 Ralph A. Cacho
Ralph A. Cacho Yi Tang
Yi Tang Yit-Heng Chooi
Yit-Heng Chooi- 1Chemical and Biomolecular Engineering Department, University of California, Los Angeles, Los Angeles, CA, USA
- 2Chemistry and Biochemistry Department, University of California, Los Angeles, Los Angeles, CA, USA
- 3Plant Sciences Division, Research School of Biology, The Australian National University, Canberra, ACT, Australia
Genomics has revolutionized the research on fungal secondary metabolite (SM) biosynthesis. To elucidate the molecular and enzymatic mechanisms underlying the biosynthesis of a specific SM compound, the important first step is often to find the genes that responsible for its synthesis. The accessibility to fungal genome sequences allows the bypass of the cumbersome traditional library construction and screening approach. The advance in next-generation sequencing (NGS) technologies have further improved the speed and reduced the cost of microbial genome sequencing in the past few years, which has accelerated the research in this field. Here, we will present an example work flow for identifying the gene cluster encoding the biosynthesis of SMs of interest using an NGS approach. We will also review the different strategies that can be employed to pinpoint the targeted gene clusters rapidly by giving several examples stemming from our work.
Introduction
Human health has been benefited from the secondary metabolites (SMs) produced by fungi. These small molecules, also known as natural products, include important clinical drugs like the antibiotic penicillins (Kardos and Demain, 2011), the cholesterol-lowering statins (Endo, 2010), the immunosuppressive cyclosporins (Britton and Palacios, 1982) and the antifungal echinocandins (Balkovec et al., 2014). Microbial SMs, including those from bacteria and fungi, continue to serve as important sources of molecules for drug discovery. For many decades, the fascinating and diverse structures of microbial SMs have inspired the organic chemists to embark on a quest to elucidate their biosynthetic pathways. Many basic insights into SM pathways were obtained by organic chemists using isotopic tracers during the 1950s (Bentley, 1999). The research shifted to the molecular biology of SM biosynthesis with the availability of tools for DNA cloning and sequencing. This is marked by several landmark papers, which described the molecular cloning of whole SM biosynthetic pathway on a contiguous stretch of DNA from actinomycete bacteria (Malpartida and Hopwood, 1984; Cortes et al., 1990; Donadio et al., 1991). Around the same period, the first fungal SM gene cluster, the penicillin biosynthetic gene cluster with the core non-ribosomal peptide synthetase (NRPS) gene encoding L-δ-(α-aminoadipoyl)-L-cysteinyl-D-valine (ACV) synthetase had been discovered in the fungus Penicillium chrysogenum (Díez et al., 1990). This is followed by the discovery of the terpenoid gene cluster encoding trichothecenes (Hohn et al., 1993), and polyketide gene clusters encoding aflatoxin/sterigmatocystin biosynthesis in Aspergillus sp. (Brown et al., 1996; Yu et al., 2004) and lovastatin biosynthesis in A. terreus (Kennedy et al., 1999). This hallmark trait of gene clustering was then observed in almost all other classes of fungal SM pathways including indole alkaloids and terpenoids (Keller et al., 2005). The tendency for the biosynthetic genes in microbial SM pathways to cluster on a chromosomal locus greatly accelerated the elucidation of enzymatic steps involved in biosynthesis of individual SM compounds using molecular biology approaches. Consequently, identification of the gene cluster that encodes the production of a given SM is now becoming the common first step toward elucidating the molecular and enzymatic basis for the biosynthesis of a given SM. Subsequent verification of the predicted gene cluster is often achieved via targeted deletion and/or heterologous expression of key biosynthetic genes. Further characterization of the biosynthetic pathway can be done by deletion of the individual biosynthetic genes in the cluster or reconstruction of the whole pathway in heterologous systems.
Targeted SM gene cluster discovery in fungi in the pre-genomic era is a tedious and time-consuming process. Traditionally, this was done by either complementation of blocked mutants by cosmid libraries (e.g. Mayorga and Timberlake, 1990; Hendrickson et al., 1999), or insertional mutagenesis followed by plasmid rescue from the blocked mutant (e.g., Yang et al., 1996; Chung et al., 2003). Both of these aforementioned methods rely on screening of blocked/complementation mutants, which work well for pigment compounds but can be cumbersome if the phenotype, cannot be easily observed or assayed. For example, in the pioneering work to identify lovastatin gene cluster, 6000 mutants were screened for restored lovastatin production by HPLC/TLC after transformation of A. terreus with a cosmid library (Hendrickson et al., 1999). Other methods for identifying key biosynthetic gene include antibody screening of cDNA expression library (e.g., Beck et al., 1990), differential display reverse transcriptase-PCR (e.g., Linnemannstons et al., 2002), suppression subtractive hybridization-PCR (O’Callaghan et al., 2003). However, these methods often lead to isolation of a single gene or partial gene cluster. Further cosmid library walking is required to obtain the whole gene cluster. Due to the relatively large genome size, genome scanning method, such as that demonstrated for discovery of enediyne antitumor antibiotic pathways in actinomycete bacteria (Zazopoulos et al., 2003), is less feasible for fungi.
Biosynthesis of polyketide SMs has been subjected to more intensive studies among other classes of SM pathways in fungi (Chooi and Tang, 2012). One of the earlier major advances in identification of fungal polyketide SM gene clusters is the development of degenerate primer PCR based on conserved ketosynthase (KS) domain of polyketide synthases (PKSs). The KS domain DNA fragments are then use as probes to identify the cosmid library clone carrying the whole or partial gene cluster. This method has been employed to localize the aflatoxin and fumonisin PKS genes (Feng and Leonard, 1995; Proctor et al., 1999). The subsequent primer sets developed to target KS domains of a non-reducing (NR-), partial-reducing (PR-), and highly-reducing polyketide synthases (HR-PKSs) are especially useful for localizing specific PKS gene clusters in cosmid libraries (Bingle et al., 1999; Nicholson et al., 2001). Pioneering work by Kroken et al. (2003) which performed a phylogenomic analysis of the PKS genes in the genomes of Gibberella, Neurospora, Cochliobolus, and Botrytis species revealed that fungal PKSs are likely to be derived from eight major lineages (Kroken et al., 2003). Nevertheless, despite the degeneracy of the primers and subdivision of fungal PKS genes in to different subclasses, some PKS genes in the genome can be still be missed by the degenerate primer PCR approach. Furthermore, it has to be bear in mind that, although most SM pathways are clustered on chromosome in fungi some of them can be split into two or three smaller subclusters, such as the pathways for dothistromin in Dothistroma septosporum (Chettri et al., 2013), tryptoquivaline in A. clavatus (Gao et al., 2011), echinocandin in Emericella rugulosa (Cacho et al., 2012), and prenylated xanthones in A. nidulans (Sanchez et al., 2011). There are also instances where the gene clusters of multiple SM pathways are intertwined together, such as the fumitremorgin, fumagillin, and pseurotin supercluster (Wiemann et al., 2013a). In such cases, the absence of whole genome sequence information can complicate the identification of the complete gene set for the target SM pathway.
Whole genome sequencing (WGS) of the target SM-producing fungus bypasses the need for the cumbersome library construction, screening, and chromosome walking. More importantly, the genome sequence can reveal the inventory of all the SM gene clusters in the producing fungus. Even though each fungus can harbor 30–50 SM gene cluster, the number is still finite and one of them must encode the SM of interest. For example, WGS of G. zeae with the Sanger sequencing method has allowed the systematic deletion of all 15 PKS genes in the fungal genome, which lead to identification of the gene cluster for zearalenone, aurofusarin, fusarin C and an unidentified black perithecial pigment (Gaffoor et al., 2005). This opens up the opportunities for detailed characterization of these pathways for zearalenone (Kim et al., 2005; Zhou et al., 2010; Lee et al., 2011), aurofusarin (Frandsen et al., 2006; Frandsen et al., 2011), and fusarin C (Niehaus et al., 2013). The development of next-generation sequencing (NGS) technologies in the last decade has dramatically lowered the cost for DNA sequencing and put the power of microbial WGS in the hand of individual laboratories (van Dijk et al., 2014). This technology revolution has energized the natural product research field and sparked some exciting NGS-based targeted SM gene cluster discovery projects in fungi (Table 1). Our work that used such NGS approach includes the discovery of SM clusters encoding viridicatumtoxin and griseofulvin (Chooi et al., 2010), tryptoquialanine (Gao et al., 2011), echinocandin (Cacho et al., 2012), asperlicin (Haynes et al., 2012), ardeemin (Haynes et al., 2013), and brefeldin (Zabala et al., 2014). Other studies that have taken the advantage of NGS have identified the SM clusters for fungal bicyclo[2.2.2]diazaoctane indole alkaloids (Li et al., 2012), equisetin (Kakule et al., 2013), and pneumocandin (Chen et al., 2013).
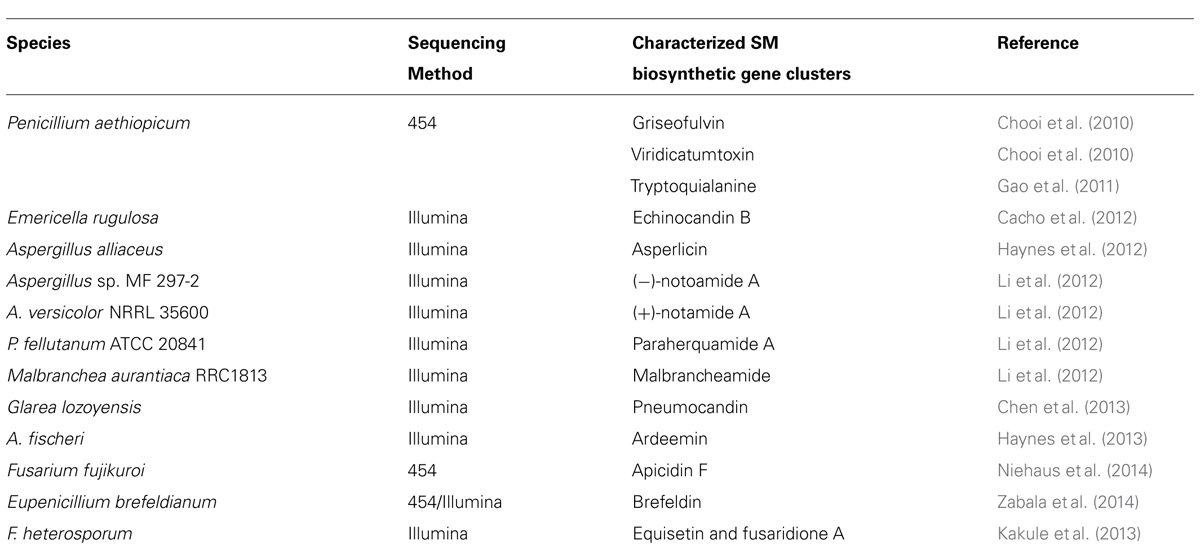
TABLE 1. Examples of biosynthetic gene clusters assigned to their respective compounds using next-generation sequencing technology.
Contemporary natural product research programs are now increasingly based on the understanding of the relationship between the SM molecules and the biosynthetic genes (Walsh and Fischbach, 2010). Although such studies are primarily motivated by the desire to understand the molecular and enzymatic basis of SM biosynthesis, the potential benefits and implications derived from such work are manifold. Firstly, the new chemical insights obtained in elucidating the metabolic pathway can be used to design more efficient total synthesis routes for complex natural products. Secondly, knowledge about the gene cluster can facilitate the metabolic engineering effort to increase the yield of useful SMs for commercial production (Pickens et al., 2011). The knowledge also forms the basis for generation of new SM analogs by mutasynthesis and combinatorial biosynthesis. A recent example is the combinatorial biosynthesis of benzenediol lactones (Xu et al., 2014b), which built on previous studies (Reeves et al., 2008; Wang et al., 2008; Zhou et al., 2010; Xu et al., 2013b). Novel biocatalysts useful for green chemistry and chemoenzymatic process development may also be discovered from SM pathways. For example, the characterization of the acyltransferase LovD in lovastatin pathway led to the development of green chemistry process for the semisynthetic cholesterol-lowering drug simvastatin (Xie et al., 2006, 2009; Gao et al., 2009; Xu et al., 2013a). Furthermore, the gene cluster information will be useful for knowledge-based genome mining for structurally-related compound in other fungi, e.g., the discovery of the immunosuppressive neosartoricin based on viridicatumtoxin biosynthesis genes (Chooi et al., 2012, 2013a). Importantly, these established links between genes and SMs are valuable knowledge that contributes to the overarching aims for (1) accurate prediction of SM structures based on DNA sequences and (2) rational design of biosynthetic pathways for synthesis of organic molecules. Finally, bridging the gaps between genes and molecules may facilitate our understanding of the natural functions of SMs using comparative genomics and transcriptomics tools (Chooi and Solomon, 2014).
It is important to note that a SM/natural product-motivated fungal genome sequencing project has different aims compared to the conventional WGS project coordinated by an international consortium of researchers associated with large sequencing centers. The major goal is to obtain the whole SM gene cluster that encode the targeted SM on a contiguous stretch of DNA sequence contig or scaffold. That means the completeness of the genome sequence coverage and whole-genome annotation is of lower priorities. The key question is how to rapidly narrow down and accurately pinpoint the correct SM cluster in the genome that encodes the production of a target SM. Getting an accurate initial prediction will significantly reduce the time spent on gene cluster verification. Below, we will provide the general work flow and guidelines for researchers who are considering adopting NGS technologies for targeted SM gene cluster discovery based on our own experience. We will also use several examples stemming from our work, two PKS pathways and two NRPS pathways, to illustrate the concepts and strategies.
Next-Generation De Novo Fungal Genome Sequencing and Assembly
The standard NGS-based targeted SM gene cluster discovery work flow used in our laboratory is presented in Figure 1. The fungal strain acquired from culture collections or her sources are first verified for production of the targeted compound before its genomic DNA was sent for sequencing. A variety of culture media and conditions can also be tested to optimize the production of the target compound. Gene deletion or disruption followed by the detection of loss of target compound production is still the most common method for initial gene cluster verification. Alternatively, if genetic transformation is proven to be difficult on the fungus, expression of the backbone biosynthetic enzymes [e.g., PKS, NRPS, terpene synthase, or dimethylallyltryptophan synthase (DMATS)] in the candidate SM gene cluster in a heterologous system may be another way to verify the gene cluster.
