e., same molecular formula but different chemical structure) of anthracene and fluoranthene, phenanthrene and pyrene, respectively, are rather formed during the catagenesis of organic matter leading to petroleum formation. P/A ratio values below 10 indicate a pyrogenic origin and F/P ratio values above 1 indicate pyrogenic sources of PAHs (Budzinski et al., 1997).
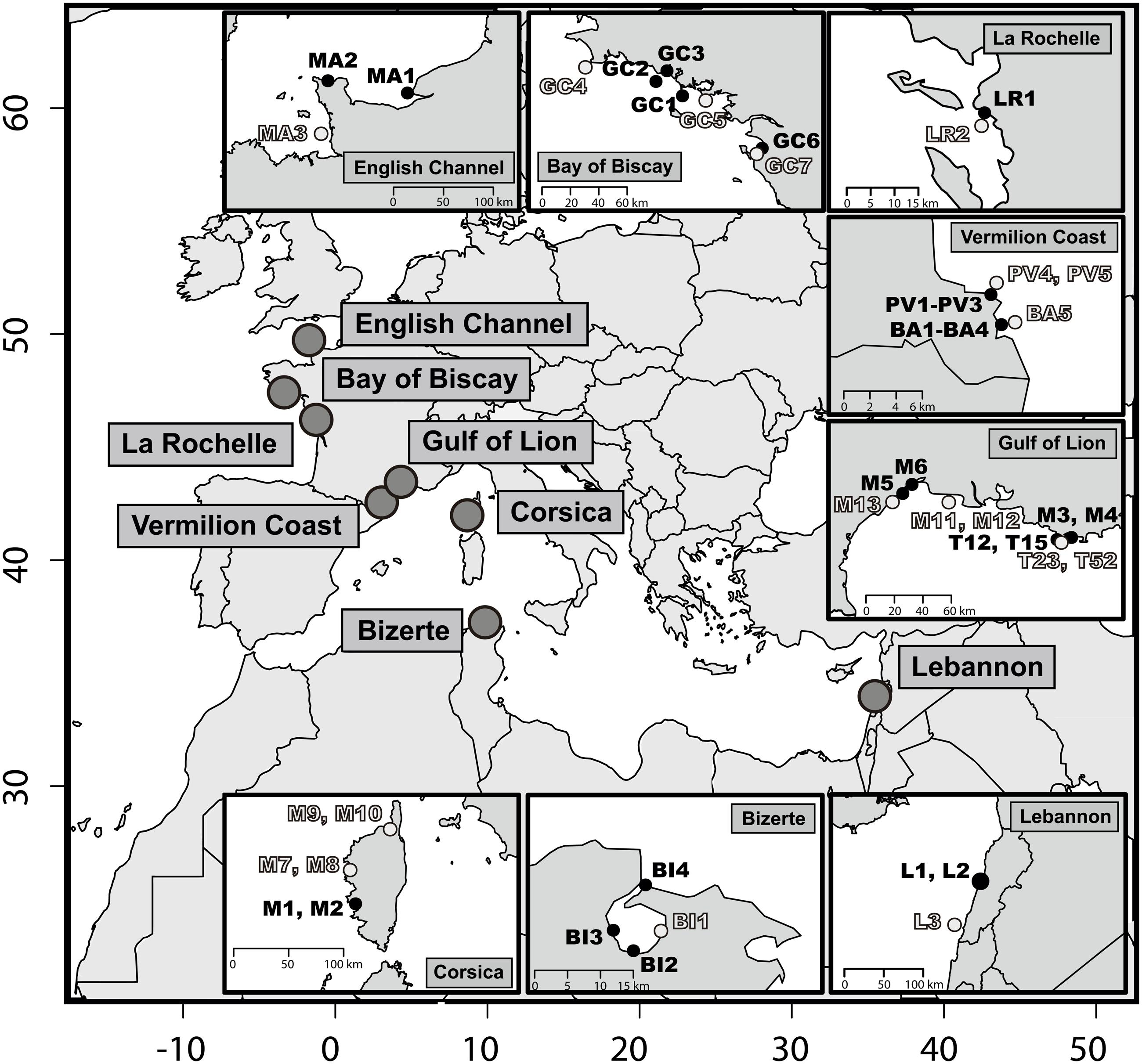
FIGURE 1. Map of the 46 sediments sampling stations. At each location a priori non-contaminated sites are in light gray and a priori contaminated sites in black. The x-axis and y-axis stand for decimal longitude and latitude, respectively.
Total genomic DNA (gDNA) was extracted from 250 mg of sediment samples using the PowerSoil DNA isolation kit (MO-BIO Laboratories Inc.). PCR amplifications were performed using primers 27f and 519r targeting the V1–V3 region of the bacterial 16S rRNA gene (Weisburg et al., 1991), primers 344f and 915r targeting the V3–V4 region of the archaeal 16S rRNA gene (Raskin et al., 1994) and primers 1560f and 2035r targeting the VR4 region of the eukaryotic 18S rRNA gene (Hardy et al., 2010). Two and half microliters of gDNA extract was used in a PCR reaction containing 4 mM MgCl2, 0.8 mM of dNTPs, 0.15 mM of each primer, and 1U of AmpliTaq Gold 360 DNA Polymerase (ThermoFisher Scientific), in a final volume of 30 μl. PCR was performed with initial denaturation at 95°C for 10 min, followed by 30 cycles at 95°C for 30 s, 53°C for 45 s and 72°C for 105 s, with a final extension of 72°C for 7 min. Pyrosequencing was performed at MR DNA - Molecular Research (Shallowater, TX, USA), using a Roche 454 GS-FLX Titanium instrument (Roche, NJ, USA). Pyrosequencing resulted in 693687, 290816, and 110044 raw reads for bacterial, archaeal and eukaryotic datasets, respectively (Supplementary Table 2). Raw reads from the 3 datasets were independently quality-based, trimmed and aligned on the May 2013 Greengenes reference alignment for prokaryotes (McDonald et al., 2012) and SILVA 119 database for eukaryotes (Quast et al., 2013). Operationnal taxonomic units (OTUs) were clustered at a 96% (Bacteria), 97% (Archaea), and 90% (Eukarya) identity threshold following the Mothur 454 Standard Operating Procedure (Schloss et al., 2009) with a few modifications. Bacterial OTUs were clustered at 96% identity threshold in order to obtain an unbiased richness compared to the full-length 16S rRNA gene (Kim et al., 2011). As the mean length of 18S rRNA gene sequences was 153 bp after trimming, taxonomic affiliations could not be defined at deep taxonomic levels for Eukarya, and eukaryotic OTUs were thus clustered at a 90% identity threshold. Eukaryotic taxonomic affiliations were determined as described by Taib et al. (2013). Sequence clustering resulted in 23280 bacterial OTUs, 4633 archaeal OTUs, and 1571 eukaryotic OTUs. All statistical analyses were performed on a random subsample of 3238, 829, and 804 sequences for bacterial, archeal and eukaryotic datasets, respectively, corresponding to the smaller number of sequences per sample in the datasets, after trimming and quality processing. The eukaryotic dataset encompassed 42 samples, and both prokaryotic datasets 46 samples. The complete data set was deposited in the NCBI Sequence Read Archive (SRA) database under study Accession no SRP063723.
Using ARB software (Ludwig et al., 2004), representative sequences of each prokaryotic OTU were aligned on the Greengenes ARB database (May 2013) and eukaryotic representative sequences were aligned on the 119 non-redundant releases in of the SILVA ARB database. Resulting phylogenetic trees were exported and used to run UniFrac weighted matrix calculation (Lozupone et al., 2006). Richness was computed using the R package phyloseq (McMurdie and Holmes, 2013). The mean phylogenetic diversity (PD) of 1000 random subsamples of each sample and phylogenetic species variability (PSV) were calculated with R.
Predictions of metagenomic functions for Bacteria and Archaea were performed using a bioinformatic tool that predicts gene family abundances based on 16S rRNA gene surveys, using a database of phylogenetically referenced genomes (PICRUSt, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States, Langille et al., 2013). PICRUSt is based on the correlation between phylogeny and functions and uses the phylogenetic proximity between public genomes and 16S rRNA OTUs. The first stage of the PICRUSt pipeline is pre-computed and consists of building a table of predicted KEGG (Kyoto Encyclopedia of Genes and Genomes; Kanehisa and Goto, 2000) Orthology (KO) counts of every taxon in the 16S reference tree, which also contains taxa with genomic data. The second stage consists of (i) normalizing the 16S OTU table by the predicted 16S rRNA gene copy number per cell, which is pre-computed in PICRUSt according to reference archaeal and bacterial genomes; and (ii) predicting metagenomic data from a biom table, which included taxonomic data from the 2013 issue of the Greengenes database and OTU table, based on the previous pre-computed files. Nearest Taxon Index (NSTI) scores, which are computed with metagenome prediction, are an indicator of the accuracy of the predictions, with lower values indicating a better accuracy. NSTIvalues were 0.19 ± 0.04 for Bacteria and 0.22 ± 0.07 for Archaea (mean ± SD, n = 46). These ranges of values are in agreement with values found for environmental samples (e.g., soil and hypersaline mats; Langille et al., 2013), indicating highly diverse communities and a lack of reference genomes for Archaea. Metagenomic prediction resulted in KO counts for each sample. Inferred KO counts were then grouped into KEGG PATHWAY maps (KEGG pathways). KEGG pathways unrelated to prokaryotic functions were manually discarded. KEGG pathways present in the datasets related to hydrocarbon biodegradation (n = 14 for Bacteria and n = 12 for Archaea) and related to organic matter metabolism and biosynthesis (n = 36) were sorted by hand according to their KEGG identification.
Although several studies have demonstrated congruencies between 16S rRNA based and functional gene phylogenies (Langille et al., 2013; Martiny et al., 2013; Aßhauer et al., 2015), we are aware that prediction approaches cannot replace whole metagenome or metatranscriptome profiling. The functional predictions made in this work are therefore considered only as an indication of the functional potential held in each community and not the ground-truth.
The similarity of samples according to PAH profiles was assessed by calculating Euclidean distances based on 11 PAH concentrations and plotted as a dendrogram based on a hierarchical classification analysis (HCA, Supplmentary Figure 2). The Wilcoxon-Mann-Whitney U test or Student’s t-Test was used to compare mean total PAH concentrations between contaminated and non-contaminated samples from each geographic region.
