 Shailendra K. Verma
Shailendra K. Verma Lalit Batra
Lalit Batra Urmil Tuteja
Urmil Tuteja
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 05 July 2016
Sec. Microbial Immunology
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.01053
Plague is one of the most dangerous infections in humans caused by Yersinia pestis, a Gram-negative bacterium. Despite of an overwhelming research success, no ideal vaccine against plague is available yet. It is well established that F1/LcrV based vaccine requires a strong cellular immune response for complete protection against plague. In our earlier study, we demonstrated that HSP70(II) of Mycobacterium tuberculosis modulates the humoral and cellular immunity of F1/LcrV vaccine candidates individually as well as in combinations in a mouse model. Here, we made two recombinant constructs caf1–lcrV and caf1–lcrV–hsp70(II). The caf1 and lcrV genes of Y. pestis and hsp70 domain II of M. tuberculosis were amplified by polymerase chain reaction. Both the recombinant constructs caf1–lcrV and caf1–lcrV–hsp70(II) were cloned in pET28a vector and expressed in Escherichia coli. The recombinant fusion proteins F1–LcrV and F1–LcrV–HSP70(II) were purified using Ni-NTA columns and formulated with alum to evaluate the humoral and cell mediated immune responses in mice. The protective efficacies of F1–LcrV and F1–LcrV–HSP70(II) were determined following challenge of immunized mice with 100 LD50 of Y. pestis through intraperitoneal route. Significant differences were noticed in the titers of IgG and it’s isotypes, i.e., IgG1, IgG2b, and IgG3 in anti- F1–LcrV–HSP70(II) sera in comparison to anti-F1–LcrV sera. Similarly, significant differences were also noticed in the expression levels of IL-2, IFN-γ and TNF-α in splenocytes of F1–LcrV–HSP(II) immunized mice in comparison to F1–LcrV. Both F1–LcrV and F1–LcrV–HSP70(II) provided 100% protection. Our research findings suggest that F1–LcrV fused with HSP70 domain II of M. tuberculosis significantly enhanced the humoral and cellular immune responses in mouse model.
Yersinia pestis (Y. pestis), etiologic agent of plague is accountable for several outbreaks throughout the world and caused the deaths of millions of people right through the history. In spite of substantial advancement in its prevention and cure, ~50,000 human cases of plague have been reported during the last two decades (WHO, 2005). Worldwide, an average of 4000 human plague cases are reported every year, with outbreaks erupting from time to time which cause mass casualties (Green et al., 2014). Therefore, now, World Health Organization (WHO) considered plague as a re-emerging disease (Schrag and Wiener, 1995; WHO, 2005).
Humans are very prone to plague infection and the etiology of the infection is reliant to the route and source of the infection, consequential in one of the three main clinical forms of plague: bubonic, septicaemic, and pneumonic (Williamson, 2009). Transmission of Y. pestis bacilli to a human begins with an accidental bite of an infected flea and causing the bubonic plague. Y. pestis occasionally reaches to the lungs or inhalation of aerosolized Y. pestis follow-on pneumonic plague. This pneumonic form of plague has the highest fatality rate with a 1–3 days incubation period. This pneumopathy is 100% lethal usually less than 3 days if no treatment is provided (Butler, 2013).
The possible use of aerosolized Y. pestis as a bio-weapon agent is also a serious risk due to its high fatality rate and transmission of human-to-human capability. Centers for Disease Control (CDC) has classified to Y. pestis in a category of biosafety level-3 (BSL-3) organisms. There are antibiotics available for the treatment of plague but recently antibiotic resistant plague bacilli have been reported (Galimand et al., 1997; Guiyoule et al., 2001). It is documented that antibiotic resistant strains of Y. pestis can be prepared in laboratory by transferring the plasmids containing antibiotic resistance genes (Guiyoule et al., 2001; Chain et al., 2004). Therefore, antibiotics are likely to be less effective in the future against intentional/natural outbreaks of plague. In such a scenario, vaccines may be one of the only left over options to control the fatality toll in humans. Therefore, it is imperative to develop an effective and safe plague vaccine.
Currently, there is no licensed vaccine available against plague. Earlier, the strain EV76 of Y. pestis (live attenuated) was used and confers protection in humans (Girard, 1963; Feodorova and Motin, 2011). However, Y. pestis has been considered a major obstacle in its use as live attenuated or killed vaccine due to its genetic instability and several risk factors (side effects) associated with it (Chain et al., 2004; Cui et al., 2014). Recently, many recombinant protein based vaccines candidates have been developed; amongst them only two vaccines, i.e., RypVaxTM and rF1VTM are the most superior in clinical trials. These vaccines mainly rely on a combination of two proteins, F1 and LcrV (Williamson et al., 1995; Heath et al., 1998). These two antigens F1 and LcrV are the main and effective targets which provide protective immunity against plague (Burrows, 1956; Girard, 1963). Recombinant vaccine candidates are generally formulated with aluminum hydroxide gel, a human compatible adjuvant and thus are strong inducers of humoral immune response but poor inducers of cellular immunity (Mannhalter et al., 1985; Dinc et al., 2014). The cellular immune response, however, plays an important role to develop protective immunity against plague (Smiley, 2008). Despite of adequate humoral immune response, F1–V-based vaccines provide poor and inconsistence protection in African Green monkeys, probably due to a weak cellular immune response.
In our earlier studies (Batra et al., 2014), we have shown that F1 and LcrV in formulation with HSP70(II) of M. tuberculosis induced an improved cellular immune response in mouse model. HSP70(II) modulated the cell mediated immunity as there was a significant difference in the expression of IFN-γ and TNF-α in mice immunized with a cocktail of F1, LcrV, and HSP70(II) in comparison to mice immunized with F1+LcrV. In continuation to our efforts to develop a more effective vaccine against plague, here we made two recombinant constructs caf1–lcrV and caf1–lcrV–hsp70(II) successfully expressed in Escherichia coli and purified the recombinants proteins by metal affinity chromatography upto homogeneity. To evaluate the protective potential and immune responses, Balb/C mice were immunized with F1–LcrV and F1–LcrV–HSP70(II). There was a significant difference in IgG response in the sera of immunized mice with F1–LcrV–HSP70(II) in comparison to F1–LcrV. The F1–LcrV–HSP70(II) induced significantly elevated levels of IL-2, IFN-γ, TNF-α in comparison to F1–LcrV vaccine candidate. Immunized mice were experimentally infected with 100 LD50 of Y. pestis (S1 strain) via intra-peritoneal route.
All the protocols for conducted experiments using mice approved by Institutional Animal Ethics Committee (IAEC) of Defence Research and Development Establishment via registration number 37/Go/C/1999/CPCSEA. All the procedures of good laboratory animal care were followed during the course of experiment. The experimental mice were maintained according to the recommendations of committee for the purpose of control and supervision of experiments on animals (CPCSEA), Govt. of India.
Escherichia coli host strain BL21 (DE3) and DH5α were purchased from Invitrogen, USA. Y. pestis (S1 strain), an Indian clinical isolate (Verma et al., 2013; Batra et al., 2014) was collected from DRDE repository (DB182YEPE1) used for challenge experiments. The expression vector pET28a+ was from Novagen, USA. Live Y. pestis cultures and challenge experiments were completed in high containment facility (BSL-3), at DRDE, Gwalior.