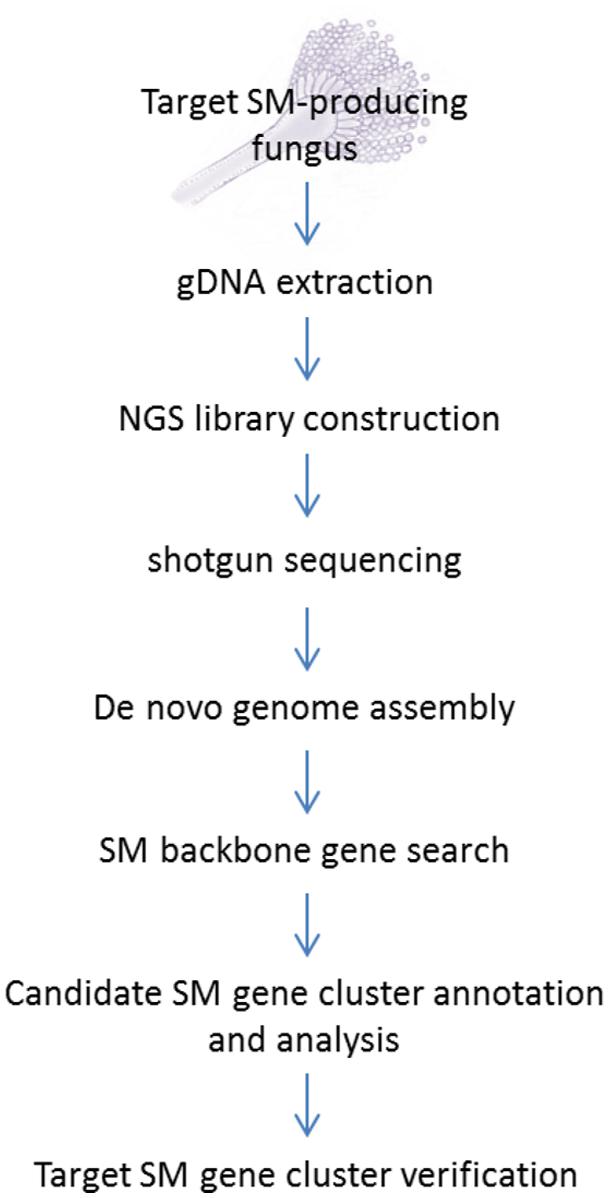
FIGURE 1. Next-generation sequencing (NGS) workflow for targeted secondary metabolite (SM) gene cluster discovery.
The next thing to consider will be the choice of sequencing method. The choice will be mainly based on the cost, sequence quality, sequence read length, speed and project throughput (the number of strains to be sequenced together). There are currently several NGS technologies on the market suitable for de novo fungal WGS, with Illumina now dominating the market (Nowrousian, 2010; van Dijk et al., 2014). We have experience in both Roche/454 FLX Titanium and Illumina HiSeq2000, but other DNA sequencing platforms may work for this purpose as well. For an overview of the different latest NGS technologies, the readers are referred to van Dijk et al. (2014). The de novo sequencing of P. aethiopicum (synonym P. lanosocoeruleum) IBT5753 genome is one of the earliest examples of SM-motivated fungal WGS undertaken by individual laboratories, which used the Roche/454 FLX Titanium platform (Chooi et al., 2010).
The introduction of Illumina HiSeq2000 (now superseded by HiSeq2500) with ∼100 bp paired end (PE) reads dramatically reduced the cost of sequencing and increased the sequencing output. The shorter read length of HiSeq2000 is compensated by the deeper coverage and the paired-end nature of the Illumina reads. The Illumina PE reads allowed the assembly of longer scaffolds with gaps, which can be filled in later using routine PCR and Sanger sequencing if any of the scaffold harbor interesting SM gene cluster. Longer PE information can be obtained by generating mate pair libraries with larger insert size (e.g., 3 kbp or 5 kbp). Indeed, it has been recently demonstrated that good fungal genome assembly can be obtained on platform with shorter sequence reads (50 bp) like SOLiD using mate pair libraries (Umemura et al., 2013b).
We first used HiSeq2000 for sequencing of the brefeldin-producing Eupenicillium brefeldianum ATCC 58665 and the echinocandin-producing E. rugulosa (Cacho et al., 2012; Zabala et al., 2014). The amount of sequence data acquired for assembly of the ∼32 Mbp E. rugulosa genome is more than sufficient despite the shorter read length (N50 = 235 kbp). In fact, we can get equally good quality assembly using half the amount of data. Now, we routinely perform HiSeq2000 sequencing multiplexed for four fungal genomic samples per lane (∼200X depth of coverage each genome) and can routinely obtain good quality assembly for the purpose of SM gene cluster discovery. Using this arrangement, the cost of sequencing per fungal strain can be lower than the cost required for cosmid/fosmid library construction, screening, and chromosome walking, not to mention the significant time savings. With the use of mate pair libraries, more fungal genomic samples can likely be included into HiSeq2000 per lane yet still yield fine assembly. One recent study shows that the optimum sequencing depth for small bacteria to medium eukaryotic genomes with 2X 100 bp PE Illumina reads is 50-100X (Desai et al., 2013).
There is more than a dozen of software available for assembly of short reads generated by NGS, some are more memory intensive than the others, and there is differences in assembly speed as well. The popular ones including Velvet (Zerbino and Birney, 2008), SOAPdenovo (Li et al., 2010a), AllPATHS (Butler et al., 2008), and ABySS (Simpson et al., 2009). With optimization, some software may allow some small-medium size genomes to be assembled on a standalone workstation (Kleftogiannis et al., 2013). There are several studies that compare the efficiency and assembly quality of different assembly software (Lin et al., 2011; Zhang et al., 2011). We have experience mostly in using the SOAPdenovo assembler developed by BGI (Li et al., 2010a). Our SOAPdenovo assemblies were run on the UCLA Hoffman2 computer cluster. The assemblies can usually be completed with 32–128 GB memory requested from the cluster, depending on the genome size and the amount of input data. For the standard 2X 100 bp PE reads, we often use a k-mer size of 63 or 79 for the SOAPdenovo assembly and were able to obtain good results. The latest version, SOAPdenovo2, promised to improve memory efficiency and assembly quality, and more optimized for the longer Illumina reads (rather than the 35–50 bp reads from older Illumina platforms; Luo et al., 2012). Many NGS providers also provide service for sequence assembly with a fee.
Locating the Target Secondary Metabolite Gene Cluster
The first task after obtaining the scaffolds generated from the assembly is to narrow down the scaffolds containing the candidate SM gene clusters. Despite the enormous structural diversity, most fungal SMs can be divided into four major classes, polyketides, non-ribosomal peptides, terpenes, and indole alkaloids, based on the limited classes of carbon building blocks they derived from (Keller et al., 2005). Thus, an efficient way of narrowing down the scaffolds that may contain the target gene cluster is to search for genes encoding backbone biosynthetic enzymes that synthesized the specific class of compounds correspond to the target SM. This can be most easily achieved by performing a TBLASTN search against a database generated from the assembled fungal genome scaffolds using the “makeblastdb” command in the NCBI stand-alone BLAST application1. Depending on the class of SM, an arbitrary chosen conserved domain of the corresponding backbone enzymes can be used as a TBLASTN query. For example, a protein query sequence of a KS domain of PKS for polyketide SMs, an adenylation (A) domain of NRPS for non-ribosomal peptide SMs, terpene synthase for terpenoid SMs and DMATS for prenylated indole alkaloids. The TBLASTN will generate a list of scaffolds containing the SM gene clusters belongs the corresponding SM classes ranked based on homology to the query sequence. Alternatively, commercial bioinformatics software programs with intuitive graphic user interface (GUI) that support BLAST on local database are also available, e.g., CLC Genomics Workbench2 (CLC Bio) and Geneious3 (Biomatters). After locating the scaffolds containing gene clusters of the target SM classes, the number of candidate scaffolds can be further narrowed down with comparative genomics analysis (See Comparative Genomics Approach for Target SM Gene Cluster Prediction) followed by gene predictions and more in-depth knowledge-based bioinformatics analysis (see Figure 2 and Section “Pinpointing Target SM Gene Cluster with Knowledge-Based Analysis – Retrobiosynthesis”).
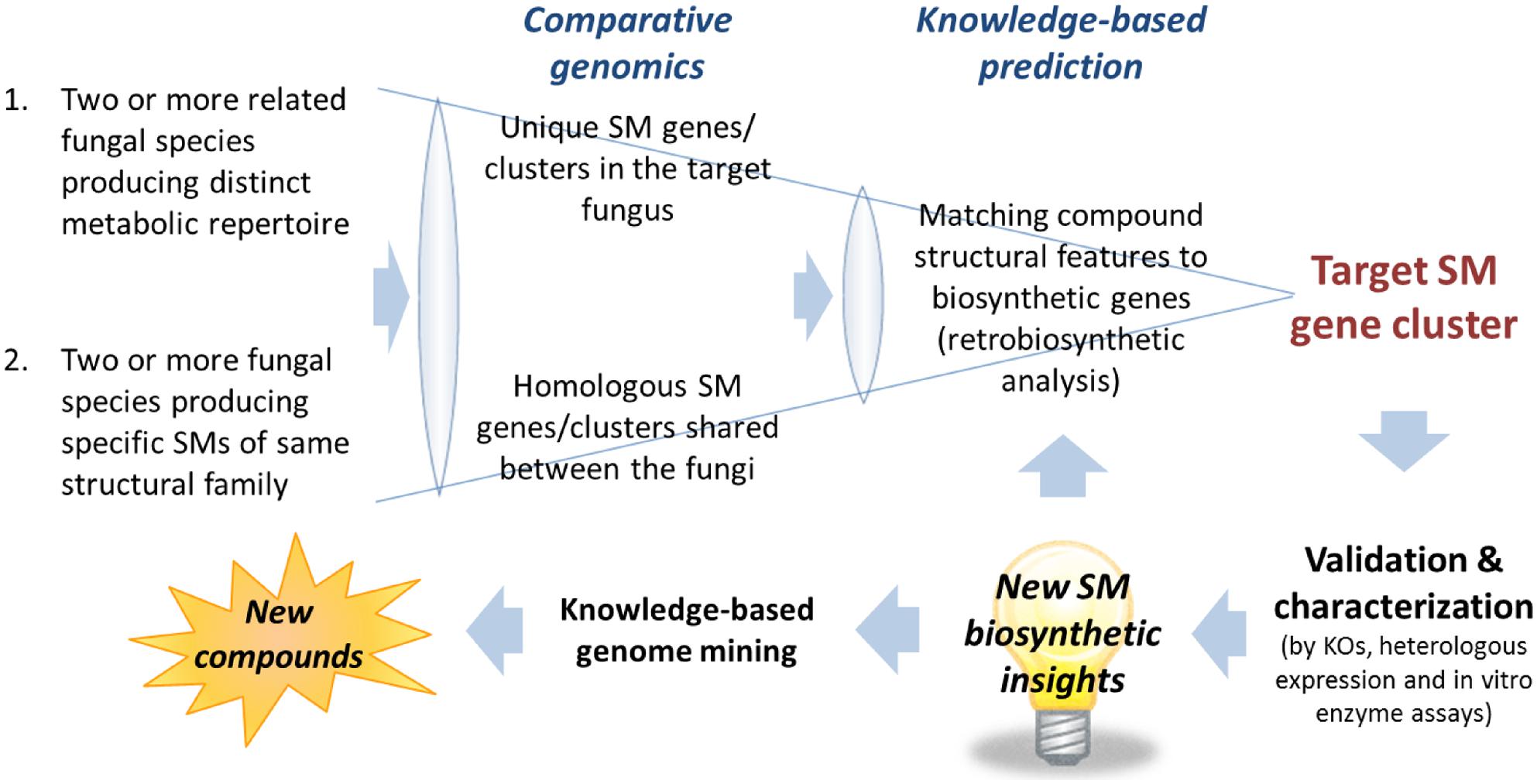
FIGURE 2. Comparative genomics and knowledge-based prediction strategies for targeted SM gene cluster discovery. New biosynthetic knowledge and insights into gene-to-molecular structure relationship would be useful for target SM gene cluster predictions and knowledge-based genome mining for discovery of novel SM compounds.
For fungal gene predictions (the locations and exon–intron structures of genes on individual scaffolds) we routinely use FGENESH (Softberry; Solovyev et al., 2006), which can yield relatively accurate predictions for fungi. The web server allows direct submission of FASTA nucleotide sequences with gene-finding parameters trained using datasets from several fungal species4. Alternatively, AUGUSTUS also provides relatively accurate fungal gene prediction5 based on training sets from various fungal species (Stanke et al., 2004). Individual protein sequences predicted in a candidate scaffold are then submitted to NCBI BLASTP server for detailed conserved domain analysis (using the integrated NCBI Conserved Domain Search feature) and homologous sequence comparison. The EBI Interproscan also offers similar conserved domain prediction and protein functional analysis. As each gene cluster usually contain 3–15 genes (>20 genes in some cases), it is often not too time consuming when the number of candidate scaffolds has been narrowed down significantly. Several SM gene cluster prediction software programs have also been developed, which can aid this process considerably (see Pinpointing Target SM Gene Cluster with Knowledge-Based Analysis – Retrobiosynthesis).
