 Xuewei Pan
Xuewei Pan Xiaoli Cui
Xiaoli Cui Fenjiao Zhang
Fenjiao Zhang Yang He
Yang He Lingyan Li
Lingyan Li Hongjiang Yang
Hongjiang Yang- Key Laboratory of Industrial Microbiology, Ministry of Education, Tianjin Key Laboratory of Industrial Microbiology, College of Biotechnology, Tianjin University of Science and Technology, Tianjin, China
Phage therapy requires the comprehensive understanding of the mechanisms underlying the host-phage interactions. In this work, to identify the genes related to Pseudomonas aeruginosa phage K8 receptor synthesis, 16 phage-resistant mutants were selected from a Tn5G transposon mutant library of strain PAK. The disrupted genetic loci were identified and they were related to O-specific antigen (OSA) synthesis, including gene wbpR, ssg, wbpV, wbpO, and Y880_RS05480, which encoded a putative O-antigen polymerase Wzy. The Lipopolysaccharide profile of the Y880_RS05480 mutant was analyzed and shown to lack the O-antigen. Therefore, the data from characterization of Y880_RS05480 by TMHMM and SDS-PAGE silver staining analysis suggest that this locus might encode Wzy. The complete phage K8 genome was characterized as 93879 bp in length and contained identical 1188-bp terminal direct repeats. Comparative genomic analysis showed that phage K8 was highly homologous to members of the genus PaP1-like phages. On the basis of our genetic findings, OSA of P. aeruginosa PAK is proven to be the receptor of phage K8. The highly conserved structural proteins among the genetic closely related phages suggest that they may recognize the same receptor.
Introduction
Pseudomonas aeruginosa is an opportunistic pathogen present in diverse environmental niches. It is also one of the most common causes of healthcare-associated infections including pneumonia, bloodstream infections, urinary tract infections, and surgical site infections, accounting for about 9% of all nosocomial infections (Emori and Gaynes, 1993; Lister et al., 2009). Antibiotics are widely used for prevention and control of the infections caused by P. aeruginosa, leading to the emergence and the increasing prevalence of multidrug-resistant P. aeruginosa clinical isolates (Karaiskos and Giamarellou, 2014). There is an urgent need to discover and develop new classes of antibiotics and alternatives to the conventional drugs. Among the potential candidate antibacterials, lytic phages can kill bacteria efficiently and specifically, bringing great promises to combat drug-resistant pathogens (Kutter et al., 2010; Chan et al., 2013).
Phages are the most abundant biological entities present in various environmental habitats of the Earth’s biosphere, providing a reliable unlimited resource for possible phage applications (Srinivasiah et al., 2008). To date, 1630 phage genomes have been sequenced1. One hundred fifty were found to be P. aeruginosa phages, including 47 Myoviridae phages, 42 Podoviridae phages, 46 Siphoviridae phages, Levivirus Pseudomonas phage PP7 and PRR1 (Olsthoorn et al., 1995; Ruokoranta et al., 2006), Inovirus Pseudomonas phage Pf1 (Holland et al., 2006) and Pf3 (Luiten et al., 1985), and 11 uncharacterized dsDNA phage such as PA11 (Kwan et al., 2006) and vB_PaeP_Tr60_Ab31 (Latino et al., 2014). P. aeruginosa phages of each family can be further grouped into genus level. Podoviridae phages mainly consist of N4-like phages, phiKMV-like phages, T7-like phages, and LUZ24-like phages; Siphoviridae phages have four major genera, including D3112-like phages, D3-like phages, Yu-like phages, and Mu-like phages; Myoviridae phages are classified into five genera such as phiKZ-like phages, PaP1-like phages, P2-like phages, PB1-like phages, and KPP10-like phages1.
Though great progress has been made in phage discovery and applications, the underlying mechanisms of host–phage interactions still remain to be elucidated. In this study, P. aeruginosa phage K8 was selected for biological characteristic analysis, phage genome annotation, and screening for host genes encoding the phage receptors.
Materials and Methods
Bacterial Strains, Phage, Plasmids, and Growth Conditions
Phage K8 was first isolated using P. aeruginosa PAK as the indicator strain from Haihe river located in Tianjin, China (Li et al., 2010). Bacterial strains were grown in LB medium at 37°C. When appropriate, the medium was supplemented with ampicillin (100 μg/ml) or gentamicin (10 μg/ml) for Escherichia coli strains cultivation; and carbenicillin (150 μg/ml) or gentamicin (100 μg/ml) for P. aeruginosa strains cultivation. P. aeruginosa SK2, SK5, SK15, SK16, SK21, SK23, SK24, SK28, SK41, SK45, SK73, SK75, SK88, SK91, SK92, and SK98 were phage K8-resistant mutants derived from the Tn5G transposon mutagenesis bank of strain PAK. Vector pUCP18 was used to express the target genes in the isolated phage-resistant mutants for complementation tests (Table 1).

TABLE 1. Strains, phage, and plasmids used in this study.
Transmission Electron Microscopy (TEM)
Phage particles were purified as described previously (Yang et al., 2010). In brief, phage K8 lysate (about 1011 pfu/ml) was treated with DNase I (5 μg/ml) and RNase A (5 μg/ml) at 37°C for 1 h. With the addition of 0.1 M NaCl, the mixture was kept on ice bath for 1 h and spun at 12000 × g for 20 min. The collected supernatant was supplemented with PEG6000 (10%) and stored at 4°C overnight before centrifuging at 12000 × g for 20 min. Phage pellet was suspended with 2% ammonium acetate (pH 7.0) and filtrated with Amicon-100 filter. The purified phages were adsorbed onto a carbon-coated copper grid for 5 min, and subsequently negatively stained with 2% phosphotungstic acid (pH 6.7) for 5 min. Morphology observation was carried out with a JEM-1400 transmission electron microscope operating at 100 kV.
Latent Period and Burst Size Analysis
Latent period and burst size of phage K8 was determined by one-step growth experiment described previously (Yang et al., 2010). Briefly, PAK cells were harvested from the 50 ml culture (OD600 at 0.6) and suspended in 0.5 ml LB medium. The suspension was mixed with 0.5 ml appropriately diluted phage K8 solution at a MOI (multiplicity of infection) of 0.0001. After adsorption for 1 min, the mixture was spun at 13000 × g for 30 s to remove free phage particles. The pellet was resuspended in 100 ml LB medium for immediate cultivation. At 5 min intervals, samples were taken and the infection centers were determined by the double-layer agar plate method (Li et al., 2010).
Screening of Phage Resistant Mutants
Tn5G transposon was used to mutagenize P. aeruginosa PAK to construct an insertional mutant library as described earlier (Nunn and Lory, 1992). After mating, a fraction of the mutant bank was mixed with the stock solution of phage K8 and incubated for 4 h with a shaking speed of 220 rpm. Aliquots were plated onto L-agar medium with 100 μg/ml gentamicin and 100 μg/ml ampicillin. The grown colonies were selected as the phage-resistant mutants. The mutants were confirmed by the spotting assay and the double-layer plate method as described previously (Li et al., 2010). The adsorption rate of the phage-resistant mutants was determined as described previously (Yang et al., 2010).
Identification of the Transposon Insertion Sites by Inverse PCR
Inverse PCR was performed as described previously (Wang et al., 1996). In brief, chromosomal DNA was isolated from the phage-resistant mutants, digested with the restriction enzyme TaqI or PstI, self-ligated, and amplified using primers OTn1 and OTn2, Tn1 and Tn2, or F1 and R1, respectively (Table 2). The PCR products was sequenced directly or cloned into pGEM-T Easy vector for sequencing. The obtained sequences were analyzed by searching the genome database of Pseudomonas strains2.
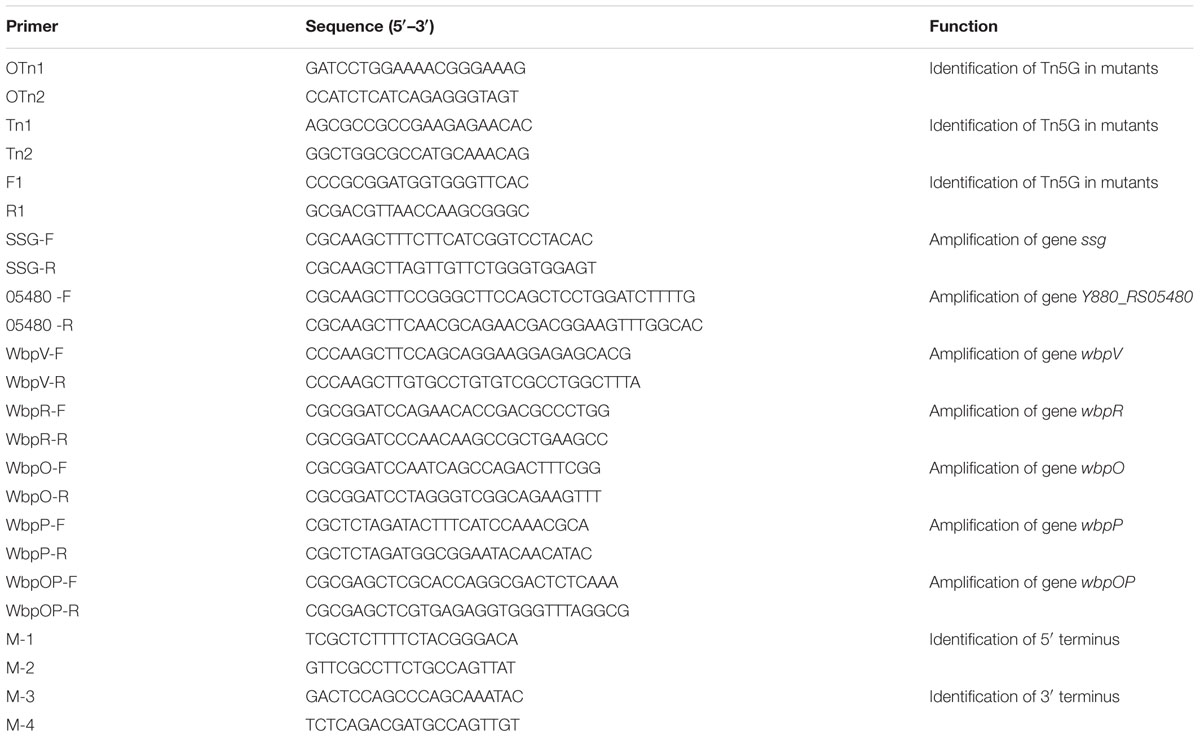
TABLE 2. Primer used in this study.
