 Ángel A. Soler-García
Ángel A. Soler-García Antonio J. De Jesús
Antonio J. De Jesús Kishana Taylor
Kishana Taylor Eric W. Brown
Eric W. Brown- Molecular Methods and Subtyping Branch, Division of Microbiology, Center for Food Safety and Applied Nutrition, US Food and Drug Administration, College Park, MD, USA
Rapid molecular typing methods are important tools in surveillance and outbreak investigations of human Salmonella infections. Here we described the development of a three-genes PCR-RFLP typing method for the differentiation of Salmonella species, subspecies and serovars using the Agilent 2100 Bioanalyzer. The fliC, gnd, and mutS genes were PCR-amplified in 160 Salmonella strains representing the two Salmonella species, six subspecies, and 41 different serovars of S. enterica subspecies enterica. PCR products were individually cut with two different restriction enzymes and the resulting 930 restriction patterns were collected using the Agilent 2100 Bioanalyzer followed by cluster analysis. Both species of Salmonella were differentiated by conventional PCR. All of S. bongori tested were gnd PCR negative due to a mismatch at the 3′-end in one the PCR primers. Salmonella subspecies were differentiated into third-teen homogeneous groups representing each of the six subspecies by cluster analysis of restriction patterns generated from the mutS gene cut with AciI. S. enterica subspecies enterica serovars were further differentiated by the combination of the three target genes and five out the six sets of restriction patterns with a discriminatory power of 0.9725 by cluster analysis. The combined RFLP results of five sets of restriction patterns allowed us to assign each of the 160 strains to one of 128 restriction types. During inoculation studies we were able to identify S. Saintpaul and Typhimurium from 24 h pre-enrichment samples using the described method. The use of fliC, gnd, and mutS PCR-RFLP with the Agilent 2100 Bioanalyzer can provide an accessible and automated alternative method for differentiation of Salmonella pathogens.
Introduction
Contaminated food consumed in the United States causes an estimated 48 million illnesses, 128,000 hospitalizations, and 3,000 deaths annually (Scallan et al., 2011a,b). Salmonella alone causes approximately 1 million foodborne infections (Scallan et al., 2011b), 19,336 hospitalizations, 378 deaths annually (CDC, 2011a) with a cost of $365 million in direct medical expenditure (CDC, 2011b). Human salmonellosis is one of the most frequently occurring food-borne diseases worldwide (Wattiau et al., 2011). Foods prepared with contaminated raw eggs, egg products, insufficiently heated poultry meat and pork have been identified as the primary sources of human Salmonella infections (Buchholz et al., 2005). Although non-typhoid Salmonella strains commonly cause self-limiting gastroenteritis, severe infections, including bacteremia and meningitis, have also been reported (Sirinavin et al., 1999). A combination of sanitary measures and surveillance programs monitoring the entire food chain (animal feed, living animals, slaughterhouses, retail sector, and restaurants) in a timely manner are essential for the detection and prevention of human Salmonella infections (Bertrand et al., 2010). Success depends upon having rapid and sensitive methods for the detection and characterization of Salmonella. Work to develop and improve these methods may lessen the disease burden caused by this pathogen.
Salmonella is divided into two different species, S. enterica and S. bongori. S. enterica itself consists of six subspecies, enterica (I), salamae (II), arizonae (IIIa), diarizonae (IIIb), houtenae (IV), and indica (VI) forming a diverse group 2,557 serovars (Tindall et al., 2005; Grimont and Weill, 2007). Of the six subspecies, only members of subspecies enterica are associated with disease in warm-blooded animals and only a small fraction of these frequently cause disease in humans and domestic animals. The classical methods for identifying and typing S. enterica isolates consist of phenotypic methods that include biochemical profiling, serotyping and phage typing (Andrews et al., 2007; Grimont and Weill, 2007). The gold standard for Salmonella serotyping is based on the scheme developed by Kuffman, White, and Le Minor (Grimont and Weill, 2007). Serotyping deciphers the antigenic makeup of the organisms by identifying the somatic (O) and flagellar (H) antigens through reactions with specific antisera and is useful for international surveillance programs (Herikstad et al., 2002). However, traditional serotyping is unable to adequately fingerprint strains, therefore molecular typing has become primary tool for understanding the evolution of Salmonella and trace clones with special traits such as antibiotic resistance (Herikstad et al., 2002; Foley et al., 2007, 2009).