Comparative Genomics Approach for Target SM Gene Cluster Prediction
Comparative genomics can be a useful approach for filtering of candidate gene clusters (Figure 2). The increasing number of sequenced fungal genomes in public databases, due in part to the lower sequencing cost enabled by NGS technologies, allows one to find a suitable genome of related sequenced organism for comparison with the organism of interest. Since the genomes of different organisms, even those belonging to the same genus, would encode different array of SMs, one can compare the SM backbone biosynthesis gene inventory of one organism with a closely related organism to rapidly narrow down the unique SM gene cluster. This approach is especially useful when there is pre-existing knowledge about the SM repertoire of the reference organism used for comparison. For example, in the search of SM gene clusters responsible for the biosynthesis of viridicatumtoxin and griseofulvin in P. aethiopicum (Chooi et al., 2010), we compared the PKS inventory of P. aethiopicum with that of P. chrysogenum based on prior knowledge from previous chemotaxonomy studies (Frisvad and Samson, 2004). Since it is known that P. chrysogenum produces neither viridicatumtoxin nor griseofulvin, we can first filter out the orthologous PKS genes shared between the two fungi (Figure 3A; Section Case Study A: Griseofulvin and Case Study B: Viridicatumtoxin). Similar the genome-wide comparison of NRPS genes in A. nidulans and E. rugulosa was used to narrow down the candidate echinocandin gene cluster (Cacho et al., 2012). It has to be bear in mind that the absence of report about the presence of a target SM compound in a specific species does not always correlate with the absence of the gene cluster in the genome as there can be strain-strain variation and many SM gene clusters could be transcriptionally silent. For example, TAN-1612, reported in another A. niger strain as BMS-192548 (Kodukula et al., 1995; Shu et al., 1995), is not detected in the sequenced A. niger ATCC 1015. However, the corresponding gene cluster can be identified and activated by transcriptional regulator overexpression (Li et al., 2011). The abilities to produce fumitremorgin by A. fumigatus (Kato et al., 2013), and gibberrellin and beauvericin by G. fujikuroi (Wiemann et al., 2013b), also vary from strain to strain. Nonetheless, such a comparative genomics approach had served as a useful first-pass filter in our hands, especially for fungal species that have been characterized chemotaxonomically, such as those species in the Aspergillus and Penicillium genera (Frisvad and Samson, 2004; Frisvad et al., 2007).
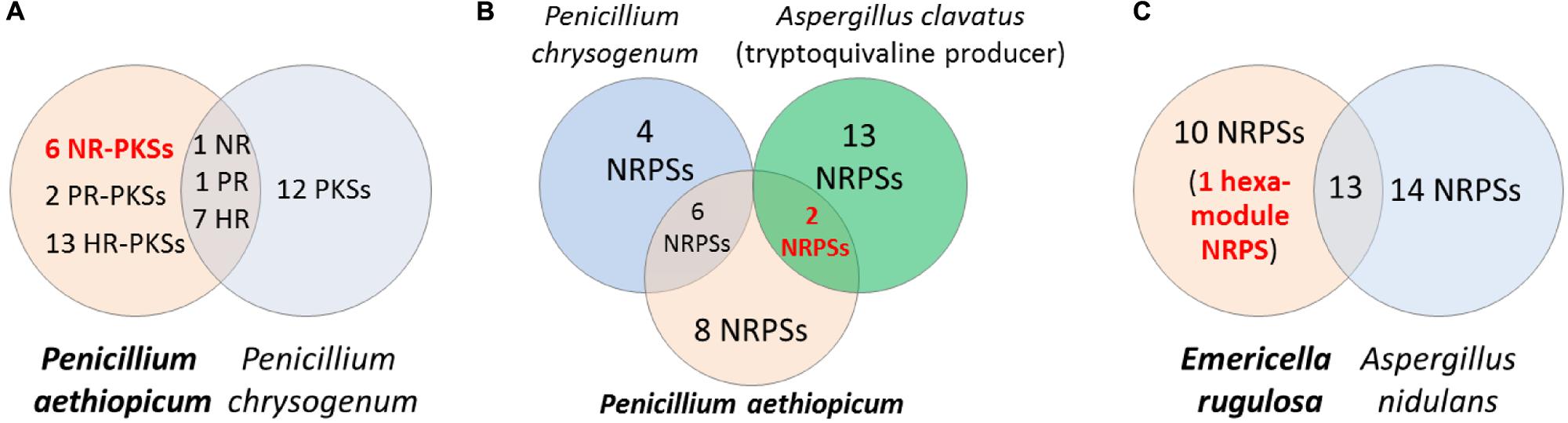
FIGURE 3. Comparative genomics approach for discovery of SM biosynthetic gene cluster. Comparison of the PKS genes in Penicillium aethiopicum and P. chrysogenum was utilized in searching for the griseofulvin and viridicatumtoxin gene cluster (A). In order to narrow down possible candidates for the tryptoquialanine gene cluster in P. aethiopicum, NRPS genes that are non-orthologous to P. chrysogenum NRPS genes but are orthologous to the NRPS genes of the tryptoquivaline producer A. clavatus were found (B). The echinocandin NRPS gene was found in Emericella rugulosa was found by searching for a hexamodule NRPS gene in E. rugulosa that is non-orthologous to A. nidulans NRPS genes (C).
Conversely, comparative genomics can also be utilized for identifying the biosynthetic gene clusters of a target group of SMs bearing structural similarities but made by less-related fungal species (Figure 2). In this case, orthologous gene clusters in two organisms that are capable of synthesizing the same family of compounds are targeted. An example of the latter strategy is demonstrated in our work on tryptoquialanine/tryptoquivaline gene cluster identification in P. aethiopicum/A. clavatus (Gao et al., 2011; Figure 3B). The strategy is also well-demonstrated in the work by the Sherman group in investigating the biosynthesis of fungal bicyclo[2.2.2]diazaoctane indole alkaloids (-)- and (+)-notoamide A, paraherquamide A, and malbrancheamide A by Aspergillus sp. MF297-2, A. versicolor NRRL35600, P. fellutanum ATCC20841, and Malbranchea aurantiaca RRC1813, respectively (Li et al., 2012). More recently, NGS-driven comparative genomics of four Stachybotrys strains from two chemotypes producing either atranones or stratoxins have revealed two unique gene clusters, which possibly encode the biosynthesis of the two respective terpenoid-derived SMs (Semeiks et al., 2014). However, the identities of the two gene clusters are yet to be verified experimentally. Such comparative genomics approach is highly compatible with the high-throughput nature of Illumina sequencing as genomic samples from multiple fungal strains can be multiplexed on a single lane of an Illumina sequencer flow cell. In the case where the target SM gene cluster is split into more than one scaffold in one of the fungal genome assemblies, either due to disruption of assembly by repetitive sequence or the pathway is encoded by multiple loci, comparative genomics of the different strains can help localize the complete biosynthetic gene sets. This approach has facilitated the identification of genes for biosynthesis of tryptoquivaline in A. clavatus, which was separated into three genomic loci, by comparing with the tryptoquialanine-producing P. aethiopicum genome (Gao et al., 2011).
Lastly, one can use comparative genomics to make an educated guess on where the boundaries of the SM gene cluster are located. Oftentimes, SM gene clusters are flanked by syntenic blocks containing highly-conserved core genes (Machida et al., 2005; Tamano et al., 2008). Similarly, we have observed this trend in our studies toward the discovery of gene clusters encoding griseofulvin, viridicatumtoxin, cytochalasin, and tryptoquialanine. All four putative SM gene clusters are flanked by syntenic block of genes with high shared identity (>85%) in closely related ascomycetes species (Chooi et al., 2010; Gao et al., 2011; Qiao et al., 2011). In fact, a study reported a motif-independent bioinformatics approach for detection of SM gene cluster based on non-syntenic blocks in fungal genomes (Umemura et al., 2013a). One of the software that is useful for identifying and visualizing syntenic regions across multiple genomes is Mauve (Darling et al., 2004). While it does not guarantee that genes that are not highly conserved to a related organism are part of the cluster, this strategy is helpful toward minimizing the number of genes subjected to further functional analysis.
Pinpointing Target SM Gene Cluster with Knowledge-Based Analysis – Retrobiosynthesis
As mentioned above, gene predictions of individual scaffolds can be performed using software like FGENESH or AUGUSTUS after narrowing down the number of candidate SM gene clusters to a handful of scaffolds. Further detailed bioinformatics analysis of the individual SM gene clusters is then needed to identify the target gene cluster. Some software programs have been developed to aid SM gene cluster predictions. Two of the popular software programs available for prediction of SM gene clusters are antiSMASH (Medema et al., 2011; Blin et al., 2013) and SMURF (Khaldi et al., 2010). SMURF is specific for predicting SM gene clusters in fungal genomes, while antiSMASH can be used for both bacteria and fungi. Guidelines and detailed protocols for using these two and other related SM biosynthetic gene prediction programs can be found in Fedorova et al. (2012). SMURF requires a protein FASTA file and a gene coordinate file as input data. On the other hand, antiSMASH can accepts single nucleotide FASTA file and can be a very useful tool for getting an initial idea of the composition of SM gene cluster on each candidate scaffold along with gene annotation suggestions. Besides providing the predicted domain architecture of multi-domain backbone enzymes (i.e., PKSs and NRPSs), the smCOG (SM Cluster of Orthologous Groups) analysis module in antiSMASH also predicts the function of probable tailoring enzymes encoded in the cluster based on conserved domain analysis. Unfortunately, the substrate prediction function of antiSMASH is yet to be as useful for fungal SM clusters compared to bacterial ones. Moreover, intron prediction using antiSMASH is not as accurate compared to FGENESH, which can potentially hamper efforts toward heterologous expression of fungal SM biosynthetic enzymes in E. coli or yeast. With increasing number of established connections between fungal SM gene clusters and molecular structures, this feature is likely to improve in the future.
Nonetheless, a good understanding of the biochemistry of SM biosynthesis and knowledge about the relationship between biosynthetic genes and molecular structures is often needed to accurately pinpoint the SM gene cluster of interest. An excellent introduction to the common building blocks, enzymes, and biochemical reactions involve in SM biosynthesis is available (Dewick, 2009). The specific question that will be asked by the researcher is “what kind (and combination) of enzymes and precursors are likely to be involved in the biosynthesis of the target SM compound?” Such analytic-deductive approach is sometimes referred as “retro-biosynthetic analysis” where the SM structure are taken apart into simpler intermediates and precursors to help determine the enzymes and biological building blocks required for the target SM biosynthesis. Here, we will focus on the general strategies that can be adopted for pinpointing target PKS and NRPS gene clusters.
Polyketides
A great majority of fungal PKSs belong to the iterative type I PKSs whereupon the different PKS catalytic domains are juxtaposed on a single large polypeptide and the single set of PKS domains performs all the necessary catalytic activity during the biosynthesis of the polyketide. Fungal iterative type I PKSs are further classified into three major classes based on the degree of β-keto reduction performed by the PKS; namely the NR-PKSs, the PR-PKSs, and the HR-PKSs. The enzymology and classification of fungal iterative type I PKSs have been reviewed extensively (Cox, 2007; Crawford and Townsend, 2010; Chooi and Tang, 2012). As a general rule, aromatic polyketide compounds are synthesized by NR-PKSs, while HR-PKSs produce aliphatic compounds. PR-PKSs, on the other hand, has been shown to produce compounds lacking a phenolic hydroxyl group at the aromatic ring on the position where a β-keto group has been reduced to alcohol on the polyketide chain before cyclization, such as 6-methylsalicylic acid (Beck et al., 1990; Fujii et al., 1996) and (R)-mellein (Chooi et al., 2015). Thus, depending on the nature of the target polyketide compound, the number of candidate scaffolds can be further narrowed down by targeting NR-, PR-, or HR-PKS using corresponding KS domain. Querying the local BLAST database consist of the assembled genomic scaffolds with a corresponding KS domain will resulted in scaffolds containing the corresponding group of PKS genes appearing on top of the TBLASTN hit list. The specific group of PKSs encoded in the candidate scaffolds can be subjected to further scrutiny to pinpoint the PKS gene responsible for biosynthesis of the target SM. Type III PKSs, which present mainly in plants but can be found in some fungi as well, but are usually very limited in number (one or two) in most fungal genomes and are known to synthesize resorcylic acid-type compounds (Hashimoto et al., 2014).
In addition to the minimal PKS domains, a typical NR-PKS also contains the starter unit:ACP transacylase (SAT; Crawford et al., 2006); the product template (PT; Crawford et al., 2008, 2009). An NR-PKS could also contain a thioesterase/Claisen-like cyclase (TE/CLC; Fujii et al., 2001; Korman et al., 2010) or a terminal reductive domain (Bailey et al., 2007) for product release. Although examples of NR-PKSs utilize an in-trans releasing domain (Awakawa et al., 2009; Li et al., 2011) and NR-PKS that do not require any releasing domain or enzyme (Cacho et al., 2013) have been characterized as well. Of these aforementioned NR-PKS domains, the PT domain, which controls the first ring cyclization of the incipient reactive polyketide backbone to form the aromatic product, was shown to be useful in predicting the product of the NR-PKS. In the work by Li et al. (2010b), sequences of PT domains from characterized NR-PKS were subjected to phylogenetic analysis resulting in the grouping of the PT domains in accordance to their respective cyclization regiospecificity. The authors validated the model by demonstrating the previously uncharacterized PT domain of An03g05440 from A. niger, predicted to catalyzed a C2-C7-type cyclization, does indeed perform the expected cyclization mode in a chimeric PKS in vitro system. Ahuja et al. (2012) has made similar observation for NR-PKSs in A. nidulans. Such PT domain analysis has facilitated the identification of viridicatumtoxin and TAN-1612 gene cluster (Chooi et al., 2010; Li et al., 2011).
Unlike NR-PKSs, HR-PKSs utilize the triad of the β-keto reductive domains ketoreductase (KR), dehydratase (DH), and enoylreductase (ER) domains to introduce complexity in the incipient polyketide during each extension cycle. However, in contrast to NR-PKSs, there is a lack of in-depth bioinformatic studies to predict the products of uncharacterized HR-PKS. While more studies in deciphering the biosynthetic rules programmed within these tailoring domains is needed to construct an accurate model of predicting HR-PKS product, important clues can be garnered about the possible product of the HR-PKS using rudimentary bioinformatic analysis. For instance, conserved domain prediction analysis tools can help reveal the presence of in-cis polyketide tailoring domains in the HR-PKS protein sequence such as a C-methyltransferase domain that would indicate α-methylation during one or more polyketide extension cycle. This feature has been exploited for identification of the PKS gene encoding the biosynthesis of the tetraketide side chain of squalestatin in a genomic FNA library using C-methyltransferase domain sequence probe (Cox et al., 2004). High sequence similarity and proximity of phylogenetic relationship with a characterized HR-PKS could, to a limited extend, suggest that the unknown PKS produce similar chain length and structure (Kroken et al., 2003).