Yersinia pestis cultures were grown on Brain Heart Infusion (BHI) agar plate and incubated at 28°C for 48 h. Genomic DNA was extracted by DNeasy Blood and Tissue kit (Qiagen, Germany) according to the manufacturer’s instruction. The genes caf1 and lcrV of Y. pestis were amplified by polymerase chain reaction (PCR). The gene hsp70(II) of M. tuberculosis was amplified by PCR from an earlier cloned plasmid (Batra et al., 2014). The descriptions of used primers are given in Table 1. To prepare the caf1–lcrV construct, first caf1 was ligated with pET28a vector using Nco I and Bam HI and later lcrV ligated in the same plasmid using BamHI and Sal I restriction enzymes. Similarly, in order to prepare the caf1–lcrV–hsp70(II) construct, lcrV reverse primer (R) was replaced with reverse primer (R′) that carrying the Sac I restriction site. First caf1 was ligated with pET28a vector using the same restriction enzymes as mentioned above followed by lcrV using Bam HI and Sac I followed by hsp70(II) using Sac I and Hind III restriction enzymes. These two ligated constructs were transformed in DH5α cells and positive clones were selected using kanamycin (50 μg/ml).

TABLE 1. List of oligos.
The individual recombinant construct corresponding to caf1–lcrV and caf1–lcrV–hsp70(II) cloned in pET28a vector was transformed in BL21(DE3) cells. The positive colonies were selected on Luria-Bertani (LB) agar plates using kanamycin (50 μg/ml). The single colony of each construct, i.e., caf1–lcrV and caf1–lcrV–hsp70(II) was inoculated individually into 5 ml of LB broth and grown at 37°C for overnight. Next day, 500 ml of LB broth was inoculated with 1% of overnight grown culture and incubated at 37°C. When the optical density (OD600) reached upto ~0.6, the cultures were induced with 1 mM isopropylthiogalactoside (IPTG) and grown for 4 h at 37°C. The cultures were pelleted by centrifugation at 10,000 × g for 10 min at 4°C. The cells were lysed in sample buffer and analyzed by SDS–PAGE for the expression. The recombinant fusion proteins F1–LcrV and F1–LcrV–HSP70(II) were purified using 8 M urea by metal affinity chromatography using Ni-NTA columns (Qiagen, USA). The purified antigens were run on 10% SDS–PAGE and anti-histidine tag antibodies (Qiagen, USA) recognized both recombinant proteins in Western blot experiments. The purified proteins were subjected under dialysis. The dialyzed proteins were concentrated using Amicon ultra centrifugal filter devices (Millipore). Protein concentrations were measured by BCA method using BCA kit (Sigma, USA).
In order to test the protective efficacy and immune responses of F1–LcrV and F1–LcrV–HSP70(II) vaccine candidates against Y. pestis, female Balb/C mice (6 week old) were collected from DRDE animal facility, Gwalior. The animals were divided in two batches; each batch was containing three groups (8 mice/group), i.e., Control group; F1–LcrV group; and F1–LcrV–HSP70(II) group (Figure 3A). Batch-I was utilized for the study of humoral immune response (IgG antibody) and challenge experiments against Y. pestis. Batch-II was utilized for the study of cell mediated immune response (cytokine profiling). Animals were immunized subcutaneously with 20 μg/mouse of each purified protein in formulation with aluminum hydroxide gel. The animals of control group received PBS only. The immunizations were given on day 0; day 14; and day 21. All the immunized animals were subjected for blood collection on day 0, 21, and 28 as shown in Figure 3B. Sera were separated from the blood samples for the study of humoral immune response and IgG isotyping.
The end point titers of IgG antibodies were measured by enzyme-linked immunosorbent assay (ELISA) in the sera of immunized and control group animals. In brief, 96 well ELISA plates were coated individually with F1–LcrV and F1–LcrV–HSP70(II) fusion proteins using 100 ng/well each antigen in 0.05 M carbonate buffer, pH 9.6. The coated plates were incubated for overnight at 4°C. After three extensive washings with 0.05% Tween 20 in PBS (PBS-T), the plates were blocked with 3% bovine serum albumin (BSA), incubated for 2 h at 37°C. Plates were washed, test sera collected from immunized and control groups after first and second boosters were serially diluted in triplicate wells (100 μl/well). The plates were incubated for 1 h at 37°C. After five extensive washings, plates were probed with anti-mouse horseradish peroxidase (HRP) labeled IgG (Sigma, USA) raised in rabbit at 1:20,000 dilutions in PBS, incubated for 1 h at 37°C. The plates were washed as earlier and the reaction was developed with o-phenylenediamine dihydrochloride as substrate and after 10 min stopped by 2N H2SO4. The optical density (OD) was measured at 490 nm by a multimode reader (Biotek, USA).
In order to characterize the IgG isotypes, microtiter plates were coated with F1–LcrV and F1–LcrV–HSP70(II) (0.1 μg/well) and blocked with BSA as mentioned above. IgG isotypes sera from animal groups viz; control, F1–LcrV and F1–LcrV–HSP70(II) collected after second booster, added at 1:1000 dilution in PBS in triplicate wells. Plates were incubated for 1 h at 37°C, washed and again incubated with goat anti-mouse isotype specific antibodies IgG1, IgG2a, IgG2b, and IgG3 (Sigma, USA) at 1:1000 dilutions for 1 h at 37°C. Detection of bound isotype specific antibodies was estimated with rabbit anti-goat IgG HRP conjugated (1:5000 dilutions). The reaction was stopped with 2N H2SO4 and optical density (OD) was measured at 490 nm.
Animal groups of batch-II were sacrificed and their spleens were removed in aseptic conditions. To measure the cytokines single cell suspension of spleen cells was prepared. The cells (1 × 106 cells/well) of each group were seeded in triplicates in a culture plate. The cells were stimulated with F1–LcrV or F1–LcrV–HSP70(II) antigens or ConA (5 μg/ml each). After 48 h, the supernatants of cultured spleen cells were collected. The induced level of IL-2; IL-4; IL-10; IFN-γ; and TNF-α were estimated by ELISA kit (BD Biosciences, USA) according to the manufacturer’s instructions. The concentrations of cytokines in picograms per milliliter (pg/ml) were measured with the help of standard curves using recombinant cytokines as standard with the ELISA kit.
Yersinia pestis (S1 strain) from the frozen stocks were streaked on BHI agar plate and incubated at 28°C for 48 h. Single colony from BHI agar plate was further inoculated in 5 ml of BHI broth and grown at 28°C for 48 h. The LD50 of Y. pestis (S1 strain, an Indian clinical isolate) has been determined earlier (Verma et al., 2013; Batra et al., 2014). To determine the protective potential of F1–LcrV and F1–LcrV–HSP70(II) vaccine candidates, animals of batch-I were infected with 100 LD50 of Y. pestis via intraperitoneal route after 1 month of last booster. Infected animals were observed for their survival for 30 days of challenge (Figure 3A).
Results were presented as the means value ± standard deviation (SD). Statistical comparisons among various immunized groups were analyzed by SigmaStat 3.5, one way ANOVA, All Pairwise Multiple Comparison Procedure (Fisher LSD Method). *P < 0.05; **P < 0.01; ***P < 0.001; #P < 0.001. Survival curves were prepared by Kaplan–Meier method using GraphPad Prism 6.0 demo version (****P < 0.0001).
