 Khaled F. A. Abdelrhman1
Khaled F. A. Abdelrhman1 Giovanni Bacci1
Giovanni Bacci1 Cecilia Mancusi2
Cecilia Mancusi2 Alessio Mengoni1*Fabrizio Serena2Alberto Ugolini1
Alessio Mengoni1*Fabrizio Serena2Alberto Ugolini1- 1Dipartimento di Biologia, Università di Firenze, Sesto Fiorentino, Italy
- 2Agenzia Regionale per la Protezione Ambientale della Toscana, Livorno, Italy
Introduction
In the last years the microbial communities (microbiota) associated with the digestive tract of animals have been subjected to wide research interest (Ley et al., 2008; Zhu et al., 2010; Huttenhower et al., 2012). The presence of functional relationship between the host and the associated microbiome (the genes and genomes of the microbiota) has been highlighted, and the new term of hologenome has been proposed to refer to the set of functions (genes) of host and microorganisms associated with it (Zilber-Rosenberg and Rosenberg, 2008). The study of model animals has revealed roles for the microbiome in adaptive immunity development and in host physiology, ranging from mate selection to skeletal biology and lipid metabolism (Ley et al., 2008; Kostic et al., 2013; Du Toit, 2016). For vertebrates, most of the studies on gut microbiota and microbiome have been performed in mammals (i.e., mouse, rat and humans) and in fishes (as the model Danio rerio) (Huttenhower et al., 2012; Kostic et al., 2013). Recently, microbiotas and microbiomes of non-model organisms have started to be investigated with the aim to shed light on animal-associated microbial diversity (Keenan et al., 2013; Mengoni et al., 2013; Cahill et al., 2016) and to potentially discover new biotechnologically important microbial strains (Papaleo et al., 2012; Sanchez et al., 2012).
Sea turtles (Testudines, Reptilia) occur in oceanic and neritic habitats, from the tropics to subarctic waters, and venture onto terrestrial habitats to nest or bask in tropical and temperate latitudes. Sea turtle populations around the world have dwindled and, in many places, continue to decline (Wallace et al., 2010). Caretta caretta L. (Loggerhead Turtle) is distributed throughout the subtropical and temperate regions of the Mediterranean Sea and Pacific, Indian, and Atlantic Oceans. Loggerhead Turtle is classified as Vulnerable A2b in the IUCN Red List (http://www.iucnredlist.org/details/3897/0). The Loggerhead Turtle plays important roles in maintaining marine ecosystem (Bjorndal and Jackson, 2002; Bolten and Witherington, 2003). These roles range from maintaining productive coral reef ecosystems to transporting essential nutrients from the oceans to beaches and coastal dunes. However, in spite of the considerable importance for the study of vertebrates, few studies only are present on microbial communities associated with sea turtles (Ferronato et al., 2009; Sarmiento-Ramírez et al., 2014; Yuan et al., 2015) and no reports on gut microbial communities.
The aim of this work is the characterization, for the first time, of the gut microbiota of the sea turtle C. caretta, to shed a preliminary light on its features with respect to other reptiles and to marine vertebrates. Both feces and intestine samples were taken to have the wider overview of gut microbiota taxonomic composition.
Links to Deposited Data
The sequences dataset (Table 1) was deposited in the GenBank database (URL: http://www.ncbi.nlm.nih.gov/bioproject/; Bioproject PRJNA314462, Biosample accessions SAMN04508196-SAMN04508205). Users can download and use the data freely for research purpose only with acknowledgment to us and quoting this paper as reference to the data.