Fungi use division of labor between NR-PKS and HR-PKS to generate compounds with an aromatic portion and highly-reduced aliphatic portion respectively (Chooi and Tang, 2012). The linear aliphatic chains generated by HR-PKSs are often used as starter units for NR-PKSs. Compounds generated by such NR/HR two-PKS systems include the resorcylic acid family of compounds (Zhou et al., 2010; Xu et al., 2013b) and asperfuranone (Chiang et al., 2009). The linear aliphatic acyl chain may also be attached to an aromatic polyketide portion as an ester, such as in the case of azanigerone (Zabala et al., 2012) and chaetoviridin (Winter et al., 2012). Thus, such structural features, if present in the target SM, will be a useful tell-tale for predicting the target SM gene cluster as there is limited (one or two) such two-PKS gene cluster(s) in filamentous fungal genomes surveyed to date. SM gene clusters encoding hybrid polyketide-nonribosomal peptide compounds (Boettger and Hertweck, 2013), such as cytochalasins (Qiao et al., 2011; Ishiuchi et al., 2013), equisetin (Kakule et al., 2013), pseurotin (Maiya et al., 2007), and fusarin C (Niehaus et al., 2013) can be identified easily in a genome as well as there is usually one or two PKS-NRPS hybrid gene(s) in most ascomycete fungal genomes. Further analysis of the tailoring enzymes encoding in the vicinity of these PKS genes can help pinpointing the exact target SM gene cluster (see Tailoring Enzymes).
Non-ribosomal peptides
Unlike iterative fungal PKSs, most fungal NRPSs are modular. Like the bacterial counterparts, fungal NRPSs are assembly-line-like protein complexes arranged in functional units known as modules (Finking and Marahiel, 2004, Strieker et al., 2010). Each NRPS module is minimally comprise of three domains: an adenylation (A) domain that selects and activates the amino acid (aa) substrate of the module, a thiolation (T) or peptidyl carrier protein (PCP) domain that serves as a covalent tether for the aa substrate or the growing peptide chain and a condensation (C) domain that catalyzes the peptide bond formation. One notable characteristic of NRPS enzymology is the ability of NRPSs to incorporate non-proteinogenic aa into the NRPS product (Walsh et al., 2013). In addition, NRPSs typically follow the collinearity rule such that the substrate specificity, the number and the linear arrangement of the module within the assembly determines the composition of the NRPS product. Thus, in most cases, the candidate NRPS-encoding genomic scaffolds can be narrowed down easily by first matching the number of modules in the NRPSs to the number of aa residues linked by peptide bonds in the target non-ribosomal peptide product. The presence of the in-line tailoring domain in the NRPS assembly line such as an epimerization (E) or N-methylation (M) domains can further indicate that the NRPS product undergoes respective modification.
There are some exceptions to the co-linearity rule for some fungal NRPSs, in which certain domains or modules on these NRPSs are used iteratively. Notable examples are the fungal siderophore NRPSs, which involved the iterative use of one of the A domains (activating an identical aa residue more than once in a complete NRPS catalytic cycle). These NRPSs harbor additional T–C partial modules that extend the non-ribosomal peptide products beyond the number of complete A–T–C modules in the NRPSs (Schwecke et al., 2006; Johnson, 2008). There are also other non-canonical NRPSs, such as those that synthesize fungal cyclooligomer depsipetides (Glinski et al., 2002; Xu et al., 2008; Sussmuth et al., 2011) and the recently identified fungisporin NRPS from P. chrysogenum (Ali et al., 2014).
Pioneering studies by the Marahiel group led to the development of the A domain 10 aa code, which aided in prediction of the substrate specificities of adenylation of uncharacterized bacterial NRPSs via sequence alignment (Conti et al., 1997; Stachelhaus et al., 1999). Other algorithms that predict adenylation domain substrate specificity and online servers that utilized these algorithms were subsequently developed and are now in widespread use for preliminary bioinformatic characterization of NRPS genes (Challis et al., 2000; Röttig et al., 2011). However, the utility of the A domain 10 aa code and NRPS analysis software programs for prediction of fungal NRPS domain is still limited for fungi compared to bacteria, as there remains a lack of sequence-substrate specificity relationship for fungal NRPSs. Nonetheless, the new NRPSpredictor2 software incorporates A domain substrate specificity prediction for fungal NRPSs and can be a useful starting point (Röttig et al., 2011). Some progress have been made in this area for the A domain 10 aa codes for anthranilate (Ames and Walsh, 2010), α-keto acids (Wackler et al., 2012), and L-tryptophan (Xu et al., 2014a). Phylogenetic and structural analysis of fungal A domains may also yield some insights into the possible substrate of the A domain and the structural class of the final product (Ames and Walsh, 2010; Bushley and Turgeon, 2010). For instance, characterization of the anthranilate-activating fungal A domain in fumiquinazoline F biosynthesis (Ames and Walsh, 2010) led to the discovery of the gene clusters of fungal anthranilic acid-containing non-ribosomal peptides (Gao et al., 2011; Haynes et al., 2012, 2013).
Tailoring enzymes
Since genes for the biosynthesis of SMs are typically clustered together, the types of tailoring reaction the polyketide or non-ribosomal backbone undergoes can be surmised based on the type of functional domains encoded in vicinity of the PKS or NRPS gene, respectively. Essentially, if the backbone biosynthetic gene analysis narrowed down the target SM gene cluster to a couple of possibilities, the combination of tailoring enzymes that matches the SM structure would allow one to confidently pinpoint the right cluster. Tailoring enzymes involved in functional-group transfer such as methyltransferases, acyltransferases, prenyltransferases, and halogenases are especially helpful in correlating a gene cluster to its corresponding compound since there is little ambiguity on what type of reaction these enzymes catalyzes. As described in Section 4 below, the co-localization of multiple methyltransferase genes with an NR-PKS gene was critical toward the discovery of the griseofulvin gene cluster, while the presence of a prenyltransferase gene and a methyltransferase gene flanking an NR-PKS gene pinpointed the correct viridicatumtoxin gene cluster (Chooi et al., 2010). Genes encoding redox enzymes, such as flavoenzymes, cytochrome P450s, and non-heme iron oxygenases, can suggest that the product of the cluster undergoes oxidative transformation. However, their role in biosynthesis can be ambiguous as these enzymes can catalyze diverse redox reactions including hydroxylation, epoxidation, oxidative cleavage, and rearrangement. This is illustrated by our work in functional elucidation of the redox enzymes in viridicatumtoxin, tryptoquialanine, and echinocandin B pathways. As described below, some of the functions of these redox enzymes turned out to be quite surprising. Thus, caution is advised on inferring the function of genes for redox enzymes based on conserved domain analysis, especially in cases where there is no known characterized enzyme that share high sequence similarity with the enzyme of interest.
Examples Illustrating the Strategies for Targeted SM Gene Cluster Discovery
Case Study A: Griseofulvin
Griseofulvin is an antifungal polyketide made by several different Penicillium species (Frisvad and Samson, 2004). To identify the griseofulvin gene cluster, a curation of the PKS genes in P. aethiopicum was performed. Using a local BLAST search of the genome using an arbitrary KS as a query, 30 putative intact PKS genes were found within the genome of P. aethiopicum. Comparison of the PKS genes in P. aethiopicum and the closely related species P. chrysogenum revealed that the former contains six NR-PKS genes that are not orthologous to P. chrysogenum NR-PKS genes (van den Berg et al., 2008; Chooi et al., 2010). Since P. chrysogenum was not known to produce griseofulvin, it was inferred that one of the six non-orthologous NR-PKS genes in P. aethiopicum was responsible for the biosynthesis of griseofulvin (Figure 3A).
Based on the distinct structural features of griseofulvin, the presence of multiple methyltransferase genes as well as a chlorinase gene in the gene cluster encoding griseofulvin is expected (Figure 4). Using these search criteria, the gsf gene cluster, the candidate gene cluster for the biosynthesis of the compound, was found. Along with the expected three S-adenosyl-methionine (SAM) methyltransferase genes (gsfB-D) and a flavin-dependent halogenase gene (gsfI) flanking the NR-PKS gene gsfA, genes for the two redox enzymes GsfE and GsfF were also found within the cluster (Figure 4). Deletion of the gsfA gene led to loss of production of griseofulvin in P. aethiopicum and thus confirming the role of the gsf cluster in the biosynthesis of the compound (Chooi et al., 2010). A subsequent paper by our group revealed the function of the genes in the cluster as well as the regioselectivity of the O-methyltransferases GsfB, C, and D (Cacho et al., 2013). A combination of gene deletion studies and reconstitution of the enzymatic reaction in the same study also revealed the role of GsfF and GsfE in the grisan ring formation and cyclohexadienone reduction, respectively.
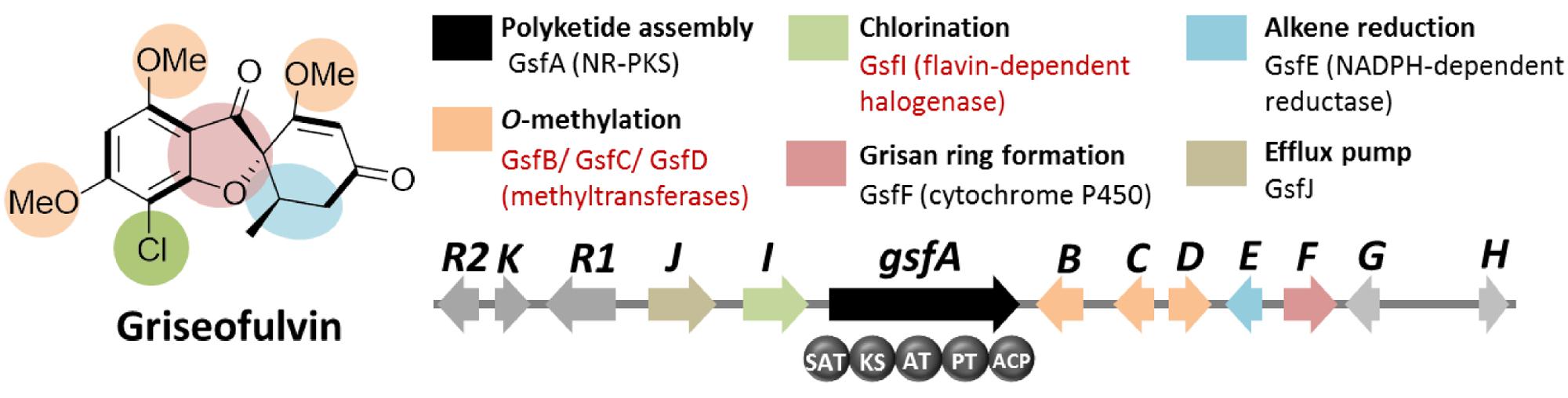
FIGURE 4. Griseofulvin biosynthetic gene clusterin P. aethiopicum. Shown are the roles of the genes in the biogenesis of the compound. The non-reducing polyketide synthase (NR-PKS) gene gsfA is found via comparison of PKS genes in P. aethiopicum and P. chrysogenum. Two structural features, O-methylation and chlorination (in red text), were used as criteria to find the candidate gene clusters.
Case Study B: Viridicatumtoxin
Viridicatumtoxin is another aromatic polyketide synthesized by P. aethiopicum. It contained a naphthacenedione core reminiscent of the core of the bacterial tetracyclines. Moreover, viridicatumtoxin also contained a cyclized geranyl moiety (Figure 5), indicative of the possible presence of a prenyltransferase gene within the cluster. As in the case of griseofulvin, viridicatumtoxin was not known to be produced by P. chrysogenum and likewise, one of the non-orthologous NR-PKS genes in P. aethiopicum was presumably responsible for the biosynthesis of the compound (Figure 3A). In addition to the aforementioned cyclized terpene group, viridicatumtoxin also contained an O-methyl group similar to what was found in griseofulvin. Using these two structural features as “landmarks,” the viridicatumtoxin biosynthetic (vrt) gene cluster is localized on a scaffold (Chooi et al., 2010). Flanking the NR-PKS gene vrtA are genes encoding a farnesyl diphosphate synthase analog (geranyl diphosphate synthase), a prenyltransferase gene vrtC and a SAM-dependent O-methyltransferase gene vrtF; all three are in agreement with the two distinctive chemical features found in the compound. Deletion of the NR-PKS gene vrtA confirmed the role of the cluster in the biogenesis of the compound (Chooi et al., 2010).
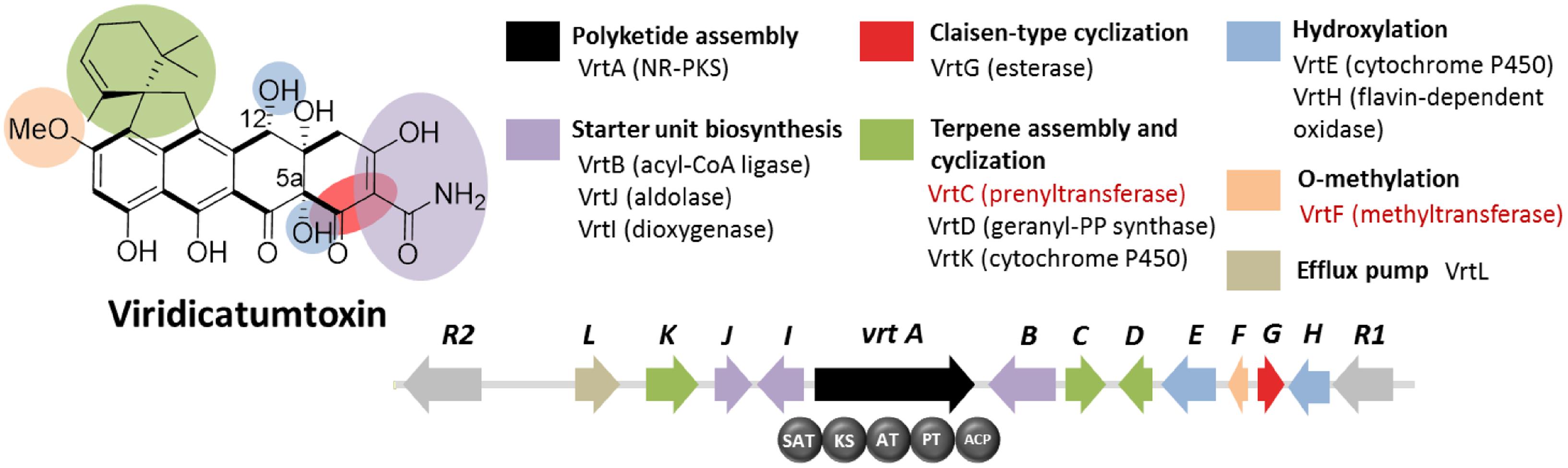
FIGURE 5. Viridicatumtoxin biosynthetic gene cluster in P. aethiopicum. As with the case for the griseofulvin, the NR-PKS gene vrtA is non-orthologous to PKS genes in P. chrysogenum. The presence of the prenyltransferase gene vrtC and the O-methyltransferase gene vrtF (in red text) led to the identification of the gene cluster.