The current gold standard for molecular typing is Pulse-Field Gel Electrophoresis (PFGE), which can provide discrimination between similar serotypes and is the basis for PulseNet surveillance (Schwartz and Cantor, 1984). However, PFGE is laborious, time-consuming, and expensive. A subtyping method should be rapid, robust, portable, and sensitive. It should be able to reliably differentiate epidemiologically unrelated strains from each other and group all isolates associated with the same source without disrupting the present classification of Salmonella into subspecies and serovars. Such a subtyping system would also need to work within budget constraints of laboratories. For these reasons, we explored ways to improve existing techniques using an accessible platform that can be widely-distributed.
Polymerase Chain Reaction-Restriction Fragment Length Polymorphism (PCR-RFLP) is a variation of RFLP in which a specific PCR product is amplified followed by restriction digestion with restriction endonucleases to generate a specific restriction banding pattern (Owen and Leeton, 1999). For adequate discrimination the amplified region or gene needs to have a variable region flanked by conserved regions to allow PCR amplification and generation of different restriction patterns after cutting restriction enzymes. Restriction patterns are analyzed using a conventional agarose gel followed by gel documentation to analyze the resolved banding patterns. PCR-RFLP has been used previously for the serotyping of Salmonella (Kilger and Grimont, 1993; Shah and Romick, 1997; Dauga et al., 1998; Kwon et al., 2000; Matsui et al., 2001; Hong et al., 2003; Kisiela et al., 2005; Albarnaz et al., 2007; Gallegos-Robles et al., 2008; Hu et al., 2009). Several targets such as ribosomal (Albarnaz et al., 2007), groEL (Hu et al., 2009), fimA (Kisiela et al., 2005) and recA (Matsui et al., 2001) genes have been used for the differentiation of Salmonella subspecies and serovars. For this purpose fliC has been the most targeted gene (Kilger and Grimont, 1993; Shah and Romick, 1997; Dauga et al., 1998; Kwon et al., 2000; Hong et al., 2003; Gallegos-Robles et al., 2008). By relying on one region of the genome or specific gene, the technique is limited in its discriminatory power, and by the possibility of ambiguous bands on a conventional agarose gel. Previous studies have demonstrated improved accuracy and reproducibility of RFLP using the 2100 Agilent Bioanalyzer for the sizing of the DNA fragments (Panaro et al., 2000; Nachamkin et al., 2001; Lu et al., 2002; Hathaway et al., 2007). In this study, we test the utility of the 2100 Agilent Bioanalyzer for differentiating Salmonella species, subspecies and serovars using PCR-RFLP of the of fliC, gnd, and mutS genes.
Materials and Methods
Salmonella Strains and Target Genes for Molecular Serotyping
The 160 reference strains included in this study represent 41 different serovars of S. enterica subsp. enterica (subsp. I). The six Salmonella subspecies belong to the Salmonella Reference Collections SAR A (72), B (72), and C (16) (Beltran et al., 1991; Boyd et al., 1993, 1996). Recently corrections have been made to certain serovars in the SAR A and B collections (Achtman et al., 2013). Figures 1A,B show the distribution of Salmonella species, subspecies and serovars. The Salmonella fliC, gnd and mutS genes were selected as candidate targets for the development of the PCR-RFLP. The fliC gene encodes for the phase 1 flagellar antigen and it is present in all Salmonellae (Mcquiston et al., 2004). For the phase 1 antigen, 52 antigenic factors and 61 serotypes (single factors or combinations of factors) have been distinguished (Li et al., 1994). The gnd gene codes for 6-phosphogluconate, an enzyme of the pentose-phosphate pathway, and is located between the rfb locus and the highly variable cld gene (Nelson and Selander, 1994; Thampapillai et al., 1994). The mutS gene, a key component of the methyl-directed mismatch repair system, acts as barrier to horizontal gene transfer by blocking recombination of diverged DNA (Brown et al., 2002, 2003).