Stepwise multiple regressions were performed on 32 samples, in order to investigate the relationships between alpha-diversity indices and environmental variables (concentrations of 11 individual PAHs, fluoranthene/pyrene and anthracene/phenanthrene ratios, temperature, salinity, and temperature of the water column, %TOC and PSD). Collinearities in the independent variables were tested before running the stepwise multiple regressions. Variables with collinearity up to 0.75 according to Spearman correlations (p < 0.05) were grouped together, and proxies of each group were used as explanatory variables (Supplementary Table 3). The PAH proxy concentration (i.e., dibenz[a,h]anthracene concentration) was log transformed, and the F/P ratio was squared transformed to improve the linearity and homoscedasticity of residuals, which were evaluated using the Shapiro-Wilk and Barlett tests, respectively. All statistical analyses were performed with R software.
Community similarity was represented by non-metric multidimensionnal scaling (NMDS) using the weighted UniFrac distance for data relating to 16S and 18S rRNA genes data and the Bray-Curtis dissimilarity for data relating to inferred KEGG pathways data (i.e., the whole prokaryotic dataset, hydrocarbons or organic matter related pathways). The phylogenetic structure was evaluated with the PSV index for each study (Helmus et al., 2007). PSV estimates PD as the variance of a trait evolving under a neutral model. The value is 1 when all species are phylogenetically unrelated (i.e., a star phylogeny) and approaches 0 as species become more related. To test statistically whether communities were composed of species that were related to each other to a greater or a lesser degree than expected, we compared the mean observed PSV with distributions of mean null values (1000 iterations) using two different randomization procedures. Null model 1 maintains species occurrence, whereas null model 2 maintains habitat species richness (Helmus et al., 2007). All these analyses were run with the R package picante (Kembel et al., 2010).
Mantel tests based on 9999 permutations were performed in order to test the correlations between Bray-Curtis matrices based on KEGG pathway composition, and UniFrac matrices. To assess the sources of variation (i.e., PAH concentrations, P/A and F/P ratios, latitude, PSD, %TOC, temperature and salinity) in both the phylogenetic and the functional distance matrix of the continental (i.e., Mediterranean and Atlantic data) and regional (i.e., Mediterranean or Atlantic data) datasets, we used a permutational multivariate analysis of variance (PerMANOVA) based on 1000 permutations (McArdle and Anderson, 2001) with the function adonis in the vegan package (Oksanen et al., 2013). Using the envfit function of the same package, significant environmental variables were fitted to the NMDS ordinations as vectors.
Biomarkers of environmental variation and hydrocarbon contamination were detected using the LEfSe algorithm (Segata et al., 2011). The first analysis step is a non-parametric Kruskal-Wallis (KW) sum-rank test allowing the detection of taxa with significant differential abundance. Biological consistency was subsequently investigated using a pairwise Wilcoxon test. Finally, Linear Discriminant Analysis (LDA) was used to estimate the effect size of each differentially abundant taxon. Alpha Values of 0.05 were used for KW and Wilcoxon tests and a threshold of 2.0 was used for logarithmic LDA scores. Significant differences between hydrocarbon-related KEGG pathways of contaminated and non-contaminated samples were tested using the t-test or the Wilcoxon test depending on the value distribution (i.e., normality of the data).
Salinity was higher at the Mediterranean sites (37.8 ± 0.8 g/l) than at the Atlantic sites (35.5 ± 2.2 g/l), and was highly correlated with latitude (rho = –0.87, p-value <0.001). Average temperature, PSD and %TOC were also significantly higher at the Mediterranean sites than at the Atlantic sites (Supplementary Figure 1). Within each geographic region, samples were collected in a priori contaminated areas like marinas and harbors, and in a priori uncontaminated areas (i.e., defined as uncontaminated prior to sampling). The concentrations of the 11 PAHs measured in this work were above detection limits for 44 of the 46 coastal sediment samples (Supplementary Table 1). As the Atlantic and Mediterranean sites were significantly different in terms of salinity, temperature, %TOC and PSD (Supplementary Figure 1), we analyzed the PAH gradient independently for each region. A HCA performed on the PAH concentrations showed that Mediterranean, and to a lesser extent Atlantic, sites were well separated according to our a priori contamination criterion (Supplementary Figure 2). PAH concentrations were significantly correlated (rho > 0.75, p < 0.05, Supplementary Table 3), allowing the calculation of the total PAH concentration for each sample and its comparison to the PAH sediment quality guidelines (SQGs) developed by Buchman (2008) (Figure 2): Probable Effect Level (PEL), Effect Range Low (ERL) and T50 (50% Toxicity Concentration). SQGs were available for 9 PAHs (fluoranthene, pyrene, anthracene, benz[a]anthracene, benzo[ghi]perylene, chrysene, dibenz[a,h]anthracene, fluorene, indeno[1,2,3-cd]pyrene, phenanthrene) among the concentrations of 11 PAHs measured for the whole sample set. The sum of these 9 PAHs (Σ PAH9) and the sum of the corresponding SQGs are shown in Figure 2. For both geographic regions, mean PAH concentrations were significantly higher in contaminated samples (p < 0.05), but the difference in PAH concentrations between non-contaminated and contaminated samples was much higher in the Mediterranean Sea (Figure 2), thus corroborating the results of the HCA. The mean of Σ PAH9 concentrations in highly contaminated samples from the Mediterranean Sea was significantly higher than the most conservative SQG with a value of 6483 μg.kg-1 (dry weight sediment; p < 0.05). In contrast, mean PAH concentrations in both non-contaminated and contaminated Atlantic samples were between PEL and ERL values, and consequently could not be distinguished regarding SQGs (Figure 2).
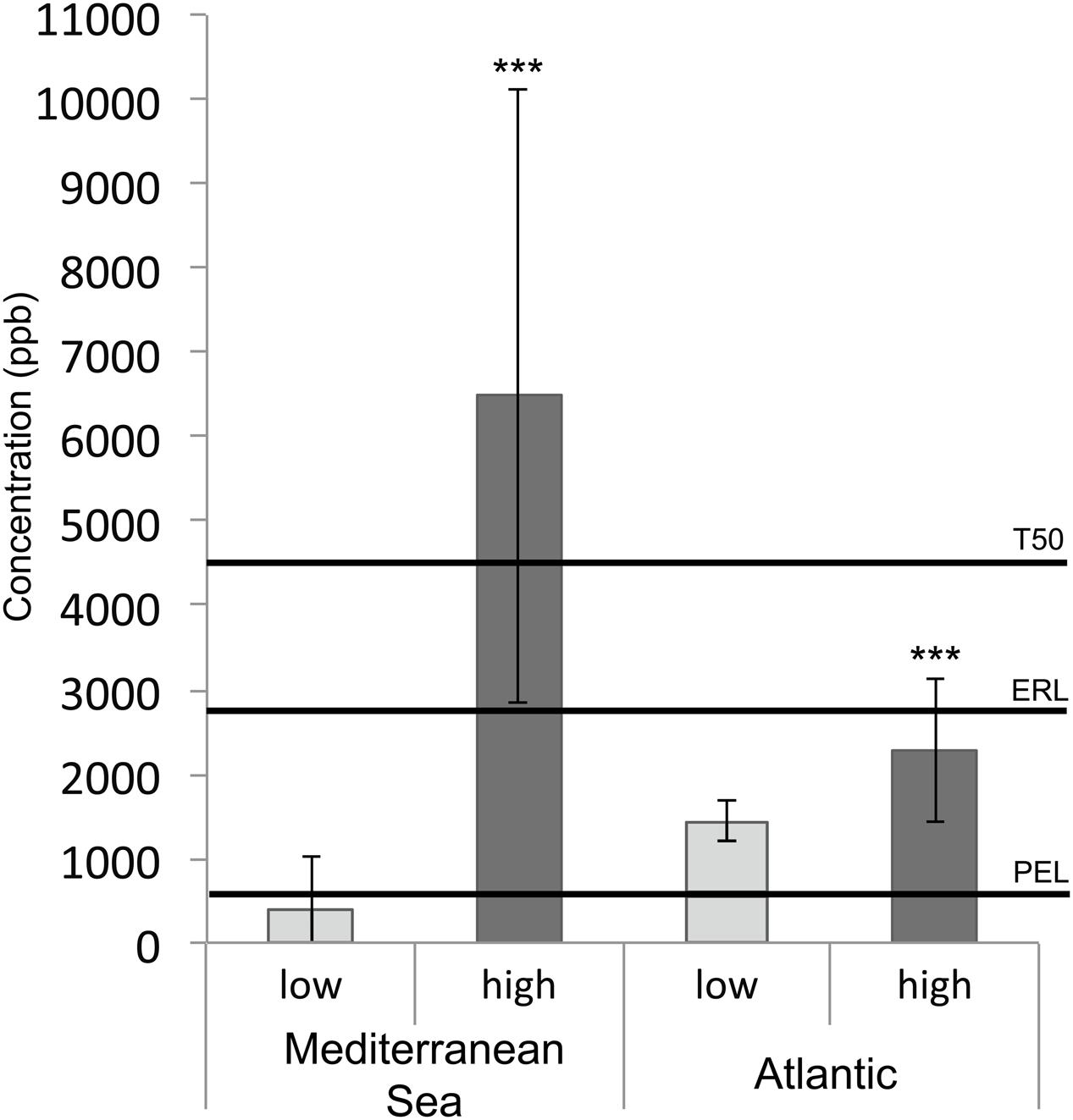
FIGURE 2. Histogram presenting high and low PAH concentrations (μg/kg dw - sum of 9 individual compounds Σ PAH9: fluoranthene, pyrene, anthracene, ben[a]anthracene, benzo[ghi]perylene, chrysene, dibenz[a,h]anthracene, fluorene, indeno[1,2,3-cd]pyrene, phenanthrene) in sediment samples from the Mediterranean Sea and Atlantic coasts. The asterisks indicate significant differences between highly contaminated and non-contaminated samples (p < 0.001). The plotted thresholds (solid line) correspond to Sediment Quality Guidelines: Probable Effect Level (PEL), Effect Range Low (ERL), and 50% toxicity concentration (T50).
Fluoranthene to pyrene (F/P) and phenanthrene to anthracene (P/A) “source”-isomeric ratios are indicators of mixed-PAH origin (Budzinski et al., 1997; Neff et al., 2005). The values of the ratios indicates whether the PAH contamination originates more from direct input (petrogenic source) or from combustion processes (pyrogenic source). The ratio values suggested that the PAH contamination origin was predominantly pyrogenic in most of the samples, with P/A values lower than 10 and F/P values above 1 (Supplementary Figure 3; Supplementary Table 1). Therefore, our sediment samples were fairly representative of sites polluted by chronic PAH contamination, mainly due to from fossil fuel combustion, for most of the coastal areas studied. However, for 14 out of 44 samples, the indications given by the two ratios were contradictory, suggesting that both petrogenic and pyrogenic sources contribute to the contamination at these locations.