Subsequent characterization of the prenyltransferase VrtC opened the doors toward the discovery of new fungal monoterpenoid biosynthetic gene cluster through genome mining (Chooi et al., 2012). In addition to the aforementioned genes, other genes encode five redox enzymes (vrtE, vrtG, vrtH, vrtI, and vrtK), a PLP-dependent threonine aldolase gene vrtJ and an acyl-CoA ligase gene vrtB were also found in the vrt gene cluster (Chooi et al., 2010). It was later found that VrtG and other related dimanganese-dependent thioesterase catalyze the Claisen-type cyclization in viridicatumtoxin and other fungal naphthacenediones (Li et al., 2011). Surprisingly, the cytochrome P450 VrtK, instead of a terpene cyclase, was shown to mediate the cyclization of the geranyl moiety found in viridicatumtoxin to afford a spirobicyclic structure fused to the tetracyclic core (Chooi et al., 2013b). Additionally, later studies revealed the role of VrtE and VrtH in the hydroxylation in the 5 and 12a positions in viridicatumtoxin, respectively (Chooi et al., 2013a,b).
Case Study C: Tryptoquialanine
Tryptoquialanine (Figure 6) is a quinazoline-containing indole alkaloid produced by P. aethiopicum. It is structurally similar to a known tremorgenic mycotoxin from A. clavatus tryptoquivaline. Due to the presence of the non-proteinogenic aa anthranilic acid in the scaffold of the peptide, it was inferred that the compound is assembled by a NRPS (Ames and Walsh, 2010). Comparative bioinformatic analysis of the NRPS genes between P. aethiopicum and P. chrysogenum revealed the presence of 10 NRPS genes in P. aethiopicum that are non-orthologous to the NRPS genes in P. chrysogenum and were therefore candidates for tryptoquialanine biosynthesis (Gao et al., 2011). On the other hand, comparative bioinformatic analysis of the NRPS genes in P. aethiopicum with NRPS genes with the tryptoquivaline-producing A. clavatus, led to the identification of two NRPS genes in P. aethiopicum that are orthologous with A. clavatus NRPS genes but not with P. chrysogenum NRPS genes (Figure 3B). The two candidate NRPS genes, found on two separated scaffolds, encode a trimodule NRPS annotated as tqaA and a single-module NRPS annotated as tqaB. Primer walking and fosmid sequencing later revealed that both NRPS genes are co-localized in one segment of the genome. This demonstrates that cosmid library can be used to complement NGS-based targeted SM gene cluster discovery. Furthermore, the discovery of tqa cluster facilitated the identification of the tryptoquivaline biosynthetic genes, which are distributed on three separated genomic loci in A. clavatus (Gao et al., 2011).
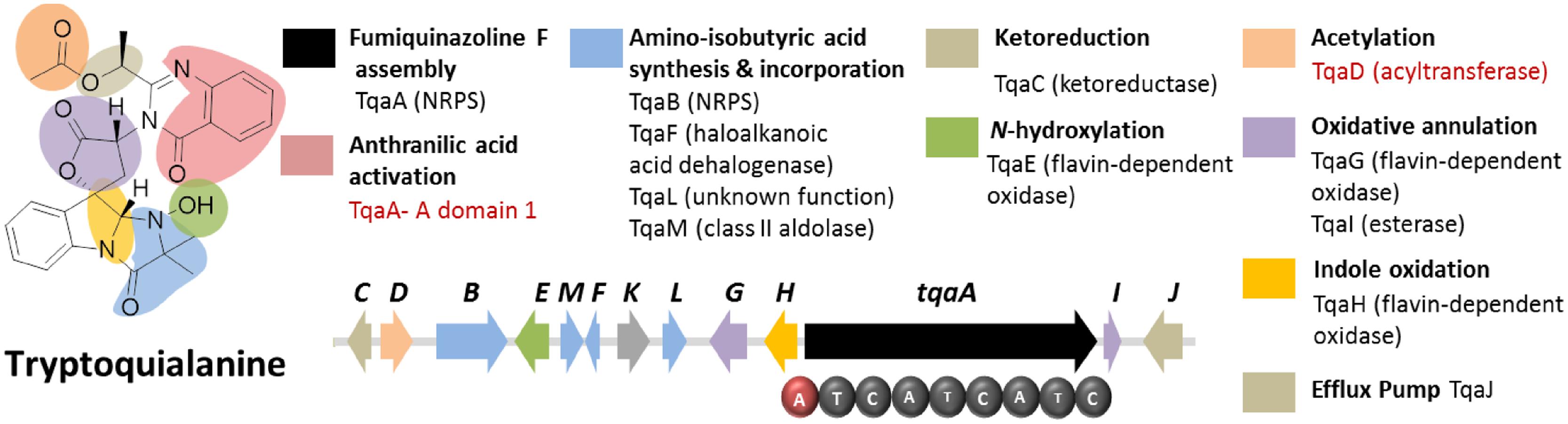
FIGURE 6. Tryptoquialanine biosynthetic gene cluster in P. aethiopicum. The presence of acyltransferase gene tqaD and the anthranilic acid-activating A domain of TqaA (in red text) were clues to identification of the cluster.
Sequence analysis of TqaA revealed high overall shared identity with Af12080, which was previously implicated to be involved in the biosynthesis of fumiquinazoline A, a related quinazoline-containing alkaloid from A. fumigatus (Ames and Walsh, 2010). Subsequent in-depth characterization of TqaA revealed its role in assembling fumiquinazoline F from anthranilate, L-tryptophan and L-alanine (Gao et al., 2012). The study also demonstrated the function of the terminal condensation domain in cyclization of the NRPS product and facilitated future genome mining of fungal cyclic non-ribosomal peptides. On the other hand, the second NRPS TqaB, based on the preliminary retrobiosynthetic analysis, presumably installs the 2-amino-isobutyric acid portion of the imidazolindolone pendant group (Gao et al., 2011). Interestingly, this reflects the arrangement of the aa building blocks, where anthranilic acid, L-tryptophan and L-alanine make up the main peptide chain while the fourth building block 2-amino-isobutyric acid is added to the modified L-tryptophan side chain.
Figure 6 shows the other genes involved in the biosynthesis of tryptoquialanine (Gao et al., 2011). Based solely on the conserved domains of the enzymes encoded by the tqaA genes, only the function of TqaD in the installation of the acetyl group in the tryptoquialanine can be confidently assigned. The function of the remaining genes in the cluster could only be assigned by targeted deletion of individual genes (Gao et al., 2011). This revealed that tqaE and tqaH are responsible for N-hydroxylation and the 2,3-epoxidation of the pendant indole ring of tryptoquialanine, respectively. tqaC encodes a short-chain dehydrogenase that reduces the ketone in tryptoquialanone. Meanwhile, tqaG and tqaI were demonstrated to encode enzymes that mediate the formation of N-deoxytryptoquialanone. Finally, knockout of tqaL and tqaM demonstrated that both genes are required for the biosynthesis of the 2-amino-isobutyric acid.
Case Study D: Echinocandin B
Echinocandin B is a cyclic lipopeptide made by the ascomycetes E. rugulosa. Due to their efficacy against a broad range of Candida species, semisynthetic derivatives of natural echinocandins such as anidulafungin, micafungin, and caspofungin are currently in use as frontline treatment against invasive candidiasis (Kett et al., 2011). Structurally, echinocandin B is consist of six aa: (4R, 5R)-4,5-dihydroxy-L-ornithine, two units of L-threonine, (3R)-3-hydroxy-L-proline, (3S, 4S)-3,4-dihydroxy-L-homotyrosine, and (3S, 4S)-3-methyl-4-hydroxy-L-proline. In addition, a linoleic acid is appended to the cyclic peptide comprising of the six-aa (Figure 7). As with the examples given above, genome mining for the echinocandin biosynthetic gene cluster is initiated with comparative genomic analysis; in this case, between the NRPS genes in E. rugulosa and A. nidulans A4 (von Dohren, 2009; Cacho et al., 2012). Since A. nidulans A4 was not known to produce echinocandin B, NRPS genes orthologous to both species can be eliminated as candidates for echinocandin synthetase. Annotation of the unique NRPS genes in E. rugulosa revealed that only one of the ten non-orthologous NRPS genes encode for a six-module NRPS (one NRPS module is minimally consist of a C, A and T domain), which was the required number for the assembly of echinocandin B based on the collinearity rule (Figure 3C). In addition, the six-module NRPS, annotated as EcdA, contained an additional C-terminal condensation domain that is expected to catalyze the cyclization of the full-length peptide product (Gao et al., 2012), in accordance with the cyclic nature of the compound.
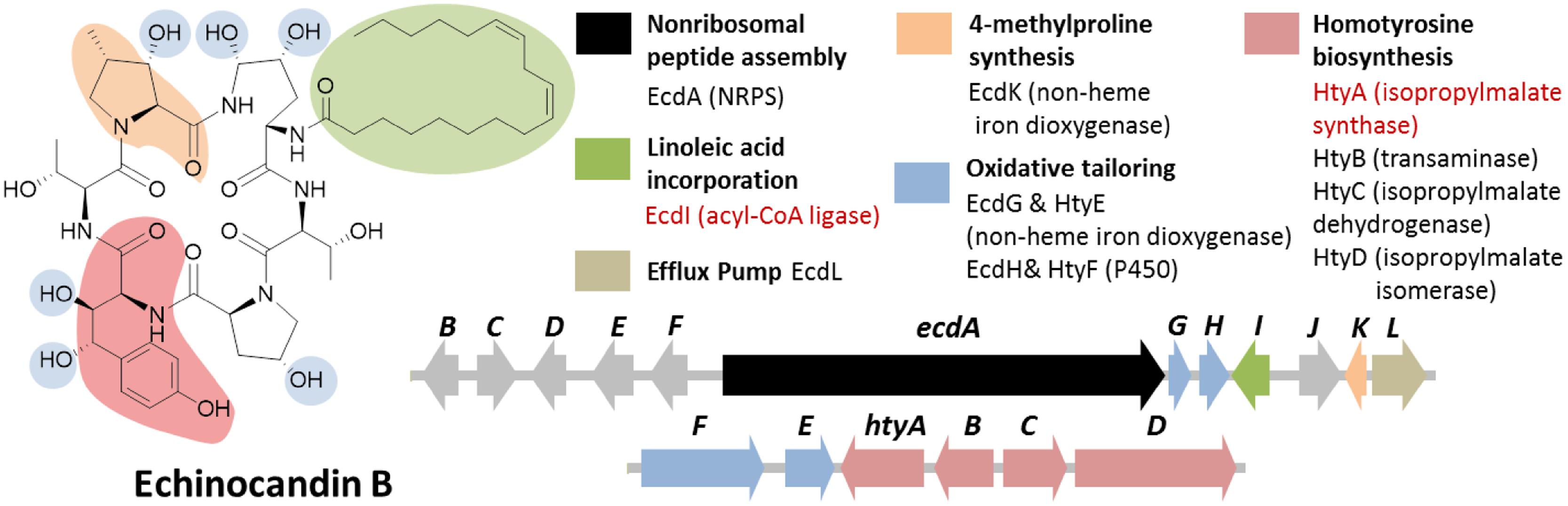
FIGURE 7. Echinocandin B biosynthetic gene cluster in E. rugulosa. The gene cluster was found in two loci (ecd and hty). The ecd cluster contained a six module NRPS gene ecdA and an acyl-CoA ligase gene ecdI (in red text), indicating that the gene cluster is involved in the biosynthesis of a lipo-hexapeptide SM. The separate homotyrosine biosynthesis hty gene cluster at a different locus was located by searching for an isopropylmalate synthase (IPMS) homolog (in red text).
In addition to the NRPS gene ecdA, other genes found in the cluster in agreement to the chemical features of the compound were also found in the cluster such as the acyl-CoA ligase gene ecdI and the redox enzymes ecdG, H and K (Figure 7; Cacho et al., 2012). The presence of ecdI, in conjunction with its co-localization with the NRPS gene ecdA, was highly indicative that the product of the ecd gene cluster is a lipopeptide since EcdI, based on its predicted conserved domain, belongs to a family of enzyme that can convert a carboxylic acid to the more reactive acyl-CoA. Enzymatic characterization of EcdI verified its role in the activation and transfer of linoleic acid to the N-terminal thiolation domain of EcdA.
While the presence of the multiple genes for redox enzymes in the locus suggested that the product of the ecd gene cluster undergoes a plethora of oxidative modification steps, the low shared identity of the protein sequences of EcdG, H and K with of then characterized protein in sequence databases hindered the deciphering of the exact roles of the enzymes solely by bioinformatic analysis. Later gene knockout and enzymatic reconstitution study revealed the regioselectivity of EcdG toward the C3 of L-homotyrosine and regioselectivity of EcdH toward C4 and C5 of L-ornithine in echinocandin B biosynthesis (Jiang et al., 2013). On the other hand, EcdK was revealed to perform the two-step oxidation of L-leucine to afford 5-hydroxy-L-leucine and γ-methyl-glutamic acid-γ-semialdehyde en route to the biosynthesis of (4R)-R-methyl-L-proline (Jiang et al., 2013).