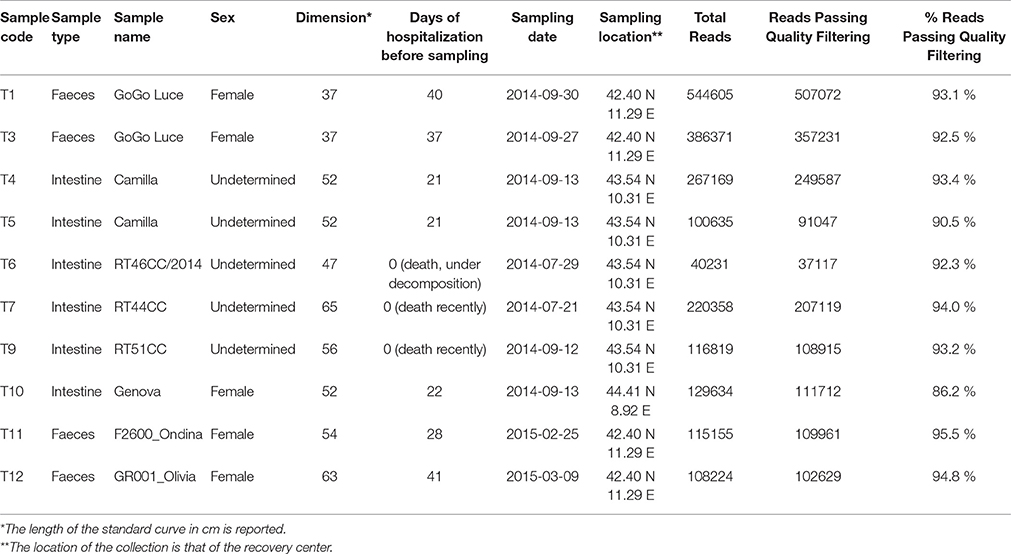
Table 1. Samples details and sequencing statistics.
Materials and Methods
Sampling and Sequence Production
Samples of feces and intestine of C. caretta were collected in the years 2014 and 2015, from different individuals stranded or accidentally caught along the Tyrrhenian sea coast in Tuscany and Liguria regions (Italy). Animals were hosted in the recovery centers associated with network of the Tuscan Observatory for Biodiversity. A total 10 samples of eight individuals was analyzed (Table 1). The samples consisted of four samples of feces (T1, T3, T11, T12) and six cloacal contents and intestine sections (colorectal) (T4, T5, T6, T7, T9, T10). Intestine sections were collected from animals stranded or dead in the recovery centers, immediately after retrieval. Faeces were collected immediately after deposition from living animals in hospitalized conditions in the recovery centers. T1 and T3 were feces from the same individual (“GoGo Luce”) collected in different days (at 37 and 40 days after hospitalization), as well as T4 and T5 were different portions of cloacal samples from the same individual (“Camilla”). All samples were immediately stored at −20°C prior of the extraction of DNA.
DNA was extracted, simultaneously for all samples, from feces, cloacal contents and gut tissues using the FastDNA™ SPIN Kit for soil (MP Biomedicals, Italy). From the extracted DNA, the bacterial V4 region of 16S rRNA genes was amplified with specific primers (515F, 806R) using the protocol reported in the 16S Metagenomic Sequencing Library Preparation protocol from Illumina (Part # 15044223 Rev. B; URL: http://www.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-libraary-prep-guide-15044223-b.pdf). PCR products were sequenced in a single run using Illumina MiSeq technology with pair-end sequencing strategy with MiSeq Reagent Kit v3. Library preparation and demultiplexing have been performed following Illumina 's standard pipeline (Caporaso et al., 2012).
Raw Data Processing and Statistical Analyses
Raw sequences were clustered into “Operation Taxonomic Units” (OTUs) following the UPARSE pipeline as previously described (Bacci et al., 2015a,b). A pre-processing step was also included using StreamingTrim (Bacci et al., 2014), to remove low-quality reads that can generate errors in downstream analyses. Read pairs were merged using PANDAseq assembler with default settings (Masella et al., 2012). Singletons were removed before the OTU clustering step, which was performed using an identity threshold of 97%. Chimeras were detected and removed by UPARSE during clustering step (“cluster_otus” command). Finally, from OTU cluster, a single representative sequence was selected and used for taxonomical identification by SINA classifier on the latest SILVA dataset available when we performed the analysis (SSURef Nr99 version 119). Reads which were attributed to chloroplast and mithocondria were removed from the OTU table. All steps were implemented with an in-house pipeline available at (https://github.com/GiBacci/o2tab).
Collected 16S rRNA sequences were taxonomically classified using the Ribosomal Database Project classifier with 80% confidence threshold, as the most informative threshold (Masella et al., 2012).
Rarefaction analysis was carried out plotting the number of observed OTUs against the number of reads at genus level (Table S1). Tabulated values were used to produce a rarefaction curve for each sample and estimate diversity values. Specific differences in community composition and the similarity among microbial communities was determined using similarity percentage (simper) analysis and Principal Component Analysis (PCA). Both analyses were performed with the modules present in PAST (PAlaeontological STatistics) ver. 3 software (Hammer et al., 2001).