Prokaryotic 16S rRNA genes were successfully amplified for all the samples, while eukaryotic 18S rRNA genes were successfully amplified for 42 out of 46 samples. Using Richness (i.e., the number of OTUs per sample) and PD indices, we investigated the effect of PAHs on the alpha-diversity of the three domains of life. Figure 3 shows the results of the stepwise multiple regression models of the two alpha-diversity indices against the explanatory variables. Temperature explained 28% and 17% of the variance of bacterial PD and archaeal PD, respectively. Salinity explained 27% of bacterial richness, and together with PSD, it explained 18% of archaeal richness. PSD significantly explained 37% of eukaryotic richness and 18% of eukaryotic PD. PAH concentrations and ratios were not found to significantly explain the variance of the alpha-diversity indices among the sites.
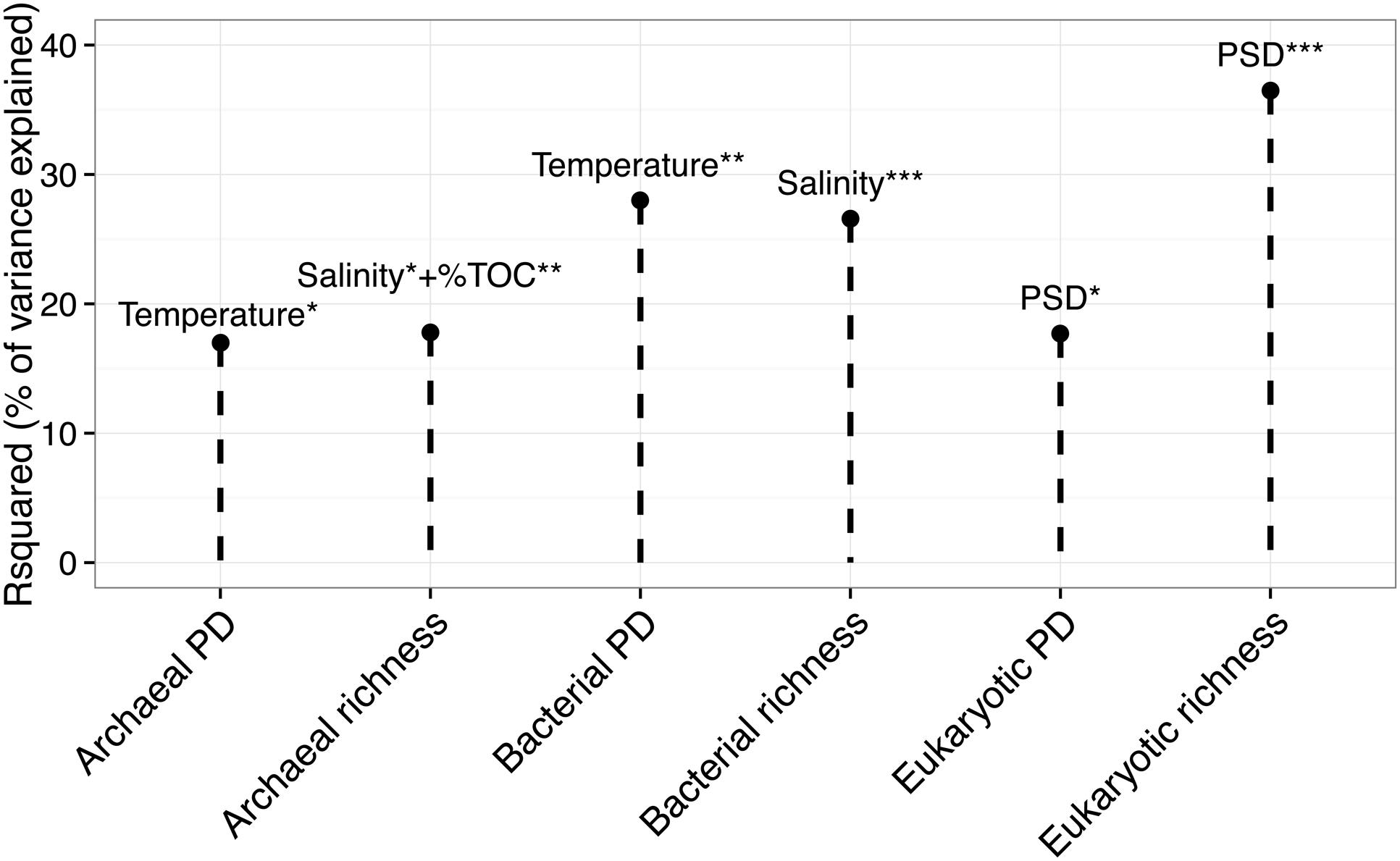
FIGURE 3. Multiple linear regression model of richness and phylogenetic diversity (PD) of the three kingdoms (x-axis) against the set of explanatory variables. The y-axis represents the determination coefficient (r-squared). Only significant regressions are shown. Asterisk corresponds to the level of significativity: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
In order to unveil a potential structuration effect of PAHs on microbial communities, these communities were sorted into an ordination plot according to their phylogenetic similarity (NMDS, Figure 4). For the three domains of life, the mean observed PSV values (0.66, 0.73, and 0.56 for Bacteria, Archaea, and Eukarya, respectively) were significantly lower (i.e., more phylogenetically clustered) than the null distribution for both model 1 (0.68, 0.74, 0.59, for Bacteria, Archaea, and Eukarya, respectively, p < 0.05) and model 2 (0.67, 0.76, and 0.57 for Bacteria, Archaea, and Eukarya, respectively, p < 0.05), indicating that sampling of phylotypes from the sequence pools was non-random. As non-random evolutionary processes were at work for both prokaryotes and eukaryotes, we tested their association with environmental parameters (i.e., salinity, temperature, %TOC particle size distribution, F/P and P/A ratios and dibenz[a,h]anthracene concentration). NMDS plots and PerMANOVA results revealed that the dissimilarities in the community phylogenetic structure of Bacteria, Archaea, and Eukarya were mainly driven by salinity (also a proxy of latitude), indicating the occurrence of different microbial communities at the regional scale (i.e., Atlantic vs Mediterranean, Figure 4; Table 1). The PerMANOVA analysis also showed that %TOC and particle size distribution (% <63 μm) significantly explained, respectively, 8.1% and 8.3% (p < 0.05) of bacterial communities phylogenetic variation, while no significant effect of these parameters was detected for the archaeal and eukaryotic domains. The habitat filter (i.e., selection of species by the local abiotic environment) therefore represented a strong structuring factor for the benthic communities of the three domains of life. The effect PAH concentrations was only detected for eukaryotic communities, for which 6.5% of the variation was explained by the PAH concentration proxy (i.e., dibenz[a,h]anthracene). When focusing on the Mediterranean or Atlantic regions, where environmental variability was lower, the structure of both bacterial and eukaryotic communities was significantly influenced by the P/A ratio (R2 = 9,3%, p < 0.05) and dibenz[a,h]anthracene (R2 = 11.8%, p < 0.05), respectively, (Table 1; Supplementary Figure 7). However, this tendency was observed only in Mediterranean samples, where the difference in PAH concentrations was maximal between polluted and unpolluted samples.
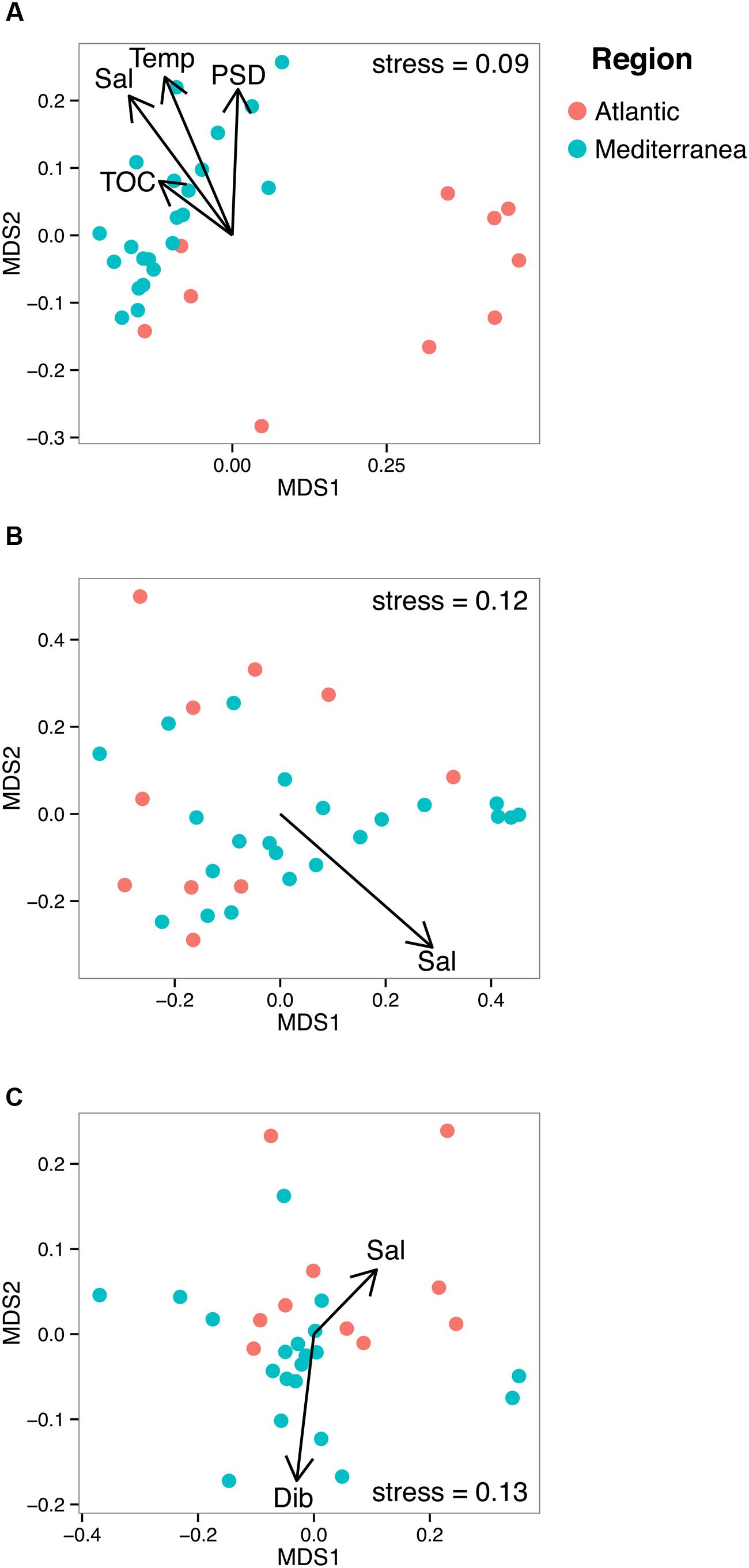
FIGURE 4. Non-metric multidimensional scaling plots of bacterial (A), archaeal (B), and eukaryotic (C) UNIFRAC matrices. The arrows represent the significant environmental variables fitted as vectors using the envfit function of the vegan package. Temp stands for temperature, Sal for salinity and Dib for Dibenz[a,h]anthracene.