Notably missing in the ecd gene cluster, however, were the genes required for the biosynthesis of L-homotyrosine, another non-proteinogenic aa building block of echinocandin B. Based on previous labeling studies on L-homotyrosine biosynthesis demonstrated that the latter originated from acetate and L-tyrosine (Adefarati et al., 1991). It was also proposed that the pathway is analogous to that of L-leucine biosynthesis, the first step of which is catalyzed by isopropylmalate synthase (IPMS). Thus, we predicted that a homolog of IPMS was involved in the first step of the biosynthesis of L-homotyrosine and found by genome mining the IPMS-like gene htyA in the E. rugulosa genome that is non-orthologous to the IPMS gene in A. nidulans A4 genome. Subsequent deletion of htyA implicated its role in the biosynthesis of L-homotyrosine (Cacho et al., 2012). Flanking the htyA gene were additional genes presumably involved in the biosynthesis of L-homotyrosine (htyB, C, and D) as well as two additional oxidase genes htyE and htyF.
Shortly after the studies on echinocandin B biosynthesis were reported, the biosynthetic gene cluster for the structurally-related pneumocandin B was described (Chen et al., 2013). The pneumocandin biosynthetic gene clusters contained gene homologs echinocandin gene cluster include ecdA, ecdI, cytochrome P450 genes (ecdH and htyF), ecdG, htyE, and ecdK and the L-homotyrosine biosynthetic genes. Interestingly, whereas the echinocandin B biosynthetic genes are located at multiple loci, the pneumocandin biosynthetic genes are situated in a single gene cluster. In addition, the pneumocandin biosynthetic gene cluster also contained a HR-PKS gene for the biosynthesis of the 10,12-dimethyl-myristic acid and an additional 2-oxoglutarate, non-heme iron-dependent oxygenase gene (GLAREA10042) which presumably is involved in the hydroxylation of L-glutamine at C3; both corresponding to the structural features of pneumocandin B but not of echinocandin B. The recent availability of several fungal genomes that encode production of the echinocandin family of lipopeptides has allowed bioinformatics comparison of these gene clusters and enables functional prediction of the unique pathway genes in correspondence to the unique structural features of individual echinocandin analogs (Bills et al., 2014).
Conclusion and Outlook
Next-generation sequencing technologies have significantly accelerated the process of targeted SM gene cluster discovery. The whole process from extraction of genomic DNA of the producing-fungus, sequencing, to initial identification of the target SM gene cluster can occur in one to three months. Using the described strategies, we can often identify the target SM gene cluster in the genome, within 2 weeks from receiving the sequencing data, with high accuracy. Compared to the traditional genomic library screening approach, which can take years beginning from the process of identifying the first biosynthetic gene to finally obtaining the complete gene cluster, such pace of discovery is unimaginable as recent as half a decade ago. The more arduous task ahead is the subsequent gene cluster verification and characterization as fungal genetic transformation protocols need to be developed for individual organisms. The development of heterologous expression systems and in vitro recombinant enzyme characterization methods greatly complement the genetic approach and can provide biosynthetic insights unattainable by traditional knockout approach. More and more fungal natural product research labs are expected to take advantage of NGS technology for identifying the gene cluster encoding the biosynthesis of SMs of interest. The increasing throughput and lowering cost for NGS would also encourage the simultaneous sequencing of multiple strains that produce a same family of compounds, which enables the use of powerful comparative genomics tools in the gene cluster identification and promotes combinatorial biosynthesis of fungal natural products.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Ralph A. Cacho is supported by the NIH Chemistry-Biology Interface Training Grant (NRSA GM-08496) and UCLA Graduate Division. Yi Tang is supported by the NIH grant 1DP1GM106413. Yit-Heng Chooi is currently supported by an Australian Research Council (ARC) Discovery Early Career Researcher Award (DECRA).
Footnotes
- ^ http://www.ncbi.nlm.nih.gov/guide/howto/run-blast-local/
- ^ http://www.clcbio.com/
- ^ http://www.geneious.com/
- ^ http://www.softberry.com/berry.phtml?topic=index&group=programs&subgroup=gfind
- ^ http://bioinf.uni-greifswald.de/webaugustus/predictiontutorial.gsp#exampledata
References
Adefarati, A. A., Giacobbe, R. A., Hensens, O. D., and Tkacz, J. S. (1991). Biosynthesis of L-671,329, an echinocandin-type antibiotic produced by Zalerion arboricola: origins of some of the unusual amino acids and the dimethylmyristic acid side chain. J. Am. Chem. Soc. 113, 3542–3545. doi: 10.1021/ja00009a048
Ahuja, M., Chiang, Y. M., Chang, S. L., Praseuth, M. B., Entwistle, R., Sanchez, J. F.,et al. (2012). Illuminating the diversity of aromatic polyketide synthases in Aspergillus nidulans. J. Am. Chem. Soc. 134, 8212–8221. doi: 10.1021/ja3016395
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ali, H., Ries, M. I., Lankhorst, P. P., Van Der Hoeven, R. A., Schouten, O. L., Noga, M.,et al. (2014). A non-canonical NRPS is involved in the synthesis of fungisporin and related hydrophobic cyclic tetrapeptides in Penicillium chrysogenum. PLoS ONE 9:e98212. doi: 10.1371/journal.pone.0098212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ames, B. D., and Walsh, C. T. (2010). Anthranilate-activating modules from fungal nonribosomal peptide assembly lines. Biochemistry 49, 3351–3365. doi: 10.1021/bi100198y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Awakawa, T., Yokota, K., Funa, N., Doi, F., Mori, N., Watanabe, H.,et al. (2009). Physically discrete beta-lactamase-type thioesterase catalyzes product release in atrochrysone synthesis by iterative type I polyketide synthase. Chem. Biol. 16, 613–623. doi: 10.1016/j.chembiol.2009.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bailey, A. M., Cox, R. J., Harley, K., Lazarus, C. M., Simpson, T. J., and Skellam, E. (2007). Characterisation of 3-methylorcinaldehyde synthase (MOS) in Acremonium strictum: first observation of a reductive release mechanism during polyketide biosynthesis. Chem. Commun. (Camb.) 39, 4053–4055. doi: 10.1039/b708614h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balkovec, J. M., Hughes, D. L., Masurekar, P. S., Sable, C. A., Schwartz, R. E., and Singh, S. B. (2014). Discovery and development of first in class antifungal caspofungin (CANCIDAS(R))–a case study. Nat. Prod. Rep. 31, 15–34. doi: 10.1039/c3np70070d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beck, J., Ripka, S., Siegner, A., Schiltz, E., and Schweizer, E. (1990). The multifunctional 6-methylsalicylic acid synthase gene of Penicillium patulum. Its gene structure relative to that of other polyketide synthases. Eur. J. Biochem. 192, 487–498. doi: 10.1111/j.1432-1033.1990.tb19252.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bentley, R. (1999). Secondary metabolite biosynthesis: the first century. Crit. Rev. Biotechnol. 19, 1–40. doi: 10.1080/0738-859991229189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bills, G., Li, Y., Chen, L., Yue, Q., Niu, X. M., and An, Z. (2014). New insights into the echinocandins and other fungal non-ribosomal peptides and peptaibiotics. Nat. Prod. Rep. 31, 1348–1375. doi: 10.1039/c4np00046c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bingle, L. E., Simpson, T. J., and Lazarus, C. M. (1999). Ketosynthase domain probes identify two subclasses of fungal polyketide synthase genes. Fungal Genet. Biol. 26, 209–223. doi: 10.1006/fgbi.1999.1115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blin, K., Medema, M. H., Kazempour, D., Fischbach, M. A., Breitling, R., Takano, E.,et al. (2013). AntiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, W204–W212. doi: 10.1093/nar/gkt449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boettger, D., and Hertweck, C. (2013). Molecular diversity sculpted by fungal PKS-NRPS hybrids. Chembiochem 14, 28–42. doi: 10.1002/cbic.201200624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Britton, S., and Palacios, R. (1982). Cyclosporin A–usefulness, risks and mechanism of action. Immunol. Rev. 65, 5–22. doi: 10.1111/j.1600-065X.1982.tb00425.x
Brown, D. W., Yu, J. H., Kelkar, H. S., Fernandes, M., Nesbitt, T. C., Keller, N. P.,et al. (1996). Twenty-five coregulated transcripts define a sterigmatocystin gene cluster in Aspergillus nidulans. Proc. Natl. Acad. Sci. U.S.A. 93, 1418–1422. doi: 10.1073/pnas.93.4.1418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bushley, K. E., and Turgeon, B. G. (2010). Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol. Biol. 10:26. doi: 10.1186/1471-2148-10-26
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Butler, J., Maccallum, I., Kleber, M., Shlyakhter, I. A., Belmonte, M. K., Lander, E. S.,et al. (2008). ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820. doi: 10.1101/gr.7337908
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cacho, R. A., Chooi, Y. H., Zhou, H., and Tang, Y. (2013). Complexity generation in fungal polyketide biosynthesis: a spirocycle-forming P450 in the concise pathway to the antifungal drug griseofulvin. ACS Chem. Biol. 8, 2322–2330. doi: 10.1021/cb400541z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cacho, R. A., Jiang, W., Chooi, Y. H., Walsh, C. T., and Tang, Y. (2012). Identification and characterization of the echinocandin B biosynthetic gene cluster from Emericella rugulosa NRRL 11440. J. Am. Chem. Soc. 134, 16781–16790. doi: 10.1021/ja307220z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Challis, G. L., Ravel, J., and Townsend, C. A. (2000). Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 7, 211–224. doi: 10.1016/S1074-5521(00)00091-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, L., Yue, Q., Zhang, X., Xiang, M., Wang, C., Li, S.,et al. (2013). Genomics-driven discovery of the pneumocandin biosynthetic gene cluster in the fungus Glarea lozoyensis. BMC Genomics 14:339. doi: 10.1186/1471-2164-14-339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chettri, P., Ehrlich, K. C., Cary, J. W., Collemare, J., Cox, M. P., Griffiths, S. A.,et al. (2013). Dothistromin genes at multiple separate loci are regulated by AflR. Fungal Genet. Biol. 51, 12–20. doi: 10.1016/j.fgb.2012.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiang, Y. M., Szewczyk, E., Davidson, A. D., Keller, N., Oakley, B. R., and Wang, C. C. (2009). A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 131, 2965–2970. doi: 10.1021/ja8088185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chooi, Y. H., Cacho, R., and Tang, Y. (2010). Identification of the viridicatumtoxin and griseofulvin gene clusters from Penicillium aethiopicum. Chem. Biol. 17, 483–494. doi: 10.1016/j.chembiol.2010.03.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chooi, Y. H., Fang, J., Liu, H., Filler, S. G., Wang, P., and Tang, Y. (2013a). Genome mining of a prenylated and immunosuppressive polyketide from pathogenic fungi. Org. Lett. 15, 780–783. doi: 10.1021/ol303435y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chooi, Y. H., Hong, Y. J., Cacho, R. A., Tantillo, D. J., and Tang, Y. (2013b). A cytochrome P450 serves as an unexpected terpene cyclase during fungal meroterpenoid biosynthesis. J. Am. Chem. Soc. 135, 16805–16808. doi: 10.1021/ja408966t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chooi, Y.-H., Krill, C., Barrow, R. A., Chen, S., Trengove, R., Oliver, R. P.,et al. (2015). An in planta-expressed polyketide synthase produces (R)-mellein in the wheat pathogen Parastagonospora nodorum. Appl. Environ. Microbiol. 81, 177–186. doi: 10.1128/AEM.02745-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chooi, Y.-H., and Solomon, P. S. (2014). A chemical ecogenomics approach to understand the roles of secondary metabolites in fungal cereal pathogens. Front. Microbiol. 5:640. doi: 10.3389/fmicb.2014.00640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chooi, Y. H., and Tang, Y. (2012). Navigating the fungal polyketide chemical space: from genes to molecules. J. Org. Chem. 77, 9933–9953. doi: 10.1021/jo301592k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chooi, Y. H., Wang, P., Fang, J., Li, Y., Wu, K., Wang, P.,et al. (2012). Discovery and characterization of a group of fungal polycyclic polyketide prenyltransferases. J. Am. Chem. Soc. 134, 9428–9437. doi: 10.1021/ja3028636
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chung, K. R., Ehrenshaft, M., Wetzel, D. K., and Daub, M. E. (2003). Cercosporin-deficient mutants by plasmid tagging in the asexual fungus Cercospora nicotianae. Mol. Genet. Genomics 270, 103–113. doi: 10.1007/s00438-003-0902-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conti, E., Stachelhaus, T., Marahiel, M. A., and Brick, P. (1997). Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183. doi: 10.1093/emboj/16.14.4174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cortes, J., Haydock, S. F., Roberts, G. A., Bevitt, D. J., and Leadlay, P. F. (1990). An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 348, 176–178. doi: 10.1038/348176a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cox, R. J. (2007). Polyketides, proteins and genes in fungi: programmed nano-machines begin to reveal their secrets. Org. Biomol. Chem. 5, 2010–2026. doi: 10.1039/b704420h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cox, R. J., Glod, F., Hurley, D., Lazarus, C. M., Nicholson, T. P., Rudd, B. A.,et al. (2004). Rapid cloning and expression of a fungal polyketide synthase gene involved in squalestatin biosynthesis. Chem. Commun. (Camb.) 21, 2260–2261. doi: 10.1039/b411973h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crawford, J. M., Dancy, B. C. R., Hill, E. A., Udwary, D. W., and Townsend, C. A. (2006). Identification of a starter unit acyl-carrier protein transacylase domain in an iterative type I polyketide synthase. Proc. Natl. Acad. Sci. U.S.A. 103, 16728–16733. doi: 10.1073/pnas.0604112103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crawford, J. M., Korman, T. P., Labonte, J. W., Vagstad, A. L., Hill, E. A., Kamari-Bidkorpeh, O.,et al. (2009). Structural basis for biosynthetic programming of fungal aromatic polyketide cyclization. Nature 461, 1139–1143. doi: 10.1038/nature08475
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crawford, J. M., Thomas, P. M., Scheerer, J. R., Vagstad, A. L., Kelleher, N. L., and Townsend, C. A. (2008). Deconstruction of iterative multidomain polyketide synthase function. Science 320, 243–246. doi: 10.1126/science.1154711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crawford, J. M., and Townsend, C. A. (2010). New insights into the formation of fungal aromatic polyketides. Nat. Rev. Microbiol. 8, 879–889. doi: 10.1038/nrmicro2465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Desai, A., Marwah, V. S., Yadav, A., Jha, V., Dhaygude, K., Bangar, U.,et al. (2013). Identification of optimum sequencing depth especially for de novo genome assembly of small genomes using next generation sequencing data. PLoS ONE 8:e60204. doi: 10.1371/journal.pone.0060204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dewick, P. M. (2009). Medicinal natural products: a biosynthetic approach. Chichester: John Wiley & Sons. doi: 10.1002/9780470742761
Díez, B., Gutiérrez, S., Barredo, J. L., van Solingen, P., van der Voort, L. H., and Martín, J. F. (1990). The cluster of penicillin biosynthetic genes. Identification and characterization of the pcbAB gene encoding the alpha-aminoadipyl-cysteinyl-valine synthetase and linkage to the pcbC and penDE genes. J. Biol. Chem. 265, 16358–16365.