Ethical Statement
Samples were collected from hospitalized animals (the feces) or dead animals (the intestine samples). All animals were kept in Authorized Recovery Centers (as defined by the Italian regulation).
Results
A total 1882390 reads of all samples of C. caretta passed quality filtering sequences (92.8% of total reads) (Table S1). After OTU assignment (Table S1) rarefaction curves obtained reached or nearly reached a plateau, indicating a satisfactory level of diversity sampling (Figure S1).
Concerning the taxonomic composition (Figure 1) feces samples were dominated by members of phyla Firmicutes (66%), Proteobacteria (23%), Bacteroidetes (6.2%). Within the phylum Firmicutes the class Clostridia was the most abundant (63.20%). The intestine samples were dominated by phyla Firmicutes (87%), Proteobacteria (4.2%) and Bacteroidetes (3.4%). Firmicutes were represented by member of the classes Clostridia (43%) and Bacilli (42.5%). This latter was entirely represented (100%) by order Lactobacillales (Table S1). While the most represented bacterial genera among intestine samples were Vagococcus with 42.3%, and among feces were Clostridium XI 21.3%, and Clostridium sensu strict 14.6% (Table S1). Principal Component Analysis on OTU representation (Figure S2) showed that most of the sample were very similar each other. However, notably the two samples of feces from the same individual (T1 and T3, taken in different times) were separated from the rest of the samples. In particular, for T1 and T3, OTU 3, OTU 4, and OTU 5 (all attributed to Clostridiales) collectively contributed for more than 30% of total variance in the differentiation from the other samples (Table S2). Indeed, T1 and T3 were taken from a young female after few days of hospitalization in the recovery center and may mirror the microbiota of a relatively healthy individual in the wild, while the other samples mainly were from animals kept in the recovery centers for longer times. However, we cannot exclude that T1 and T3 microbiota may represent a phase of rapid changes in gut microbiota due to the change in diet (i.e., artificial feeding in the recovery center), which then may bring to a more stable and homogenous microbiota (present in the other samples) after more days. Sampling of more individuals (healthy) would be needed to clarify this issue.
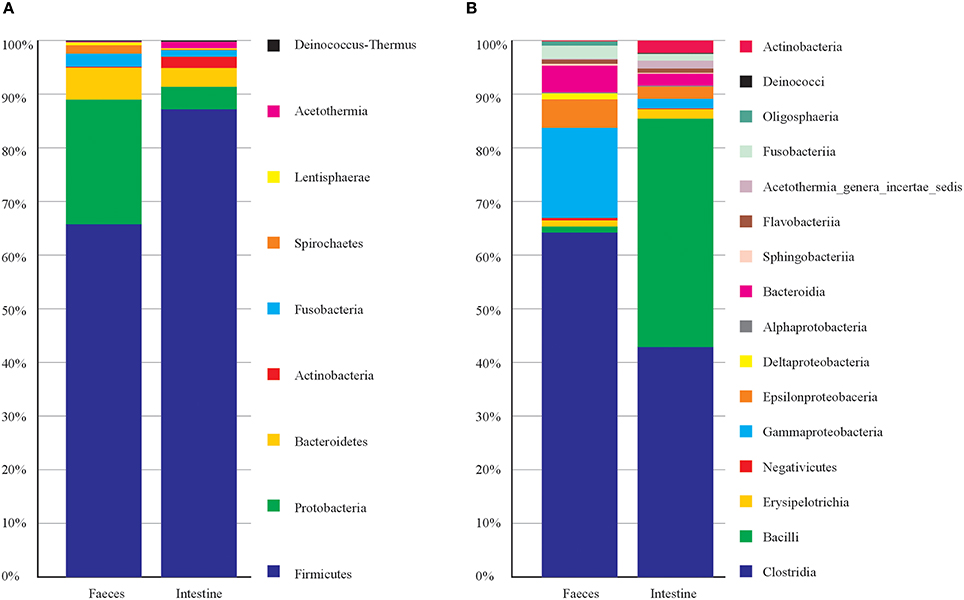
Figure 1. Taxonomic composition of C. caretta gut and faeces microbiota at different taxonomic levels. The percentage of occurrence of each taxon is reported as cumulative bar chart. (A) Phylum; (B) Class. The legend shows the list of taxa from top to bottom of the bars.