The primers were designed to amplify the target genes disrupted in the phage-resistant mutants. The PCR products were subsequently cloned into the multiple cloning sites of plasmid pUCP18. The recombinant plasmids were transformed into the phage-resistant mutants. Sensitivity to phage K8 was tested in the transformants with the spotting and the double-layer method (Li et al., 2010).
Transmembrane Helices Prediction and Lipopolysaccharide (LPS) Profile Analysis
The transmembrane helices of the putative O-antigen polymerase encoded by gene Y880_RS05480 was analyzed by the software TMHMM 2.03 (Krogh et al., 2001).
Lipopolysaccharide (LPS) was extracted using the hot water-phenol method as described previously (Westphal and Jann, 1965). In brief, PAK cells in 100 ml culture (OD600 at 1.0) were harvested and subjected to the treatments of hot water and phenol sequentially. The residual phenol was removed by dialysis in water. The LPS solution was concentrated by dialysis in 40% PEG6000 solution. DNase I (10 μg/ml) and RNase A (100 μg/ml) were added to remove the residual nucleic acid in the LPS samples. LPS was analyzed by 12% SDS-PAGE and visualized by the silver staining method as described previously (Fomsgaard et al., 1990).
Biofilm Assay
Biofilm production was assessed in wild-type strain PAK and the phage-resistant mutants as described previously (Brencic et al., 2009). In brief, LB medium was diluted three times and used for biofilm production. The cultivation was carried out in 96 well plates and incubated at 37°C for 48 h. Biofilm was quantitatively measured with the crystal violet staining method (Djordjevic et al., 2002).
Phage Genome Sequencing and Annotation
Purified phage particles were subjected to genomic DNA extraction according to the method described previously (Yang et al., 2010). Genome sequencing was carried out by Hiseq Illumina 2500 in GENEWIZ, Inc., China4. The adaptor sequences were removed with the software Trimmomatic v0.30 (Bolger et al., 2014). A total of 7803122 reads and 773403396 bp were obtained as clean data without any uncertain bases. The sequences were assembled with the software Velvet_v1.12.10 (Zerbino and Birney, 2008). DNA Master was used for the phage genome annotation5 by searching against the non-redundant protein database (nr) from NCBI (Wheeler et al., 2003). The software tRNAscan-SE v1.21 was used to predict tRNA genes (Schattner et al., 2005). GC content was determined with the software DNAStar. The GC skew was analyzed by the software DNAPlotter (Carver et al., 2009).
Identification of Phage Genome Termini
Sequencing depth was analyzed across the assembled genome to find the high-frequency sequences (HFSs) which might represent the phage genome termini (Li et al., 2014). Restriction enzyme cleavage sites and restriction mapping in linear or circular genome sequences were simulated using the software DNA Master. The fragments containing the possible 3′ or 5′ terminus of the K8 genome were purified by the agarose gel electrophoresis. The purified DNA fragments were treated with the Klenow fragment and T4 DNA ligase sequentially. PCR was performed with the specific primers (Table 2) and the PCR products were sequenced to identify the genome termini. The assembled genome was curated and the complete genome sequence was deposit in GenBank in NCBI with the accession number KT736033.
Comparative Genomic Analysis
The homology search of the K8 genome sequence was performed against the NCBI nucleotide database. Four phage genome sequences were selected for comparison analysis by the software Mauve, including PaP1, JG004, PAK_P2, and vB_PaeM_C2-10_Ab1 (Darling et al., 2004). The tail fiber proteins of phage K8 were analyzed and the phylogenetic trees was constructed with MEGA5 (Tamura et al., 2011).
Results
Characteristics of Phage K8
Purified phage K8 particles were negatively stained using 2% phosphotungstic acid and observed by TEM. The obtained images showed that phage K8 has an icosahedral head structure connected with a contractile tail. The phage head was about 76.0 nm in diameter and the tail was about 122.0 nm in length (Figure 1A). The observed morphology indicated that phage K8 should be tentatively classified as a member of Myoviridae family. The progeny production of phage K8 was characterized by one-step growth experiment with a MOI of 0.0001. As inferred from the triphasic curve, the latent period was about 20 min and the burst size was about 46.3 pfu/infection center (Figure 1B).
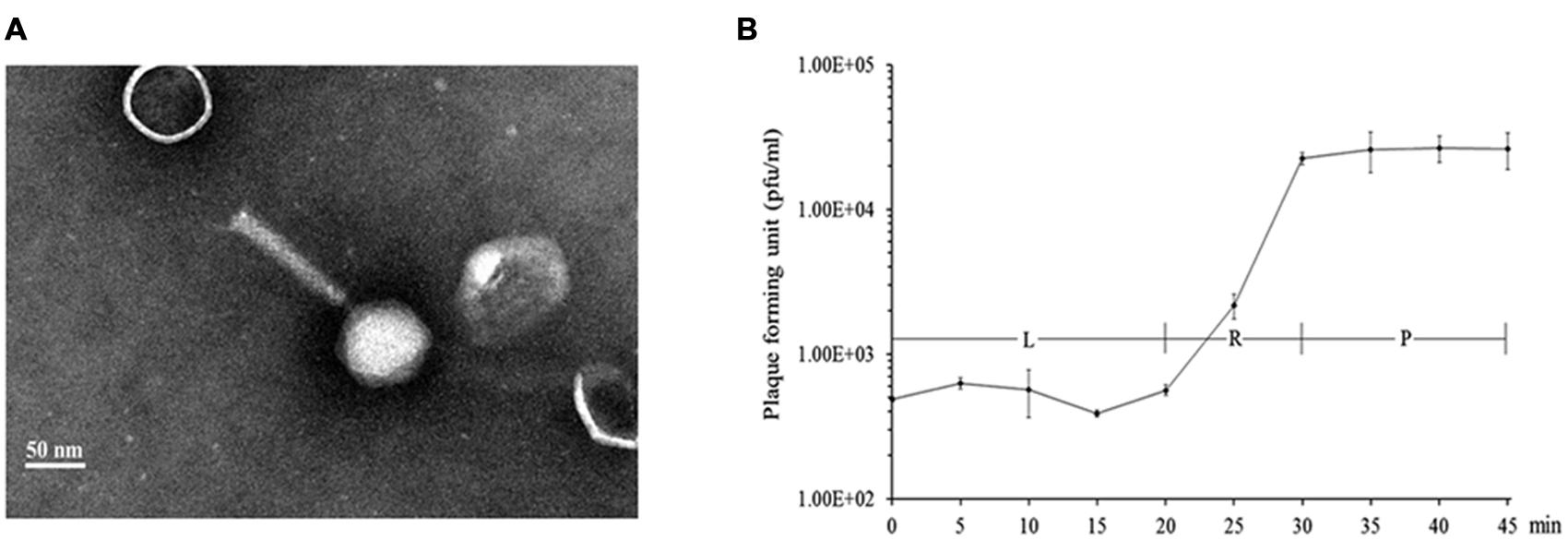
FIGURE 1. Characteristics of phage K8. (A) Electron microscopic image of negatively stained phage K8 particle. The scale bar represents 50 nm. (B) One-step growth curve of phage K8. L, latent phase; R, rise phase; P, plateau phase.
Identification of Phage Receptor Related Genes
A random Tn5G transposon library of P. aeruginosa PAK was constructed to identify the host genes involved in the phage infection process. A total of 16 phage K8 resistant mutants were isolated. With inverse PCR, five different genes were identified disrupted in the mutated strains, including two mutants with the inactivated gene identical to wbpR gene of strain LESB58, four mutants with the inactivated gene identical to wbpV gene of strain PA96, 1 mutant with the inactivated gene identical to ssg gene of strain PAO1 (Veeranagouda et al., 2011), two mutants with the inactivated gene identical to wbpO gene of strain PA96, and seven mutants with the inactivated gene Y880_RS05480 encoding a probable O-antigen polymerase with 22.1% identity to Wzy (AIG62435) of E. coli at the amino acid sequence level (Figure 2 and Supplementary Table S1).