In order to prepare F1–LcrV and F1–LcrV–HSP70(II) proteins, caf1 of 513 bp and lcrV 981 bp of Y. pestis and hsp70(II) of M. tuberculosis were amplified by PCR (Figures 1 and 2) with the help of primers manipulated with compatible restriction site (Table 1). First, caf1 and lcrV were linked with help of Bam HI restriction enzymes (Figure 1A(c)). The caf1–lcrV construct was carrying NcoI and Sal I restriction sites at 3′ and 5′, respectively. Similarly, to prepare the caf1–lcrV–hsp70(II), caf1, lcrV, and hsp70(II) were linked with help of Bam HI and Sac I restriction sites (Figure 2A(d)). The caf1–lcrV–hsp70(II) construct was carrying Nco I and Hind III restriction sites at 3′ and 5′, respectively. The DNA constructs, caf1–lcrV of ~1500 bp encoding F1–LcrV a bivalent protein (~54 kDa) and caf1–lcrV–hsp70(II) of ~2130 bp encoding F1–LcrV–HSP70(II) a trivalent protein (~79 kDa) were cloned in pET28a+ vector using Nco I/Sal I and Nco I/Hind III restriction sites. The individual pET vector carrying the caf1–lcrV and caf1–lcrV–hsp70(II) was transformed in DH5α cells and the positive clones were selected on LB agar plates with kanamycin. The in-frames of the cloned constructs were validated by nucleotide sequencing (Chromous Biotech, India).
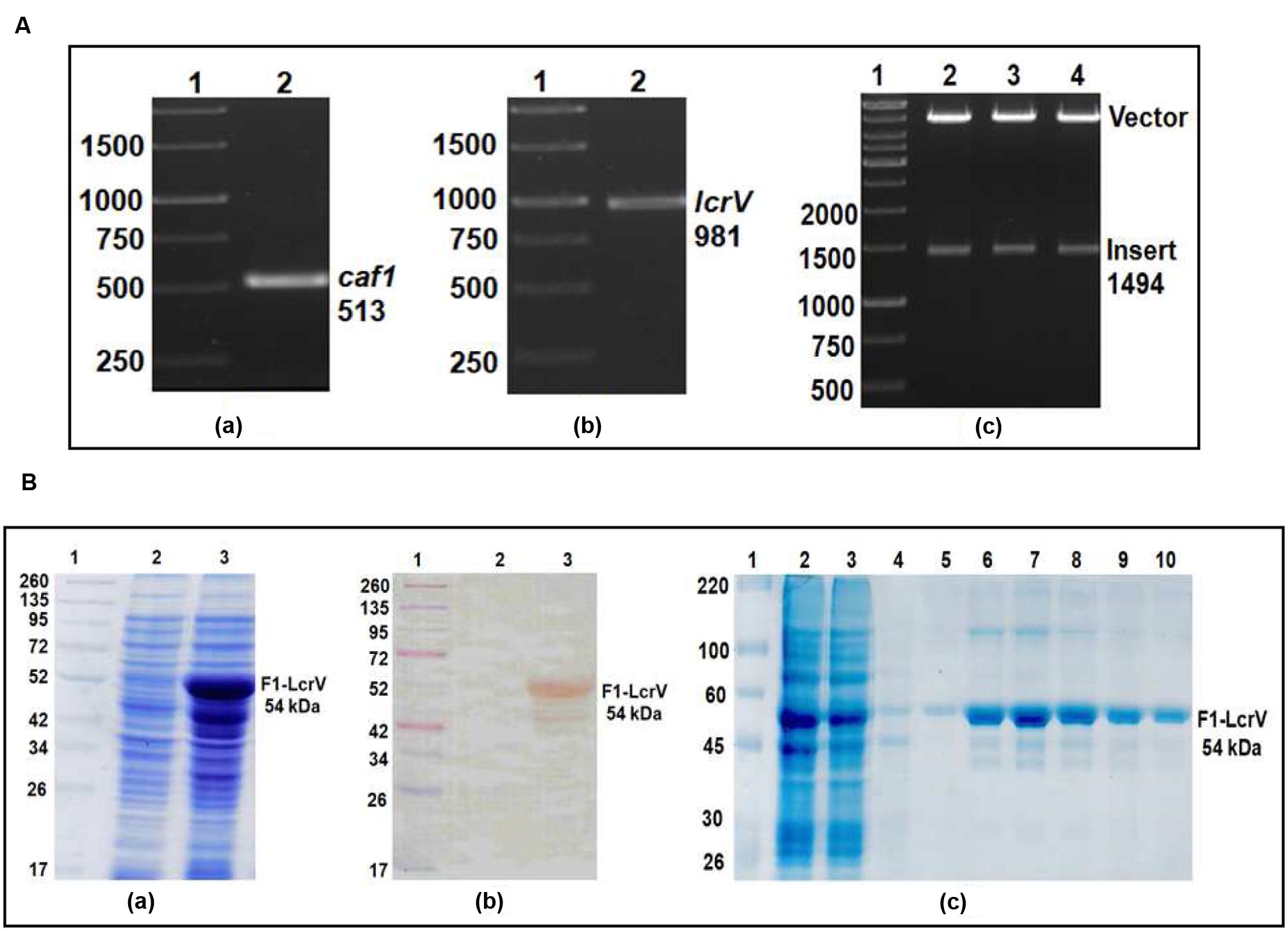
FIGURE 1. (A) PCR amplification of caf1 and lcrV genes of Yersinia pestis & cloning of caf1–lcrV in pET vector. caf1 gene (a); Lane 1, 1Kb DNA ladder; Lane 2, caf1 gene product of 513 bp. lcrV gene (b); Lane 1, 1Kb DNA ladder; Lane 2, lcrV gene product of 981 bp. Screening of positive clones of caf1–lcrV fusion product after ligation in pET28a vector (c); Lane 1, 1 Kb DNA ladder; Lane 2–4, released inserts (caf1–lcrV) of ~1500 bp after restriction digestion from pET vector. (B) Recombinant F1–LcrV fusion protein expression profile and purification. (a) SDS–PAGE analysis of F1–LcrV protein expression. Lane 1, Pre-stained protein marker; Lane 2, Uninduced Escherichia coli cell lysate clone; Lane 3, Induced E. coli cell lysate (b) Western blot analysis of F1–LcrV protein showing reaction with anti HIS antibody. Lane 1, Pre-stained protein marker; Lane 2, Uninduced E. coli cell lysate clone; Lane 3, Induced E. coli cell lysate. (c) Purification of F1–LcrV protein. Lane 1, Protein marker; Lane 2, Cell lysate; Lane 3, Flow through; Lane 4, Wash; Lane 5–10, Eluted fractions of protein showing F1–LcrV protein of 54 kDa.