Donadio, S., Staver, M. J., Mcalpine, J. B., Swanson, S. J., and Katz, L. (1991). Modular organization of genes required for complex polyketide biosynthesis. Science 252, 675–679. doi: 10.1126/science.2024119
Endo, A. (2010). A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86, 484–493. doi: 10.2183/pjab.86.484
Fedorova, N. D., Moktali, V., and Medema, M. H. (2012). Bioinformatics approaches and software for detection of secondary metabolic gene clusters. Methods Mol. Biol. 944, 23–45. doi: 10.1007/978-1-62703-122-6_2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, G. H., and Leonard, T. J. (1995). Characterization of the polyketide synthase gene (pksL1) required for aflatoxin biosynthesis in Aspergillus parasiticus. J. Bacteriol. 177, 6246–6254.
Finking, R., and Marahiel, M. A. (2004). Biosynthesis of nonribosomal peptides1. Annu. Rev. Microbiol. 58, 453–488. doi: 10.1146/annurev.micro.58.030603.123615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frandsen, R. J., Nielsen, N. J., Maolanon, N., Sorensen, J. C., Olsson, S., Nielsen, J.,et al. (2006). The biosynthetic pathway for aurofusarin in Fusarium graminearum reveals a close link between the naphthoquinones and naphthopyrones. Mol. Microbiol. 61, 1069–1080. doi: 10.1111/j.1365-2958.2006.05295.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frandsen, R. J., Schutt, C., Lund, B. W., Staerk, D., Nielsen, J., Olsson, S.,et al. (2011). Two novel classes of enzymes are required for the biosynthesis of aurofusarin in Fusarium graminearum. J. Biol. Chem. 286, 10419–10428. doi: 10.1074/jbc.M110.179853
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frisvad, J. C., Larsen, T. O., De Vries, R., Meijer, M., Houbraken, J., Cabanes, F. J.,et al. (2007). Secondary metabolite profiling, growth profiles and other tools for species recognition and important Aspergillus mycotoxins. Stud. Mycol. 59, 31–37. doi: 10.3114/sim.2007.59.04
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frisvad, J. C., and Samson, R. A. (2004). Polyphasic taxonomy of Penicillium subgenus Penicillium - A guide to identification of food and air-borne terverticillate Penicillia and their mycotoxins. Stud. Mycol. 49, 1–173.
Fujii, I., Ono, Y., Tada, H., Gomi, K., Ebizuka, Y., and Sankawa, U. (1996). Cloning of the polyketide synthase gene atX from Aspergillus terreus and its identification as the 6-methylsalicylic acid synthase gene by heterologous expression. Mol. Gen. Genet. 253, 1–10. doi: 10.1007/s004380050289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fujii, I., Watanabe, A., Sankawa, U., and Ebizuka, Y. (2001). Identification of Claisen cyclase domain in fungal polyketide synthase WA, a naphthopyrone synthase of Aspergillus nidulans. Chem. Biol. 8, 189–197. doi: 10.1016/S1074-5521(00)90068-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gaffoor, I., Brown, D. W., Plattner, R., Proctor, R. H., Qi, W., and Trail, F. (2005). Functional analysis of the polyketide synthase genes in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Eukaryot. Cell 4, 1926–1933. doi: 10.1128/EC.4.11.1926-1933.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, X., Chooi, Y. H., Ames, B. D., Wang, P., Walsh, C. T., and Tang, Y. (2011). Fungal indole alkaloid biosynthesis: genetic and biochemical investigation of the tryptoquialanine pathway in Penicillium aethiopicum. J. Am. Chem. Soc. 133, 2729–2741. doi: 10.1021/ja1101085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, X., Haynes, S. W., Ames, B. D., Wang, P., Vien, L., Walsh, C. T.,et al. (2012). Cyclization of fungal nonribosomal peptides catalyzed by a terminal condensation-like domain. Nat. Chem. Biol. 8, 823–830. doi: 10.1038/nchembio.1047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, X., Xie, X., Pashkov, I., Sawaya, M. R., Laidman, J., Zhang, W.,et al. (2009). Directed evolution and structural characterization of a simvastatin synthase. Chem. Biol. 16, 1064–1074. doi: 10.1016/j.chembiol.2009.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glinski, M., Urbanke, C., Hornbogen, T., and Zocher, R. (2002). Enniatin synthetase is a monomer with extended structure: evidence for an intramolecular reaction mechanism. Arch. Microbiol. 178, 267–273. doi: 10.1007/s00203-002-0451-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hashimoto, M., Nonaka, T., and Fujii, I. (2014). Fungal type III polyketide synthases. Nat. Prod. Rep. 31, 1306–1317. doi: 10.1039/c4np00096j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haynes, S. W., Gao, X., Tang, Y., and Walsh, C. T. (2012). Assembly of asperlicin peptidyl alkaloids from anthranilate and tryptophan: a two-enzyme pathway generates heptacyclic scaffold complexity in asperlicin E. J. Am. Chem. Soc. 134, 17444–17447. doi: 10.1021/ja308371z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haynes, S. W., Gao, X., Tang, Y., and Walsh, C. T. (2013). Complexity generation in fungal peptidyl alkaloid biosynthesis: a two-enzyme pathway to the hexacyclic MDR export pump inhibitor ardeemin. ACS Chem. Biol. 8, 741–748. doi: 10.1021/cb3006787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hendrickson, L., Davis, C. R., Roach, C., Nguyen, D. K., Aldrich, T., Mcada, P. C.,et al. (1999). Lovastatin biosynthesis in Aspergillus terreus: characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. Chem. Biol. 6, 429–439. doi: 10.1016/S1074-5521(99)80061-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hohn, T. M., Mccormick, S. P., and Desjardins, A. E. (1993). Evidence for a gene cluster involving trichothecene-pathway biosynthetic genes in Fusarium sporotrichioides. Curr. Genet. 24, 291–295. doi: 10.1007/BF00336778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ishiuchi, K., Nakazawa, T., Yagishita, F., Mino, T., Noguchi, H., Hotta, K.,et al. (2013). Combinatorial generation of complexity by redox enzymes in the chaetoglobosin A biosynthesis. J. Am. Chem. Soc. 135, 7371–7377. doi: 10.1021/ja402828w
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiang, W., Cacho, R. A., Chiou, G., Garg, N. K., Tang, Y., and Walsh, C. T. (2013). EcdGHK are three tailoring iron oxygenases for amino acid building blocks of the echinocandin scaffold. J. Am. Chem. Soc. 135, 4457–4466. doi: 10.1021/ja312572v
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johnson, L. (2008). Iron and siderophores in fungal-host interactions. Mycol. Res. 112, 170–183. doi: 10.1016/j.mycres.2007.11.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kakule, T. B., Sardar, D., Lin, Z., and Schmidt, E. W. (2013). Two related pyrrolidinedione synthetase loci in Fusarium heterosporum ATCC 74349 produce divergent metabolites. ACS Chem. Biol. 8, 1549–1557. doi: 10.1021/cb400159f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kardos, N., and Demain, A. L. (2011). Penicillin: the medicine with the greatest impact on therapeutic outcomes. Appl. Microbiol. Biotechnol. 92, 677–687. doi: 10.1007/s00253-011-3587-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kato, N., Suzuki, H., Okumura, H., Takahashi, S., and Osada, H. (2013). A point mutation in ftmD blocks the fumitremorgin biosynthetic pathway in Aspergillus fumigatus strain Af293. Biosci. Biotechnol. Biochem. 77, 1061–1067. doi: 10.1271/bbb.130026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keller, N. P., Turner, G., and Bennett, J. W. (2005). Fungal secondary metabolism - from biochemistry to genomics. Nat. Rev. Microbiol. 3, 937–947. doi: 10.1038/nrmicro1286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kennedy, J., Auclair, K., Kendrew, S. G., Park, C., Vederas, J. C., and Hutchinson, C. R. (1999). Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 284, 1368–1372. doi: 10.1126/science.284.5418.1368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kett, D. H., Shorr, A. F., Reboli, A. C., Reisman, A. L., Biswas, P., and Schlamm, H. T. (2011). Anidulafungin compared with fluconazole in severely ill patients with candidemia and other forms of invasive candidiasis: support for the 2009 IDSA treatment guidelines for candidiasis. Crit. Care 15, R253. doi: 10.1186/cc10514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khaldi, N., Seifuddin, F. T., Turner, G., Haft, D., Nierman, W. C., Wolfe, K. H.,et al. (2010). SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 47, 736–741. doi: 10.1016/j.fgb.2010.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, Y. T., Lee, Y. R., Jin, J., Han, K. H., Kim, H., Kim, J. C.,et al. (2005). Two different polyketide synthase genes are required for synthesis of zearalenone in Gibberella zeae. Mol. Microbiol. 58, 1102–1113. doi: 10.1111/j.1365-2958.2005.04884.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kleftogiannis, D., Kalnis, P., and Bajic, V. B. (2013). Comparing memory-efficient genome assemblers on stand-alone and cloud infrastructures. PLoS ONE 8:e75505. doi: 10.1371/journal.pone.0075505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kodukula, K., Arcuri, M., Cutrone, J. Q., Hugill, R. M., Lowe, S. E., Pirnik, D. M.,et al. (1995). BMS-192548, a tetracyclic binding inhibitor of neuropeptide Y receptors, from Aspergillus niger WB2346. I. Taxonomy, fermentation, isolation and biological activity. J. Antibiot. (Tokyo) 48, 1055–1059. doi: 10.7164/antibiotics.48.1055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Korman, T. P., Crawford, J. M., Labonte, J. W., Newman, A. G., Wong, J., Townsend, C. A.,et al. (2010). Structure and function of an iterative polyketide synthase thioesterase domain catalyzing Claisen cyclization in aflatoxin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 6246–6251. doi: 10.1073/pnas.0913531107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kroken, S., Glass, N. L., Taylor, J. W., Yoder, O. C., and Turgeon, B. G. (2003). Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc. Natl. Acad. Sci. U.S.A. 100, 15670–15675. doi: 10.1073/pnas.2532165100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S., Son, H., Lee, J., Lee, Y. R., and Lee, Y. W. (2011). A putative ABC transporter gene, ZRA1, is required for zearalenone production in Gibberella zeae. Curr. Genet. 57, 343–351. doi: 10.1007/s00294-011-0352-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z.,et al. (2010a). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y., Xu, W., and Tang, Y. (2010b). Classification, prediction, and verification of the regioselectivity of fungal polyketide synthase product template domains. J. Biol. Chem. 285, 22764–22773. doi: 10.1074/jbc.M110.128504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, S., Anand, K., Tran, H., Yu, F., Finefield, J. M., Sunderhaus, J. D.,et al. (2012). Comparative analysis of the biosynthetic systems for fungal bicyclo[2.2.2]diazaoctane indole alkaloids: the (+)/(-)-notoamide, paraherquamide and malbrancheamide pathways. Medchemcomm. 3, 987–996. doi: 10.1039/C2MD20029E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y., Chooi, Y. H., Sheng, Y., Valentine, J. S., and Tang, Y. (2011). Comparative characterization of fungal anthracenone and naphthacenedione biosynthetic pathways reveals an alpha-hydroxylation-dependent Claisen-like cyclization catalyzed by a dimanganese thioesterase. J. Am. Chem. Soc. 133, 15773–15785. doi: 10.1021/ja206906d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, Y., Li, J., Shen, H., Zhang, L., Papasian, C. J., and Deng, H. W. (2011). Comparative studies of de novo assembly tools for next-generation sequencing technologies. Bioinformatics 27, 2031–2037. doi: 10.1093/bioinformatics/btr319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Linnemannstons, P., Schulte, J., Del Mar Prado, M., Proctor, R. H., Avalos, J., and Tudzynski, B. (2002). The polyketide synthase gene pks4 from Gibberella fujikuroi encodes a key enzyme in the biosynthesis of the red pigment bikaverin. Fungal Genet. Biol. 37, 134–148. doi: 10.1016/S1087-1845(02)00501-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J.,et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. doi: 10.1186/2047-217X-1-18
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Machida, M., Asai, K., Sano, M., Tanaka, T., Kumagai, T., Terai, G.,et al. (2005). Genome sequencing and analysis of Aspergillus oryzae. Nature 438, 1157–1161. doi: 10.1038/nature04300
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maiya, S., Grundmann, A., Li, X., Li, S. M., and Turner, G. (2007). Identification of a hybrid PKS/NRPS required for pseurotin A biosynthesis in the human pathogen Aspergillus fumigatus. Chembiochem 8, 1736–1743. doi: 10.1002/cbic.200700202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malpartida, F., and Hopwood, D. A. (1984). Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature 309, 462–464. doi: 10.1038/309462a0
Mayorga, M. E., and Timberlake, W. E. (1990). Isolation and molecular characterization of the Aspergillus nidulans wA gene. Genetics 126, 73–79.
