 Yan Yang1
Yan Yang1 Ruurd T. Zijlstra
Ruurd T. Zijlstra Michael G. Gänzle
Michael G. Gänzle- 1Department of Agricultural, Food and Nutritional Science, University of Alberta, Edmonton, AB, Canada
- 2School of Food and Pharmaceutical Engineering, Hubei University of Technology, Wuhan, China
Lactobacillus reuteri is used as probiotic culture in food and feed applications; however, strain specific properties of L. reuteri that mediate probiotic activity remain unknown. This study aimed to determine effects of feed fermentation with exopolysaccharide and reutericyclin producing L. reuteri on the transition of the gut microbiome of piglets after weaning. The reutericyclin and reuteran producing L. reuteri TMW1.656 was compared to the reutericyclin negative and levan producing L. reuteri LTH5794 and unfermented controls. Both strains were fermented at conditions supporting exopolysaccharide formation, or at conditions not supporting exopolysaccharide formation. Fecal microbiota were characterized by partial sequencing of 16S rRNA genes, and by quantitative PCR targeting clostridial toxins. The transition to solid food resulted in a transient increase of Proteobacteria to 12% of total bacteria, and increased bacterial diversity by increasing the abundance of anaerobic fiber fermenting Firmicutes. Three weeks after weaning, Prevotella and Lactobacillus were among the dominant bacterial genera. Feed fermentation with L. reuteri affected the abundance of few bacterial taxa and particularly reduced the abundance of Enterobacteriaceae (P < 0.05) when compared to unfermented controls. Reutericyclin producing L. reuteri increased the abundance of Dialister spp. and Mitsuokella spp. (P < 0.05) but did not influence the abundance of clostridial toxins in the feces. In conclusion, data on the contribution of specific metabolic activities of L. reuteri to probiotic activity will facilitate the strain selection for probiotic applications in food and feed.
Introduction
Lactobacillus reuteri is a host-specific intestinal symbiont of humans and vertebrate animals (Walter, 2008; Frese et al., 2011). L. reuteri is commercially applied in cereal fermentations (Brandt, 2014) and as probiotic culture (Tuohy et al., 2003). Strain- or lineage specific metabolic traits of L. reuteri mediate host-specific colonization (Frese et al., 2014; Wilson et al., 2014), and contribute to its competitiveness in cereal fermentations and improved bread quality (Galle et al., 2012; Lin and Gänzle, 2014; Zhao et al., 2015). Strain specific properties of L. reuteri that mediate probiotic activity, however, remain unknown.
Metabolic traits that were suggested to mediate probiotic activity of L. reuteri include acid resistance (Teixeira et al., 2014), histamine decarboxylation (Spinler et al., 2014), exopolysaccharide production (Chen et al., 2014), and antimicrobial activity against pathogens (Gänzle, 2004; Rea et al., 2014). Reuteran and levan from L. reuteri prevented adhesion of enterotoxigenic Escherichia coli (ETEC) in vitro, and reduced mucosa-adherent ETEC in a swine model (Wang et al., 2010; Chen et al., 2014). The production of reuterin and reutericyclin by L. reuteri were proposed to provide protection against Salmonella Typhimurium and Clostridium difficile, respectively (Hurdle et al., 2011; De Weirdt et al., 2012). However, only two studies performed in rodent models demonstrate that antimicrobial compounds from lactic acid bacteria are active in vivo. Bacteriocin production by probiotic lactic acid bacteria reduced infection by Listeria monocytogenes (Corr et al., 2007), and reduced colonization by vancomycin resistant enterococci (Millette et al., 2008). Evidence for activity of antimicrobial metabolites of probiotics against autochtonous microbiota is inconclusive. The bacteriocin producing L. salivarius Abp118 altered the gut microbiome of swine and mice when compared to controls that did not receive probiotics. Changes induced by the bacteriocin producing strain, however, were not different from those induced by a bacteriocin-negative derivative of the same strain (Riboulet-Bisson et al., 2012).
Probiotic applications of L. reuteri and related organisms specifically targeted piglets (Konstantinov et al., 2008). Gut microbiota of pigs undergo a transition after weaning (Konstantinov et al., 2006; Lallès et al., 2007). The microbiome of suckling pigs is dominated by lactobacilli (Konstantinov et al., 2006) while strict anaerobic Firmicutes and Bacteroidetes dominate the microbiome of adult pigs (Lamendella et al., 2011; Riboulet-Bisson et al., 2012). The abundance of lactobacilli decreases after weaning (Konstantinov et al., 2006), providing opportunity for overgrowth of pathogens. Probiotics may decrease the abundance of pathogens (Zhang et al., 2010; Bednorz et al., 2013). Supplementation with L. amylovorus reduced levels of ETEC in the intestine but did not alter the hindgut microbiome (Konstantinov et al., 2008; Su et al., 2008).
This study aimed to determine the effect of feed fermentation with L. reuteri on the development of the gut microbiome in weanling piglets. The experimental design aimed to determine the contribution of viable L. reuteri, exopolysaccharide formation by L. reuteri, and reutericyclin formation by L. reuteri on the evolution of the gut microbiome in weanling piglets. The fecal microbiome was characterized by high throughput sequencing of 16S rRNA genes, and by quantitative PCR (qPCR) specifically targeting C. difficile, C. perfringens, and toxins produced by these organisms.
Materials and Methods
Feed Fermentation and Diet Preparation
Wheat flour was provided by University of Alberta Swine Research and Technology Centre, mixed with an equal amount of tap water, and inoculated with approximately 107 CFU g−1 of the levan-producing L. reuteri LTH5794 or the reuteran-producing L. reuteri TMW1.656. A more detailed account of the feed fermentation and the control experiments ensuring the identity of fermentation microbiota with the inoculum is provided by Yang et al. (2015). Feed fermentation was carried out with addition of 10% (w/w flour) sucrose to support levan or reuteran formation during fermentation, or addition of 5% (w/w flour) glucose and 5% (w/w flour) fructose, which do not support reuteran or levan formation by L. reuteri but result in a formation of comparable levels of lactic and acetic acids. A chemically acidified control was prepared with 5% (w/w flour) fructose, 5% glucose (w/w flour), and addition of lactic acid (80%) and glacial acidic acid in a ratio of 4:1 (v/v) to acidify the feed to a pH of 3.8 (Table 1). Basal diets were mixed with 20–50% fermented or acidified wheat to produce the experimental feeds. Control diet was obtained by a mixture of basal diet and unfermented wheat. All diets were formulated to meet or exceed nutrient recommendation of National Research Council Canada (NRC) (2012) for 5–10 kg pigs. Titanium dioxide (TiO2) was added to each of the test diets as an indigestible marker.
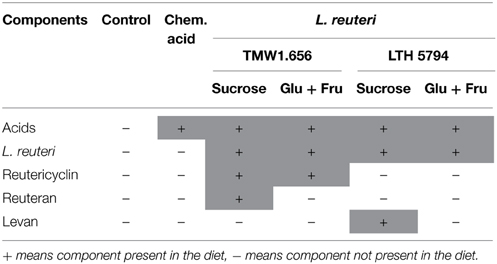
Table 1. Experimental diets used in this study.