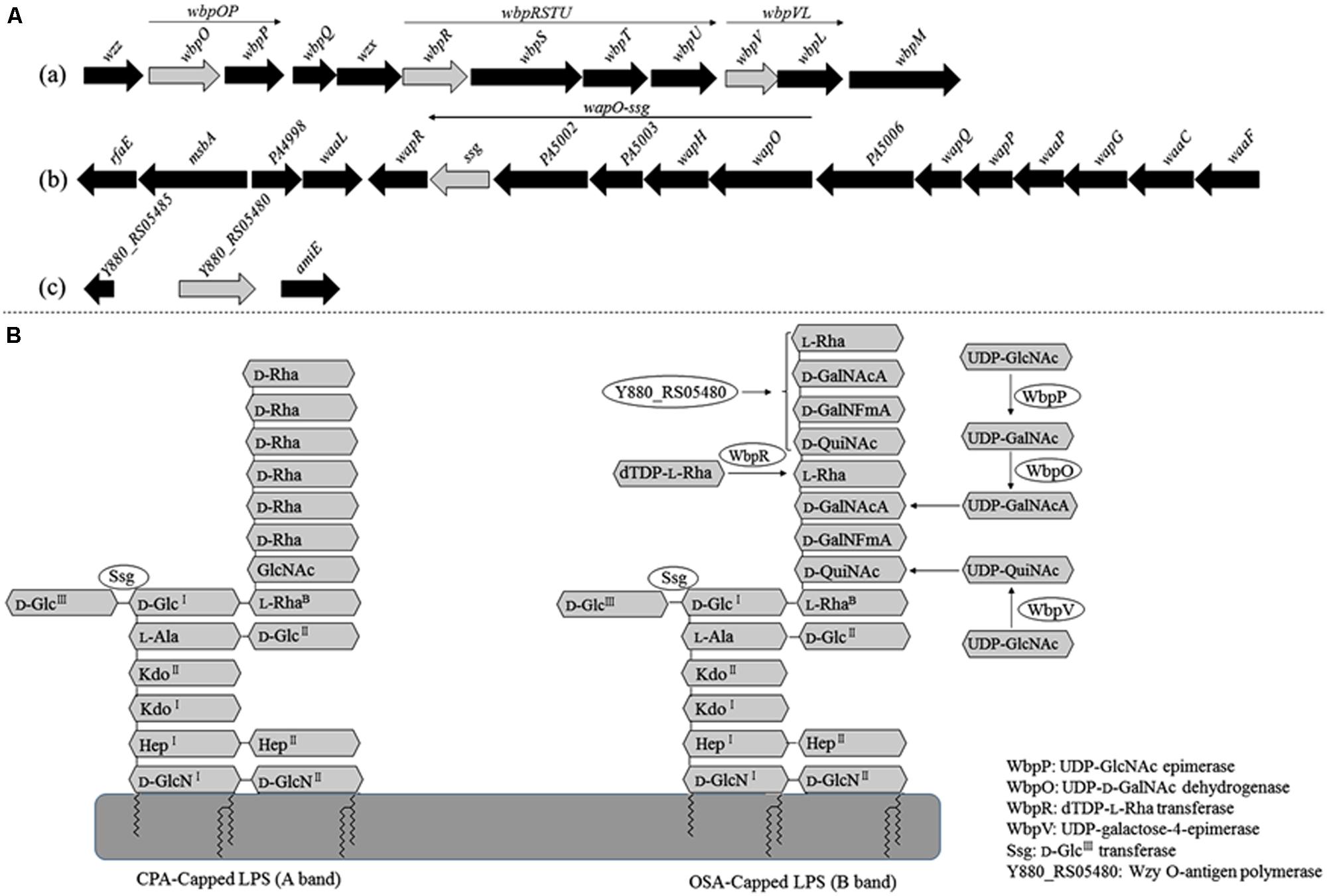
FIGURE 2. Analysis of the genes related to the synthesis of phage K8 receptor. (A) The genetic loci identified in phage resistant mutants. Light gray arrows represent the disrupted genes by Tn5G transposon. (a) The wbp gene cluster of PAK related to O-specific antigen (OSA) Lipopolysaccharide (LPS) synthesis, including wbpOP operon, wbpVL operon, and wbpRUST operon. (b) The 17 genes cluster of PAO1 involved in core oligosaccharide (OS) moiety biosynthesis, including wapO-ssg operon. (c) Y880_RS05480 gene in PAK genome, encoding a Wzy O-antigen α-polymerase. (B) The scheme of LPS structure and the enzymes required by LPS biosynthesis.
Adsorption Rate Analysis and Confirmation of the Phage Resistant Mutants
The adsorption ability of the phage-resistant mutants was analyzed. Compared with the parent strain PAK, the relative adsorption rates of phage K8 to the mutants were between 28.5 and 73.7%, implying that the phage receptors were impaired in these mutants (Figure 3A). Complementation test was carried out for each mutant. Gene wbpR, wbpV, ssg, and Y880_RS05480 were cloned and complementation successfully restored the sensitivity to phage K8 in the corresponding mutants, while gene wbpO and wbpP were both required for the phage sensitivity restoration in the wbpO mutant (Figure 3B). The results indicated that the disrupted genes in the mutants were responsible for the phage resistance phenotype.
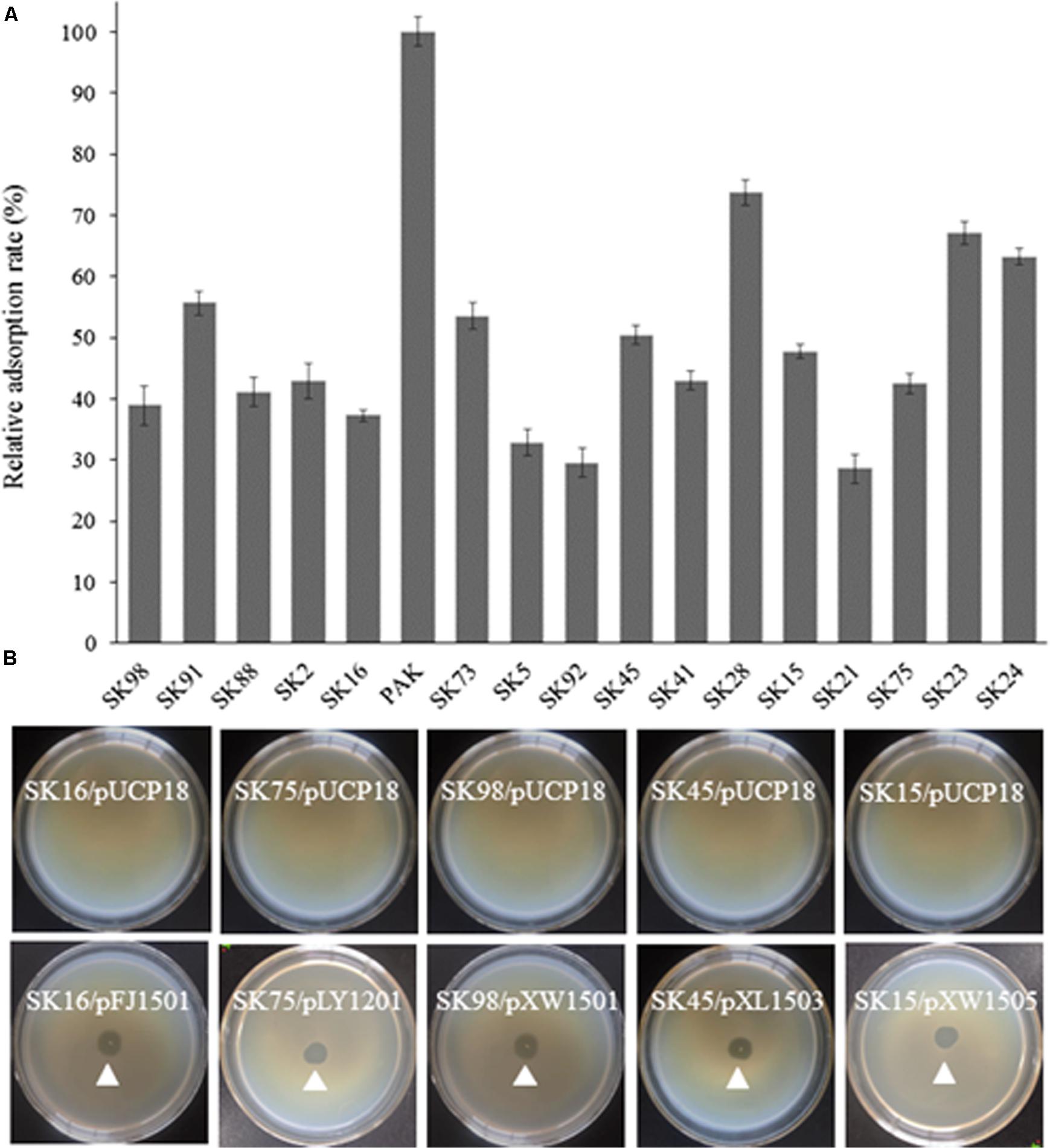
FIGURE 3. Characterization of the isolated phage resistant mutants. (A) The relative adsorption rate of phage resistant mutant. The parent strain PAK is used as control. (B) Phage sensitivity test of the isolated mutants. The mutants SK16, SK75, SK98, SK45, and SK15 have disrupted genes wbpV, Y880_RS05480, ssg, wbpR, and wbpO, respectively. Plasmid pFJ1501 carries wbpV gene; pLY1201 carries Y880_RS05480 gene; pXW1501 carries ssg gene; pXL1503 carries wbpR gene; and pXW1505 carries wbpOP genes.
Function Analysis of Gene Y880_RS05480
Lipopolysaccharide (LPS) is comprised of two forms of O-antigen, the common polysaccharide antigen (CPA) and the O-specific antigen (OSA). Wzy O-antigen polymerases are essential for O-antigen biosynthesis. They exhibit the low sequence conservation among the P. aeruginosa strains with the different serotypes. Currently Wzy proteins are not identified in O6, O7, and O8 strains of P. aeruginosa despite the fact that they produce normal O-antigen on the surface of their cells (Islam and Lam, 2014). The inactivated gene Y880_RS05480 encodes a hypothetical protein sharing 22.1% identity to the Wzy O-antigen polymerase (AIG62435) of E. coli. With the TMHMM prediction service, this hypothetical protein displayed a large periplasmic loop in its C-terminal and 11 transmembrane helices (Figure 4A), similar to the topology of the Wzy proteins found in the P. aeruginosa serotype O5 strain PAO1 and the other serotype strains (Islam and Lam, 2014).