Finally, we inspected which taxa of the microbiota mostly contribute to differentiate feces vs. intestine. Results obtained after Simper analysis showed that the genera mostly contributing to differences were Vagococcus (Bacilli, Enteroccoaceae) with 11.92%, Robinsoniella (Class Clostridia) with 6.29%, this latter represented more in intestine samples, Clostridium XI (Class Clostridia) with 7.37 % and represented more in feces samples (Table S2).
Conclusions
This first investigation on the gut microbiota of C. caretta showed a pattern of taxa which include well know members colonizing vertebrate guts. In particular the most abundant phyla found (Firmicutes and Bacteroidetes) are also abundant in the human gut (Ley et al., 2008) as well as in other land vertebrates and reptiles (Costello et al., 2010; Keenan et al., 2013). However, especially in the feces samples, Gammaproteobacteria were particularly present (more than 15% of total reads) including member of Oceanospirillales, Alteromonadaceae, Pseudomonadaceae, Enterobacteriaceae. Moreover, as suggested by T1 and T3 samples, quite important differences in the microbiota could be detected, which may be related to the influence of hospitalization in most of the sampled animals.
The presented data could be used for comparative analyses of vertebrate gut microbiotas.
Author Contributions
KA performed the experiments. GB helped in data analysis. FS, CM performed sampling. AU conceived the work. AM coordinated the work and drafted the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was performed under the project MICROMAR, GoGreen Mare 2014 CAL, Toscany Region (funds assigned to A. Ugolini). KA was supported by a grant from the Libyan Embassy in Italy. We acknowledge the contribution of Valentina Crobe for technical assistance in sample collection and DNA extraction.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01060
Figure S1. Rarefaction analysis on sequencing data of C. caretta gut microbiota.
Figure S2. Principal Component Analysis of OTU abundance in each sample. The percentage of variance displayed by the first two components is reported.
Table S1. Occurrence of Operative Taxonomic Units (OTUs). The number of reads for each OTU is reported for each sample. The taxonomy of each OTU is also reported.
Table S2. Results of SIMPER analysis on taxa occurrence. The taxonomic attribution of OTUs, the percentage of contribution and the cumulative contribution are reported.
References
Bacci, G., Bani, A., Bazzicalupo, M., Ceccherini, M. T., Galardini, M., Nannipieri, P., et al. (2015a). Evaluation of the performances of Ribosomal Database Project (RDP) classifier for taxonomic assignment of 16S rRNA metabarcoding sequences generated from Illumina-Solexa, N. G. S. J. Genomics 3, 36–39. doi: 10.7150/jgen.9204
Bacci, G., Bazzicalupo, M., Benedetti, A., and Mengoni, A. (2014). StreamingTrim 1.0: a Java software for dynamic trimming of 16S rRNA sequence data from metagenetic studies. Mol. Ecol. Resour. 14, 426–434. doi: 10.1111/1755-0998.12187
Bacci, G., Pagoto, E., Passaponti, M., Vannocci, P., Ugolini, A., and Mengoni, A. (2015b). Composition of supralittoral sediments bacterial communities in a Mediterranean island. Ann. Microbiol. 65, 1–13. doi: 10.1007/s13213-014-0829-8
Bjorndal, K. A., and Jackson, J. B. (2002). 10 Roles of sea turtles in marine ecosystems: reconstructing the past. Biol. Sea Turtles 2, 259. doi: 10.1201/9781420040807.ch10
Bolten, A. B., and Witherington, B. E. (2003). Loggerhead Sea Turtles, Smithsonian Books. Washington, DC: Smithsonian Books.
Cahill, P. L., Fidler, A. E., Hopkins, G. A., and Wood, S. A. (2016). Geographically conserved microbiomes of four temperate water tunicates. Environ. Microbiol. Rep. doi: 10.1111/1758-2229.12391. [Epub ahead of print].