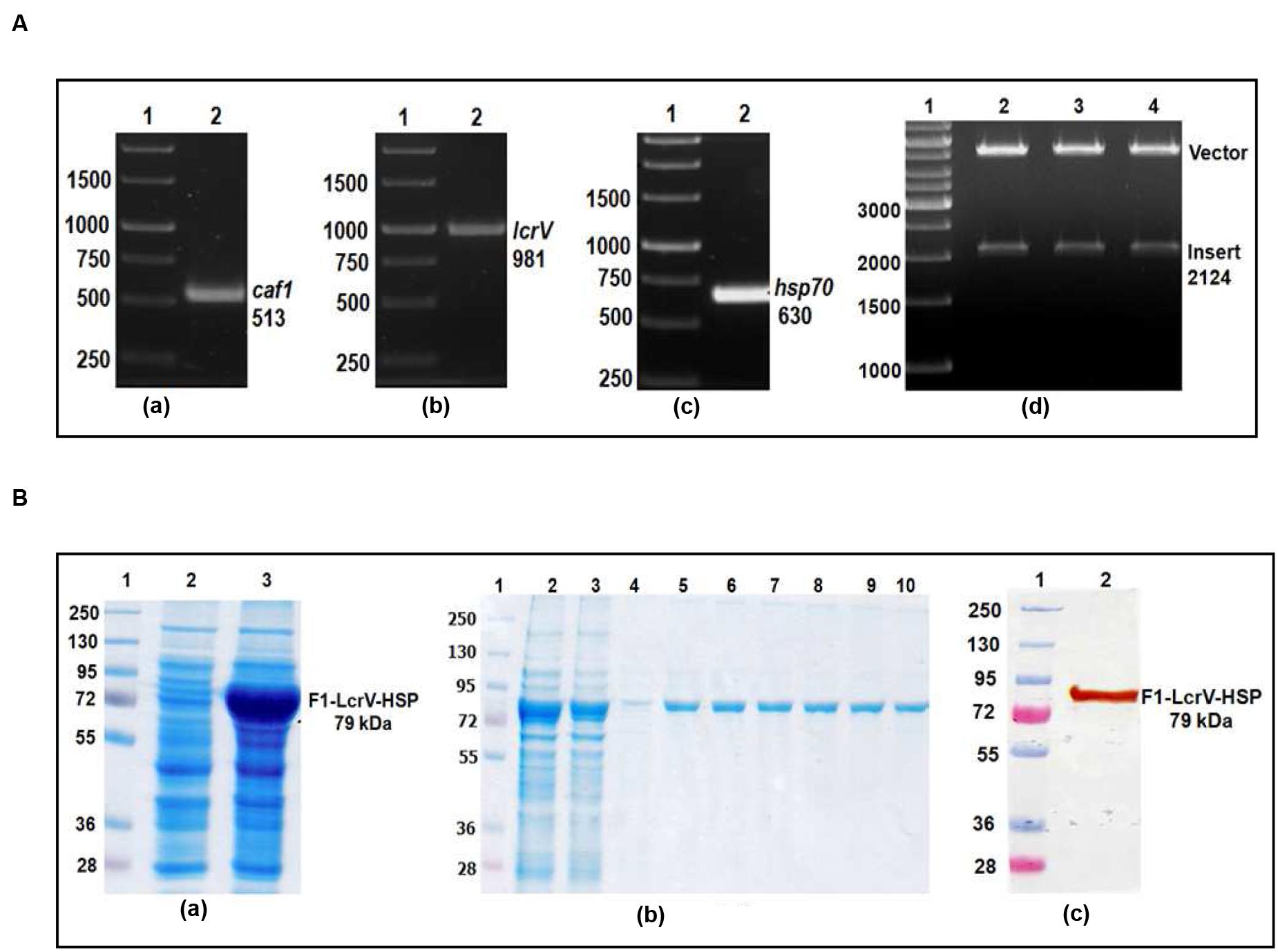
FIGURE 2. (A) PCR amplification of caf1, lcrV genes of Y. pestis and hsp70(II) of Mycobacterium tuberculosis & cloning of caf1–lcrV–hsp70(II) in pET vector. caf1 gene (a); Lane 1, 1 Kb DNA ladder; Lane 2, caf1 gene product of 513 bp: lcrV gene (b); Lane 1, 1 Kb DNA ladder; Lane 2, lcrV gene product of 981 bp: hsp70(II) gene (c); Lane 1, 1 Kb DNA ladder; Lane 2, hsp70(II) gene product of 630 bp. Screening of positive clones of caf1–lcrV–hsp70(II) fusion product after ligation in pET28a vector (d); Lane 1, 1 Kb DNA ladder; Lane 2–4, released inserts (caf1–lcrV) of 2124 bp after restriction digestion from pET vector. (B) Recombinant F1–LcrV-HSP70(II) trivalent fusion protein expression profile & purification. (a) SDS–PAGE analysis of F1–LcrV–HSP70(II) protein expression: Lane 1, Protein marker; Lane 2, Uninduced E. coli cell lysate clone; Lane 3, Induced E. coli cell lysate. (b) Purification of F1–LcrV–HSP70(II) protein: Lane 1, Protein markers; Lane 2, Cell lysate; Lane 3, Flow through; Lane 4, Wash; Lane 5–10, Eluted fractions of purified protein showing F1–LcrV–HSP70(II) fusion protein of 79 kDa. (c) Western blot analysis of purified F1–LcrV–HSP70(II) protein showing reaction with anti HIS antibody: Lane 1, Pre-stained protein marker; Lane 2, purified F1–LcrV–HSP70(II) protein.
One of the positive clones corresponding to caf1–lcrV and caf1–lcrV–hsp70(II) was individually transformed in E. coli host BL-21 (DE3). For the expression of F1–LcrV and F1–LcrV–HSP70(II), 500 ml LB broth was inoculated to each clone and the cultures were induced with IPTG. The expressed proteins F1–LcrV and F1–LcrV–HSP70(II) were analyzed by SDS–PAGE. The proteins SDS–PAGE profiles of un-induced and IPTG-induced cultures for F1–LcrV and F1–LcrV–HSP70(II) are shown in Figures 1B(a) and 2B(a), respectively. In Western blot experiments, anti-histidine antibody recognized F1–LcrV and F1–LcrV–HSP70(II) protein bands corresponding to their molecular weights as shown in Figures 1B(b) and 2B(c). In order to purify the fusion proteins, i.e., F1–LcrV and F1–LcrV–HSP70(II), the recombinant constructs were engineered to bear the 6X-Histine tag at C-terminus. Both the cultures were lysed using native conditions and both the proteins were found present in pellet fractions. The purification was carried out under denaturing conditions by solubilizing the pellet of each culture separately in 8 M urea. Both the proteins were purified by Ni-NTA affinity chromatography using earlier published protocol (Verma et al., 2013). The purified F1–LcrV and F1–LcrV–HSP70(II) proteins were analyzed by SDS–PAGE as shown in Figures 1B(c) and 2B(b), respectively. Both purified proteins were concentrated and the obtained yield of F1–LcrV and F1–LcrV–HSP70(II) was 2.0 and 2.5 mg/L of shake flask cultures, respectively.
To measure the IgG endpoint titers, sera were collected 7 days after first and second injections, respectively, from all the immunized animals as shown Figure 3B. The cut-off value for the assays was calculated as the mean OD (+2 SD) from sera of control group assayed at 1:200 dilutions. The IgG endpoint titers were calculated as reciprocal of the highest serum dilution giving an OD more than the cut-off. The IgG endpoint titer to F1–LcrV was observed 1.28 × 105 and 2.56 × 105 in the sera of F1–LcrV group after first and second booster, respectively. The IgG endpoint titer to F1–LcrV–HSP70(II) was observed 2.56 × 105 and 5.12 × 105 in the sera of F1–LcrV–HSP70(II) group after first and second booster, respectively. A significant difference (#P < 0.001) was observed in the endpoint titer of F1–LcrV–HSP70(II) immunized sera in comparison to F1–LcrV immunized sera (Figure 4A).
FIGURE 3. Representation of animal groups and the schedule of vaccination activities. (A) Prepared groups of Balb/C mice (8/group) for vaccination studies with F1–LcrV and F1–LcrV–HSP70(II) in formulation with alum. (B) Schematic representation of vaccination schedule, blood collection for humoral and cell mediated studies and challenge experiments.