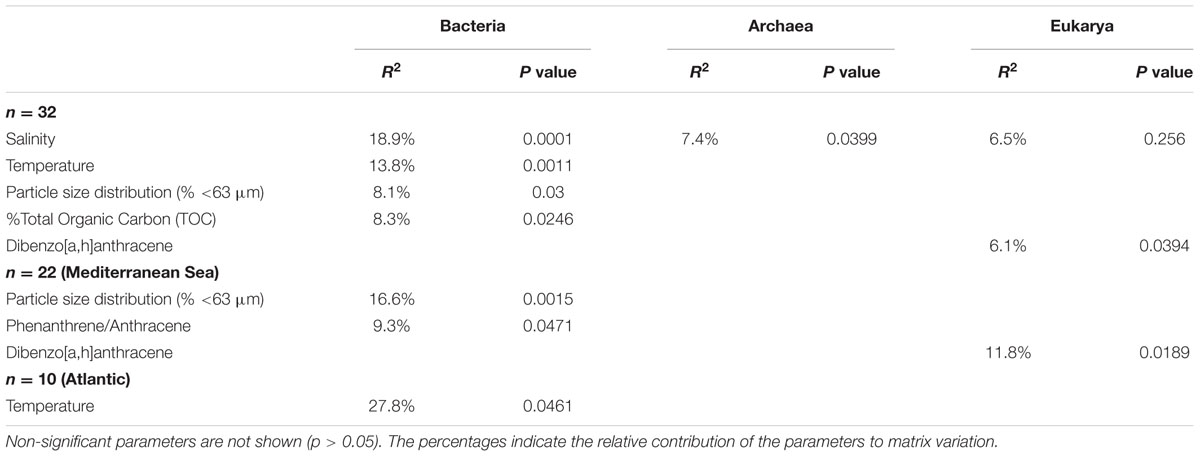
TABLE 1. PerMANOVA results for environmental parameters against UNIFRAC matrices.
The composition of the microbial communities was typical of marine benthic habitats with a domination of bacterial communities by Deltaproteobacteria (particularly of the Desulfobacterales order, Supplementary Figure 5) and Gammaproteobacteria, while archaeal communities were dominated by Halobacteria and Thermoplasmata and eukaryotic communities were dominated by Annelida, Arthropoda, and Dinophyceae (Supplementary Figures 4 and 5). We used the LEfSe algorithm in order to detect differentially abundant taxa between geographic regions and levels of contamination (i.e., the finding of environmental and hydrocarbon contamination biomarkers). LEfSe detected 78 taxa showing statistically significant and biologically consistent differences between geographic regions (Figure 5), with entire high taxonomic levels (i.e., aaa phylum level, and their respective taxonomic levels below) found as characteristic of Mediterranean (Proteobacteria, Chloroflexi, Acidobacteria, Actinobacteria, Crenarchaeota, and Opisthokonta) or Atlantic sediments (Firmicutes and Holozoa). In agreement with the PerMANOVA results showing an absence of influence of PAH contamination on the prokaryotic community structure at the continental scale, biomarkers of PAH contamination (i.e., with the presence and abundance statistically associated with the environmental condition) were rare with only 16 taxa detected. Among them, only Archaea of the Miscellaneous Crenarchaeotic Group (MCG) and eukaryotes from unclassified Dinophyceae to the superphylum Alveolata were found to be overrepresented in contaminated sediments (Figure 5).
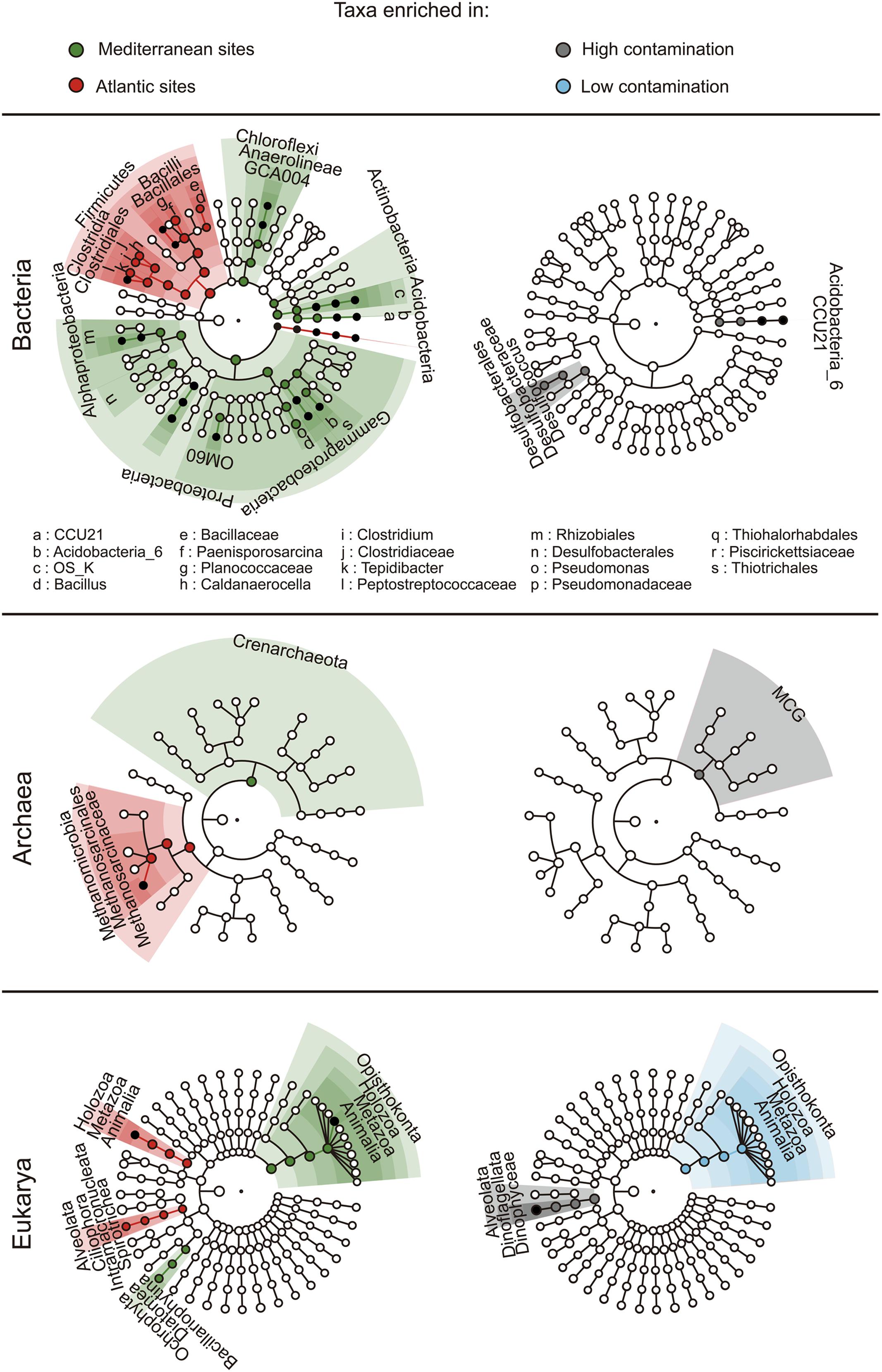
FIGURE 5. LefSe analysis of the geographic origin (left panel) and the level of contamination (right panel). The cladograms show taxa enriched (i.e., with significant differential abundance) within the environmental groups of samples (i.e., Mediterranean Sea and Atlantic; high and low contamination levels). The roots of the cladograms stand for the domain, and concentric circles represent the following taxonomic levels until the tips standing for genera. Biomarkers are shaded according to the groups of samples. The dot diameter is proportional to the abundance of the corresponding taxa.
The functional diversity of prokaryotic communities was reconstructed using PICRUSt. Overall, 60 and 55% of level 3 KEGG pathways (196 and 179 of 328 pathways) were present in the bacterial and archaeal datasets, respectively. Sources of variation related to environmental conditions in the Bray-Curtis dissimilarity matrices calculated on KEGG pathway abundances were explored by means of a PerMANOVA analysis (Table 2) and NMDS (Supplementary Figure 6). Similarly to the phylogenetic beta-diversity, the habitat filter represented a strong structuring factor for the reconstructed functional beta-diversity. Indeed, salinity, temperature and %TOC were the principal environmental variables shaping the structure of prokaryotic predicted functions whether we considered all functions, functions involved in organic matter degradation or functions related to hydrocarbon metabolism (Table 2). This is in agreement with the good correlation observed between the phylogenetic and functional distance matrices for Bacteria (R2 = 0.79, p = 0.0001) and Archaea (R2 = 0.66, p = 0.0001). No significant influences of the environmental parameters were detected when the Mediterranean and the Atlantic regions were considered independently. We then focused on the specific predicted KEGG pathway related to PAH degradation. Comparison of mean occurrences of the PAH degradation pathway between contaminated and non-contaminated samples in each geographic region (Mediterranean Sea and Atlantic) resulted in no significant difference for either Archaea or Bacteria (Figure 4). Overall, prokaryotic reconstructed metabolic potentialities were thus highly influenced by habitat, while the analysis did not detect any impact of PAH contamination.