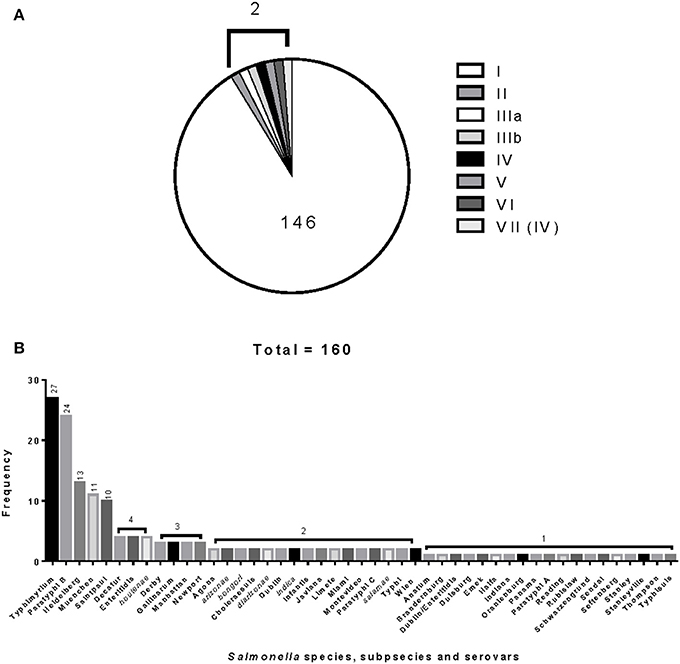
Figure 1. Distribution and frequencies Salmonella species, subspecies and serovars. The 160 Salmonella strains tested belong to three different Salmonella Reference (SAR) collections A, B and C. (A) Describes the distribution and frequency of Salmonella species and subspecies. (B) Shows the distribution and frequencies of the Salmonella species, subspecies, and serovars.
Preparation of DNA and PCR Amplification of fliC, gnd and mutS Genes
Isolates were grown in Tryptic Soy agar (TSA) plates (Difco, BD, Sparks, MD). A single colony was grown in a shaking incubator overnight at 37°C in Brain Heart Infusion broth (BHIB) (Difco). One ml of the broth culture was transferred to a 1.5 ml microcentrifuge tube and centrifuged at 12,000 rpm for 3 min. Total genomic DNA was isolated using the Promega Wizard Genomic DNA Purification kit (Promega Corporation, Madison, WI). The primers used for the amplification of the selected genes are described in Table S1. DNA amplification by PCR was performed in a reaction volume of 50 μl consisting of 25 μl of Qiagen Hot StarTaq Plus master mix (Qiagen, Valencia, CA), 20 μM primer mix, 10 ng of total genomic DNA and volume was completed with molecular biology grade water. Initial denaturation was carried out for 5 min at 95°C. Thirty cycles of amplification were performed in a DNA Engine Tetrad2 Peltier Thermal Cycler (Bio-Rad, Hercules, CA). Each cycle consisted of three steps: denaturation for 30 sec at 94°C, annealing for 30 s at 60°C, and extension for 1 min at 72°C. An additional step of extension for 7 min at 72°C was performed at the end of the amplification to complete extension of the primers. Amplification products were detected by resolving 1 μl of the PCR product using the Agilent DNA 7500 kit and the 2100 Agilent Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA).