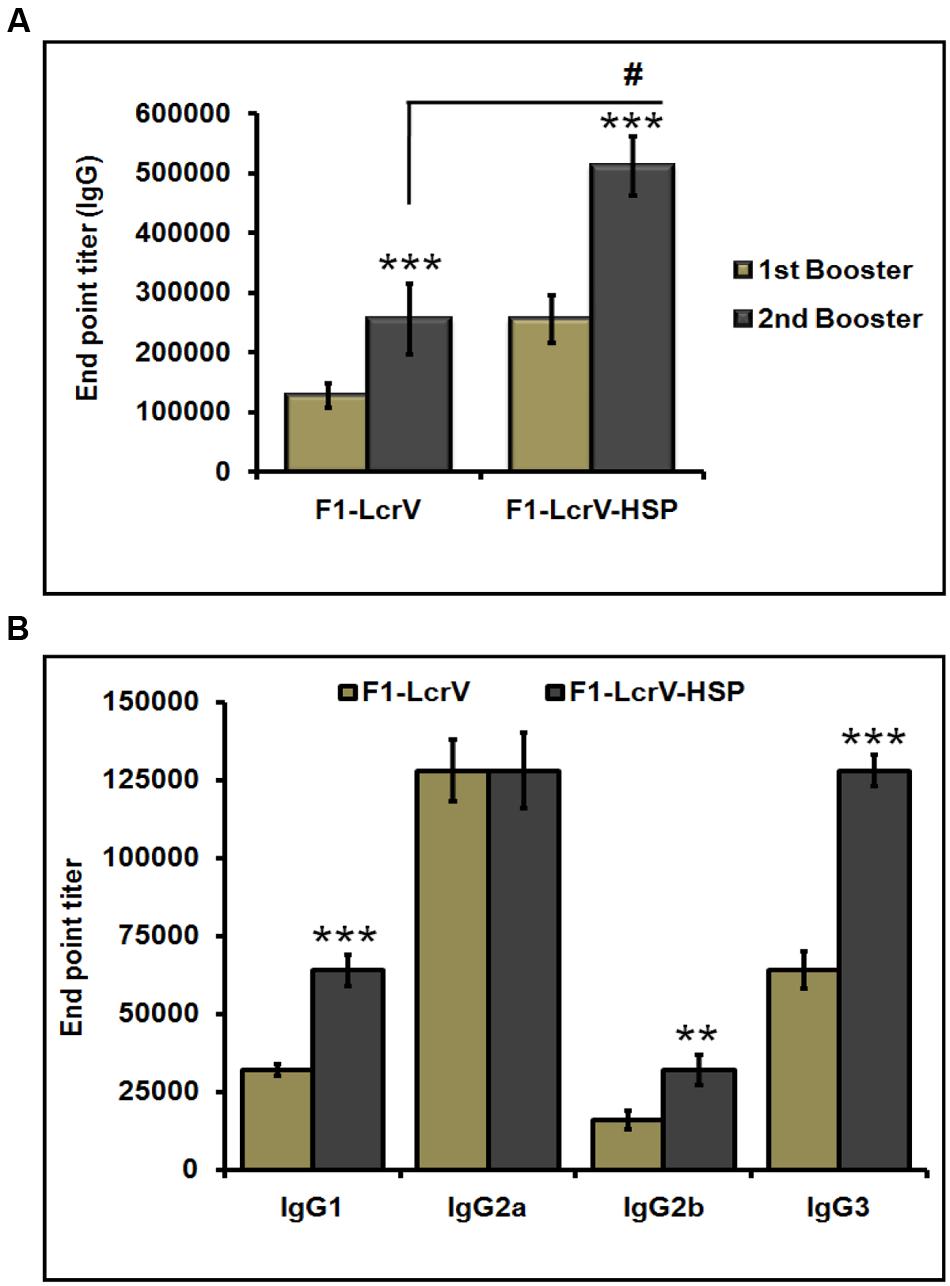
FIGURE 4. End point titers of IgG antibody and IgG isotypes in the sera of immunized mice. (A) IgG end point titers were estimated by ELISA. Serum samples were collected after first and second boosters from vaccinated animal groups, i.e., F1–LcrV and F1–LcrV–HSP70(II)). (B) The IgG isotypes; IgG1, IgG2a, IgG2b, and IgG3 were measured by ELISA. End point titers of IgG isotypes in sera samples collected after second booster from vaccinated mice with F1–LcrV and F1–LcrV–HSP70(II) antigens. Analysis was done by one way ANOVA, all Pairwise Multiple Comparison Procedure (Fisher LSD Method). ** P < 0.01; *** P < 0.001; #P < 0.001.
The endpoint titers of IgG isotypes were also determined similarly as described above. IgG isotypes were measured by ELISA specific to F1–LcrV and F1–LcrV–HSP70(II) in sera samples collected after 7 days of second boosters from all the immunized groups. Significantly elevated levels of IgG1, IgG2b and IgG3 were observed to F1–LcrV–HSP70(II) (Figure 4B) in comparison to F1–LcrV group whereas; there was no difference in the expression of IgG2b to both the groups.
The expression levels of cytokines, i.e., IL-2, IL-4, IL-10, IFN-γ, and TNF-α of all immunized animal groups were measured in supernatants of splenocytes stimulated with required antigen/s. A significance difference (***P < 0.001) was noticed in the expression levels of IL-2 (Figure 5A), IFN-γ (Figure 5B), and TNF-α (Figure 5C) in F1–LcrV and F1–LcrV–HSP70(II) immunized animal groups in comparison to control group. There was no significant difference in the expression levels of IL-4 and IL-10 (data not shown). The splenocytes from all animal groups were induced with ConA and responded non-specifically. We also observed a significant difference (#P < 0.001) in the expression of IL-2, IFN-γ and TNF-α in F1–LcrV–HSP70(II) immunized group in comparison to F1–LcrV group.