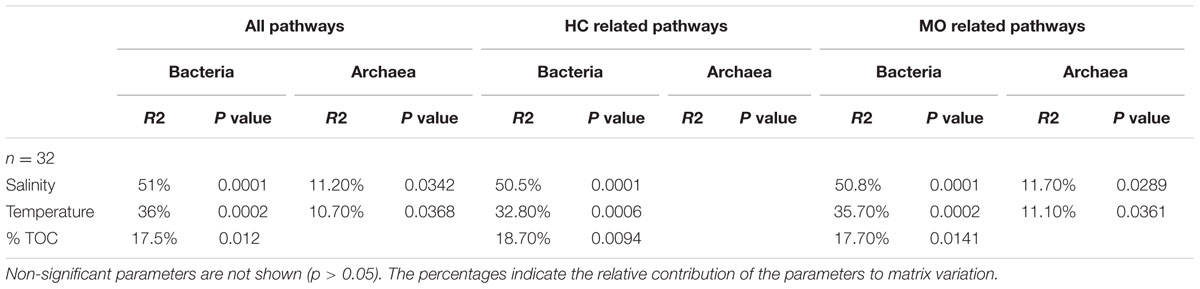
TABLE 2. PerMANOVA results for environmental parameters against Bray-Curtis matrices of all reconstructed bacterial metabolic pathways, metabolic pathways (from the KEGG database) related to organic matter metabolism (OM, n = 113 for bacteria and n = 101 for archaea), and hydrocarbons (HC, n = 14 for Bacteria and n = 10 for Archaea).
In one of the largest surveys performed so far on coastal sediments, we adopted here a global approach, as opposed to experimental reductionist approaches, in which we characterized the microbial communities from the three domains of life, across a large PAH contamination gradient. This approach aimed to disentangle the impact of chronic PAH exposure from that of the habitat on microbial communities considering both the PD and the reconstructed metabolic potentialities.
Microbial communities from coastal environments impacted by transitory input of hydrocarbons have been extensively studied during the past few years, especially following the Deepwater Horizon oil spill (Bik et al., 2012; Dubinsky et al., 2013; Kimes et al., 2013; Mahmoudi et al., 2013; Newton et al., 2013; Lamendella et al., 2014). An increase in the relative abundance of hydrocarbonoclastic bacteria following an oil spill is a very consistent result across studies (for reviews see Head et al., 2006; Kimes et al., 2014; King et al., 2015). The emergence of specialized taxa leads to the reduction of diversity within samples (Atlas et al., 2015). However, these profound changes in community composition are transitory (Ager et al., 2010; Mahmoudi et al., 2013) and a recovery of alpha-diversity has been reported in chronically or long-term hydrocarbon polluted areas (Gillan et al., 2005; Roesch et al., 2007). Generally, a decline in habitat health or a loss in ecosystem services results in an erosion of the richness or evenness of communities (Allison and Martiny, 2008). In contrast to what is observed in acute contamination events, we showed that the richness and PD of microbial communities in contrasted sedimentary coastal environments were not influenced by the chronic hydrocarbon pollution. This is typical of coastal microbial communities facing chronic pollution, and particularly bacterial communities (Hernandez-Raquet et al., 2006; Païssé et al., 2008; Zhang et al., 2008).
In contrast to prokaryote alpha-diversity, eukaryotic richness and PD were shown here to be highly related to particle size distribution, with higher richness and PD in coarser sediment. A greater richness in coarser sediments has been consistently observed in meiofauna and macrofauna assemblages, in both deep-sea and coastal sediments (Rodríìguez et al., 2003; McLachlan and Dorvlo, 2005; Netto et al., 2005; Hack et al., 2007; Leduc et al., 2012). Alternatively, no erosion of alpha-diversity indices could be attributed to PAH contamination, as previously shown by the comparisons between contaminated and non-contaminated sites (Chariton et al., 2010). However, as the sorption of PAHs on sediment matrices was not evaluated here, we cannot exclude the hypothesis that acute bioavailability of PAHs in finer sediments (Rockne et al., 2002; Ghosh et al., 2003) contributes, through an increase in toxicity, to the decline of eukaryotic richness in these finer sediments.
Gillan et al. (2005) demonstrated that alpha-diversity alone is a poor indicator of ecosystem stress in chronically polluted systems, as the proliferation of new tolerant species can lead to the recovery of the diversity. Here, beta-diversity analyses revealed that the drivers of community structures where different depending on the scale of the dataset used. At the continental scale, and regardless of the domain of life, PAHs did not affect the microbial community composition compared to the selection pressure exerted by the natural environmental filter. In contrast, when considering the regional scale (i.e., the Mediterranean coast), the PAH contamination became the main driver of eukaryotic and bacterial communities variability, as the environmental filter was reduced and the range of PAH concentrations was larger. It is, however, important to note that most of the variance in community structure remained unexplained indicating that other regional environmental variables like nutrients or other contaminants (metals, antibiotics, etc.), may play an important role in shaping communities (Sun et al., 2012, 2013, Quero et al., 2015, Xie et al., 2016). Nonetheless, this result tends to agree with small-scales studies (i.e., covering few meters), which recorded changes in eukaryotic and bacterial community structures across similar PAH gradients (Païssé et al., 2008; Chariton et al., 2010; Païssé et al., 2010; Said et al., 2010; Sauret et al., 2015), although the changes were more subtle in the present work. Overall, the discrepancy between continental or regional datasets patterns gives an answer to a fundamental question raised by Nogales et al. (2011) by providing evidence that results from small scale studies may not be transposable to larger scales. Future risk assessment studies should therefore pay a particular attention to the scale of description when investigating the effect of contaminants on biological communities.
Although we acknowledge that functional assessment of microbial communities should involve the measurement of process rates (i.e., hydrocarbon degradation, prokaryotic production, enzymatic activities, etc.) (Rocca et al., 2015), we found that the subtle change in bacterial community composition was not reflected on predicted functions, indicating some degree of functional redundancy. Fundamental ecosystem services insured by sediment prokaryotic communities such as organic matter remineralization or hydrocarbon degradation did not seem to be altered by elevated PAH concentrations (Figure 6). Sun et al. (2012, 2013) found that PAHs were not as influential on bacterial communities as metal contaminants probably because of their lower toxicity due to their potential role as carbon source (Païssé et al., 2010), and potentially because of their high adsorption rate on sediment matrix, causing a decline of their bioavailability (Kraaij et al., 2002; Ghosh et al., 2003). Alternatively, a large diversity of known hydrocarbon degraders exists in the environment, particularly in Bacteria but also in Archaea and Eukarya (Al-Mailem et al., 2010; Prince, 2010; Tapilatu et al., 2010), and many of them have not yet been characterized (Nogales et al., 2011). Deltaproteobacteria and Gammaproteobacteria, which dominated unpolluted and polluted sites in this work are well known to be dominant clades in marine sediments and to encompass a large number of hydrocarbon degrader strains (Rojo, 2009; Kostka et al., 2011; Acosta-González et al., 2013). Archaea from the class Halobacteria, which dominated archaeal communities are also known hydrocarbon degraders (Al-Mailem et al., 2010). The diversity of prokaryotic degraders found here suggests that the benthic communities were naturally well adapted to face chronic hydrocarbon contamination, facilitating their resistance /resilience to this stress.
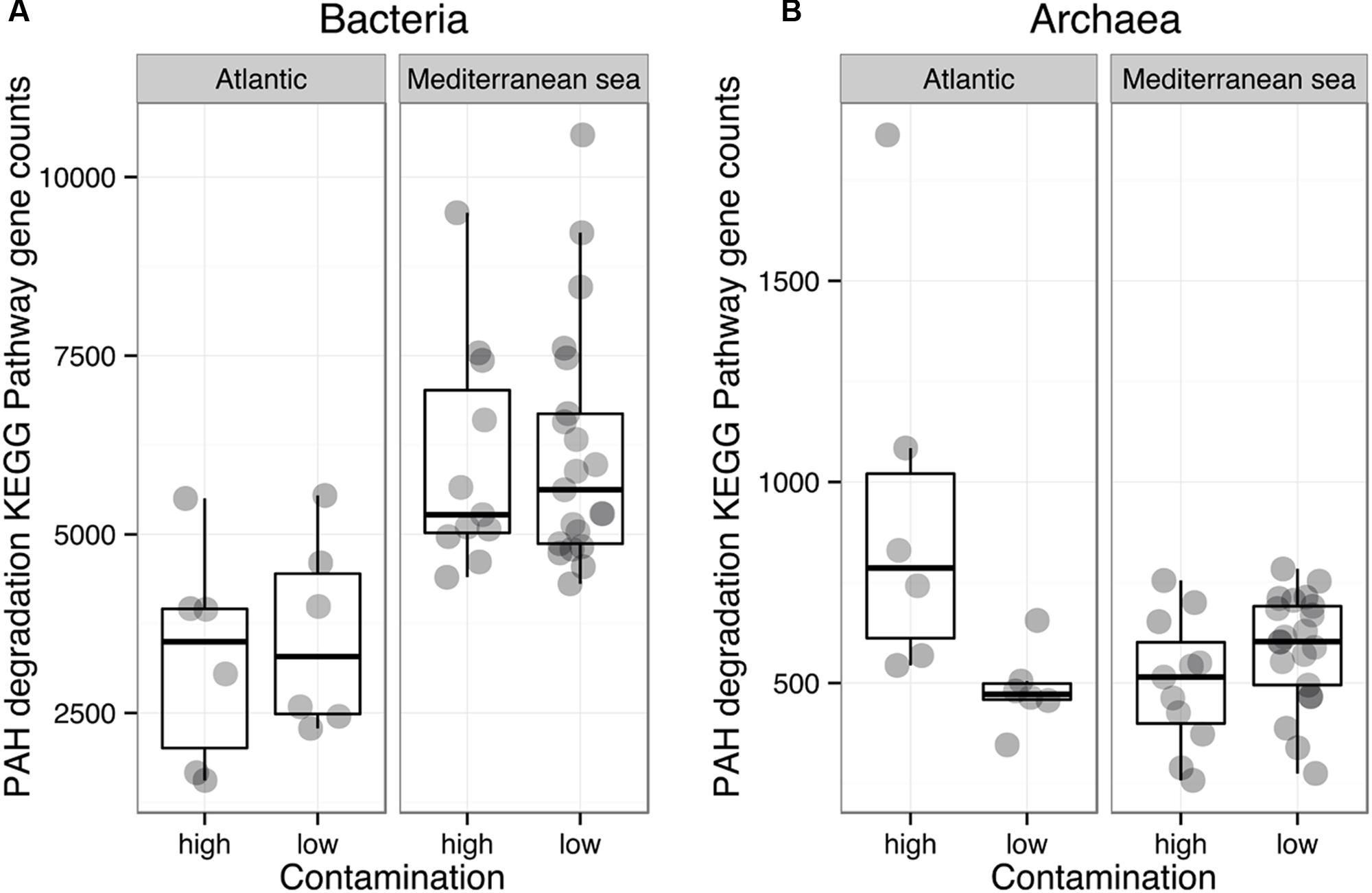
FIGURE 6. Comparison of the mean abundances of bacterial (A) and archaeal (B) KEGG pathways related to the polyaromatic hydrocarbons (PAHs) degradation, within the Mediterranean and Atlantic geographic subsets.