Restriction Digestion and Validation of PCR-RFLP Database for Sequenced Salmonella Genomes
We used the In silico (http://insilico.ehu.es) database to virtually test PCR primers and select possible restriction enzymes to test experimentally during RFLP (Bikandi et al., 2004; Roberts et al., 2007). The Salmonella database consisted of 27 genomes representing 15 species (S. bongori), subspecies (S. enterica subsp. arizonae) and S. enterica serovars: Agona, Choleraesuis, Dublin, Enteritidis, Gallinarum-Pullorum (2), Heidelberg (2), Newport, Paratyphi A (2), Paratyphi B, Paratyphi C, Schwarzengrund, Typhi (3) and Typhimurium (8). Enzymes showing the most number of different restriction patterns among the 27 Salmonella genomes were selected for pilot experiments.
The PCR products were cut using the following restriction enzymes: fliC was cut with HhaI and Sau3AI; gnd with AciI and AluI; and mutS with AciI and HaeII. Restriction enzymes from New England Biolabs (Ipswich, MA) and Fermentas (Glen Burnie, MD) were used during the development of the molecular typing method. Single digestions were done by mixing 5 μl of the selected PCR product and 2.5U of NEB endonucleases or 1 Fast digest unit of the Fermentas endonucleases in final volume of 10 μl. NEB endonuclease mixtures were incubated for 1 h at 37°C. Fast digest mixtures were incubated for 10 min at 37°C. After incubation, DNA digestion was terminated by heat inactivation at 65°C for 20 or 10 min depending on the enzyme used or by the addition of 20 mM EDTA. In selected experiments, restriction digestions were cleaned using the MinElute Reaction Cleanup kit (Qiagen). Restriction digestions were repeated two to three times to test reproducibility of the restriction patterns. Restriction patterns were analyzed using the Agilent DNA 1000 kit and the 2100 Agilent Bioanalyzer (Agilent Technologies, Inc.).
RFLP Cluster Analysis, Sequencing of fliC, gnd, mutS, and MLST Housekeeping Genes
Data files containing RFLP patterns from the 2100 Agilent Bioanalyzer were exported as data set tables in CSV format. These were then imported into BioNumerics version 6.6 (Applied Maths, Inc., Austin, TX). The relationships between restriction patterns were calculated by cluster analysis for each and/or combination of restriction patterns using Ward and DICE coefficient with optimization of 1% and tolerance of 0.25%. Ward and Dice were used as recommended by the Guidelines for the validation and application of typing methods for use in bacterial epidemiology (Van Belkum et al., 2007). All the nucleotide sequencing was performed in both directions through MCLAB (San Francisco, CA) and assembled into single complete sequences using the CLC Main Workbench software version 6.8.2 (Aarhus, DK). The fliC, gnd, and mutS genes were sequenced using the primers in Table S1 in all Salmonella collections. The mutS in the SAR B; mutS and gnd in the SAR C collections were obtained from GenBank (NCBI) in FASTA format (Brown et al., 2002, 2003). Sequences for the seven MLST housekeeping genes, aroC, dnaN, hemD, hisD, purE, sucA, and thrA for SARA and B collections were obtained from the NCBI database (Bell et al., 2011) and the MLST Databases at the ERI, University College Cork, respectively. MLST was performed on the 16 Salmonella strains composing the SAR C collection (Boyd et al., 1996; Maiden et al., 1998; Enright and Spratt, 1999). All primers sequences of the seven MLST genes, for amplification and sequencing are described in Table S1. These primers contain M13/pUCR forward (5′-CCCAGTCACGACGTTGTAAAACG-3′) and reverse (5′-AGCGGATAACAATTTCACACAGGAA-3′) universal sequencing priming sites.