Animals and Experimental Design
This animal trial was approved by the University of Alberta Animal Care and Use Committee under the guidelines of the Canadian Council on Animal Care and was conducted at the University of Alberta Swine Research and Technology Centre, Edmonton, AB, Canada. A total of 36 crossbred castrated weaning male pigs (~21 d of age) were selected and housed in a temperature-controlled room (28 ± 2.5°C). Pigs were divided into six consecutive and similar blocks with six pigs per block and one pig per pen (0.5 × 1.22 m). The experiment was designed to indicate whether the bacterial metabolites lactic and acetic acid, reuteran, levan, or reutericyclin influence the evolution of gut microbiota (Table 1). One pig per block was assigned to one of the six diets for a total of six observations per diet (Yang et al., 2015). Pigs were offered ad libitum food and water intake allowing for adequate growth. Meals in mash form were provided in equal amount twice daily (at 8 a.m. and 4 p.m.). Fresh fecal samples were collected from the pen floor in a sterile plastic bag at weaning, and 1, 2, or 3 weeks after weaning. A total of 137 samples obtained were collected and stored at −20°C. Frozen samples were thawed, mixed aseptically by spatula and 2–3 g subsamples were stored at −80°C.
DNA Extraction
Bacterial DNA was extracted from fecal samples using QIAamp® DNA stool Mini kit (50) (Qiagen, Inc., Valencia, CA, USA), following the manufacturer's instructions. Fecal DNA was quantified by Nano-Drop spectrophotometer system ND-1000 (Thermo Fisher Scientific Inc., Wilmington, USA). DNA quality was assessed by determining the ratio of absorbance at 260 and 280 nm. Only DNA samples that had 260:280 nm ratios higher than 1.8 were used for further analysis.
PCR Primers and Probes
Primers and probes used in this study are listed in Table 2. Sequences of genes coding for clostridial toxins (α-, β-, β2-, entero-, and ι-toxin) collected from GeneBank (http://www.ncbi.nlm.nih.gov/genbank). The sequence data were aligned with CLUSTAL-W (Thompson et al., 1994) to identify conserved sequences. Primers and probes were designed to target the conserved toxin sequences. The specificity of primer sequences was checked by Basic Local Alignment Search Tool (BLAST) (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). Primers and probes were synthesized by Integrated DNA Technologies (Coralville, IA, U.S.A.).
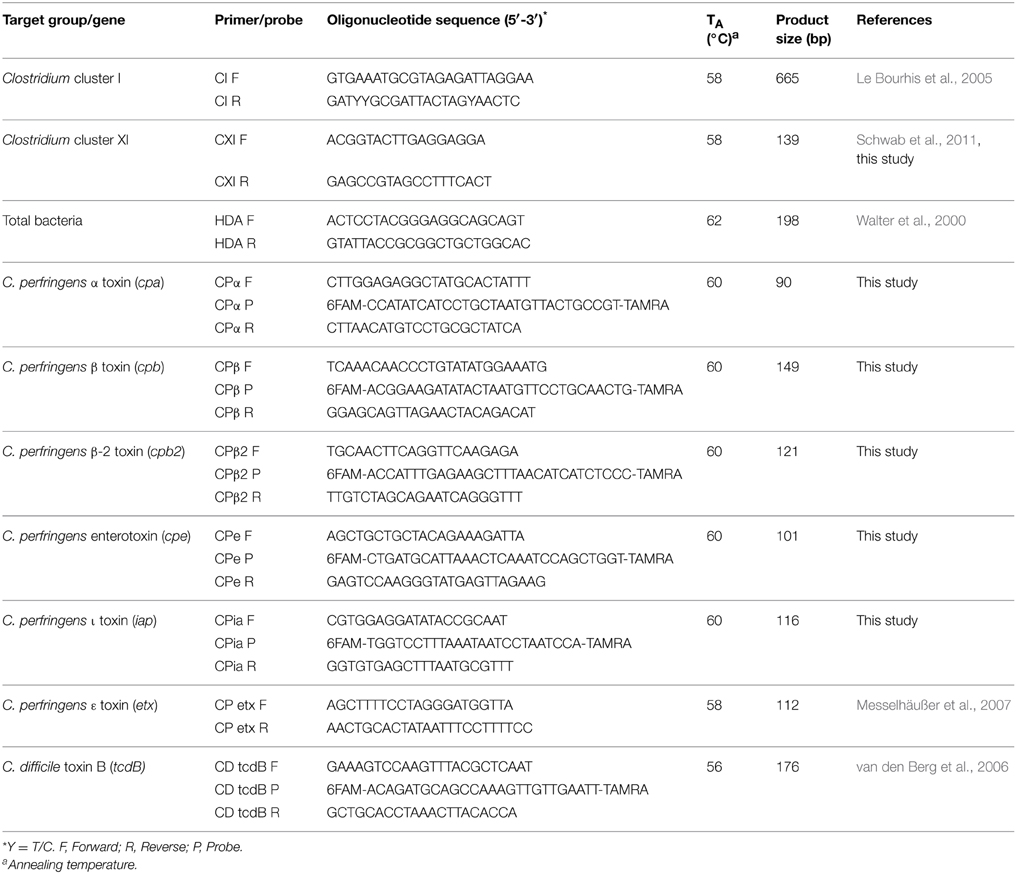
Table 2. Oligonucleotide sequences of the primers and probes used in this study.
Quantification of Clostridia and their Toxins by qPCR
Quantitative PCR (qPCR) was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using methodology described earlier (Metzler-Zebeli et al., 2010). To obtain positive controls for primers and probes targeting clostridial toxins, gBlocks® Gene Fragments were designed and synthesized by Integrated DNA Technologies. Standard curves for quantification of toxin genes were generated with 10-fold serial dilutions of purified PCR amplicons, which were amplified from gBlocks® Gene Fragments with the same primer pair and probe. For quantification of eubacteria and Clostridium clusters, standard curves were generated with amplicons that were amplified from serial dilutions of fecal DNA. The concentration of amplicons was determined by Nano-Drop spectrophotometer system ND-1000.
Fecal DNA was diluted to a concentration of 100 mg/L and analyzed in duplicate in a MicroAmp Fast Optical 96-well reaction plate sealed with MicroAmp Optical Adhesive Film (Applied Biosystems). Genes coding for 16S rRNA and the ε-toxin were amplified with the Quanti Fast SYBR Green master mix (Applied Biosystems). Taqman Fast master mix (Applied Biosystems) was used for detection of other toxins with probes. qPCR reaction contained 12.5 μL master mix, 4 μL of 10 μM primer solution in water, 2 μL of template DNA, and 6.5 μL nuclease-free water. Amplification of target sequences was achieved in 40 PCR cycles with primer annealing temperatures as shown in Table 2. Specific amplification of the target DNA was verified by melting curve analysis where applicable and by determination of the size of the amplicons by agarose gel electrophoresis.
Sequencing of 16S rRNA Sequence Tags and Sequence Data Analysis
High throughput sequencing of 16S rRNA sequence tags was performed by the University of Minnesota Genomics Center (Minneapolis, MN, USA) on a Illumina MiSeq. The V1–V3 regions of the 16S rRNA gene was amplified using primers Meta_V1_27F (TCGTCGGCA GCGTCAGATGTGTATAAGAGACAG AGAGTTTGATCMTGGCTCAG) and Meta_V3_534R (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG ATTACCGCGG CTGCTGG). The bold part of each primer is complementary to the eukaryotic 16S sequences while upstream sequences corresponded to Illumina adapters that are required for sequencing and multiplexing. Paired-end sequencing was performed according to the manufacturer's instructions.