The identification of indicator species or biomarkers (i.e., a species or taxa lineage that define a trait or characteristic of the environment) is conceptually straightforward and has been widely used in ecological studies concerning both macro-organisms (Dufrêne and Legendre, 1997; De Cáceres et al., 2010; Pelletier et al., 2010) and microorganisms (Auguet et al., 2009; Chariton et al., 2010; Fillol et al., 2016). Here, community composition of the three domains of life was more stable across the PAH concentrations gradient than between geographic regions. Accordingly, fewer taxa indicative of PAH contamination were found, as compared to the taxa indicative of the geographic regions (Figure 5).
For Bacteria, the genus Desulfococcus and the taxonomic levels above (i.e., Desulfobacteraceae and Desulfobacterales) were identified as indicators of low PAH contaminations. The Desulfobacterales are particularly interesting, since the class to whom they belong (i.e., Deltaproteobacteria) is dominant in both polluted and pristine benthic marine ecosystems (Païssé et al., 2008; Leloup et al., 2009; Said et al., 2010; Korlević et al., 2015). This class also includes most of the sulfate-reducing genera, which play a crucial role in the anaerobic degradation of organic matter (Leloup et al., 2009) but also in hydrocarbon degradation (Suárez-Suárez et al., 2011; Stauffert et al., 2013). Members of the Desulfobacterales are dominant in the core microbiota of estuarine sediments (Sun et al., 2013). Therefore, a decline of the bacteria belonging to this order may negatively influence the rate of the processes they are involved in, and ultimately results in an important erosion of ecosystem health. However, genes involved in organic matter degradation pathways were not altered by high PAH levels potentially indicating a replacement by more tolerant SRB contributing to the functional redundancy of the system.
For Archaea, one taxon, the MCG was significantly linked to high levels of PAH contamination. MCG is a dominant and core archaeal taxa in sediment worldwide (Lloyd et al., 2013; Fillol et al., 2016). Recent findings from metagenomic data (Meng et al., 2014) and single cell genomics (Lloyd et al., 2013) suggested that members of this taxon might be involved in the degradation of aromatic compounds and detrital proteins, respectively. The former function may confer to members of the MCG lineage the capacity to use PAHs as source of carbon. This would be in agreement with their higher abundance in contaminated sediments.
Eukaryotic communities were the most impacted by PAHs both in terms of diversity and structure. Consequently, we were able to find more biomarkers related to PAH contamination for eukaryotic communities, than for Bacteria or Archaea. Taxa going from Opisthokonta to Animalia were significantly more abundant in non-polluted sediment, which is consistent with a potential toxicity of PAHs causing typically a decrease of macro-biota richness, characterized by the loss of sensitive species (Johnston and Roberts, 2009). In contrast, Eukaryotes from unclassified Dinophyceae taxa, and above until the superphylum Alveolata, were found to be overrepresented in contaminated sediments. Taxa belonging to the Dinophyceae class (dinoflagellates) were also detected as potential indicators of contaminated estuarine sediments (Chariton et al., 2010). We currently do not know whether the positive selection of Dinophyceae by PAHs is due to their capacity to metabolize hydrocarbons, or to their opportunistic ability to replace sensitive taxa. In any case, the increase of Dinophyceae in contaminated sediments may be of paramount importance since harmful algal blooms of benthic dinoflagellates cause major social, economical, environmental and health problems (European Environment Agency, 2006; Hoppenrath et al., 2013).
Overall, our results showed that chronic PAH contamination was a marginal driver for coastal sediment community diversity, structure, and prokaryotic predicted functions compared to the habitat filter represented by salinity, latitude, particle size distribution and TOC. Nonetheless, the influence of the PAH contamination was greater especially for Eukarya at the scale of the Mediterranean coast. We provide evidence that results from studies focusing on the local scale may not be transposable to larger scales. Despite the weak response of prokaryotic and eukaryotic communities to PAH, we were able to identify potential biomarkers of chronic contamination. Owing to their higher sensitivity to PAHs, eukaryotes showed the greatest potential as biomarkers in Mediterranean and Atlantic sediments among the three domains of life, both at the regional and continental scales.
J-CA, JG, and RD conceived and designed the study. MJ ran the experiments. MJ, J-CA, JG, NT, and DD analyzed the resulting data. J-FG, JT, OS, HA, and CG provided samples, chemical and physical analysis. MJ and J-CA wrote the manuscript. J-FG, JT, HA, CG, and RD revised the manuscript.
This work was supported by a PhD grant from the French Ministry of Higher Education and Research to MJ, and by a CNRS INSU EC2CO “Microbien” and “ECODYN” program. Chemical and pyrosequencing analysis on sediments from Toulon bay were financially supported by CARTOCHIM (funded by “Toulon-Provence-Méditerranée (TPM),” the “Région PACA,” and “l’Agence de l’Eau Rhône-Méditerranée et Corse”) and MERMEX-WP3-C3A (part of the international IMBER project) research projects.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the two reviewers, who helped to improve the manuscript. We also thank Sally Ferguson (Alba Traduction) for carefully checking the English language.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01303
Acosta-González, A., Rosselló-Móra, R., and Marqués, S. (2013). Characterization of the anaerobic microbial community in oil-polluted subtidal sediments: aromatic biodegradation potential after the prestige oil spill. Environ. Microbiol. 15, 77–92. doi: 10.1111/j.1462-2920.2012.02782.x
Ager, D., Evans, S., Li, H., Lilley, A. K., and van der Gast, C. J. (2010). Anthropogenic disturbance affects the structure of bacterial communities. Environ. Microbiol. 12, 670–678. doi: 10.1111/j.1462-2920.2009.02107.x
Allison, S. D., and Martiny, J. B. H. (2008). Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. U.S.A. 105, 11512–11519. doi: 10.1073/pnas.0801925105
Al-Mailem, D. M., Sorkhoh, N. A., Al-Awadhi, H., Eliyas, M., and Radwan, S. S. (2010). Biodegradation of crude oil and pure hydrocarbons by extreme halophilic archaea from hypersaline coasts of the Arabian Gulf. Extremophiles 14, 321–328. doi: 10.1007/s00792-010-0312-9
Atlas, R. M., Stoeckel, D. M., Faith, S. A., Minard-Smith, A., Thorn, J. R., and Benotti, M. J. (2015). Oil biodegradation and oil-degrading microbial populations in marsh sediments impacted by oil from the deepwater horizon well blowout. Environ. Sci. Technol. 49, 8356–8366. doi: 10.1021/acs.est.5b00413
Auguet, J.-C., Barberan, A., and Casamayor, E. O. (2009). Global ecological patterns in uncultured Archaea. ISME J. 4, 182–190. doi: 10.1038/ismej.2009.109
Azoury, S., Tronczyński, J., Chiffoleau, J.-F., Cossa, D., Nakhlé, K., Schmidt, S., et al. (2013). Historical records of mercury, lead, and polycyclic aromatic hydrocarbons depositions in a dated sediment core from the Eastern Mediterranean. Environ. Sci. Technol. 47, 7101–7109. doi: 10.1021/es4005637
Aßhauer, K. P., Wemheuer, B., Daniel, R., and Meinicke, P. (2015). Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884. doi: 10.1093/bioinformatics/btv287
Bik, H. M., Halanych, K. M., Sharma, J., and Thomas, W. K. (2012). Dramatic shifts in benthic microbial Eukaryote communities following the Deepwater Horizon Oil Spill. PLoS ONE 7:e38550. doi: 10.1371/journal.pone.0038550
Borja, A., Barbone, E., Basset, A., Borgersen, G., Brkljacic, M., Elliott, M., et al. (2011). Response of single benthic metrics and multi-metric methods to anthropogenic pressure gradients, in five distinct European coastal and transitional ecosystems. Mar. Pollut. Bull. 62, 499–513. doi: 10.1016/j.marpolbul.2010.12.009
Buchman, M. F. (2008). Screening Quick Reference Tables (SQuiRTs). NOAA OR&R Report 08-1. Seattle, WA: US National Oceanic and Atmospheric Administration.
Budzinski, H., Jones, I., Bellocq, J., Piérard, C., and Garrigues, P. (1997). Evaluation of sediment contamination by polycyclic aromatic hydrocarbons in the Gironde estuary. Mar. Chem. 58, 85–97. doi: 10.1016/S0304-4203(97)00028-5
Burgess, R. M., Ryba, S. A., and Cantwell, M. G. (2000). Importance of organic carbon quantity on the variation of K oc in marine sediments. Toxicol. Environ. Chem. 77, 9–29. doi: 10.1080/02772240009358935
Chariton, A. A., Court, L. N., Hartley, D. M., Colloff, M. J., and Hardy, C. M. (2010). Ecological assessment of estuarine sediments by pyrosequencing eukaryotic ribosomal DNA. Front. Ecol. Environ. 8:233–238. doi: 10.1890/090115
Coulon, F., Chronopoulou, P.-M., Fahy, A., Païssé, S., Goñi-Urriza, M., Peperzak, L., et al. (2012). Central role of dynamic tidal biofilms dominated by aerobic hydrocarbonoclastic bacteria and diatoms in the biodegradation of hydrocarbons in coastal mudflats. Appl. Environ. Microbiol. 78, 3638–3648. doi: 10.1128/AEM.00072-12
Creer, S., Fonseca, V. G., Porazinska, D. L., Giblin-Davis, R. M., Sung, W., Power, D. M., et al. (2010). Ultrasequencing of the meiofaunal biosphere: practice, pitfalls and promises. Mol. Ecol. 19, 4–20. doi: 10.1111/j.1365-294X.2009.04473.x
Danovaro, R., Gambi, C., Dell’Anno, A., Corinaldesi, C., Fraschetti, S., Vanreusel, A., et al. (2008). Exponential decline of deep-sea ecosystem functioning linked to benthic biodiversity loss. Curr. Biol. 18, 1–8. doi: 10.1016/j.cub.2007.11.056
De Cáceres, M., Legendre, P., and Moretti, M. (2010). Improving indicator species analysis by combining groups of sites. Oikos 119, 1674–1684. doi: 10.1111/j.1600-0706.2010.18334.x
Dias, A. C. F., Dini-Andreote, F., Taketani, R. G., Tsai, S. M., Azevedo, J. L., de Melo, I. S., et al. (2011). Archaeal communities in the sediments of three contrasting mangroves. J. Soils Sediments 11, 1466–1476. doi: 10.1007/s11368-011-0423-7
Dubinsky, E. A., Conrad, M. E., Chakraborty, R., Bill, M., Borglin, S. E., Hollibaugh, J. T., et al. (2013). Succession of hydrocarbon-degrading bacteria in the aftermath of the Deepwater Horizon Oil Spill in the Gulf of Mexico. Environ. Sci. Technol. 47, 10860–10867. doi: 10.1021/es401676y
Dufrêne, M., and Legendre, P. (1997). Species assemblages and indicator species:the need for a flexible asymmetrical approach. Ecol. Monogr. 67, 345–366. doi: 10.1890/0012-9615(1997)067[0345:SAAIST]2.0.CO;2
European Environment Agency (2006). Priority Issues in the Mediterranean Environment. Copenhagen: European Environmental Agency.