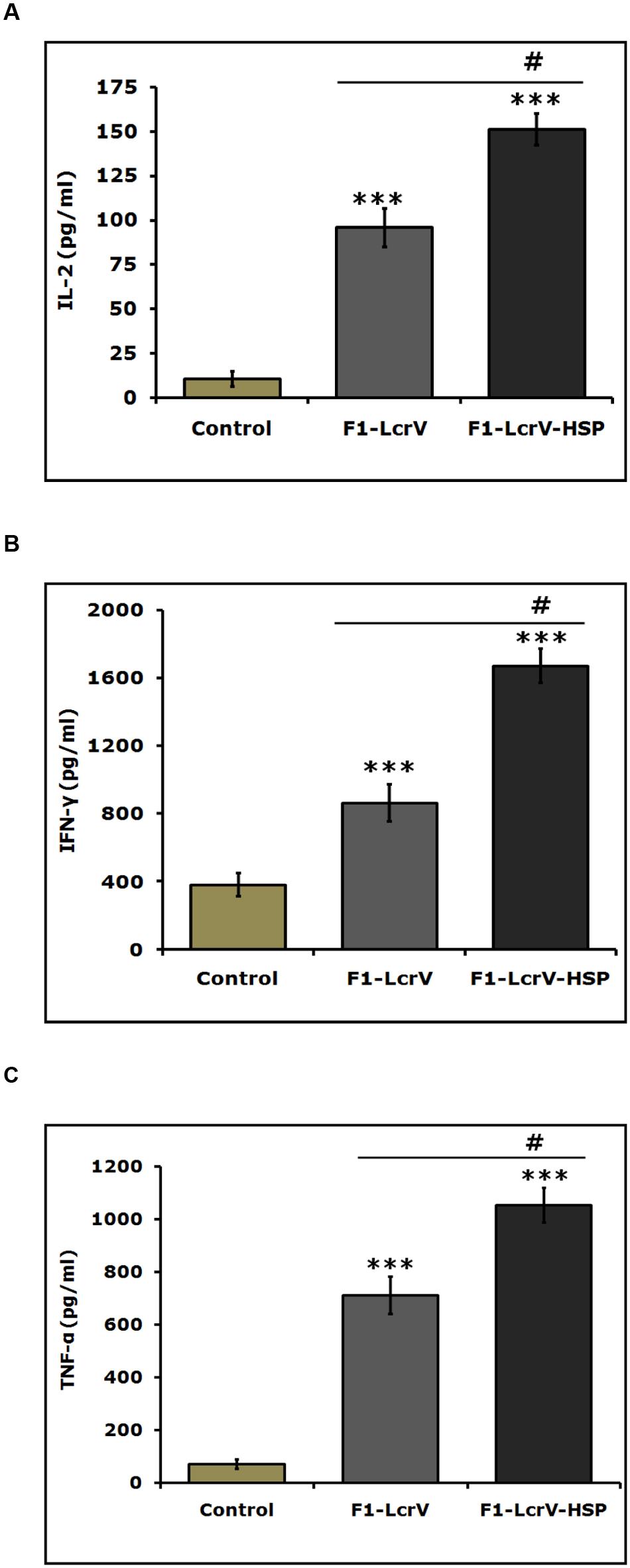
FIGURE 5. Estimation of cytokines in vaccinated animal groups. Splenocytes isolated from immunized BALB/c mice with F1–LcrV and F1–LcrV–HSP70(II) including control group. Splenocytes were stimulated with F1–LcrV and F1–LcrV–HSP70(II) antigens (5 μg/ml each) and after 48 h, the expression levels of cytokines were measured. The expression levels of cytokines (A) IL-2, (B) IFN-γ, and (C) TNF-α were calculated in picograms per milliliter (pg/ml) as shown in graphs. Data represent the average of 8 mice/group ± SD of the results determined. One way ANOVA was applied for analysis. *** P < 0.001; #P < 0.001.
To study the protective efficacy, the immunized animals were challenged with 100 LD50 of Y. pestis (S1 strain) including control group. Survivals of the animals were monitored for 30 days post challenge (Figure 6). Both vaccine candidates F1–LcrV and F1–LcrV–HSP70(II) provided 100% protection from the Y. pestis challenged mice (****P < 0.0001). There was no protection observed in control groups.
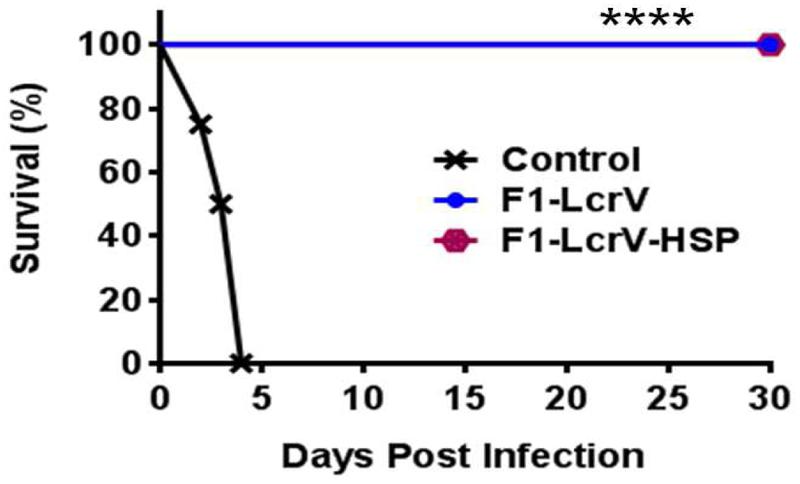
FIGURE 6. Vaccination with F1–LcrV and F1–LcrV–HSP70(II) protects mice against Y. pestis (S1 strain). The vaccinated and control group animals were experimentally infected via i.p. route with 100 LD50 of virulent Y. pestis. The survivals of animals were monitored for 30 days after challenge. The survival curve of both the vaccine candidates F1–LcrV and F1–LcrV–HSP70(II) was determined by Kaplan Meier’s method to calculate percentage survivals (**** P < 0.0001).
Plague is a historic and lethal infectious disease which is so far prevalent in many regions of the recent world and caused by Y. pestis, a potential bio-warfare agent. After the attacks of anthrax in 2001 in USA, it became preferential to stockpile the safe and ideal vaccines against anthrax and plague to protect a mass population against these deadly dangerous bioterror attacks. Nevertheless, there is no plague vaccine has been approved worldwide so far. The possible explanations comprise inconsistence protection, weak stability, poor immunogenicity and the problems in formulation strategies. Modern antigens designing strategies and new vaccine proposals will be helpful to conquer these problems. These new strategies would be of immense attention to develop an effective plague vaccine. In this study, we generated a new trivalent fusion protein that provided full protection against plague in a mouse model.
The F1, a capsular protein and LcrV, a type III secretion protein are the surface-exposed antigens of Y. pestis and have been the leading antigens to develop a subunit vaccine against plague for many years (Anderson et al., 1998; Heath et al., 1998; Williamson, 2009). The vaccine based on these two candidates (F1/LcrV) is poorly immunogenic and mainly induces humoral immune response. Despite of generating high titers of anti-F1/LcrV specific IgG antibodies, some vaccinated animals succumbed to challenge (Bashaw et al., 2007). The protection conferred by live attenuated Salmonella expressing LcrV antigen in mouse model did not show any correlation with LcrV antibody titers (Garmory et al., 2003). Similarly, vaccination of non-human primates specially, African Green monkeys with recombinant F1–LcrV fusion protein impart partial and inconsistent protection against plague and the level of protection did not correlate with the generated humoral immune response either F1 or LcrV antibody titers (Bashaw et al., 2007; Rosenzweig et al., 2011; Williamson and Oyston, 2013). These conclusions, strongly advocate that antibodies titers alone cannot sufficiently envisage the efficacy of F1/LcrV based vaccines.
In order to improve the efficacy of F1/LcrV-based vaccines, several approaches were undertaken like genetic modification of the antigens, e.g., by mutating cysteine amino acid in rF1/V (Goodin et al., 2007) or by deleting the immunosuppressive region of LcrV (DeBord et al., 2006), use of alternate adjuvants like alum, poly-L-lactide microspheres, Monophosphoryl lipids, TiterMax, and flagellin as an molecular adjuvant (Honko et al., 2006; Jones et al., 2006; Feodorova and Motin, 2012), delivery platforms (Garmory et al., 2003; Palin et al., 2007; Yang et al., 2007) and formulations with additional antigen/s that might play the role of an immunomodulator/s or an immunoregulator/s to enhance the immune response.