The QIIME pipeline (MacQIIME 1.8.0 20140103 OS10.6) (Caporaso et al., 2010) was used to analyze the sequences of 16S rRNA genes. PANDAseq (Masella et al., 2012) was used for quality filtering and assembly of the two ends of each read into contigs. Pairs with miscalled or uncalled bases in the overlapping region were discarded. Operational Taxonomic Units (OTUs) were generated using the UPARSE workflow (Edgar, 2013). Briefly, all sequences were merged into a single file, and the library name was used for multiplexing. To minimize computing time, sequences were dereplicated and sorted by abundance. Unique sequences in the data set were discarded. Sequences were clustered into OTUs by USEARCH (Edgar, 2010) using the Greengenes reference database (release October 2013), and a 97% similarity threshold. UCHIME (Edgar, 2010; Edgar et al., 2011) was used for filtering of chimeric sequences. OTUs with abundance below 0.005% of the total number of sequences were discarded (Bokulich et al., 2013).
Downstream analyses including taxonomy assignments, and alpha and beta diversity estimations were conducted using the QIIME workflow core_diversity_analysis.py, with a sampling depth of 7939 (Navas-Molina et al., 2013). This analysis was conducted with default parameters: taxonomy was assigned using Ribosomal Database Project (RDP) Classifier V2 (Wang et al., 2007), alpha diversity was estimated by Phylogenetic Diversity (PD) Whole Tree, Chao 1 and Observed Species indices (Colwell et al., 2012), beta-diversity was estimated through UniFrac distances (Vázquez-Baeza et al., 2013).
Statistical Methods
Data analyses of relative abundance and qPCR results were performed in SAS, (version 9.3, SAS Institute, 2012). The gene copy numbers of Clostridium cluster I and XI were converted into percentage of total bacteria gene counts for analysis. Mixed Procedure (Proc MIXED) was used based on randomized complete block design with repeated measurement. In the model, diet and week and diet × week were considered as fixed effects, while block was considered as random effect and pigs were considered as experimental unit. Comparisons of treatments were determined by contrast of target groups (SAS version 9.3). For relative abundance analysis, data obtained at weaning were used as covariate when comparing the difference between combination groups to assess the effects of L. reuteri and its metabolites exopolysaccharides and reutericyclin.
To test hypotheses, p < 0.05 was considered significant, after Bonferroni-adjustment. Normality of all variables was tested by Kolmogorov-Smirnoff test (Young, 1977). Results are presented as means ± standard deviation. Alpha- and beta- diversity were analyzed in MacQIIME v1.8.
Results
Effects of Diet on Animal Health
All pigs remained healthy during the experiment period and diarrhea or any clinical signs of disease were not observed. Prior analyses of samples obtained in the same study reported the strain-specific quantification of L. reuteri and ETEC but not the overall composition of the hindgut microbiome (Yang et al., 2015).
Diversity of the Fecal Microbiome of Piglets
During the feeding trial, 137 fecal samples were collected and a total of 5,292,722 sequences with a minimum of 7939 sequences per sample were obtained. A total of 7434 OTUs were identified, representing 6 phyla, 27 families, 42 genera, and 49 species. Phyla in fecal samples and their abundance included Bacteroidetes (40.8–45.8%), Firmicutes (35.8–45.1%), Proteobacteria (0.9–12.9%), Tenericutes (0.7–5.2%), Spirochaetes (1.4–3.1%), and Planctomycetes (0–1.3%).
Alpha and beta diversity analyses revealed that the effects of treatments were small when compared to the differences occurring over time. In Alpha diversity metrics (within sample diversity), species richness of samples taken at week 3 was significantly higher (p < 0.001) than the diversity of samples taken earlier in the experiment (Figure 1) but differences between diets were not significant (p > 0.05). Beta diversity (between sample diversity) also demonstrated that samples taken at different times differed (p < 0.0001) (Figure 2). The distances between samples within each week were always smaller than the between week comparisons (Figure 2).
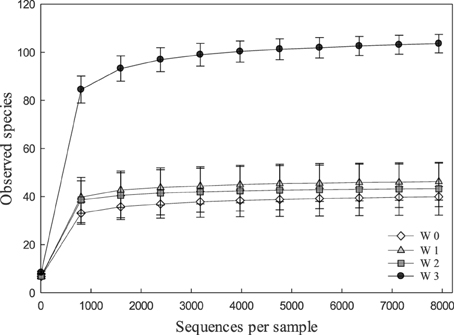
Figure 1. Rarefaction curves indicating the effect of the number of partial sequences of 16S rRNA genes that were analyzed on the number of OUT's in fecal microbiota of pigs. Rarefaction curves were calculated in QIIME with the sample depth of 7939 sequences per sample from 137 fecal samples obtained at weaning (week 0, n = 29), or at week 1 (n = 36), week 2 (n = 36), and week 3 (n = 36) after weaning.
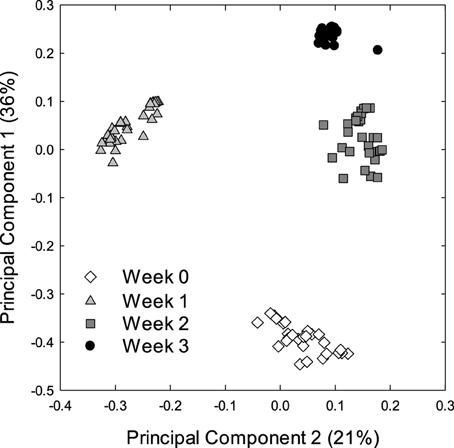
Figure 2. Principle coordinate analysis (PCA) of the bacterial microbiota of piglets. The PCA plot was generated using the unweighted UniFrac distance metric. Each dot represents an individual sample collected at the start of weaning week 0 (n = 29) or after week 1 (n = 36), week 2 (n = 36), and week 3 (n = 36) of weaning.
Transition of Bacteria over Time and Prevalent Bacterial Genera
The fecal microbiome was characterized by analysis of the relative abundance of bacterial taxa at the phylum level (Figure 3) and at the genus level (Table 3). The proportion of Bacteriodetes and Firmicutes remained unchanged over the study period. Major shifts were observed in the Proteobacteria, Tenericutes, and Planctomycetes (Figure 3). Proteobacteria peaked at week 1 and 2 and decreased again at week 3; Tenericutes and Planctomycetes increased over time (Figure 3). Within the Bacteroidetes, Prevotella and the unassigned S24-7 genus increased while Bacteroides and other unknown genera decreased (Table 3). The overall increase of microbial diversity (Figure 1) was largely attributed to increased abundance and diversity of bacterial taxa in the Firmicutes. Changes in the Proteobacteria were mainly attributable to Enterobacteriaceae (compare Table 3 and Figure 3).
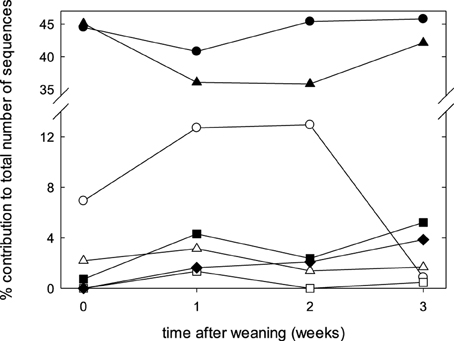
Figure 3. Composition of fecal microbiota at the phylum level at weaning, and during the first 3 weeks after weaning. Data represent the median proportions of each phylum as determined by the RDP (Ribosomal Database Project) classifier. The phyla are represented by symbols as follows: ●, Bacteroidetes; ▴, Firmicutes;  , Proteobacteria; Δ, Spirochaetes; ■, Tenericutes;
, Proteobacteria; Δ, Spirochaetes; ■, Tenericutes;  , Planctomycetes; ♦, unassigned.