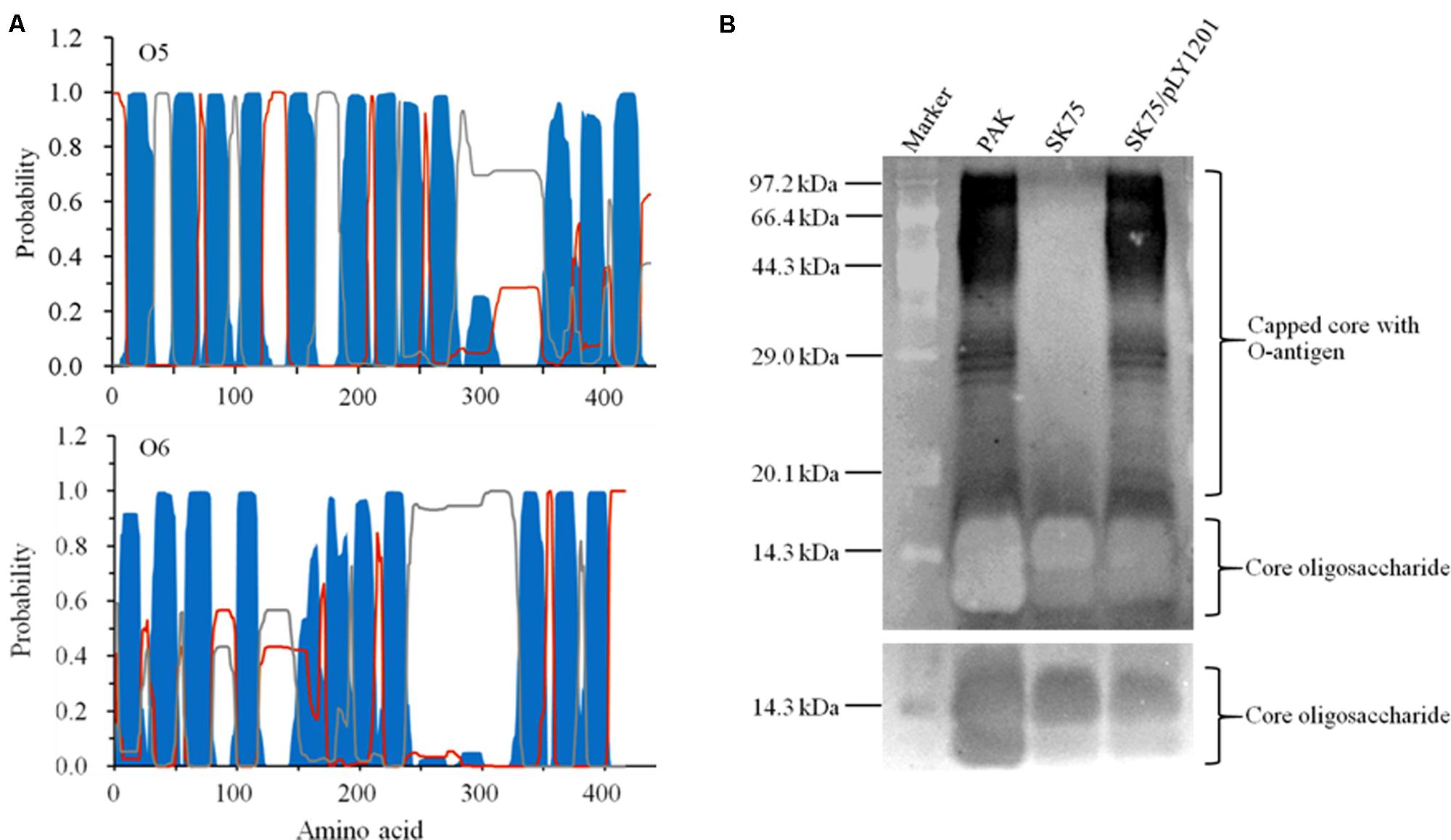
FIGURE 4. Characterization of the protein encoded by Y880_RS05480 gene. (A) Prediction of the transmembrane structures by TMHMM 2.0. Blue: the transmembrane helices. Gray: the periplasmic loops. Red: the cytoplasmic loops. The upper panel displays the structure of Wzy O-antigen polymerase of the serotype O5 strain PAO1 and the lower panel displays the structure of the hypothetical protein Y880_RS05480 of the serotype O6 strain PAK. (B) LPS profiles analysis with the silver-stained SDS-PAGE gel. M: the protein standard marker. PAK: the wild-type strain. SK75: the Y880_RS05480 mutant. pLY1201: the recombinant plasmid carries Y880_RS05480 gene. The upper panel displays the long-exposure photography for visualizing the O-antigen, while the lower panel displays the short-exposure photography for visualizing the core oligosaccharide.
The LPS profiles were analyzed in the strains PAK, the Y880_RS05480 mutant SK75, and the mutant carrying the intact gene Y880_RS05480 (SK75/pLY1201). The Y880_RS05480 mutant SK75 was devoid of the O-antigen with high molecular weight, whereas the wild-type strain PAK and the mutant carrying the intact the Y880_RS05480 gene produced the normal pattern of O-antigen LPS, including O-antigen and core oligosaccharide (Figure 4B). The results indicate that the product of gene Y880_RS05480 may have a similar function in the serotype O6 strain PAK as the Wzy O-antigen polymerases in their corresponding strains.
Biofilm Production Assay
Strains with different LPS phenotypes produced different amounts of biofilm under various conditions (Murphy et al., 2014; Ruhal et al., 2015). The phage-resistant mutants were analyzed for biofilm production after 48 h incubation. The mutants yielded 1.5–11.5 times biofilm compared with the wild-type strain PAK, and the ssg mutant produced the highest level of biofilm (Figure 5; Veeranagouda et al., 2011). When the mutants were complemented with their corresponding genes, the resulted strains produced significantly less amount of biofilm (Figure 5). The results indirectly indicated the phage-resistant mutants had altered LPS profiles.
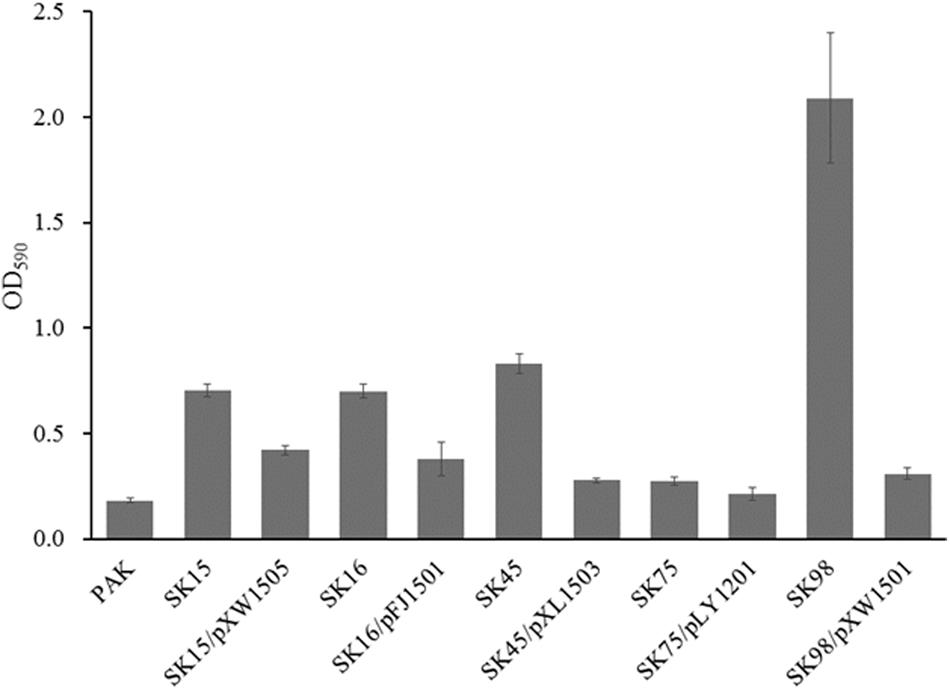
FIGURE 5. Biofilm production assay. The mutants SK16, SK75, SK98, SK45, and SK15 have the disrupted genes wbpV, wzy (Y880_RS05480), ssg, wbpR, and wbpO, respectively. Plasmid pFJ1501 carries wbpV gene; pLY1201 carries wzy (Y880_RS05480) gene; pXW1501 carries ssg gene; pXL1503 carries wbpR gene; and pXW1505 carries wbpOP genes.
Identification of the K8 Genome Termini
Two 102-bp HFSs were found with the sequencing depths 62.8 times over the average level in the assembled phage K8 genome, possibly representing the termini of phage genome (Li et al., 2014). Based on this prediction, the restriction mapping of the enzyme NotI and NdeI was simulated, respectively (Figures 6A,B). Enzyme NotI digestion produced one specific 7.5-kb fragment containing 3′ terminus (Figure 6B). Enzyme NdeI digestion produced a 3.5-kb fragment instead of the proposed 2.3-kb fragment, indicating that the 5′ terminus included a piece of unknown DNA fragment of about 1.2-kb was absent from the draft K8 genome (Figure 6B). The resultant 7.5 and 3.5-kb fragments were further found including identical 1188-bp sequences, demonstrating that the K8 genome has the identical terminal direct repeats. The 1.0-kb PCR product was also analyzed and was part of the 3′ terminus possibly amplified from the phage genome fragments (Figure 6C).