PCR cycling conditions were as follows. Initial denaturation was carried out for 5 min at 95°C. Thirty-five cycles of amplification were performed in a DNA Engine Tetrad2 Peltier Thermal Cycler (Bio-Rad). Each cycle consisted of three steps: denaturation for 1 min at 94°C, annealing for 1 min at 55°C, and extension for 1 min at 72°C. An additional step of extension for 5 min at 72°C was performed at the end of the amplification to allow complete extension of the primers. Amplification products were detected by resolving 1 μl of the PCR product using the Agilent DNA 1000 kit and the 2100 Agilent Bioanalyzer (Agilent Technologies, Inc.).
The fliC, gnd, and mutS genes were aligned using BioEdit version 7.1.11, and trimmed with GeneStudio version 2.2.0.0. The seven housekeeping genes sequences were aligned with allele templates from the MLST Database, and then aligned and trimmed as described above. Then, the sequences were queried to the MLST Database website for allele number assignment. Concatenated analyses of fliC, gnd, and mutS; and the seven housekeeping genes were conducted using MEGA software version 5.0.5, using neighbor-joining method with Tamura-Nei distance and 1000 bootstrapping replicates (Felsenstein, 1985; Saitou and Nei, 1987; Tamura et al., 2011).
Preparation of Salmonella Inocula, Artificial Inoculation and Analysis of Food Commodities
Salmonella enterica serovars Newport, Saintpaul, and Typhimurium were selected for artificial contamination of alfalfa sprouts, jalapeno peppers and tomatoes, respectively (CDC, 2006, 2008, 2010). These serovars have been previously implicated in outbreaks related to these food commodities. Salmonella inocula for artificial contamination of produce were prepared as described before (Zhang et al., 2011). Alfalfa sprouts, jalapeno peppers and tomatoes were obtained from local supermarkets. These food commodities were processed as described before (Zhang et al., 2011). Briefly, for each Salmonella serovar and their corresponding produce, two 25 g portions of food were placed aseptically into a sterile Seward stomacher bag (Seward, United Kingdom). The two portions were designated as A, for no inoculation and B, for high-level inoculation (105 CFU/ml). The jalapeno peppers and tomatoes were chopped aseptically in a blender into sizes similar to what is present in regular, chunky salsa and then weighed before being placed into preenrichment bags. One ml of the selected Salmonella serovar at the indicated concentration was added to the 25 g produce portion. For the no inoculated control one ml of the MDR buffer was used. Bags were massaged gently by hand for 1 min and kept at 4°C for 2 h. For enrichment, 225 ml of universal enrichment broth (Difco) were added to the bags. Bags were then shaken vigorously by hand for 30 s, and incubated (without shaking) at 35 ± 1°C for 24 ± 1 h.
One ml aliquots were taken from each bag (A and B) for DNA extraction, serial dilutions further microbiological testing. Four different DNA extraction methods were tested. First, a 1 ml sample was heated at 100°C for 12 min and then centrifuged for 2 min at 16,000× g (Eppendorf, New York). One ml samples were centrifuged and the pellets resuspended in 100 μl of sterile distilled water and boiled, or DNA was extracted using either the Epicenter Quick DNA extraction (Madison, WI) and the Promega Wizard Genomic DNA Purification following the instructions of their manufacturers. Samples were stored at −20°C. Microbiological analysis of 24 h per-enrichment samples was as previously described (Andrews et al., 2007; Zhang et al., 2011). Identification and confirmation of Salmonella were done using Biolog GEN III plates (Biolog, Inc.; Hayward, CA). Salmonella serotyping was done following the standard protocol for molecular determination of serotype in Salmonella based on the Bioplex technology (Fitzgerald et al., 2007; Mcquiston et al., 2011).