, Planctomycetes; ♦, unassigned.
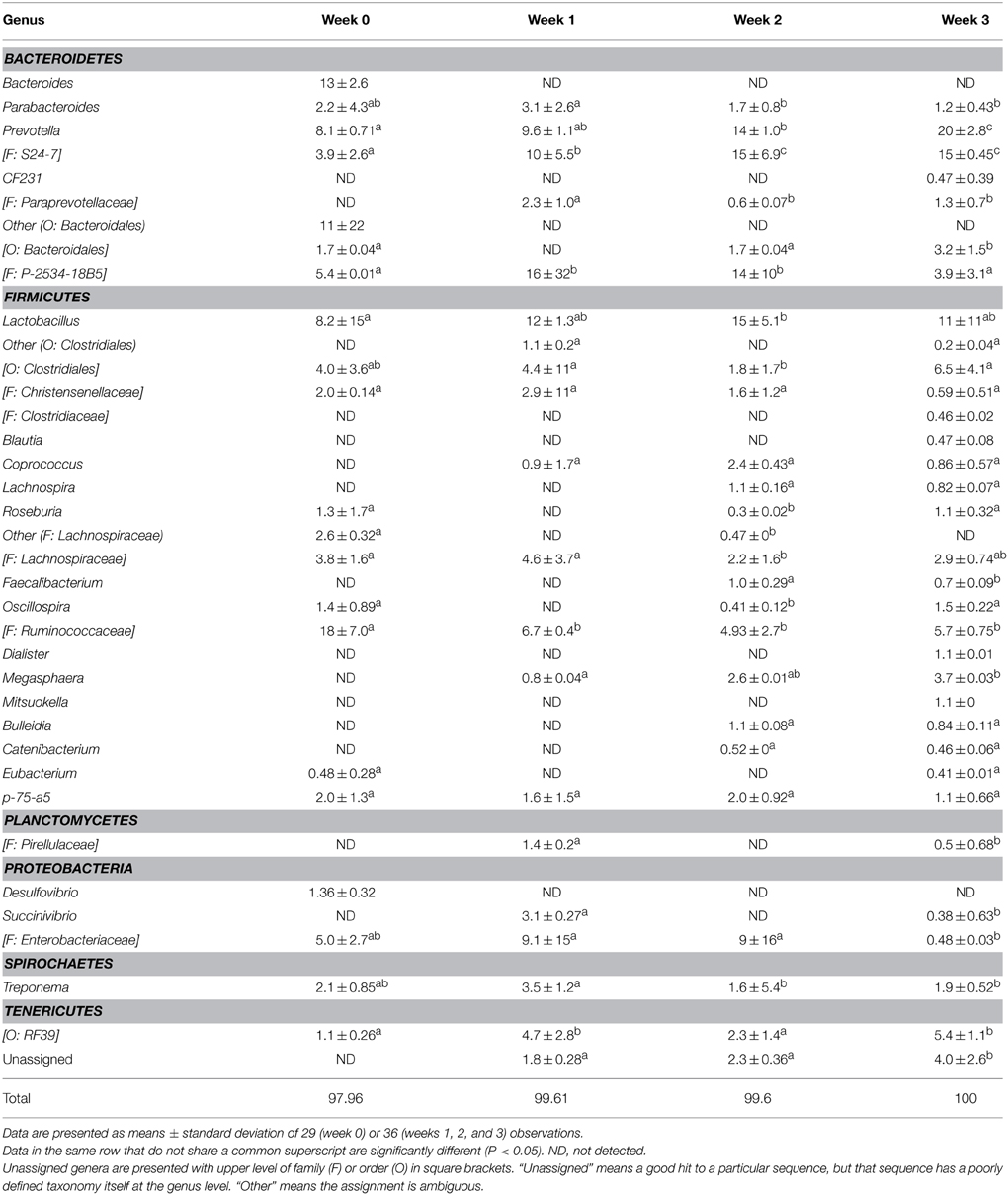
Table 3. Relative abundance (%) of bacterial genera in fecal microbiota of pigs at weaning (week 0) and at week 1, 2, or 3 after weaning, determined by Illumina sequencing of 16S rRNA tags.
Clostridia Clusters and Toxins Quantified by qPCR
To determine effects of feed fermentation on the Clostridium cluster I and XI, these organisms and toxins produced by C. difficile and C. perfringens were quantified using qPCR (Table 4 and data not shown). Both Clostridium clusters were relatively abundant in fecal samples; changes over time or changes within diets, however, were not significant (p > 0.05; data not shown). The α- and β-2 toxins from C. perfringens were detected in samples collected at week 0 and in a few samples from week 1 but not in samples taken at later times. Differences in the abundance of toxins in samples from animals fed different diets were not significant (p > 0.05). The abundance of other clostridial toxins, namely the β-, entero-, ι-, and ε-toxin of C. perfringens and the C. difficile toxin B, was below the detection limit of 3.6 log(copy number/g) in all samples (Table 4).
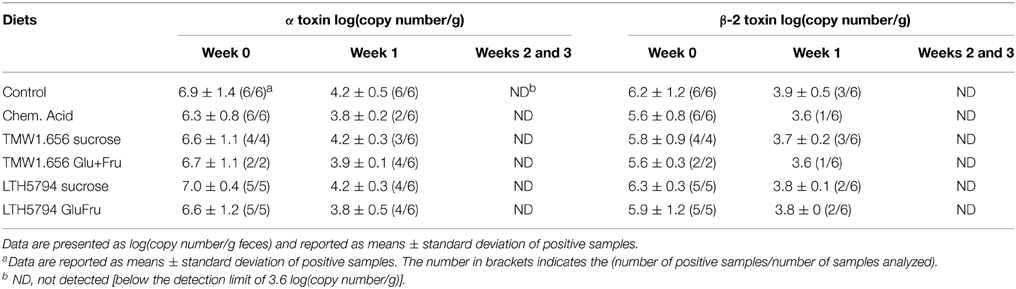
Table 4. Quantification of alpha and beta-2 toxins of C. perfringens in fecal samples collected at weaning (week 0) and at week 1, 2, 3 after weaning.
Effects of Treatments on Bacteria Species
Analysis of diet-induced changes accounted for the individual differences between animals in the same group by using data from each pig at week 0 as covariate (Table 5). To analyse the effect of specific feed components or metabolites that were present in several diets, diets were grouped as follows: Diets containing L. reuteri (groups 3, 4, 5, and 6) or not (group 1 and 2); diets containing reutericyclin (groups 3 and 4) or not (groups 1, 2, 5, and 6), and diets containing exopolysaccharides (groups 3 and 5) or not (groups 1, 2, 4, and 6). Moreover, the impact of feed fermented with L. reuteri TMW1.656 was compared to L. reuteri LTH5794 (Table 5).