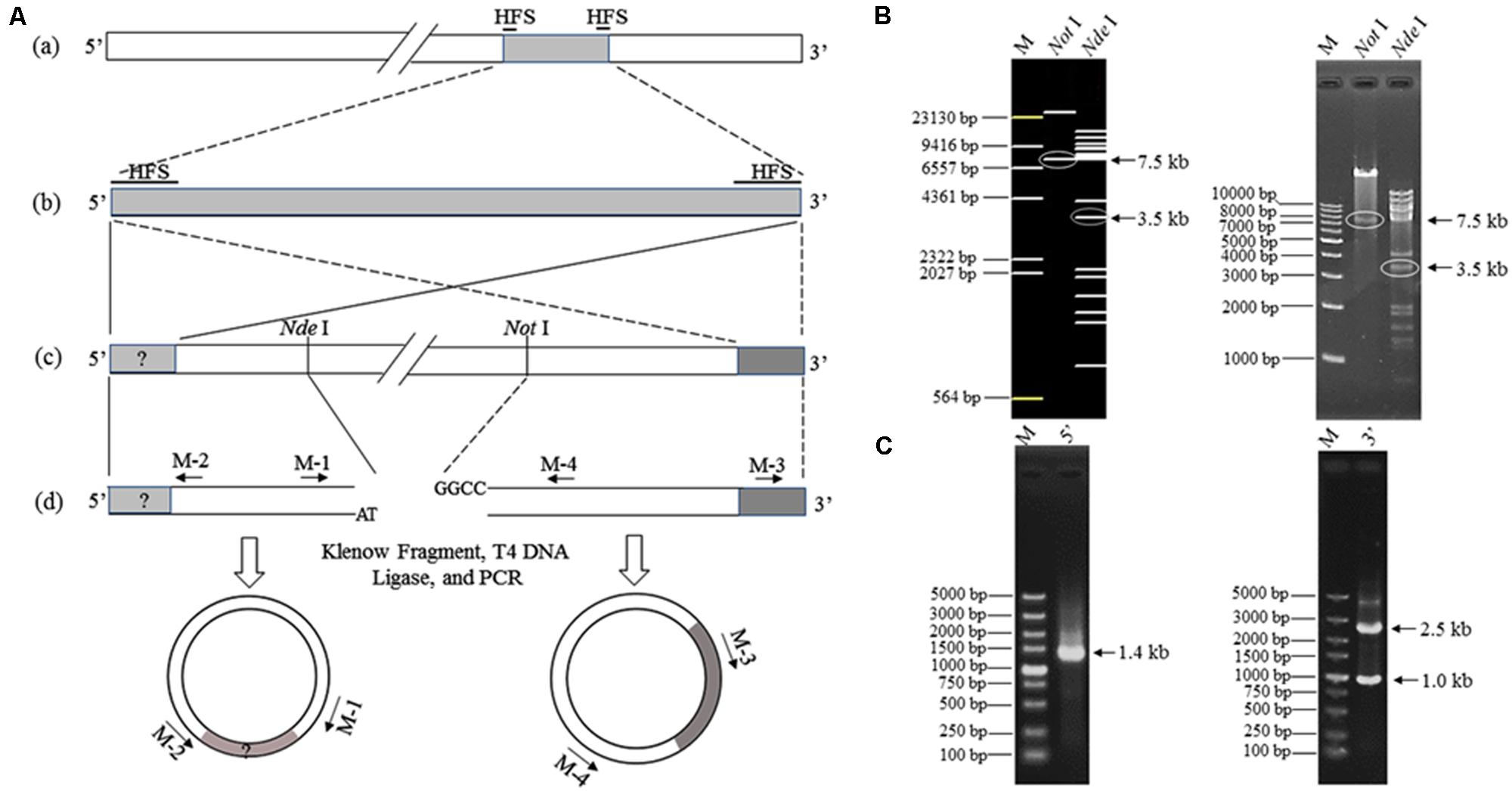
FIGURE 6. Termini identification of phage K8 genome. (A) The scheme for termini identification. (a) The assembled K8 genome of 92769 bp. HFSs refers to high-frequency sequences of 102 bp. (b) The two HFSs and the internal sequence. (c) The curated K8 genome with the proposed termini. Restriction enzymes NdeI and NotI are selected to digest the K8 genome for obtaining the fragment containing the 5′ or 3′ terminus. (d) Primers M-1, M-2, M-3, and M-4 are used for amplification of the termini. (B) Electrophoresis of the K8 genome digested by the restriction enzyme NdeI or NotI, respectively. The left panel is from the in silico simulation and the right panel was from the restriction enzyme digestions. The arrows point at the target fragments. (C) The left panel represents the PCR product containing the proposed 5′ terminus; the right panel represents the PCR product containing the proposed 3′ terminus. M: DNA markers.
Genome Structure and Annotation of Phage K8
The curated K8 genome has 93879 bp in length. The G+C content of the K8 genome is 49.35%. The abundance ratio of guanine to cytosine was analyzed by GC Plotter. The result showed that an asymmetric nucleotide composition was located near the virtual junction region between the termini of the K8 genome (Figure 7). The asymmetry might correspond to the DNA replication origin and the putative replication initiation site of phage K8 genome (Necsulea and Lobry, 2007).
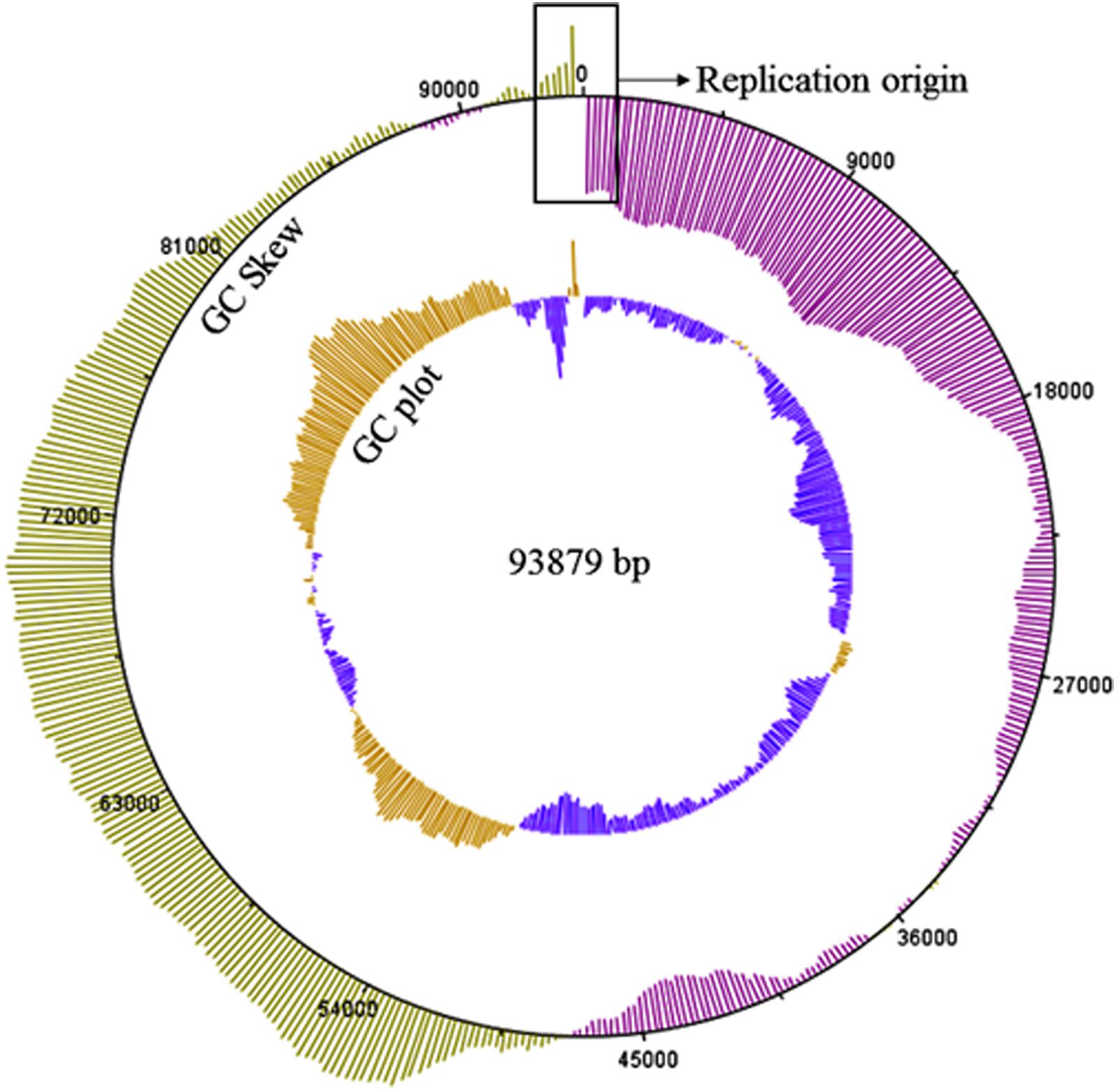
FIGURE 7. GC skew and GC plot of the K8 genome. The outer circle represents the value of the GC skew, green for positive and pink for negative. The inner circle represents the value of GC plot, yellow for G+C content above the average level 49.35% of the K8 genome and blue for G+C content below the average level of the K8 genome. The sequences in the rectangular box stand for the putative replication origin of the K8 genome.
The K8 genome has 179 predicted protein-coding genes distinctively arranged in five major clusters (Figure 8). (i) Genes in the cluster I mainly encoded proteins related to nucleotide metabolism, most of them shared great similarities with their homologs except for gene 087 encoding the pyrophosphatase that only shares 43.7% similarity with that of Burkholderia phage AH2 (Figure 8 and Supplementary Table S2). (ii) Cluster II has 10 genes encoding structural proteins and unclassified structural proteins. All proteins shared great similarities of 98.6–100% to their counterparts of phage PaP1 (Figure 8 and Supplementary Table S2) (Lu et al., 2013). (iii) Cluster III genes mainly encoded proteins related to DNA replication, transcription, recombination, and modification processes (Figure 8 and Supplementary Table S2). (iv) Genes in the cluster IV and V encoded proteins with unknown functions, and each cluster was adjacent to the termini of the K8 genome, respectively (Figure 8 and Supplementary Table S2). Thirteen tRNA genes were organized in one minor cluster between cluster I and II (Figure 8 and Supplementary Table S2).