Heat shock proteins (HSPs) are best known to modulate the immune response (Hauser and Chen, 2003; Pockley, 2003; Robert, 2003; Binder, 2006) and stimulating effective T-cell responses to model antigens (Rico et al., 1998) and pathogen-derived antigens as well (Chen et al., 2000; Huang et al., 2000). Recently, domain II of heat shock protein, HSP70(II) of M. tuberculosis has been proven for inducing the T-cell response by many scientists (Massa et al., 2005; Dhakal et al., 2013). Recombinant fusion constructs Ovalbumin-HSP70(II) elicit CD8+ cytotoxic T lymphocytes specific to ovalbumin (Huang et al., 2000). Suzue and Young (1996) proved that HSP70(II) of M. tuberculosis augment the humoral and cellular immune response to the p24 protein of HIV-1. Subsequently, Wang et al. (2002), reported that the C-terminal portion (amino acids 359–610) of the mHSP70 mainly elicits Th-1 type of cytokines (IL-2, TNF-α, and IFN-γ) and higher serum IgG2a and IgG3 isotypes of antibodies.
In our earlier studies (Batra et al., 2014), we have characterized heat shock protein 70 domain II, HSP70(II) of M. tuberculosis as an immunomodulator to augment the immune response of F1 and LcrV vaccine candidates. It has been proven in our earlier studies that HSP70(II) not only enhances the protective efficacy of F1 and LcrV but also modulates the cellular immune responses and significantly increases the expression levels of IL-2, IFN-γ, and TNF-α in mouse model. HSP70(II) significantly increased the IFN-γ secreting CD4+ and CD8+ T cells in the mice immunized with a cocktail of F1, LcrV, and HSP70(II) comparison to mice immunized with F1and LcrV. In the present study to reduce the labor, cost, and time, for generation of individual protein, we made two fusion constructs, F1–LcrV and F1–LcrV–HSP70(II) and evaluated in mice. We were able to demonstrate that fusion construct of F1–LcrV–HSP70(II) could provide a significant increase in the production of IgG antibody in comparison to F1–LcrV. A significant difference was also noticed in the titers of IgG isotypes, i.e., IgG1, IgG2b, and IgG3 in F1–LcrV–HSP70(II) vaccinated group in comparison to F1–LcrV. We also noticed the significant difference in the expression of IL-2, IFN-γ and TNF-α by the splenocytes in F1–LcrV–HSP70(II) immunized animal group in comparison to F1–LcrV. Both recombinant fusion proteins F1–LcrV and F1–LcrV–HSP70(II) provided full protection against virulent Y. pestis in a mouse model. The results generated with these two fusion proteins were in agreement with our earlier findings (Batra et al., 2014).
The study is worthwhile as the fusion of HSP70(II) with F1–LcrV significantly enhanced the humoral (IgG and its isotypes; IgG1, IgG2b, and IgG3) and cellular (IL-2, IFN-γ, and TNF-α) immune responses. These research findings open up new avenues for vaccine development against plague, however, the findings further need to be evaluated in non-human primates.
SV and UT were involved in designing the experiments, data analysis and manuscript preparation. Experiments performed by SV, UT, and LB.
The authors acknowledge funding from DRDE, Govt. of India.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are thankful to Director, Defence Research and Development Establishment (DRDE), Ministry of Defence, Govt. of India for providing the necessary facilities.
Anderson, G. W. Jr., Heath, D. G., Bolt, C. R., Welkos, S. L., and Friedlander, A. M. (1998). Short- and long-term efficacy of single-dose subunit vaccines against Yersinia pestis in mice. Am. J. Trop. Med. Hyg. 58, 793–799.
Bashaw, J., Norris, S., Weeks, S., Trevino, S., Adamovicz, J. J., and Welkos, S. (2007). Development of in vitro correlate assays of immunity to infection with Yersinia pestis. Clin. Vaccine Immunol. 14, 605–616. doi: 10.1128/CVI.00398-06
Batra, L., Verma, S. K., Nagar, D. P., Saxena, N., Pathak, P., Pant, S. C., et al. (2014). HSP70 domain II of Mycobacterium tuberculosis modulates immune response and protective potential of F1 andLcrV antigens of Yersinia pestis in a mouse model. PLoS Negl. Trop. Dis. 8:e3322. doi: 10.1371/journal.pntd.0003322
Binder, R. J. (2006). Heat shock protein vaccines: from bench to bedside. Int. Rev. Immunol. 25, 353–375. doi: 10.1080/08830180600992480
Burrows, T. W. (1956). An antigen determining virulence in Pasteurella pestis. Nature 177, 426–427. doi: 10.1038/177426b0
Butler, T. (2013). Plague gives surprises in the first decade of the 21st century in the United States and worldwide. Am. J. Trop. Med. Hyg. 89, 788–793. doi: 10.4269/ajtmh.13-0191
Chain, P. S., Carniel, E., Larimer, F. W., Lamerdin, J., Stoutland, P. O., Regala, W. M., et al. (2004). Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U.S.A. 101, 13826–13831. doi: 10.1073/pnas.0404012101
Chen, C. H., Wang, T. L., Hung, C. F., Yang, Y., Young, R. A., Pardoll, D. M., et al. (2000). Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70(II) gene. Cancer Res. 60, 1035–1042.
Cui, Y., Yang, X., Xiao, X., Anisimov, A. P., Li, D., Yan, Y., et al. (2014). Genetic variations of live attenuated plague vaccine strains (Yersinia pestis EV76 lineage) during laboratory passages in different countries. Infect. Genet. Evol. 26, 172–179. doi: 10.1016/j.meegid.2014.05.023
DeBord, K. L., Anderson, D. M., Marketon, M. M., Overheim, K. A., DePaolo, R. W., Ciletti, N. A., et al. (2006). Immunogenicity and protective immunity against bubonic plague and pneumonic plague by immunization of mice with the recombinant V10 antigen, a variant of LcrV. Infect. Immun. 74, 4910–4914. doi: 10.1128/IAI.01860-05
Dhakal, J., Brah, G. S., Agrawal, R. K., Pawar, H. N., Kaur, D., and Verma, R. (2013). Over expression of gene encoding heat shock protein 70 from Mycobacterium tuberculosis and its evaluation as vaccine adjuvant. Indian J. Med. Microbiol. 31, 123–129. doi: 10.4103/0255-0857.115222
Dinc, G., Pennington, J. M., Yolcu, E. S., Lawrenz, M. B., and Shirwan, H. (2014). Improving the Th1 cellular efficacy of the lead Yersinia pestis rF1-V subunit vaccine using SA-4-1BBL as a novel adjuvant. Vaccine 32, 5035–5040. doi: 10.1016/j.vaccine.2014.07.015
Feodorova, V. A., and Motin, V. L. (2011). “Plague vaccines,” in Vaccines against Bacterial Biothreat Pathogens, eds V. A. Feodorova and V. L. Motin (Kerala: Research Signpost), 175–233.
Feodorova, V. A., and Motin, V. L. (2012). Plague vaccines: current developments and future perspectives. Emerg. Microbes. Infect. 1:e36. doi: 10.1038/emi.2012.34
Galimand, M., Guiyoule, A., Gerbaud, G., Rasoamanana, B., Chanteau, S., Carniel, E., et al. (1997). Multidrug resistance in Yersinia pestis mediated by a transferable plasmid. N. Engl. J. Med. 337, 677–680. doi: 10.1056/NEJM199709043371004
Garmory, H. S., Griffin, K. F., Brown, K. A., and Titball, R. W. (2003). Oral immunisation with live aroA attenuated Salmonella enterica serovar Typhimurium expressing the Y. pestis V antigen protects mice against plague. Vaccine 21, 3051–3057. doi: 10.1016/S0264-410X(03)00112-9
Girard, G. (1963). Immunity in plague. Acquisitions supplied by 30 years of work on the “Pasteurella Pestis Ev” (Girard and Robic) Strain. Biol. Med. (Paris) 52, 631–731.