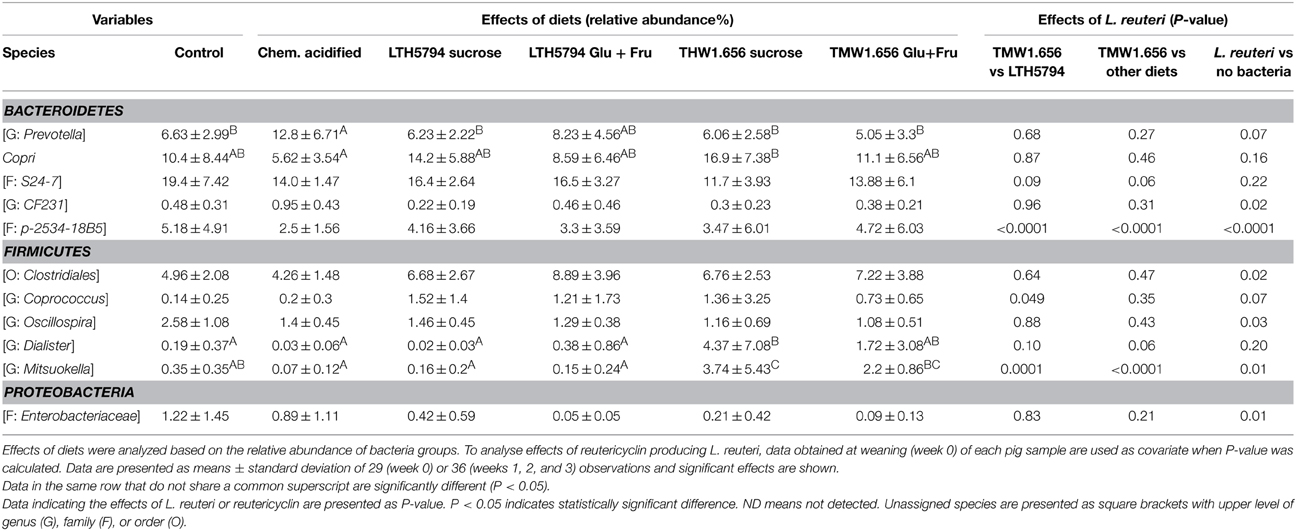
Table 5. Effects of diets and reutericyclin producing L. reuteri on the composition of fecal microbiota of pigs 3 weeks after weaning, as determined by Illumina sequencing of 16S rRNA tags.
Significant differences between individual diets pertain to few bacterial taxa in the phyla Bacteroidetes and Firmicutes (Table 5). Any L. reuteri strain altered the abundance of 6 bacterial taxa when compared to the control diets (Table 5). These changes particularly included a reduced number of the family Enterobacteriacae. Diets containing reutericyclin significantly changed the abundance of a Mitsuokella species and a family in the phylum Bacteroidetes (Table 5); these differences were also significant when the reutericyclin negative strain L. reuteri LTH5794 was compared to the reutericyclin positive L. reuteri TMW1.656 (Table 5). The presence or absence of exopolysaccharides had no significant (p > 0.05) influence on any bacterial taxon.
Discussion
The study investigated the effect of L. reuteri fermented diets on the development of intestinal microbiota of pigs after weaning. The experimental design aimed to determine the contribution of specific metabolites, i.e., reuteran, levan, and reutericyclin, on the evolution of the intestinal microbiome. The present study is the first employing high throughput sequencing to document the transition of the microbiome of piglets. In contrast, the development of the infant microbiota after birth is well documented (Koenig et al., 2011; La Rosa et al., 2014). The infant microbiome is characterized by low diversity and stability. Important determinants of the infant gut microbiome include the mode of delivery, type of feeding, antibiotic use, and the gestational age of the mother (Penders et al., 2006; Koenig et al., 2011). From birth to weaning, both human infants and piglets experience a succession of Lactobacillus spp. in the gut due to the consumption of milk (Tannock et al., 1990; Roger et al., 2010). The gradual adaptation of the diet in infants typically avoids major problems that are associated with a shifting microbiome. The sudden change of diet after weaning, however, often causes dysbiosis and diarrheal diseases in piglets, and is thus a major concern in pig production (Lallès et al., 2007).
Bacteroidetes and Firmicutes dominated the intestinal microbiome in piglets, in keeping with past studies on the swine microbiome (Leser et al., 2002; Lamendella et al., 2011; Riboulet-Bisson et al., 2012). The present study additionally documents that strict anaerobes such as Ruminococcaceae, Bacteroides, and Prevotella were the dominant bacteria at weaning. After weaning, Lactobacillus and Prevotella spp. replaced Ruminococcaceae and Bacteroides as the most abundant bacterial genera. Dietary changes modulate the gut microbiome of pigs (Lu et al., 2014). Bacteria belonging to Bacteroides-Prevotella-Porphyromonas play an important role in fiber degradation, and are stimulated by fermentable non-starch polysaccharides in pig diets (Metzler-Zebeli et al., 2010; Ivarsson et al., 2014). Human studies have linked the diversity of different plant fibers in whole grains to an increased diversity of the gut microbiome, particularly in the genera Roseburia, Bifidobacterium, Eubacterium, and Dialister (Martínez et al., 2013). Accordingly, low carbohydrate diets resulted in a substantial and diet-dependent reduction of Firmicutes (Duncan et al., 2008). The increase of bacterial diversity after weaning was particularly attributable to an increased diversity in the phylym Firmicutes and may thus be linked to the presence of whole wheat in the piglets' diet, which accounted for 20% of the diet after weaning and for 50% diet after week 1 (Yang et al., 2015).
The strains of L. reuteri used in the present study are rodent-lineage allochthones to the pig intestine (Su et al., 2012; Frese et al., 2014). Major changes in the intestinal microbiota were attributable to the presence of probiotic L. reuteri and its metabolites. The most prominent change attributable to the presence of L. reuteri was the reduced abundance of Enterobacteriaceae. This result conforms to prior reports obtained with diverse probiotic cultures (De Angelis et al., 2007; Konstantinov et al., 2008; Bednorz et al., 2013; Valdovska et al., 2014) and indicates that successful competition with Enterobacteriaceae is not a specific property of L. reuteri. Remarkably, this study also demonstrated that the abundance of several members of the Firmicutes and Bacteroidetes was influenced by L. reuteri.
Exopolysaccharides did not influence the composition of gut microbiota. Levan and reuteran are not digested by pancreatic digestive enzymes and selectively fermented by hindgut microbiota (Korakli et al., 2002; van Bueren et al., 2015). However, the exopolysaccharides levels in the feed used in this study, 1–3 g/kg feed (Yang et al., 2015), are low when compared to other studies reporting prebiotic intervention (Valdovska et al., 2014). Fermented feed containing reuteran, however, specifically reduced the abundance of enterotoxigenic E. coli (ETEC) in weanling piglets (Yang et al., 2015). The lack of any effect of reuteran on the overall composition of the gut microbiome (this study) coupled to the specific reduction of ETEC colonization (Yang et al., 2015) supports the hypothesis that effects of reuteran are mediated by a specific reduction of ETEC adhesion rather than a prebiotic effect (Chen et al., 2014).