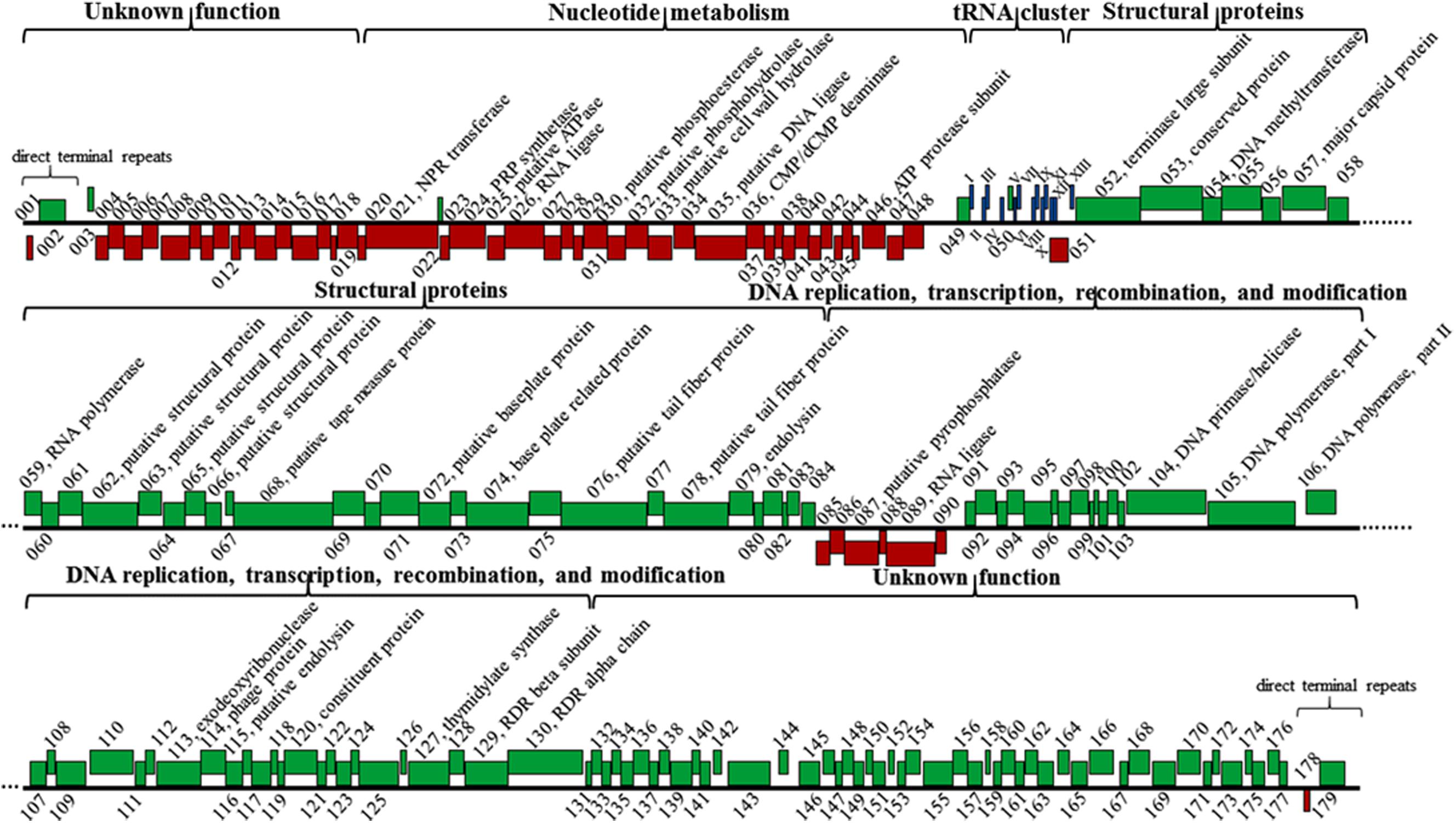
FIGURE 8. Genomic structure of phage K8. Red boxes represent genes on the minus strand. Green boxes represent genes on the plus strand. Roman numerals I-XIII refer to the genes encoding tRNAGln, tRNAArg, tRNALys, tRNALeu, tRNAIle, tRNAAsp, tRNACys, tRNAAsn, tRNAPro, tRNAGly, tRNAPhe, tRNAGlu, and tRNAThr, respectively. NPR, nicotinamide phosphoribosyl; PRP, phosphoribosyl pyrophosphate; RDR, ribonucleotide diphosphate reductase.
Three endolysins encoding genes were identified, including the putative cell wall hydrolase (gene 033) belonging to the hydrolase-2 family located within the cluster I region; the endoylsin (gene 079) identical to that of phage PaP1 located within the cluster II region; and the putative endolysin (gene 115) located within cluster III sharing 40.8% identity with that of Pseudomonas phage LU11 (Adriaenssens et al., 2012). However, no holin encoding gene was identified in phage K8 genome (Figure 8 and Supplementary Table S2).
Comparative Genomic Analysis
Homology of the K8 genome sequence was searched in NCBI. The result showed that the K8 genome has high similarities (>90%) and coverage (>90%) with phage PaP1, JG004, PAK_P2, vB_PaeM_C2-10_Ab1, PAK_P4, and PAK_P1. Comparative genomic analysis was further performed with the software Mauve (Supplementary Figure S1). Though the K8 genome was highly homologous to the reference genomes, genetic differences were found within the phage group. Compared to the K8 genome, PaP1 has six genes absent in its genome (Lu et al., 2013), JG004 has 10 genes absent in its genome (Garbe et al., 2011), PAK_P2 has 12 genes absent in its genome (Henry et al., 2015), and vB_PaeM_C2-10_Ab1 has 10 genes absent in its genome (Essoh et al., 2013). All absent genes were located within the gene clusters IV and V with unknown functions except for gene 093 which was positioned in middle of the K8 genome (Supplementary Table S2). The tail fiber proteins can act as the ligands to recognize the phage receptors during the infection process. The phylogenetic relationship was investigated among the 18 most homologous tail fiber proteins of P. aeruginosa phages including K8. The proteins were grouped into four clades on the basis of homology. The analysis showed that the phylogenetic distance of the tail fiber proteins was not correlated with the geographic locations where the phages were isolated (Figure 9).
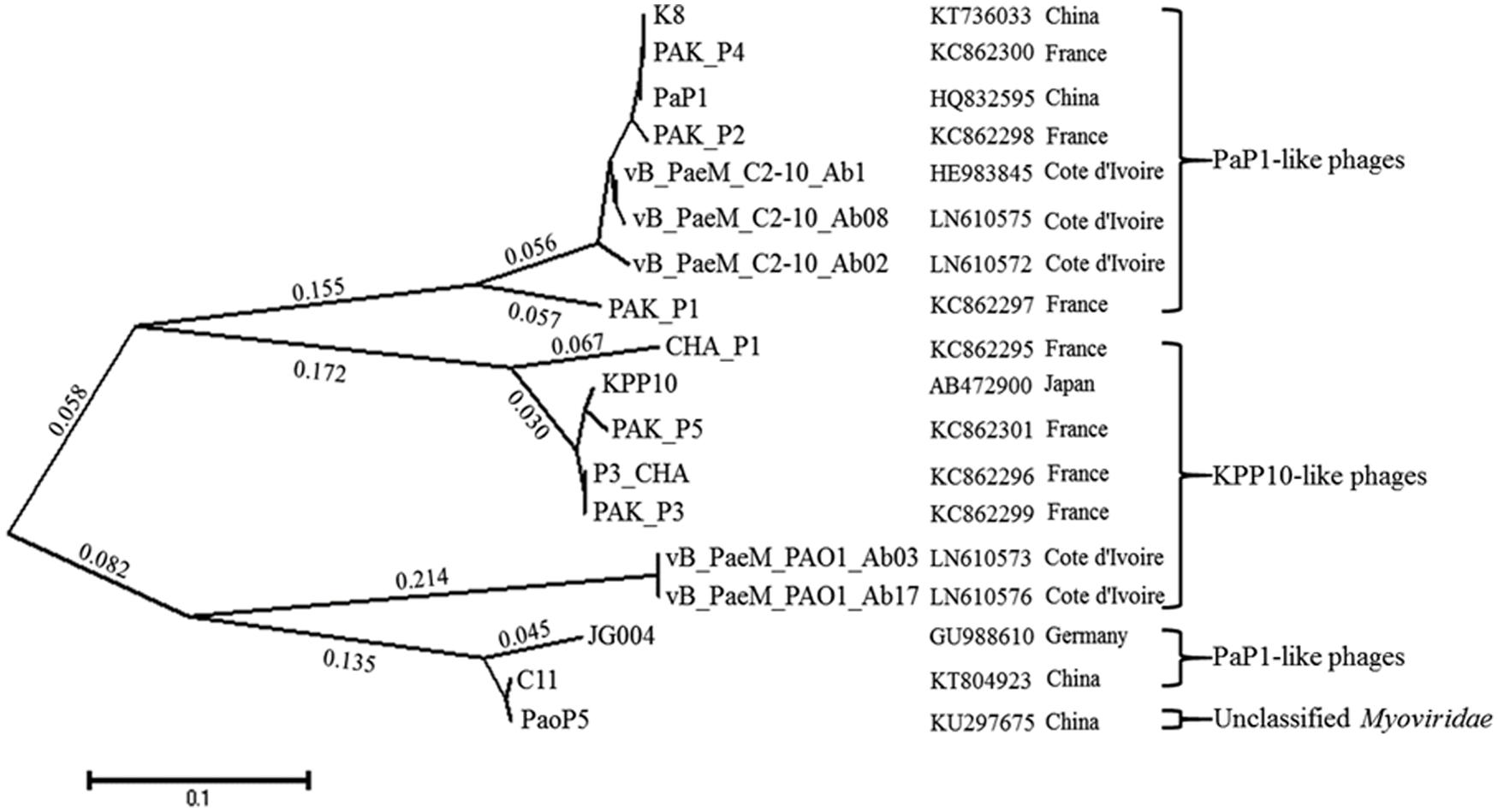
FIGURE 9. The phylogenetic tree of the tail fiber proteins with MEGA5. Seventeen tail fiber proteins which share 47–100% amino acid sequence identity to the putative tail fiber protein (076) of phage K8 are included. Pseudomonas aeruginosa phage PaoP5 is unclassified. The geographic locations represent the sites where the phages are isolated.
Discussion
Pseudomonas aeruginosa phages that have been identified so far are comprised of at least 24 genera classified into Podoviridae, Myoviridae, and Siphoviridae families (Sepulveda-Robles et al., 2012). Phage K8 exhibits an icosahedral head structure with a contractile tail and is classified into the Myoviridae family. Its genome is highly homologous to that of phage PaP1 and their major capsid proteins are identical, suggesting that phage K8 is a new member of the PaP1-like phages (Lu et al., 2013) or PAK_P1-like phages genus (Henry et al., 2015). To date, the genus includes 18 phages besides phage K8 (Essoh et al., 2015). Though these phages were isolated from France, Germany, Côte d’Ivoire, Chongqing (China), and Tianjin (China), the phage genomes share great similarities. The result is consistent with the findings that P. aeruginosa phages of specific genera are genetically closely related and can be readily isolated from environmental samples globally (Ceyssens and Lavigne, 2010).
The terminal structure of the dsDNA phage genomes has at least five major types, including the linear genomes with 5′-protruded cohesive ends (Tan et al., 2007); the linear genomes with 3′-protruded cohesive ends (Zeigler, 2013); the linear genomes with terminal direct repeats (Pajunen et al., 2001); the genomes with circular permutation and terminal redundancy with specific pac recognition sites (Alonso et al., 1997); and the genomes with circular permutation and terminal redundancy without specific pac recognition sites (Miller et al., 2003). Many P. aeruginosa phages have similar direct terminal repeats with lengths ranging from 184 to 1238 bp, including PaP1-like phages, KPP10-like phages, and some Podoviridae phages (Ceyssens et al., 2006; Henry et al., 2015). The direct terminal repeats are highly conserved among the PaP1-like phages genus and may be related to the patterns of viral genome replication in these phages.