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Costello, E. K., Gordon, J. I., Secor, S. M., and Knight, R. (2010). Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J. 4, 1375–1385. doi: 10.1038/ismej.2010.71
Du Toit, A. (2016). Microbiome: restoring healthy growth in infants. Nat. Rev. Microbiol. 14:191. doi: 10.1038/nrmicro.2016.31
Ferronato, B. O., Marques, T. S., Souza, F. L., Verdade, L. M., and Matushima, E. R. (2009). Oral bacterial microbiota and traumatic injuries of free-ranging Phrynops geoffroanus (Testudines, Chelidae) in southeastern Brazil. Phyllomedusa 8, 19–25. doi: 10.11606/issn.2316-9079.v8i1p19-25
Hammer, Ø., Harper, D. A. T., and Ryan, P. D. (2001). PAST: paleontological statistics software package for education and data analysis. Palaeontologia Electronica 41, 9. Available online at: http://palaeo-electronica.org/2001_1/past/issue1_01.htm
Huttenhower, C., Gevers, D., Knight, R., Abubucker, S., Badger, J. H., Chinwalla, A. T., et al. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Keenan, S. W., Engel, A. S., and Elsey, R. M. (2013). The alligator gut microbiome and implications for archosaur symbioses. Sci. Rep. 3:2877. doi: 10.1038/srep02877
Kostic, A. D., Howitt, M. R., and Garrett, W. S. (2013). Exploring host–microbiota interactions in animal models and humans. Genes Dev. 27, 701–718. doi: 10.1101/gad.212522.112
Ley, R. E., Lozupone, C. A., Hamady, M., Knight, R., and Gordon, J. I. (2008). Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788. doi: 10.1038/nrmicro1978
Masella, A., Bartram, A., Truszkowski, J., Brown, D., and Neufeld, J. (2012). PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13:31. doi: 10.1186/1471-2105-13-31
Mengoni, A., Focardi, A., Bacci, G., and Ugolini, A. (2013). High genetic diversity and variability of bacterial communities associated with the sandhopper Talitrus saltator (Montagu) (Crustacea, Amphipoda). Estuarine Coastal Shelf Sci. 131, 75–82. doi: 10.1016/j.ecss.2013.08.011
Papaleo, M. C., Fondi, M., Maida, I., Perrin, E., Lo Giudice, A., Michaud, L., et al. (2012). Sponge-associated microbial Antarctic communities exhibiting antimicrobial activity against Burkholderia cepacia complex bacteria. Biotechnol. Adv. 30, 272–293. doi: 10.1016/j.biotechadv.2011.06.011
Sanchez, L. M., Wong, W. R., Riener, R. M., Schulze, C. J., and Linington, R. G. (2012). Examining the fish microbiome: vertebrate-derived bacteria as an environmental niche for the discovery of unique marine natural products. PLoS ONE 7:e35398. doi: 10.1371/journal.pone.0035398
Sarmiento-Ramírez, J. M., Van Der Voort, M., Raaijmakers, J. M., and Diéguez-Uribeondo, J. (2014). Unravelling the microbiome of eggs of the endangered sea turtle eretmochelys imbricata identifies bacteria with activity against the emerging pathogen fusarium falciforme. PLoS ONE 9:e95206. doi: 10.1371/journal.pone.0095206
Wallace, B. P., DiMatteo, A. D., Hurley, B. J., Finkbeiner, E. M., Bolten, A. B., Chaloupka, M. Y., et al. (2010). Regional management units for marine turtles: a novel framework for prioritizing conservation and research across multiple scales. PLoS ONE 5:e15465. doi: 10.1371/journal.pone.015465
Yuan, M. L., Dean, S. H., Longo, A. V., Rothermel, B. B., Tuberville, T. D., and Zamudio, K. R. (2015). Kinship, inbreeding and fine-scale spatial structure influence gut microbiota in a hindgut-fermenting tortoise. Mol. Ecol. 24, 2521–2536. doi: 10.1111/mec.13169
Zhu, B., Wang, X., and Li, L. (2010). Human gut microbiome: the second genome of human body. Protein Cell 1, 718–725. doi: 10.1007/s13238-010-0093-z
Keywords: microbial communities, gut, Loggerhead Turtle, Caretta caretta, 16S rRNA gene, microbiome, Vagoccoccus
Citation: Abdelrhman KFA, Bacci G, Mancusi C, Mengoni A, Serena F and Ugolini A (2016) A First Insight into the Gut Microbiota of the Sea Turtle Caretta caretta. Front. Microbiol. 7:1060. doi: 10.3389/fmicb.2016.01060
Received: 06 June 2016; Accepted: 23 June 2016;
Published: 07 July 2016.
Edited by:
David Berry, University of Vienna, AustriaReviewed by:
David William Waite, University of Queensland, New ZealandJan Slapeta, University of Sydney, Australia
Copyright © 2016 Abdelrhman, Bacci, Mancusi, Mengoni, Serena and Ugolini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessio Mengoni, YWxlc3Npby5tZW5nb25pQHVuaWZpLml0