Bioinformatic analyses of the metagenome of intestinal microbiota suggested that the ecology of colonic microbiota is shaped by competition for substrates rather than the production of antimicrobial compounds (Walter and Ley, 2011; Zheng et al., 2015). Accordingly, bacteriocin producing L. salivarius did not induce significant changes in the gut microbiome of pigs when compared to an isogenic bacteriocin-negative strain (Riboulet-Bisson et al., 2012). Medication of grower pigs with in feed antibiotics (Chlortetracycline, sulfamethazine, and penicillin), however, caused much more substantial changes of the colonic microbiome than was observed in this study for reutericyclin (Looft et al., 2014). Reutericyclin is a unique antimicrobial compound with broad spectrum of activity against Gram-positive bacteria (Gänzle et al., 2000; Gänzle, 2004; Hurdle et al., 2011; Lin et al., 2015). Reutericyclin is produced during growth of L. reuteri in wheat sourdough (Gänzle and Vogel, 2002), suggesting that feed fermentation with L. reuteri TMW1.656 delivers active concentrations of reutericyclin to the swine gut. We hypothesized that reutericyclin may reduce the abundance of the Clostridium clusters I and XI. The pathogenic species C. perfringens (cluster I) and C. difficile (cluster XI), are sensitive to reutericyclin (Hurdle et al., 2011; Hofstetter et al., 2013). However, neither the abundance of Clostridium cluster I and XI nor the abundance of genes coding for clostridial toxins was influenced by reutericyclin-producing L. reuteri. The abundance of Dialister and Mitsuokella increased upon feeding of the reutericyclin producing L. reuteri TMW1.656, possibly as a consequence of the inhibition of reutericyclin-sensitive competitors.
In conclusion, the present study monitored the transition of fecal microbiota of weanling piglets, and determined the impact of L. reuteri and its metabolites on this transition of intestinal microbiota. After weaning, bacterial diversity increased, mainly due to an increase of bacterial taxa in the phylum Firmicutes. Weaning was also associated by a transient increase of Enterobacteriaceae, which corresponds to the susceptibility of weanling piglets to infection by enteric pathogens. The gut microbiome of weanling piglets was not influenced by the inclusion of organic acids in the diet; however, the presence of viable L. reuteri, reutericyclin, and reuteran all affected the gut microbiome. Probiotic L. reuteri altered the abundance of several bacterial taxa, notably Enterobacteriaceae including E. coli. The reutericyclin producing strain significantly increased the abundance of two strict anaerobic members of the Firmicutes, while reuteran affected the colonization with ETEC but none of the numerically dominant members of fecal microbiota (Yang et al., 2015, this study). Data on the contribution of specific metabolic activities of L. reuteri to probiotic activity will facilitate the strain selection for probiotic feed applications in animal production. The study also novel opens novel avenues to reduce the incidence of childhood diarrhea in developing countries (Thapar and Sanderson, 2004) by application of probiotic cultures, or by food fermentations with probiotic L. reuteri (Sekwati-Monang and Gänzle, 2011).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Alberta Livestock and Meat Agency is acknowledged for funding (Grant No. 2013R002R). MG acknowledges the Canada Research Chair program for funding. YY is grateful to Dr. Juan Jovel and Dr. Petya T. Koleva for the help in sequencing data analysis.
References
Bednorz, C., Guenther, S., Oelgeschläger, K., Kinnemann, B., Pieper, R., Hartmann, S., et al. (2013). Feeding the probiotic Enterococcus faecium strain NCIMB 10415 to piglets specifically reduces the number of Escherichia coli pathotypes that adhere to the gut mucosa. Appl. Environ. Microbiol. 79, 7896–7790. doi: 10.1128/aem.03138-13
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Brandt, M. J. (2014). Starter cultures for cereal based foods. Food Microbiol. 37, 41–43. doi: 10.1016/j.fm.2013.06.007
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, X. Y., Woodward, A., Zijlstra, R. T., and Gänzle, M. G. (2014). Exopolysaccharides synthesized by Lactobacillus reuteri protect against enterotoxigenic Escherichia coli in piglets. Appl. Environ. Microbiol. 80, 5752–5760. doi: 10.1128/AEM.01782-14
Colwell, R. K., Chao, A., Gotelli, N. J., Lin, S. Y., Mao, C. X., Chazdon, R. L., et al. (2012). Models and estimators linking individual-based and sample-based rarefaction, extrapolation and comparison of assemblages. J. Plant Ecol. 5, 3–21. doi: 10.1093/jpe/rtr044
Corr, S. C., Li, Y., Riedel, C. U., O'Toole, P. W., Hill, C., and Gahan, C. G. M. (2007). Bacteriocin production as a mechanism for the anti-infective activity of Lactobacillus salivarius UCC118. Proc. Nat. Acad. Sci. U.S.A. 104, 7617–7621. doi: 10.1073/pnas.0700440104
De Angelis, M., Siragusa, S., Caputo, L., Ragni, A., Burzigotti, R., and Gobbetti, M. (2007). Survival and persistence of Lactobacillus plantarum 4.1 and Lactobacillus reuteri 3S7 in the gastrointestinal tract of pigs. Vet. Microbiol. 123, 133–144. doi: 10.1016/j.vetmic.2007.02.022
De Weirdt, R., Crabbé, A., Roos, S., Vollenweider, S., Lacroix, C., van Pijkeren, J. P., et al. (2012). Glycerol supplementation enhances L. reuteri's protective effect against S. Typhimurium colonization in a 3-D model of colonic epithelium. PLoS ONE 7:e37116. doi: 10.1371/journal.pone.0037116
Duncan, S. H., Lobley, G. E., Holtrop, G., Ince, J., Johnstone, A. M., Louis, P., et al. (2008). Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 32, 1720–1724. doi: 10.1038/ijo.2008.155
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Frese, S. A., Benson, A. K., Tannock, G. W., Loach, D. M., Kim, J., Zhang, M., et al. (2011). The evolution of host specialization in the vertebrate gut symbiont Lactobacillus reuteri. PLoS Genet. 7:e1001314. doi: 10.1371/journal.pgen.1001314
Frese, S. A., Mackenzie, D. A., Peterson, D. A., Schmaltz, R., Fangman, T., Zhou, Y., et al. (2014). Molecular characterization of host-specific biofilm formation in a vertebrate gut symbiont. PLoS Genet. 9:e1004057. doi: 10.1371/journal.pgen.1004057
Galle, S., Schwab, C., Dal Bello, F., Coffey, A., Gänzle, M. G., and Arendt, E. K. (2012). Influence of in-situ synthesized exopolysaccharides on the quality of gluten-free sorghum sourdough bread. Int. J. Food Microbiol. 155, 105–112. doi: 10.1016/j.ijfoodmicro.2012.01.009
Gänzle, M. G. (2004). Reutericyclin: biological activity, mode of action, and potential applications. Appl. Microbiol. Biotechnol. 64, 326–332. doi: 10.1007/s00253-003-1536-8
Gänzle, M. G., Höltzel, A., Walter, J., Jung, G., and Hammes, W. P. (2000). Characterization of reutericyclin produced by Lactobacillus reuteri LTH2584. Appl. Environ. Microbiol. 66, 4325–4333. doi: 10.1128/AEM.66.10.4325-4333.2000
Gänzle, M. G., and Vogel, R. F. (2002). Contribution of reutericyclin production to the stable persistence of Lactobacillus reuteri in an industrial sourdough fermentation. Int. J. Food Microbiol. 80, 31–45. doi: 10.1016/S0168-1605(02)00146-0
Hofstetter, S., Gebhardt, D., Ho, L., Gänzle, M., and McMullen, M. L. (2013). Effects of nisin and reutericyclin on resistance of endospores of Clostridium spp. to heat and high pressure. Food Microbiol. 34, 46–51. doi: 10.1016/j.fm.2012.11.001