Diverse receptors of P. aeruginosa phages have been identified. Phage PA1Ø, MPK7, B3, and D3112 use type IV pili as the receptor for infection (Roncero et al., 1990; Kim et al., 2012; Bae and Cho, 2013). Phage phiCTX and H22 use core oligosaccharide of LPS as the receptor (Temple et al., 1986; Yokota et al., 1994). Phage FIZ15 and D3 use LPS O-antigen as the receptor (Kuzio and Kropinski, 1983; Vaca-Pacheco et al., 1999). Phage A7 use CPA as the receptor (Rivera et al., 1992). Phage PIK receptor in LPS contains D-mannose, L-rhamnose, and D-glucosamine and may be the heteropolymer O-antigen OSA (Patel and Rao, 1983). For phage JG004, a series of genes related to LPS pathway have been identified involved in the receptor synthesis (Garbe et al., 2011).
Lipopolysaccharide is described as a molecule with three domains, lipid A, core oligosaccharide, and O-antigen. P. aeruginosa PAK simultaneously synthesizes two different forms of O-antigen. CPA is a homopolymer of D-rhamnose (D-Rha). OSA is a heteropolymer composing of repeating units of D-QuiNAc, D-GalNAcA, D-GalNFmA, and L-Rha (Belanger et al., 1999). In this study, all the disrupted genes related to the phage receptor synthesis play a key role in LPS biosynthesis in P. aeruginosa PAK. WbpR is a putative dTDP-L-Rha transferase, adding the fourth residue L-Rha to the repeating unit of OSA in O6 strains (Figure 2; Belanger et al., 1999). Gene wbpV encodes a UDP-galactose-4-epimerase involved in the pathway of UDP-QuiNAc synthesis. UDP-QuiNAc is further added to the repeating unit of OSA as the first residue D-QuiNAc by the glycosyltransferase WbpL (Rocchetta et al., 1999). Gene wbpP is located downstream of gene wbpO within the same operon, encoding the epimerase converting UDP-GlcNAc to UDP-GalNAc (Creuzenet et al., 2000). Gene wbpO encodes the dehydrogenase converting UDP-GalNAc to UDP-GalNAcA. UDP-GalNAcA is further added as the third residue of the repeating unit of OSA (Figure 2; Zhao et al., 2000). The cluster of 17 genes of P. aeruginosa has been found involved in core oligosaccharide (OS) moiety biosynthesis. Among them, gene ssg encodes a glycosyltransferase and is responsible for the transfer of α-D-GlcIII to OS moiety (Figure 2; Lam et al., 2011; Veeranagouda et al., 2011). Both CPA and OSA are lost in the ssg mutant strain (Fernandez et al., 2013). P. aeruginosa serotype O6 strains are able to synthesize long-chain O antigen. However, no wzy gene homolog is identified within the wbp gene cluster for O-antigen synthesis in O6 strains. In this work, though the protein encoded by the gene Y880_RS05480 displays less similarities with the known Wzy polymerases, the Y880_RS05480 mutant isn’t able to produce the O-antigen with the high molecular weight, indicating the Wzy-dependent pathway existed in O6 strain PAK for LPS synthesis (Islam and Lam, 2014).
Conclusion
Five genes wbpR, wbpV, wbpO, ssg, and wzy are identified as inactivated in the phage-resistant mutants. Gene Y880_RS05480 is first proved to function as the Wzy O-antigen polymerases. In combination, the results indicate that OSA should be the receptor of phage K8.
Author Contributions
XP performed the bioinformatic analysis and experiments and wrote the manuscript. XC, FZ, and LL carried out the plasmid constructions. YH performed the bioinformatic analysis. HY designed the experiments and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work is supported by The National Natural Science Foundation of China (Grant No. 31370205 and 30970114).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00252
FIGURE S1 | Comparative genomic analyses of Pseudomonas aeruginosa phages. Ab1: vB_PaeM_C2-10_Ab1. The coordinate rulers display the size of the corresponding genomes. The height of the red and green ribbons is correlated with the level of similarities between every two genomes. Different colors indicate the inconsistent genomic organizations among the phages.
Footnotes
- ^ http://www.ncbi.nlm.nih.gov/genomes
- ^ www.pseudomonas.com
- ^ http://www.cbs.dtu.dk/services/TMHMM/
- ^ http://www.genewiz.com/
- ^ http://cobamide2.bio.pitt.edu
References
Adriaenssens, E. M., Mattheus, W., Cornelissen, A., Shaburova, O., Krylov, V. N., Kropinski, A. M., et al. (2012). Complete genome sequence of the giant Pseudomonas phage Lu11. J. Virol. 86, 6369–6370. doi: 10.1128/JVI.00641-12
Alonso, J. C., Luder, G., Stiege, A. C., Chai, S., Weise, F., and Trautner, T. A. (1997). The complete nucleotide sequence and functional organization of Bacillus subtilis bacteriophage SPP1. Gene 204, 201–212. doi: 10.1016/S0378-1119(97)00547-7
Bae, H. W., and Cho, Y. H. (2013). Complete genome sequence of Pseudomonas aeruginosa Podophage MPK7, which requires type IV Pili for infection. Genome Announc 1:e744. doi: 10.1128/genomeA.00744-13
Belanger, M., Burrows, L. L., and Lam, J. S. (1999). Functional analysis of genes responsible for the synthesis of the B-band O antigen of Pseudomonas aeruginosa serotype O6 lipopolysaccharide. Microbiology 145(Pt 12), 3505–3521. doi: 10.1099/00221287-145-12-3505
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bradley, T. J., and Khan, N. H. (1974). The production of extracellular lipids by Pseudomonas aeruginosa NCTC 2000 in stationary liquid media containing macrogols. J. Pharm. Pharmacol. 26, 900–902. doi: 10.1111/j.2042-7158.1974.tb09201.x
Brencic, A., Mcfarland, K. A., Mcmanus, H. R., Castang, S., Mogno, I., Dove, S. L., et al. (2009). The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol. Microbiol. 73, 434–445. doi: 10.1111/j.1365-2958.2009.06782.x
Carver, T., Thomson, N., Bleasby, A., Berriman, M., and Parkhill, J. (2009). DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25, 119–120. doi: 10.1093/bioinformatics/btn578
Ceyssens, P. J., and Lavigne, R. (2010). Bacteriophages of Pseudomonas. Future Microbiol 5, 1041–1055. doi: 10.2217/fmb.10.66
Ceyssens, P. J., Lavigne, R., Mattheus, W., Chibeu, A., Hertveldt, K., Mast, J., et al. (2006). Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: establishment of the phiKMV subgroup within the T7 supergroup. J. Bacteriol. 188, 6924–6931. doi: 10.1128/JB.00831-06
Chan, B. K., Abedon, S. T., and Loc-Carrillo, C. (2013). Phage cocktails and the future of phage therapy. Future Microbiol. 8, 769–783. doi: 10.2217/fmb.13.47
Creuzenet, C., Belanger, M., Wakarchuk, W. W., and Lam, J. S. (2000). Expression, purification, and biochemical characterization of WbpP, a new UDP-GlcNAc C4 epimerase from Pseudomonas aeruginosa serotype O6. J. Biol. Chem. 275, 19060–19067. doi: 10.1074/jbc.M001171200
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Djordjevic, D., Wiedmann, M., and Mclandsborough, L. A. (2002). Microtiter plate assay for assessment of Listeria monocytogenes biofilm formation. Appl. Environ. Microbiol. 68, 2950–2958. doi: 10.1128/AEM.68.6.2950-2958.2002
Emori, T. G., and Gaynes, R. P. (1993). An overview of nosocomial infections, including the role of the microbiology laboratory. Clin. Microbiol. Rev. 6, 428–442.
Essoh, C., Blouin, Y., Loukou, G., Cablanmian, A., Lathro, S., Kutter, E., et al. (2013). The susceptibility of Pseudomonas aeruginosa strains from cystic fibrosis patients to bacteriophages. PLoS ONE 8:e60575. doi: 10.1371/journal.pone.0060575
Essoh, C., Latino, L., Midoux, C., Blouin, Y., Loukou, G., Nguetta, S. P., et al. (2015). Investigation of a Large Collection of Pseudomonas aeruginosa Bacteriophages Collected from a Single Environmental Source in Abidjan, Cote d’Ivoire. PLoS ONE 10:e0130548. doi: 10.1371/journal.pone.0130548
Fernandez, L., Alvarez-Ortega, C., Wiegand, I., Olivares, J., Kocincova, D., Lam, J. S., et al. (2013). Characterization of the polymyxin B resistome of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57, 110–119. doi: 10.1128/AAC.01583-12
Fomsgaard, A., Freudenberg, M. A., and Galanos, C. (1990). Modification of the silver staining technique to detect lipopolysaccharide in polyacrylamide gels. J. Clin. Microbiol. 28, 2627–2631.
Garbe, J., Bunk, B., Rohde, M., and Schobert, M. (2011). Sequencing and characterization of Pseudomonas aeruginosa phage JG004. BMC Microbiol. 11:102. doi: 10.1186/1471-2180-11-102
Henry, M., Bobay, L. M., Chevallereau, A., Saussereau, E., Ceyssens, P. J., and Debarbieux, L. (2015). The search for therapeutic bacteriophages uncovers one new subfamily and two new genera of Pseudomonas-infecting Myoviridae. PLoS ONE 10:e0117163. doi: 10.1371/journal.pone.0117163
Holland, S. J., Sanz, C., and Perham, R. N. (2006). Identification and specificity of pilus adsorption proteins of filamentous bacteriophages infecting Pseudomonas aeruginosa. Virology 345, 540–548. doi: 10.1016/j.virol.2005.10.020
Islam, S. T., and Lam, J. S. (2014). Synthesis of bacterial polysaccharides via the Wzx/Wzy-dependent pathway. Can. J. Microbiol. 60, 697–716. doi: 10.1139/cjm-2014-0595
Karaiskos, I., and Giamarellou, H. (2014). Multidrug-resistant and extensively drug-resistant Gram-negative pathogens: current and emerging therapeutic approaches. Expert Opin. Pharmacother. 15, 1351–1370. doi: 10.1517/14656566.2014.914172
Kim, S., Rahman, M., Seol, S. Y., Yoon, S. S., and Kim, J. (2012). Pseudomonas aeruginosa bacteriophage PA1O requires type IV pili for infection and shows broad bactericidal and biofilm removal activities. Appl. Environ. Microbiol. 78, 6380–6385. doi: 10.1128/AEM.00648-12
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kutter, E., De Vos, D., Gvasalia, G., Alavidze, Z., Gogokhia, L., Kuhl, S., et al. (2010). Phage therapy in clinical practice: treatment of human infections. Curr. Pharm. Biotechnol. 11, 69–86. doi: 10.2174/138920110790725401
Kuzio, J., and Kropinski, A. M. (1983). O-antigen conversion in Pseudomonas aeruginosa PAO1 by bacteriophage D3. J. Bacteriol. 155, 203–212.