Goodin, J. L., Nellis, D. F., Powell, B. S., Vyas, V. V., Enama, J. T., Wang, L. C., et al. (2007). Purification and protective efficacy of monomeric and modified Yersinia pestis capsular F1-V antigen fusion proteins for vaccination against plague. Protein Expr. Purif. 53, 63–79. doi: 10.1016/j.pep.2006.12.018
Green, M. H., Jones, L., Little, L. K., Schamiloglu, U., and Sussman, G. D. (2014). Yersinia pestis and the three plague pandemics. Lancet Infect. Dis. 14:918. doi: 10.1016/S1473-3099(14)70878-3
Guiyoule, A., Gerbaud, G., Buchrieser, C., Galimand, M., Rahalison, L., Chanteau, S., et al. (2001). Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerg. Infect. Dis. 7, 43–48. doi: 10.3201/eid0701.010106
Hauser, H., and Chen, S. Y. (2003). Augmentation of DNA vaccine potency through secretory heat shock protein-mediated antigen targeting. Methods 31, 225–231. doi: 10.1016/S1046-2023(03)00136-1
Heath, D. G., Anderson, G. W. Jr., Mauro, J. M., Welkos, S. L., Andrews, G. P., Adamovicz, J., et al. (1998). Protection against experimental bubonic and pneumonic plague by a recombinant capsular F1-V antigen fusion protein vaccine. Vaccine 16, 1131–1137. doi: 10.1016/S0264-410X(98)80110-2
Honko, A. N., Sriranganathan, N., Lees, C. J., and Mizel, S. B. (2006). Flagellin is an effective adjuvant for immunization against lethal respiratory challenge with Yersinia pestis. Infect. Immun. 74, 1113–1120. doi: 10.1128/IAI.74.2.1113-1120.2006
Huang, Q., Richmond, J. F., Suzue, K., Eisen, H. N., and Young, R. A. (2000). In vivo cytotoxic T lymphocyte elicitation by mycobacterial heat shock protein 70 fusion proteins maps to a discrete domain and is CD4+ T cell independent. J. Exp. Med. 191, 403–408. doi: 10.1084/jem.191.2.403
Jones, T., Adamovicz, J. J., Cyr, S. L., Bolt, C. R., Bellerose, N., Pitt, L. M., et al. (2006). Intranasal Protollin/F1-V vaccine elicits respiratory and serum antibody responses and protects mice against lethal aerosolized plague infection. Vaccine 24, 1625–1632. doi: 10.1016/j.vaccine.2005.09.052
Mannhalter, J. W., Neychev, H. O., Zlabinger, G. J., Ahmad, R., and Eibl, M. M. (1985). Modulation of the human immune response by the non-toxic and non-pyrogenic adjuvant aluminium hydroxide: effect on antigen uptake and antigen presentation. Clin. Exp. Immunol. 61, 143–151.
Massa, C., Melani, C., and Colombo, M. P. (2005). Chaperon and adjuvant activity of hsp70: different natural killer requirement for cross-priming of chaperoned and bystander antigens. Cancer Res. 65, 7942–7949. doi: 10.1158/0008-5472.CAN-05-0377
Palin, A., Chattopadhyay, A., Park, S., Delmas, G., Suresh, R., Senina, S., et al. (2007). An optimized vaccine vector based on recombinant vesicular stomatitis virus gives high-level, long-term protection against Yersinia pestis challenge. Vaccine 25, 741–750. doi: 10.1016/j.vaccine.2006.08.010
Pockley, A. G. (2003). Heat shock proteins as regulators of the immune response. Lancet 362, 469–476. doi: 10.1016/S0140-6736(03)14075-5
Rico, A. I., Del Real, G., Soto, M., Quijada, L., Martinez-A, C., Alonso, C., et al. (1998). Characterization of the immunostimulatory properties of Leishmania infantum HSP70 by fusion to the Escherichia coli maltose-binding protein in normal and nu/nu BALB/c mice. Infect. Immun. 66, 347–352.
Robert, J. (2003). Evolution of heat shock protein and immunity. Dev. Comp. Immunol. 27, 449–464. doi: 10.1016/S0145-305X(02)00160-X
Rosenzweig, J. A., Jejelowo, O., Sha, J., Erova, T. E., Brackman, S. M., Kirtley, M. L., et al. (2011). Progress on plague vaccine development. Appl. Microbiol. Biotechnol. 91, 265–286. doi: 10.1007/s00253-011-3380-6
Schrag, S. J., and Wiener, P. (1995). Emerging infectious diseases: what are the relative roles of ecology and evolution? Trends Ecol. Evol. 10, 319–324. doi: 10.1016/S0169-5347(00)89118-1
Smiley, S. T. (2008). Current challenges in the development of vaccines for pneumonic plague. Exp. Rev. Vaccines. 7, 209–221. doi: 10.1586/14760584.7.2.209
Suzue, K., and Young, R. A. (1996). Adjuvant-free HSP70(II) fusion protein system elicits humoral and cellular responses to HIV-1 p24. J. Immunol. 156, 873–879.
Verma, S. K., Batra, L., Athmaram, T. N., Pathak, P., Katram, N., Agrawal, G. S., et al. (2013). Characterization of immune responses to yersinia pestis (indian isolate) infection in mouse model. Clin. Cell. Immunol. 4:151. doi: 10.4172/2155-9899.1000151
Wang, Y., Kell, C. G., Singh, M., McGowan, E. G., Carrara, A. S., Bergmeier, L. A., et al. (2002). Stimulation of Th1-polarizing cytokines, C-C chemokines, maturation of dendritic cells, and adjuvant function by the peptide binding fragment of heat shock protein 70. J. Immunol. 169, 2422–2429. doi: 10.4049/jimmunol.169.5.2422
Williamson, E. D., Eley, S. M., Griffin, K. F., Green, M., Russell, P., Leary, S. E., et al. (1995). A new improved sub-unit vaccine for plague: the basis of protection. FEMS Immunol. Med. Microbiol. 3–4, 223–230.
Williamson, E. D., and Oyston, P. C. (2013). Protecting against plague: towards a next-generation vaccine. Clin. Exp. Immunol. 172, 1–8. doi: 10.1111/cei.12044
Keywords: plague, Yersinia pestis, HSP70(II), cytokine, F1–LcrV, F1–LcrV–HSP70(II)
Citation: Verma SK, Batra L and Tuteja U (2016) A Recombinant Trivalent Fusion Protein F1–LcrV–HSP70(II) Augments Humoral and Cellular Immune Responses and Imparts Full Protection against Yersinia pestis. Front. Microbiol. 7:1053. doi: 10.3389/fmicb.2016.01053
Received: 27 April 2016; Accepted: 23 June 2016;
Published: 05 July 2016.
Edited by:
Alexandre Morrot, Federal University of Rio de Janeiro, BrazilReviewed by:
Celio Geraldo Freire De Lima, Federal University of Rio de Janeiro, BrazilCopyright © 2016 Verma, Batra and Tuteja. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shailendra K. Verma, dmVybWFza0BkcmRlLmRyZG8uaW4= c2t2ZHJkZUB5YWhvby5jby5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.