Hurdle, J. G., Heathcott, A. E., Yang, L., Yan, B., and Lee, R. E. (2011). Reutericyclin and related analogues kill stationary phase Clostridium difficile at achievable colonic concentrations. J. Antimicrob. Chemother. 66, 1773–1776. doi: 10.1093/jac/dkr201
Ivarsson, E., Roos, S., Liu, H. Y., and Lindberg, J. E. (2014). Fermentable non-starch polysaccharides increases the abundance of Bacteroides-Prevotella-Porphyromonas in ileal microbial community of growing pigs. Animal 8, 1777–1787. doi: 10.1017/S1751731114001827
Koenig, J. E., Spor, A., Scalfone, N., Fricker, A. D., Stombaugh, J., Knight, R., et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4578–4585. doi: 10.1073/pnas.1000081107
Konstantinov, S. R., Awati, A. A., Williams, B. A., Miller, B. G., Jones, P., Stokes, C. R., et al. (2006). Post-natal development of the porcine microbiota composition and activities. Environ. Microbiol. 8, 1191–1199. doi: 10.1111/j.1462-2920.2006.01009.x
Konstantinov, S. R., Smidt, H., Akkermans, A. D., Casini, L., Trevisi, P., Mazzoni, M., et al. (2008). Feeding of Lactobacillus sobrius reduces Escherichia coli F4 levels in the gut and promotes growth of infected piglets. FEMS Microbiol. Ecol. 66, 599–607. doi: 10.1111/j.1574-6941.2008.00517.x
Korakli, M., Gänzle, M. G., and Vogel, R. F. (2002). Metabolism by bifidobacteria and lactic acid bacteria of polysaccharides from wheat and rye and exopolysaccharides produced by Lactobacillus sanfranciscensis. J. Appl. Microbiol. 92, 958–965. doi: 10.1046/j.1365-2672.2002.01607.x
Lallès, J. P., Bosi, P., Smidt, H., and Stokes, C. R. (2007). Weaning - a challenge to gut physiologists. Livestock Sci. 108, 82–93. doi: 10.1016/j.livsci.2007.01.091
Lamendella, R., Domingo, J. W., Ghosh, S., Martinson, J., and Oerther, D. B. (2011). Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 11:103. doi: 10.1186/1471-2180-11-103
La Rosa, P. S., Warner, B. B., Zhou, Y., Weinstock, G. M., Sodergren, E., Hall-Moore, C. M., et al. (2014). Patterned progression of bacterial populations in the premature infant gut. Proc. Natl. Acad. Sci. U.S.A. 111, 12522–12527. doi: 10.1073/pnas.1409497111
Le Bourhis, A. G., Saunier, K., Dore, J., Carlier, J. P., Chamba, J. F., Popoff, M. R., et al. (2005). Development and validation of PCR primers to assess the diversity of Clostridium spp. in cheese by temporal temperature gradient gel electrophoresis. Appl. Environ. Microbiol. 71, 29–38. doi: 10.1128/AEM.71.1.29-38.2005
Leser, T. D., Amenuvor, J. Z., Jensen, T. K., Lindecrona, R. H., Boye, M., and Møller, K. (2002). Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl. Environ. Microbiol. 68, 673–690. doi: 10.1128/AEM.68.2.673-690.2002
Lin, X. B., and Gänzle, M. G. (2014). Effect of lineage-specific metabolic traits of Lactobacillus reuteri on sourdough microbial ecology. Appl. Environ. Microbiol. 80, 5782–5789. doi: 10.1128/AEM.01783-14
Lin, X. B., Lohans, C. T., Duar, R., Zheng, J., Vederas, J. C., Walter, J., et al. (2015). Genetic determinants of reutericyclin biosynthesis in Lactobacillus reuteri. Appl. Environ. Microbiol. 81, 2032–2041. doi: 10.1128/AEM.03691-14
Looft, T., Allen, H. K., Cantarel, B. L., Levine, U. Y., Bayles, D. O., Alt, D. P., et al. (2014). Bacteria, phages and pigs: the effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 8, 1566–1576. doi: 10.1038/ismej.2014.12
Lu, X. M., Lu, P. Z., and Zhang, H. (2014). Bacterial communities in manures of piglets and adult pigs bred with different feeds revealed by 16S rDNA 454 pyrosequencing. Appl. Microbiol. Biotechnol. 9, 2657–2665. doi: 10.1007/s00253-013-5211-4
Martínez, I., Lattimer, J. M., Hubach, K. L., Case, J. A., Yang, J., Weber, C. G., et al. (2013). Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 7, 269–280. doi: 10.1038/ismej.2012.104
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G., and Neufeld, J. D. (2012). PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13:31. doi: 10.1186/1471-2105-13-31
Messelhäußer, U., Zucker, R., Elmer-Englhard, D., Busch, U., Hörmansdorfer, S., Pudich, U., et al. (2007). Nachweis und Charakterisierung von Clostridium perfringens mittels real-time-PCR. JVL 2, 194–197. doi: 10.1007/s00003-007-0173-z
Metzler-Zebeli, B. U., Hooda, S., Pieper, R., Zijlstra, R. T., van Kessel, A. G., Mosenthin, R., et al. (2010). Nonstarch polysaccharides modulate bacterial microbiota, pathways for butyrate production, and abundance of pathogenic Escherichia coli in the pig gastrointestinal tract. Appl. Environ. Microbiol. 76, 3692–3701. doi: 10.1128/AEM.00257-10
Millette, M., Cornut, G., Dupont, C., Shareck, F., Archambault, D., and Lacroix, M. (2008). Capacity of human nisin- and pediocin-producing lactic acid bacteria to reduce intestinal colonization by vancomycin-resistant enterococci. Appl. Environ. Microbiol. 74, 1997–2003. doi: 10.1128/AEM.02150-07
Navas-Molina, J. A., Peralta-Sanchez, J. M., Gonzalez, A., McMurdie, P. J., Vazquez-Baeza, Y., Xu, Z., et al. (2013). Advancing our understanding of the human microbiome using QIIME. Meth. Enzymol. 531, 371–444. doi: 10.1016/B978-0-12-407863-5.00019-8
Penders, J., Thijs, C., Vink, C., Stelma, F. F., Snijders, B., Kummeling, I., et al. (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521. doi: 10.1542/peds.2005-2824
Rea, M. C., Alemayehu, D., Casey, P. G., O'Connor, P. M., Lawlor, P. G., Walsh, M., et al. (2014). Bioavailability of the anti-clostridial bacteriocin thuricin CD in gastrointestinal tract. Microbiology 160, 439–445. doi: 10.1099/mic.0.068767-0
Riboulet-Bisson, E., Sturme, M. H., Jeffery, I. B., O'Donnell, M. M., Neville, B. A., Forde, B. M., et al. (2012). Effect of Lactobacillus salivarius bacteriocin Abp118 on the mouse and pig intestinal microbiota. PLoS ONE 7:e31113. doi: 10.1371/journal.pone.0031113
Roger, L. C., Costabile, A., Holland, D. T., Hoyles, L., and McCartney, A. L. (2010). Examination of faecal Bifidobacterium populations in breast- and formula-fed infants during the first 18 months of life. Microbiology 156, 3329–3341. doi: 10.1099/mic.0.043224-0
Schwab, C., Cristescu, B., Northrup, J. M., Stenhouse, G. B., and Gänzle, M. G. (2011). Diet and environment shape fecal bacterial microbiota composition and enteric pathogen load of grizzly bears. PLoS ONE 6:e27905. doi: 10.1371/journal.pone.0027905
Sekwati-Monang, B., and Gänzle, M. G. (2011). Microbiological and chemical characterisation of ting, a sorghum-based sourdough product from Botswana. Int. J. Food Microbiol. 150, 115–121. doi: 10.1016/j.ijfoodmicro.2011.07.021
Spinler, J. K., Sontakke, A., Hollister, E. B., Venable, S. F., Oh, P. L., Balderas, M. A., et al. (2014). From prediction to function using evolutionary genomics: human-specific ecotypes of Lactobacillus reuteri have diverse probiotic functions. Genome Biol. Evol. 6, 1772–1789. doi: 10.1093/gbe/evu137
Su, M. S., Oh, P. L., Walter, J., and Gänzle, M. G. (2012). Intestinal origin of sourdough Lactobacillus reuteri isolates as revealed by phylogenetic, genetic, and physiological analysis. Appl. Environ. Microbiol. 78, 6777–6780. doi: 10.1128/AEM.01678-12
Su, Y., Yao, W., Perez-Gutierrez, O. N., Smidt, H., and Zhu, W. Y. (2008). 16S ribosomal RNA-based methods to monitor changes in the hindgut bacterial community of piglets after oral administration of Lactobacillus sobrius S1. Anaerobe 14, 78–86. doi: 10.1016/j.anaerobe.2007.12.004
Tannock, G. W., Fuller, R., and Pedersen, K. (1990). Lactobacillus succession in the piglet digestive tract demonstrated by plasmid profiling. Appl. Environ. Microbiol. 56, 1310–1316.