Kwan, T., Liu, J., Dubow, M., Gros, P., and Pelletier, J. (2006). Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J. Bacteriol. 188, 1184–1187. doi: 10.1128/JB.188.3.1184-1187.2006
Lam, J. S., Taylor, V. L., Islam, S. T., Hao, Y., and Kocincova, D. (2011). Genetic and functional diversity of Pseudomonas aeruginosa Lipopolysaccharide. Front. Microbiol. 2:118. doi: 10.3389/fmicb.2011.00118
Latino, L., Essoh, C., Blouin, Y., Vu Thien, H., and Pourcel, C. (2014). A novel Pseudomonas aeruginosa bacteriophage, Ab31, a chimera formed from temperate phage PAJU2 and P. putida lytic phage AF: characteristics and mechanism of bacterial resistance. PLoS ONE 9:e93777. doi: 10.1371/journal.pone.0093777
Li, L., Yang, H., Lin, S., and Jia, S. (2010). Classification of 17 newly isolated virulent bacteriophages of Pseudomonas aeruginosa. Can. J. Microbiol. 56, 925–933. doi: 10.1139/w10-075
Li, S., Fan, H., An, X., Jiang, H., Chen, Y., and Tong, Y. (2014). Scrutinizing virus genome termini by high-throughput sequencing. PLoS ONE 9:e85806. doi: 10.1371/journal.pone.0085806
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. doi: 10.1128/CMR.00040-09
Lu, S., Le, S., Tan, Y., Zhu, J., Li, M., Rao, X., et al. (2013). Genomic and proteomic analyses of the terminally redundant genome of the Pseudomonas aeruginosa phage PaP1: establishment of genus PaP1-like phages. PLoS ONE 8:e62933. doi: 10.1371/journal.pone.0062933
Luiten, R. G., Putterman, D. G., Schoenmakers, J. G., Konings, R. N., and Day, L. A. (1985). Nucleotide sequence of the genome of Pf3, an IncP-1 plasmid-specific filamentous bacteriophage of Pseudomonas aeruginosa. J. Virol. 56, 268–276.
Miller, E. S., Kutter, E., Mosig, G., Arisaka, F., Kunisawa, T., and Ruger, W. (2003). Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 67, 86–156. doi: 10.1128/MMBR.67.1.86-156.2003
Murphy, K., Park, A. J., Hao, Y., Brewer, D., Lam, J. S., and Khursigara, C. M. (2014). Influence of O polysaccharides on biofilm development and outer membrane vesicle biogenesis in Pseudomonas aeruginosa PAO1. J. Bacteriol. 196, 1306–1317. doi: 10.1128/JB.01463-13
Necsulea, A., and Lobry, J. R. (2007). A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol. Evol. 24, 2169–2179. doi: 10.1093/molbev/msm148
Nunn, D. N., and Lory, S. (1992). Components of the protein-excretion apparatus of Pseudomonas aeruginosa are processed by the type IV prepilin peptidase. Proc. Natl. Acad. Sci. U.S.A. 89, 47–51. doi: 10.1073/pnas.89.1.47
Olsthoorn, R. C., Garde, G., Dayhuff, T., Atkins, J. F., and Van Duin, J. (1995). Nucleotide sequence of a single-stranded RNA phage from Pseudomonas aeruginosa: kinship to coliphages and conservation of regulatory RNA structures. Virology 206, 611–625. doi: 10.1016/S0042-6822(95)80078-6
Pajunen, M. I., Kiljunen, S. J., Soderholm, M. E., and Skurnik, M. (2001). Complete genomic sequence of the lytic bacteriophage phiYeO3-12 of Yersinia enterocolitica serotype O:3. J. Bacteriol. 183, 1928–1937. doi: 10.1128/JB.183.6.1928-1937.2001
Patel, I. R., and Rao, K. K. (1983). Studies on the Pseudomonas aeruginosa PAO1 bacteriophage receptors. Arch. Microbiol. 135, 155–157. doi: 10.1007/BF00408026
Rivera, M., Chivers, T. R., Lam, J. S., and Mcgroarty, E. J. (1992). Common antigen lipopolysaccharide from Pseudomonas aeruginosa AK1401 as a receptor for bacteriophage A7. J. Bacteriol. 174, 2407–2411.
Rocchetta, H. L., Burrows, L. L., and Lam, J. S. (1999). Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 63, 523–553.
Roncero, C., Darzins, A., and Casadaban, M. J. (1990). Pseudomonas aeruginosa transposable bacteriophages D3112 and B3 require pili and surface growth for adsorption. J. Bacteriol. 172, 1899–1904.
Ruhal, R., Antti, H., Rzhepishevska, O., Boulanger, N., Barbero, D. R., Wai, S. N., et al. (2015). A multivariate approach to correlate bacterial surface properties to biofilm formation by lipopolysaccharide mutants of Pseudomonas aeruginosa. Colloids Surf B Biointerf. 127, 182–191. doi: 10.1016/j.colsurfb.2015.01.030
Ruokoranta, T. M., Grahn, A. M., Ravantti, J. J., Poranen, M. M., and Bamford, D. H. (2006). Complete genome sequence of the broad host range single-stranded RNA phage PRR1 places it in the Levivirus genus with characteristics shared with Alloleviviruses. J. Virol. 80, 9326–9330. doi: 10.1128/JVI.01005-06
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Schweizer, H. P. (1991). Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene 97, 109–121. doi: 10.1016/0378-1119(91)90016-5
Sepulveda-Robles, O., Kameyama, L., and Guarneros, G. (2012). High diversity and novel species of Pseudomonas aeruginosa bacteriophages. Appl. Environ. Microbiol. 78, 4510–4515. doi: 10.1128/AEM.00065-12
Srinivasiah, S., Bhavsar, J., Thapar, K., Liles, M., Schoenfeld, T., and Wommack, K. E. (2008). Phages across the biosphere: contrasts of viruses in soil and aquatic environments. Res. Microbiol. 159, 349–357. doi: 10.1016/j.resmic.2008.04.010
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tan, Y., Zhang, K., Rao, X., Jin, X., Huang, J., Zhu, J., et al. (2007). Whole genome sequencing of a novel temperate bacteriophage of P. aeruginosa: evidence of tRNA gene mediating integration of the phage genome into the host bacterial chromosome. Cell Microbiol. 9, 479–491. doi: 10.1111/j.1462-5822.2006.00804.x
Temple, G. S., Ayling, P. D., and Wilkinson, S. G. (1986). Isolation and characterization of a lipopolysaccharide-specific bacteriophage of Pseudomonas aeruginosa. Microbios 45, 81–91.
Vaca-Pacheco, S., Paniagua-Contreras, G. L., García-González, O., and De La Garza, M. (1999). The Clinically Isolated FIZ15 bacteriophage causes lysogenic conversion in Pseudomonas aeruginosa PAO1. Curr. Microbiol. 38, 239–243. doi: 10.1007/PL00006794
Veeranagouda, Y., Lee, K., Cho, A. R., Cho, K., Anderson, E. M., and Lam, J. S. (2011). Ssg, a putative glycosyltransferase, functions in lipo- and exopolysaccharide biosynthesis and cell surface-related properties in Pseudomonas alkylphenolia. FEMS Microbiol. Lett. 315, 38–45. doi: 10.1111/j.1574-6968.2010.02172.x
Wang, J., Mushegian, A., Lory, S., and Jin, S. (1996). Large-scale isolation of candidate virulence genes of Pseudomonas aeruginosa by in vivo selection. Proc. Natl. Acad. Sci. U.S.A. 93, 10434–10439. doi: 10.1073/pnas.93.19.10434
Westphal, O., and Jann, K. (1965). “Bacterial lipopolysaccharides. Extraction with phenol-water and further applications of the procedure,” in Methods in Carbohydrate Chemistry, eds R. Whistler and M. Wolfan (New York, NY: Academic press).
Wheeler, D. L., Church, D. M., Federhen, S., Lash, A. E., Madden, T. L., Pontius, J. U., et al. (2003). Database resources of the National Center for biotechnology. Nucleic Acids Res. 31, 28–33. doi: 10.1093/nar/gkg033
Yang, H., Liang, L., Lin, S., and Jia, S. (2010). Isolation and characterization of a virulent bacteriophage AB1 of Acinetobacter baumannii. BMC Microbiol. 10:131. doi: 10.1186/1471-2180-10-131
Yokota, S., Hayashi, T., and Matsumoto, H. (1994). Identification of the lipopolysaccharide core region as the receptor site for a cytotoxin-converting phage, phi CTX, of Pseudomonas aeruginosa. J. Bacteriol. 176, 5262–5269.
Zeigler, D. R. (2013). Complete genome sequence of Bacillus subtilis phage {varphi}105. Genome Announc. 1. doi: 10.1128/genomeA.00641-13
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Keywords: Pseudomonas phage K8, phage receptor, O-specific antigen (OSA), wzy gene, genome annotation
Citation: Pan X, Cui X, Zhang F, He Y, Li L and Yang H (2016) Genetic Evidence for O-Specific Antigen as Receptor of Pseudomonas aeruginosa Phage K8 and Its Genomic Analysis. Front. Microbiol. 7:252. doi: 10.3389/fmicb.2016.00252
Received: 26 December 2015; Accepted: 15 February 2016;
Published: 02 March 2016.
Edited by:
William Michael McShan, University of Oklahoma Health Sciences Center, USAReviewed by:
Joseph S. Lam, University of Guelph, CanadaScott Van Nguyen, United States Department of Agriculture, USA
Copyright © 2016 Pan, Cui, Zhang, He, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongjiang Yang, aG9uZ2ppYW5neWFuZ0B0dXN0LmVkdS5jbg==