Falkowski, P. G., Fenchel, T., and Delong, E. F. (2008). The microbial engines that drive earth’s biogeochemical cycles. Science 320, 1034–1039. doi: 10.1126/science.1153213
Fillol, M., Auguet, J.-C., Casamayor, E. O., and Borrego, C. M. (2016). Insights in the ecology and evolutionary history of the Miscellaneous Crenarchaeotic group lineage. ISME J. 10, 665–677. doi: 10.1038/ismej.2015.143
Ghosh, U., Zimmerman, J. R., and Luthy, R. G. (2003). PCB and PAH speciation among particle types in contaminated harbor sediments and effects on PAH bioavailability. Environ. Sci. Technol. 37, 2209–2217. doi: 10.1021/es020833k
Gillan, D. C., Danis, B., Pernet, P., Joly, G., and Dubois, P. (2005). Structure of sediment-associated microbial communities along a heavy-metal contamination gradient in the marine environment. Appl. Environ. Microbiol. 71, 679–690. doi: 10.1128/AEM.71.2.679-690.2005
Hack, L. A., Tremblay, L. A., Wratten, S. D., Lister, A., and Keesing, V. (2007). Benthic meiofauna community composition at polluted and non-polluted sites in New Zealand intertidal environments. Mar. Pollut. Bull. 54, 1801–1812. doi: 10.1016/j.marpolbul.2007.07.009
Halpern, B. S., Walbridge, S., Selkoe, K. A., Kappel, C. V., Micheli, F., D’Agrosa, C., et al. (2008). A global map of human impact on marine ecosystems. Science 319, 948–952. doi: 10.1126/science.1149345
Hardy, C. M., Krull, E. S., Hartley, D. M., and Oliver, R. L. (2010). Carbon source accounting for fish using combined DNA and stable isotope analyses in a regulated lowland river weir pool. Mol. Ecol. 19, 197–212. doi: 10.1111/j.1365-294X.2009.04411.x
Head, I. M., Jones, D. M., and Röling, W. F. (2006). Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 4, 173–182. doi: 10.1038/nrmicro1348
Helmus, M. R., Bland, T. J., Williams, C. K., and Ives, A. R. (2007). Phylogenetic measures of biodiversity. Am. Nat. 169, E68–E83. doi: 10.1086/511334
Hernandez-Raquet, G., Budzinski, H., Caumette, P., Dabert, P., Le Ménach, K., Muyzer, G., et al. (2006). Molecular diversity studies of bacterial communities of oil polluted microbial mats from the Etang de Berre (France). FEMS Microbiol. Ecol. 58, 550–562. doi: 10.1111/j.1574-6941.2006.00187.x
Hoppenrath, M., Chomérat, N., Horiguchi, T., Schweikert, M., Nagahama, Y., and Murray, S. (2013). Taxonomy and phylogeny of the benthic Prorocentrum species (Dinophyceae)—A proposal and review. Harmful Algae 27, 1–28. doi: 10.1016/j.hal.2013.03.006
Johnston, E. L., and Roberts, D. A. (2009). Contaminants reduce the richness and evenness of marine communities: a review and meta-analysis. Environ. Pollut. 157, 1745–1752. doi: 10.1016/j.envpol.2009.02.017
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: r tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Kim, M., Morrison, M., and Yu, Z. (2011). Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J. Microbiol. Methods 84, 81–87. doi: 10.1016/j.mimet.2010.10.020
Kimes, N. E., Callaghan, A. V., Aktas, D. F., Smith, W. L., Sunner, J., Golding, B., et al. (2013). Metagenomic analysis and metabolite profiling of deep–sea sediments from the Gulf of Mexico following the Deepwater Horizon oil spill. Front. Microbiol. 4:50. doi: 10.3389/fmicb.2013.00050
Kimes, N. E., Callaghan, A. V., Suflita, J. M., and Morris, P. J. (2014). Microbial transformation of the Deepwater Horizon oil spill—past, present, and future perspectives. Front. Microbiol. 5:603. doi: 10.3389/fmicb.2014.00603
King, G. M., Kostka, J. E., Hazen, T. C., and Sobecky, P. A. (2015). Microbial responses to the Deepwater Horizon Oil Spill: from Coastal Wetlands to the deep sea. Annu. Rev. Mar. Sci. 7, 377–401. doi: 10.1146/annurev-marine-010814-015543
Korlević, M., Zucko, J., Dragić, M. N., Blažina, M., Pustijanac, E., Zeljko, T. V., et al. (2015). Bacterial diversity of polluted surface sediments in the northern Adriatic Sea. Syst. Appl. Microbiol. 38, 189–197. doi: 10.1016/j.syapm.2015.03.001
Kostka, J. E., Prakash, O., Overholt, W. A., Green, S. J., Freyer, G., Canion, A., et al. (2011). Hydrocarbon-degrading bacteria and the bacterial community response in gulf of Mexico beach sands impacted by the deepwater horizon oil spill. Appl. Environ. Microbiol. 77, 7962–7974. doi: 10.1128/AEM.05402-11
Kraaij, R., Seinen, W., Tolls, J., Cornelissen, G., and Belfroid, A. C. (2002). Direct evidence of sequestration in sediments affecting the bioavailability of hydrophobic organic chemicals to benthic deposit-feeders. Environ. Sci. Technol. 36, 3525–3529. doi: 10.1021/es0102787
Lamendella, R., Strutt, S., Borglin, S. E., Chakraborty, R., Tas, N., Mason, O. U., et al. (2014). Assessment of the Deepwater Horizon oil spill impact on Gulf coast microbial communities. Aquat. Microbiol. 5, 130. doi: 10.3389/fmicb.2014.00130
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Leahy, J. G., and Colwell, R. R. (1990). Microbial degradation of hydrocarbons in the environment. Microbiol. Rev. 54, 305–315.
Leduc, D., Rowden, A. A., Probert, P. K., Pilditch, C. A., Nodder, S. D., Vanreusel, A., et al. (2012). Further evidence for the effect of particle-size diversity on deep-sea benthic biodiversity. Deep Sea Res. Part Oceanogr. Res. Pap. 63, 164–169. doi: 10.1016/j.dsr.2011.10.009
Leloup, J., Fossing, H., Kohls, K., Holmkvist, L., Borowski, C., and Jørgensen, B. B. (2009). Sulfate-reducing bacteria in marine sediment (Aarhus Bay, Denmark): abundance and diversity related to geochemical zonation. Environ. Microbiol. 11, 1278–1291. doi: 10.1111/j.1462-2920.2008.01855.x
Lloyd, K. G., Schreiber, L., Petersen, D. G., Kjeldsen, K. U., Lever, M. A., Steen, A. D., et al. (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature 496, 215–218. doi: 10.1038/nature12033
Lozupone, C., Hamady, M., and Knight, R. (2006). UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371. doi: 10.1186/1471-2105-7-371
Lozupone, C. A., and Knight, R. (2007). Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U. S. A. 104, 11436–11440. doi: 10.1073/pnas.0611525104
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Mahmoudi, N., Porter, T. M., Zimmerman, A. R., Fulthorpe, R. R., Kasozi, G. N., Silliman, B. R., et al. (2013). Rapid degradation of Deepwater Horizon Spilled Oil by indigenous microbial communities in Louisiana Saltmarsh sediments. Environ. Sci. Technol. 47, 13303–13312. doi: 10.1021/es4036072
Martiny, A. C., Treseder, K., and Pusch, G. (2013). Phylogenetic conservatism of functional traits in microorganisms. ISME J. 7, 830–838. doi: 10.1038/ismej.2012.160
McArdle, B. H., and Anderson, M. J. (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82, 290–297. doi: 10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
McLachlan, A., and Dorvlo, A. (2005). Global patterns in Sandy Beach macrobenthic communities. J. Coast. Res. 21, 674–687. doi: 10.2112/03-0114.1
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Meng, J., Xu, J., Qin, D., He, Y., Xiao, X., and Wang, F. (2014). Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses. ISME J. 8, 650–659. doi: 10.1038/ismej.2013.174
National Research Council (US) Committee on Oil in the Sea (2003). Oil in the Sea III: Inputs, Fates, and Effects. Washington, DC: National Academies Press.
Neff, J. M., Stout, S. A., and Gunster, D. G. (2005). Ecological risk assessment of polycyclic aromatic hydrocarbons in sediments: identifying sources and ecological hazard. Integr. Environ. Assess. Manag. 1, 22–33. doi: 10.1897/IEAM_2004a-016.1
Netto, S. A., Gallucci, F., and Fonseca, G. F. C. (2005). Meiofauna communities of continental slope and deep-sea sites off SE Brazil. Deep Sea Res. Part Oceanogr. Res. Pap. 52, 845–859. doi: 10.1016/j.dsr.2004.11.009
Newton, R. J., Huse, S. M., Morrison, H. G., Peake, C. S., Sogin, M. L., and McLellan, S. L. (2013). Shifts in the microbial community composition of Gulf Coast Beaches following beach oiling. PLoS ONE 8:e74265. doi: 10.1371/journal.pone.0074265
Nogales, B., Aguiló-Ferretjans, M. M., Martín-Cardona, C., Lalucat, J., and Bosch, R. (2007). Bacterial diversity, composition and dynamics in and around recreational coastal areas. Environ. Microbiol. 9, 1913–1929. doi: 10.1111/j.1462-2920.2007.01308.x
Nogales, B., Lanfranconi, M. P., Piña-Villalonga, J. M., and Bosch, R. (2011). Anthropogenic perturbations in marine microbial communities. FEMS Microbiol. Rev. 35, 275–298. doi: 10.1111/j.1574-6976.2010.00248.x
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R. B., et al. (2013). Package “vegan.” R Packag. Ver. 254, 20–28.