Teixeira, J. S., Seeras, A., Sanchez-Maldonado, A. F., Zhang, C., Su, M. S., and Gänzle, M. G. (2014). Glutamine, glutamate, and arginine-based acid resistance in Lactobacillus reuteri. Food Microbiol. 42, 172–180. doi: 10.1016/j.fm.2014.03.015
Thapar, N., and Sanderson, I. R. (2004). Diarrhoea in children: an interface between developing and developed countries. Lancet 363, 641–653. doi: 10.1016/S0140-6736(04)15599-2
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Tuohy, K. M., Probert, H. M., Smejkal, C. W., and Gibson, G. R. (2003). Using probiotics and prebiotics to improve gut health. Drug Discov. Today 8, 692–700. doi: 10.1016/S1359-6446(03)02746-6
Valdovska, A., Jemeljanovs, A., Pilmane, M., Zitare, I., Konosonoka, I. H., and Lazdins, M. (2014). Alternative for improving gut microbiota: use of Jerusalem artichoke and probiotics in diet of weaned piglets. Pol. J. Vet. Sci. 17, 61–69. doi: 10.2478/pjvs-2014-0008
van Bueren, A. L., Saraf, A., Martens, E. C., and Dijkhuizen, L. (2015). Differential metabolism of exopolysaccharides from probiotic lactobacilli by the human gut symbiont Bacteroides thetaiotaomicron. Appl. Environ. Microbiol. 81, 3973–3983. doi: 10.1128/AEM.00149-15
van den Berg, R. J., Kuijper, E. J., van Coppenraet, L. E. S. B., and Claas, E. C. J. (2006). Rapid diagnosis of toxinogenic Clostridium difficile in faecal samples with internally controlled real-time PCR. Clin. Microbiol. Infect. 12, 184–186. doi: 10.1111/j.1469-0691.2005.01301.x
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A., and Knight, R. (2013). EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2:16. doi: 10.1186/2047-217X-2-16
Walter, J. (2008). Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl. Environ. Microbiol. 74, 4985–4996. doi: 10.1128/AEM.00753-08
Walter, J., and Ley, R. (2011). The human gut microbiome: ecology and recent evolutionary changes. Annu. Rev. Microbiol. 65, 411–429. doi: 10.1146/annurev-micro-090110-102830
Walter, J., Tannock, G. W., Tilsala-Timisjarvi, A., Rodtong, S., Loach, D. M., Munro, K., et al. (2000). Detection and identification of gastrointestinal Lactobacillus species by using denaturing gradient gel electrophoresis and species-specific PCR primers. Appl. Environ. Microbiol. 66, 297–303. doi: 10.1128/AEM.66.1.297-303.2000
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang, Y., Gänzle, M. G., and Schwab, C. (2010). Exopolysaccharide synthesized by Lactobacillus reuteri decreases the ability of enterotoxigenic Escherichia coli to bind to porcine erythrocytes. Appl. Environ. Microbiol. 76, 4863–4866. doi: 10.1128/AEM.03137-09
Wilson, C. M., Loach, D., Lawley, B., Bell, T., Sims, I. M., O'Toole, P. W., et al. (2014). Lactobacillus reuteri 100-23 modulates urea hydrolysis in the murine stomach. Appl. Environ. Microbiol. 80, 6104–6013. doi: 10.1128/AEM.01876-14
Yang, Y., Galle, S., Le, M. H. A., Zijlstra, R., and Gänzle, M. G. (2015). Feed fermentation with reuteran- and levan producing Lactobacillus reuteri reduced colonization of weanling pigs with enterotoxigenic Escherichia coli. Appl. Environ. Microbiol. doi: 10.1128/AEM.01525-15. [Epub ahead of print].
Young, I. T. (1977). Proof without prejudice - use of Kolmogorov-Smirnov test for analysis of histograms from flow systems and other sources. J. Histochem. Cytochem. 25, 935–941. doi: 10.1177/25.7.894009
Zhang, L., Xu, Y. Q., Liu, H. Y., Lai, T., Ma, J. L., Wang, J. F., et al. (2010). Evaluation of Lactobacillus rhamnosus GG using an Escherichia coli K88 model of piglet diarrhoea: effects on diarrhoea incidence, faecal microflora and immune responses. Vet. Microbiol. 141, 142–148. doi: 10.1016/j.vetmic.2009.09.003
Zhao, C., Kinner, M., Wismer, W., and Gänzle, M. G. (2015). Effect of glutamate-accumulation during sourdough fermentation with L. reuteri on the taste of bread and sodium-reduced bread. Cereal Chem. 92, 224–230. doi: 10.1094/CCHEM-07-14-0149-R
Keywords: enterterotoxigenic Escherichia coli, ETEC, pigs, feed fermentation, reutericyclin, exopolysaccharides, Lactobacillus reuteri, probiotic
Citation: Yang Y, Zhao X, Le MHA, Zijlstra RT and Gänzle MG (2015) Reutericyclin producing Lactobacillus reuteri modulates development of fecal microbiota in weanling pigs. Front. Microbiol. 6:762. doi: 10.3389/fmicb.2015.00762
Received: 01 June 2015; Accepted: 13 July 2015;
Published: 28 July 2015.
Edited by:
Maria De Angelis, University of Bari Aldo Moro, ItalyReviewed by:
Zhao Chen, Clemson University, USAAmit Kumar Tyagi, The University of Texas MD Anderson Cancer Center, USA
Copyright © 2015 Yang, Zhao, Le, Zijlstra and Gänzle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael G. Gänzle, Department of Agricultural, Food and Nutritional Science, University of Alberta, 4-10 Ag/For Centre, Edmonton, AB T6G 2P5, Canada,bWdhZW56bGVAdWFsYmVydGEuY2E=