Medema, M. H., Blin, K., Cimermancic, P., De Jager, V., Zakrzewski, P., Fischbach, M. A.,et al. (2011). AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346. doi: 10.1093/nar/gkr466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicholson, T. P., Rudd, B. A., Dawson, M., Lazarus, C. M., Simpson, T. J., and Cox, R. J. (2001). Design and utility of oligonucleotide gene probes for fungal polyketide synthases. Chem. Biol. 8, 157–178. doi: 10.1016/S1074-5521(00)90064-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niehaus, E. M., Janevska, S., Von Bargen, K. W., Sieber, C. M., Harrer, H., Humpf, H. U.,et al. (2014). Apicidin F: characterization and genetic manipulation of a new secondary metabolite gene cluster in the rice pathogen Fusarium fujikuroi. PLoS ONE 9:e103336. doi: 10.1371/journal.pone.0103336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niehaus, E. M., Kleigrewe, K., Wiemann, P., Studt, L., Sieber, C. M., Connolly, L. R., et al. (2013). Genetic manipulation of the Fusarium fujikuroi fusarin gene cluster yields insight into the complex regulation and fusarin biosynthetic pathway. Chem. Biol. 20, 1055–1066. doi: 10.1016/j.chembiol.2013.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nowrousian, M. (2010). Next-generation sequencing techniques for eukaryotic microorganisms: sequencing-based solutions to biological problems. Eukaryot. Cell 9, 1300–1310. doi: 10.1128/EC.00123-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Callaghan, J., Caddick, M. X., and Dobson, A. D. (2003). A polyketide synthase gene required for ochratoxin A biosynthesis in Aspergillus ochraceus. Microbiology 149, 3485–3491. doi: 10.1099/mic.0.26619-0
Pickens, L. B., Tang, Y., and Chooi, Y. H. (2011). Metabolic engineering for the production of natural products. Annu. Rev. Chem. Biomol. Eng. 2, 211–236. doi: 10.1146/annurev-chembioeng-061010-114209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Proctor, R. H., Desjardins, A. E., Plattner, R. D., and Hohn, T. M. (1999). A polyketide synthase gene required for biosynthesis of fumonisin mycotoxins in Gibberella fujikuroi mating population A. Fungal Genet. Biol. 27, 100–112. doi: 10.1006/fgbi.1999.1141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiao, K., Chooi, Y. H., and Tang, Y. (2011). Identification and engineering of the cytochalasin gene cluster from Aspergillus clavatus NRRL 1. Metab. Eng. 13, 723–732. doi: 10.1016/j.ymben.2011.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reeves, C. D., Hu, Z., Reid, R., and Kealey, J. T. (2008). Genes for the biosynthesis of the fungal polyketides hypothemycin from Hypomyces subiculosus and radicicol from Pochonia chlamydosporia. Appl. Environ. Microbiol. 74, 5121–5129. doi: 10.1128/AEM.00478-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Röttig, M., Medema, M. H., Blin, K., Weber, T., Rausch, C., and Kohlbacher, O. (2011). NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 39, W362–W367. doi: 10.1093/nar/gkr323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanchez, J. F., Entwistle, R., Hung, J. H., Yaegashi, J., Jain, S., Chiang, Y. M.,et al. (2011). Genome-based deletion analysis reveals the prenyl xanthone biosynthesis pathway in Aspergillus nidulans. J. Am. Chem. Soc. 133, 4010–4017. doi: 10.1021/ja1096682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwecke, T., Gottling, K., Durek, P., Duenas, I., Kaufer, N. F., Zock-Emmenthal, S.,et al. (2006). Nonribosomal peptide synthesis in Schizosaccharomyces pombe and the architectures of ferrichrome-type siderophore synthetases in fungi. Chembiochem 7, 612–622. doi: 10.1002/cbic.200500301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Semeiks, J., Borek, D., Otwinowski, Z., and Grishin, N. V. (2014). Comparative genome sequencing reveals chemotype-specific gene clusters in the toxigenic black mold Stachybotrys. BMC Genomics 15:590. doi: 10.1186/1471-2164-15-590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shu, Y. Z., Cutrone, J. Q., Klohr, S. E., and Huang, S. (1995). BMS-192548, a tetracyclic binding inhibitor of neuropeptide Y receptors, from Aspergillus niger WB2346. II. Physico-chemical properties and structural characterization. J. Antibiot. (Tokyo) 48, 1060–1065. doi: 10.7164/antibiotics.48.1060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Solovyev, V., Kosarev, P., Seledsov, I., and Vorobyev, D. (2006). Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7(Suppl. 1), S10.1–S10.2. doi: 10.1186/gb-2006-7-s1-s10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stachelhaus, T., Mootz, H. D., and Marahiel, M. A. (1999). The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6, 493–505. doi: 10.1016/S1074-5521(99)80082-9
Stanke, M., Steinkamp, R., Waack, S., and Morgenstern, B. (2004). AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312. doi: 10.1093/nar/gkh379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strieker, M., Tanovic, A., and Marahiel, M. A. (2010). Nonribosomal peptide synthetases: structures and dynamics. Curr. Opin. Struct. Biol 20, 234–240. doi: 10.1016/j.sbi.2010.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sussmuth, R., Muller, J., Von Dohren, H., and Molnar, I. (2011). Fungal cyclooligomer depsipeptides: from classical biochemistry to combinatorial biosynthesis. Nat. Prod. Rep. 28, 99–124. doi: 10.1039/c001463j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamano, K., Sano, M., Yamane, N., Terabayashi, Y., Toda, T., Sunagawa, M.,et al. (2008). Transcriptional regulation of genes on the non-syntenic blocks of Aspergillus oryzae and its functional relationship to solid-state cultivation. Fungal Genet. Biol. 45, 139–151. doi: 10.1016/j.fgb.2007.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Umemura, M., Koike, H., Nagano, N., Ishii, T., Kawano, J., Yamane, N.,et al. (2013a). MIDDAS-M: Motif-independent de novo detection of secondary metabolite gene clusters through the integration of genome sequencing and transcriptome data. PLoS ONE 8:e84028. doi: 10.1371/journal.pone.0084028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Umemura, M., Koyama, Y., Takeda, I., Hagiwara, H., Ikegami, T., Koike, H.,et al. (2013b). Fine de novo sequencing of a fungal genome using only SOLiD short read data: verification on Aspergillus oryzae RIB40. PLoS ONE 8:e63673. doi: 10.1371/journal.pone.0063673
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van den Berg, M. A., Albang, R., Albermann, K., Badger, J. H., Daran, J. M., Driessen, A. J.,et al. (2008). Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat. Biotechnol. 26, 1161–1168. doi: 10.1038/nbt.1498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Dijk, E. L., Auger, H., Jaszczyszyn, Y., and Thermes, C. (2014). Ten years of next-generation sequencing technology. Trends Genet. 30, 418–426. doi: 10.1016/j.tig.2014.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
von Dohren, H. (2009). A survey of nonribosomal peptide synthetase (NRPS) genes in Aspergillus nidulans. Fungal Genet. Biol. 46(Suppl. 1), S45–S52. doi: 10.1016/j.fgb.2008.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wackler, B., Lackner, G., Chooi, Y. H., and Hoffmeister, D. (2012). Characterization of the Suillus grevillei quinone synthetase GreA supports a nonribosomal code for aromatic alpha-keto acids. Chembiochem 13, 1798–1804. doi: 10.1002/cbic.201200187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walsh, C. T., and Fischbach, M. A. (2010). Natural products version 2.0: connecting genes to molecules J. Am. Chem. Soc. 132, 2469–2493. doi: 10.1021/ja909118a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walsh, C. T., O’brien, R. V., and Khosla, C. (2013). Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem. Int. Ed. Engl. 52, 7098–7124. doi: 10.1002/anie.201208344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, S., Xu, Y., Maine, E. A., Wijeratne, E. M., Espinosa-Artiles, P., Gunatilaka, A. A.,et al. (2008). Functional characterization of the biosynthesis of radicicol, an Hsp90 inhibitor resorcylic acid lactone from Chaetomium chiversii. Chem. Biol. 15, 1328–1338. doi: 10.1016/j.chembiol.2008.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wiemann, P., Guo, C. J., Palmer, J. M., Sekonyela, R., Wang, C. C., and Keller, N. P. (2013a). Prototype of an intertwined secondary-metabolite supercluster. Proc. Natl. Acad. Sci. U.S.A. 110, 17065–17070. doi: 10.1073/pnas.1313258110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wiemann, P., Sieber, C. M., Von Bargen, K. W., Studt, L., Niehaus, E. M., Espino, J. J.,et al. (2013b). Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathog. 9:e1003475. doi: 10.1371/journal.ppat.1003475
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Winter, J. M., Sato, M., Sugimoto, S., Chiou, G., Garg, N. K., Tang, Y.,et al. (2012). Identification and characterization of the chaetoviridin and chaetomugilin gene cluster in Chaetomium globosum reveal dual functions of an iterative highly-reducing polyketide synthase. J. Am. Chem. Soc. 134, 17900–17903. doi: 10.1021/ja3090498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xie, X., Meehan, M. J., Xu, W., Dorrestein, P. C., and Tang, Y. (2009). Acyltransferase mediated polyketide release from a fungal megasynthase. J. Am. Chem. Soc. 131, 8388–8389. doi: 10.1021/ja903203g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xie, X., Watanabe, K., Wojcicki, W. A., Wang, C. C., and Tang, Y. (2006). Biosynthesis of lovastatin analogs with a broadly specific acyltransferase. Chem. Biol. 13, 1161–1169. doi: 10.1016/j.chembiol.2006.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, W., Chooi, Y. H., Choi, J. W., Li, S., Vederas, J. C., Da Silva, N. A.,et al. (2013a). LovG: the thioesterase required for dihydromonacolin L release and lovastatin nonaketide synthase turnover in lovastatin biosynthesis. Angew. Chem. Int. Ed. Engl. 52, 6472–6475. doi: 10.1002/anie.201302406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Espinosa-Artiles, P., Schubert, V., Xu, Y. M., Zhang, W., Lin, M.,et al. (2013b). Characterization of the biosynthetic genes for 10,11-dehydrocurvularin, a heat shock response-modulating anticancer fungal polyketide from Aspergillus terreus. Appl. Environ. Microbiol. 79, 2038–2047. doi: 10.1128/AEM.03334-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, W., Gavia, D. J., and Tang, Y. (2014a). Biosynthesis of fungal indole alkaloids. Nat. Prod. Rep. 31, 1474–1487. doi: 10.1039/c4np00073k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Zhou, T., Zhang, S., Espinosa-Artiles, P., Wang, L., Zhang, W.,et al. (2014b). Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases. Proc. Natl. Acad. Sci. U.S.A. 111, 12354–12359. doi: 10.1073/pnas.1406999111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Orozco, R., Wijeratne, E. M., Gunatilaka, A. A., Stock, S. P., and Molnar, I. (2008). Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem. Biol. 15, 898–907. doi: 10.1016/j.chembiol.2008.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, G., Rose, M. S., Turgeon, B. G., and Yoder, O. C. (1996). A polyketide synthase is required for fungal virulence and production of the polyketide T-toxin. Plant Cell 8, 2139–2150. doi: 10.1105/tpc.8.11.2139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, J., Chang, P. K., Ehrlich, K. C., Cary, J. W., Bhatnagar, D., Cleveland, T. E.,et al. (2004). Clustered pathway genes in aflatoxin biosynthesis. Appl. Environ. Microbiol. 70, 1253–1262. doi: 10.1128/AEM.70.3.1253-1262.2004
Zabala, A. O., Chooi, Y. H., Choi, M. S., Lin, H. C., and Tang, Y. (2014). Fungal polyketide synthase product chain-length control by partnering thiohydrolase. ACS Chem. Biol. 9, 1576–1586. doi: 10.1021/cb500284t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zabala, A. O., Xu, W., Chooi, Y. H., and Tang, Y. (2012). Characterization of a silent azaphilone gene cluster from Aspergillus niger ATCC 1015 reveals a hydroxylation-mediated pyran-ring formation. Chem. Biol. 19, 1049–1059. doi: 10.1016/j.chembiol.2012.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zazopoulos, E., Huang, K., Staffa, A., Liu, W., Bachmann, B. O., Nonaka, K.,et al. (2003). A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat. Biotechnol. 21, 187–190. doi: 10.1038/nbt784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, W., Chen, J., Yang, Y., Tang, Y., Shang, J., and Shen, B. (2011). A practical comparison of de novo genome assembly software tools for next-generation sequencing technologies. PLoS ONE 6:e17915. doi: 10.1371/journal.pone.0017915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, H., Qiao, K., Gao, Z., Meehan, M. J., Li, J. W., Zhao, X.,et al. (2010). Enzymatic synthesis of resorcylic acid lactones by cooperation of fungal iterative polyketide synthases involved in hypothemycin biosynthesis. J. Am. Chem. Soc. 132, 4530–4531. doi: 10.1021/ja100060k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: filamentous fungi, secondary metabolites, gene clusters, next generation sequencing, genome mining
Citation: Cacho RA, Tang Y and Chooi Y-H (2015) Next-generation sequencing approach for connecting secondary metabolites to biosynthetic gene clusters in fungi. Front. Microbiol. 5:774. doi: 10.3389/fmicb.2014.00774
Received: 20 October 2014; Accepted: 17 December 2014;
Published online: 14 January 2015.
Edited by:
Jonathan Palmer, United States Department of Agriculture – Forest Service, USAReviewed by:
Philipp Wiemann, University of Wisconsin–Madison, USALiliana Losada, J. Craig Venter Institute, USA
Copyright © 2015 Cacho, Tang and Chooi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yit-Heng Chooi, Plant Sciences Division, Research School of Biology, The Australian National University, Linnaeus Way, Canberra, Acton, ACT 2601, Australia e-mail:eWguY2hvb2lAYW51LmVkdS5hdQ==