Page, D. S., Boehm, P. D., Douglas, G. S., Bence, A. E., Burns, W. A., and Mankiewicz, P. J. (1999). Pyrogenic polycyclic aromatic hydrocarbons in sediments record past human activity: a case study in Prince William sound, Alaska. Mar. Pollut. Bull. 38, 247–260. doi: 10.1016/S0025-326X(98)00142-8
Païssé, S., Coulon, F., Goñi-Urriza, M., Peperzak, L., McGenity, T. J., and Duran, R. (2008). Structure of bacterial communities along a hydrocarbon contamination gradient in a coastal sediment. FEMS Microbiol. Ecol. 66, 295–305. doi: 10.1111/j.1574-6941.2008.00589.x
Païssé, S., Goñi-Urriza, M., Coulon, F., and Duran, R. (2010). How a bacterial community originating from a contaminated coastal sediment responds to an oil input. Microb. Ecol. 60, 394–405. doi: 10.1007/s00248-010-9721-7
Pelletier, M. C., Gold, A. J., Heltshe, J. F., and Buffum, H. W. (2010). A method to identify estuarine macroinvertebrate pollution indicator species in the Virginian biogeographic province. Ecol. Indic. 10, 1037–1048. doi: 10.1016/j.ecolind.2010.03.005
Pérez-Pantoja, D., González, B., and Pieper, D. H. (2010). “Aerobic degradation of aromatic hydrocarbons,” in Handbook of Hydrocarbon and Lipid Microbiology, ed. K. N. Timmis (Berlin: Springer), 799–837.
Pernice, M. C., Logares, R., Guillou, L., and Massana, R. (2013). General patterns of diversity in major marine microeukaryote lineages. PLoS ONE 8:e57170. doi: 10.1371/journal.pone.0057170
Prince, R. C. (2010). “Eukaryotic hydrocarbon degraders,” in Handbook of Hydrocarbon and Lipid Microbiology, ed. K. N. Timmis (Berlin: Springer), 2065–2078.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Quero, G. M., Cassin, D., Botter, M., Perini, L., and Luna, G. M. (2015). Patterns of benthic bacterial diversity in coastal areas contaminated by heavy metals, polycyclic aromatic hydrocarbons (PAHs) and polychlorinated biphenyls (PCBs). Front. Microbiol. 6:1053. doi: 10.3389/fmicb.2015.01053
Raskin, L., Stromley, J. M., Rittmann, B. E., and Stahl, D. A. (1994). Group-specific 16S rRNA hybridization probes to describe natural communities of methanogens. Appl. Environ. Microbiol. 60, 1232–1240.
Ravindra, K., Sokhi, R., and Van Grieken, R. (2008). Atmospheric polycyclic aromatic hydrocarbons: source attribution, emission factors and regulation. Atmos. Environ. 42, 2895–2921. doi: 10.1016/j.atmosenv.2007.12.010
Rocca, J. D., Hall, E. K., Lennon, J. T., Evans, S. E., Waldrop, M. P., Cotner, J. B., et al. (2015). Relationships between protein-encoding gene abundance and corresponding process are commonly assumed yet rarely observed. ISME J. 9, 1693–1699. doi: 10.1038/ismej.2014.252
Rockne, K. J., Shor, L. M., Young, L. Y., Taghon, G. L., and Kosson, D. S. (2002). Distributed sequestration and release of PAHs in weathered sediment: the role of sediment structure and organic carbon properties. Environ. Sci. Technol. 36, 2636–2644. doi: 10.1021/es015652h
Rodríìguez, J. G., Lastra, M., and López, J. (2003). Meiofauna distribution along a gradient of sandy beaches in northern Spain. Estuar. Coast. Shelf Sci. 58, 63–69. doi: 10.1016/S0272-7714(03)00039-8
Roesch, L. F. W., Fulthorpe, R. R., Riva, A., Casella, G., Hadwin, A. K. M., Kent, A. D., et al. (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 1, 283–290.
Rojo, F. (2009). Degradation of alkanes by bacteria. Environ. Microbiol. 11, 2477–2490. doi: 10.1111/j.1462-2920.2009.01948.x
Röling, W. F. M., Brito, I. R. C., de Swannell, R. P. J., and Head, I. M. (2004). Response of Archaeal communities in beach sediments to spilled oil and bioremediation. Appl. Environ. Microbiol. 70, 2614–2620. doi: 10.1128/AEM.70.5.2614-2620.2004
Rosano-Hernández, M. C., Ramírez-Saad, H., and Fernández-Linares, L. (2012). Petroleum-influenced beach sediments of the Campeche Bank, Mexico: diversity and bacterial community structure assessment. J. Environ. Manage. 95, S325–S331. doi: 10.1016/j.jenvman.2011.06.046
Said, O. B., Goñi-Urriza, M., Bour, M. E., Aissa, P., and Duran, R. (2010). Bacterial community structure of sediments of the Bizerte lagoon (Tunisia), a Southern Mediterranean coastal anthropized lagoon. Microb. Ecol. 59, 445–456. doi: 10.1007/s00248-009-9585-x
Sanni, G. O., Coulon, F., and McGenity, T. J. (2015). Dynamics and distribution of bacterial and archaeal communities in oil-contaminated temperate coastal mudflat mesocosms. Environ. Sci. Pollut. Res. Int. 22, 15230–15247. doi: 10.1007/s11356-015-4313-1
Sauret, C., Christaki, U., Moutsaki, P., Hatzianestis, I., Gogou, A., and Ghiglione, J.-F. (2012). Influence of pollution history on the response of coastal bacterial and nanoeukaryote communities to crude oil and biostimulation assays. Mar. Environ. Res. 79, 70–78. doi: 10.1016/j.marenvres.2012.05.006
Sauret, C., Tedetti, M., Guigue, C., Dumas, C., Lami, R., Pujo-Pay, M., et al. (2015). Influence of PAHs among other coastal environmental variables on total and PAH-degrading bacterial communities. Environ. Sci. Pollut. Res. Int. 23, 4242–4256. doi: 10.1007/s11356-015-4768-0
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Stauffert, M., Cravo-Laureau, C., Jézéquel, R., Barantal, S., Cuny, P., Gilbert, F., et al. (2013). Impact of oil on bacterial community structure in bioturbated sediments. PLoS ONE 8:e65347. doi: 10.1371/journal.pone.0065347
Stauffert, M., Duran, R., Gassie, C., and Cravo-Laureau, C. (2014). Response of Archaeal communities to oil spill in bioturbated mudflat sediments. Microb. Ecol. 67, 108–119. doi: 10.1007/s00248-013-0288-y
Suárez-Suárez, A., López-López, A., Tovar-Sánchez, A., Yarza, P., Orfila, A., Terrados, J., et al. (2011). Response of sulfate-reducing bacteria to an artificial oil-spill in a coastal marine sediment. Environ. Microbiol. 13, 1488–1499. doi: 10.1111/j.1462-2920.2011.02451.x
Sun, M. Y., Dafforn, K. A., Brown, M. V., and Johnston, E. L. (2012). Bacterial communities are sensitive indicators of contaminant stress. Mar. Pollut. Bull. 64, 1029–1038. doi: 10.1016/j.marpolbul.2012.01.035
Sun, M. Y., Dafforn, K. A., Johnston, E. L., and Brown, M. V. (2013). Core sediment bacteria drive community response to anthropogenic contamination over multiple environmental gradients. Environ. Microbiol. 15, 2517–2531. doi: 10.1111/1462-2920.12133
Taib, N., Mangot, J.-F., Domaizon, I., Bronner, G., and Debroas, D. (2013). Phylogenetic affiliation of SSU rRNA genes generated by massively parallel sequencing: new insights into the freshwater protist diversity. PLoS ONE 8:e58950. doi: 10.1371/journal.pone.0058950
Tapilatu, Y. H., Grossi, V., Acquaviva, M., Militon, C., Bertrand, J.-C., and Cuny, P. (2010). Isolation of hydrocarbon-degrading extremely halophilic archaea from an uncontaminated hypersaline pond (Camargue, France). Extremophiles 14, 225–231. doi: 10.1007/s00792-010-0301-z
Tessier, E., Garnier, C., Mullot, J.-U., Lenoble, V., Arnaud, M., Raynaud, M., et al. (2011). Study of the spatial and historical distribution of sediment inorganic contamination in the Toulon bay (France). Mar. Pollut. Bull. 62, 2075–2086. doi: 10.1016/j.marpolbul.2011.07.022
Tronczynski, J. (2005). Analyse de contaminants organiques (PCB, OCP, HAP) dans les sédiments marins. Plouzané: IFREMER.
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703.
Wenning, R. J. (2005). Use of Sediment Quality Guidelines and Related Tools for the Assessment of Contaminated Sediments. Pensacola, FL: SETAC Press.
Widdel, F., and Rabus, R. (2001). Anaerobic biodegradation of saturated and aromatic hydrocarbons. Curr. Opin. Biotechnol. 12, 259–276. doi: 10.1016/S0958-1669(00)00209-3
Xie, Y., Wang, J., Wu, Y., Ren, C., Song, C., Yang, J., et al. (2016). Using in situ bacterial communities to monitor contaminants in river sediments. Environ. Pollut. 212, 348–357. doi: 10.1016/j.envpol.2016.01.031
Keywords: microbial communities, PAH, chronic contamination, coastal sediment, functional diversity
Citation: Jeanbille M, Gury J, Duran R, Tronczynski J, Ghiglione J-F, Agogué H, Saïd OB, Taïb N, Debroas D, Garnier C and Auguet J-C (2016) Chronic Polyaromatic Hydrocarbon (PAH) Contamination Is a Marginal Driver for Community Diversity and Prokaryotic Predicted Functioning in Coastal Sediments. Front. Microbiol. 7:1303. doi: 10.3389/fmicb.2016.01303
Received: 17 May 2016; Accepted: 08 August 2016;
Published: 19 August 2016.
Edited by:
Belinda Ferrari, University of New South Wales, AustraliaReviewed by:
Dennis A. Bazylinski, University of Nevada, Las Vegas, USACopyright © 2016 Jeanbille, Gury, Duran, Tronczynski, Ghiglione, Agogué, Saïd, Taïb, Debroas, Garnier and Auguet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathilde Jeanbille, bWF0aGlsZGUuamVhbmJpbGxlQHVuaXYtcGF1LmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.