All primers and probes used in this study were purchased from IDT (Coralville, IA) and are given in Table S1. Real-time PCR was done as described before (Deer et al., 2010). Briefly, qPCRs were done using the QuantiFast Multiplex PCR using all the DNA templates following the recommended protocol (Qiagen). Each 25 μl reaction contained 1× Master Mix (HotStarTaq Plus DNA Polymerase, QuantiFast Multiplex PCR Buffer, and dNTP mix), 400 nmol/l IAC primers, 200 nmol/l IAC probe and 1 μl DNA IAC template (1 · 1 pg/μl). For the multiplex reactions, invA primers, invA_176F and invA_291R; and probe invA_Tx_208 were added at 200 and 150 nmol/l, respectively. The qPCR conditions were as follows: 95°C for 5 min (for polymerase activation) and 40 cycles of 95°C for 45 s and 60°C for 45 s with fluorescence acquisition for both, Cy5 and Texas Red, following each 60°C step. All qPCR assays were run in CFX96 Real-Time System (Bio-Rad). The term Cq is equivalent to the original CT (threshold cycle) terminology according to the Minimum Information for Publication of Quantitative Real- Time PCR Experiments (MIQE) guidelines (Bustin et al., 2009, 2010). Conventional PCR for fliC, gnd, and mutS was done as described in the Materials and Methods Section.
Results
PCR-Amplification of fliC, gnd and mutS Salmonella Genes and Validation of in silico PCR Amplification Tool
Virtual PCR was done using the designed fliC, gnd, and mutS specific gene primers against the Salmonella database (Table S1). The virtual analysis showed all (100%) of the Salmonella strains were PCR positive for the fliC gene (Table 1). However, the virtual PCR simulation predicted negative results for three out of 27 (11%) database strains for the gnd and mutS genes: S. bongori str. NCTC 12419, S. enterica subsp. arizonae 62:z4,z23:- and S. enterica subsp. enterica serovar Newport str. SL254 (Table 1). Experimental PCR confirmed fliC amplification in all the tested strains. Contrary to the PCR database predictions, S. bongori (2) and S. enterica subsp. arizonae (2) were PCR positive for the mutS gene. As previously reported, one out of the three S. enterica subsp. enterica serovar Newport (SAR B37) was PCR negative for mutS (Brown et al., 2003). In agreement with the In silico database, 100% of the S. bongori (2) were PCR negative for the gnd gene experimentally (Table 1 and Figure 2).
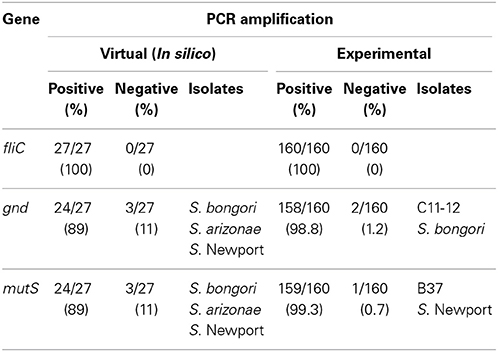
Table 1. PCR-amplification of fliC, gnd, and mutS genes in Salmonella species, subspecies and serovars.

Figure 2. Absence of gnd gene in S. bongori strains. The gnd gene was PCR-amplified as described in the Materials and Methods from eight different strains of S. bongori. PCR products were resolved using the Agilent 7500 DNA kit and the Agilent 2100 Bioanalyzer. Negative Control 1 corresponds to PCR reagents mix + water as a template; and Negative Control 2 corresponds to DNA from a known gnd negative bacterial strain using our PCR conditions.
Differentiation of S. enterica from S. bongori
To confirm our experimental results, we tested six more strains of S. bongori belonging to the Systems and Assays for Food Examination (SAFE) Reference Collection (Mcquiston et al., 2008): 94-0708 (V 48:i:-), 95-0123 (V 40:z35:-), 96-0233 (V 44:z39:-), CNM-256 (V 60:z41:-), CNM262 (V 66:z41:-), 95-0321(V 48:z35:-), and repeated strains SAR C11 and 12 for PCR amplification of the gnd gene. One hundred % of the of S. bongori were PCR negative for the gnd gene under our experimental PCR conditions (Figure 2). These data validate the predicted results from the In silico website with respect to S. bongori. A possible cause for the lack of amplification of the gnd gene in S. bongori strains can be primer-template mismatches. Mismatches located in the 3′-end region of a primer have significantly larger effects on priming efficiency than mismatches located at the 5′-end (Beard et al., 2004; Johnson and Beese, 2004; Stadhouders et al., 2010). We identified mistmatches in the 3′-end of the gnd F-1 primer (Table S1) by aligning 16 gnd Salmonella gene sequences from the SAR C Reference Collection (Boyd et al., 1996) (Figure S1). This mismatch resulted in the differentiation of S. bongori strains from the other Salmonella specie and subspecies. To confirm the specificity of these results, with conducted an exclusivity PCR test (defined here as the lack of the signal or negative reaction on closely related non-Salmonella strains) against a panel of 20 different gram-positive and gram-negative bacterial strains (Table S2). In this test, 100% of the exclusivity strains tested were PCR negative for the fliC and gnd genes. Five out 20 (25%) showed lower levels of mutS PCR product as compared to a Salmonella positive control. A PCR profile with positive fliC and mutS genes in combination with a negative PCR amplification of the gnd gene suggests the presence of S. bongori, while S. enterica will exhibit a positive PCR profile for the three genes. These data suggest that the two species of Salmonella can be differentiated by PCR using the described three PCR amplification profile.
Selection of Restriction Enzymes for Experimental RFLP and Validation of Salmonella RFLP Database for Sequenced Genomes
To select the restriction enzymes to be used for the RFLP experimentally, we conducted virtual RFLP of fliC, gnd, and mutS genes for 27 Salmonella sequenced strains in the In silico database. Analysis of the banding patterns showed several enzymes that produced four or more different restriction patterns per specific gene tested. Pairs of such restriction enzymes were chosen specifically for each of the three genes to generate experimental restriction patterns: fliC gene, HhaI and Sau3AI; gnd, AciI and AluI; and mutS, AciI and HaeII.
Given the fact that we had four out the 27 Salmonella strains with complete genomes in the database, we decided to validate the predicted RFLP patterns. We used the data obtained from the following sequenced available genomes: S. enterica subsp. arizonae 62:z4,z23:- (SAR C5), serovar Paratyphi A str. ATCC 9150 (SAR B42), Paratyphi C str. RKS4595 (SAR B49) and Typhimurium str. LT2 (SAR A2). The predicted restriction patterns were compared to the experimental data generated using the 2100 Agilent Bioanalyzer (Table S3). The degree of agreement between the predicted number of fragments and the total size, and our experimental RFLP for the four Salmonella strains was evaluated. All (100%) of the simulated restriction patterns were different as compared to the experimental ones. The differences in total size of the predicted and the experimental fragments varied from 0.6 to 10.4%.
Processing and Reproducibility of DNA Restriction Patterns
Restriction patterns were resolved using the Agilent DNA 1000 kit (Agilent Technologies). This kit reports a sizing accuracy of ±10%, depending upon the fragment size range, and a sizing resolution that varies from ±5 bp, ±5% and ±10% in the fragments ranging from 25–100, 100–500, and 500–1000 bp, respectively (Agilent Technologies). When resolving restricted DNA using the Bioanalyzer, adding EDTA and/or using heat inactivation of the restriction enzymes is recommended to avoid possible degradation of the internal DNA marker (Agilent Technologies). We tested the effect of adding 20 mM EDTA, heat inactivation, and the use of a commercially-available method for cleaning restriction digestion reactions on the resolution of restriction fragments and reproducibility of restriction patterns in the 2100 Agilent Bioanalyzer. No significant differences in the number of restriction fragments or the sizes obtained among treatments were observed (Figure 3). Although minor differences were detected among fragment sizes between 2 and 5 bp, no degradation of the 1500 and 15 bp internal markers were observed (Figure 3). Given these results, we chose the use of 20 mM EDTA for the inactivation of the restriction enzymes. Although heat inactivation is a cheaper alternative, this step adds 10–20 min to the procedure depending on the restriction enzyme in use.
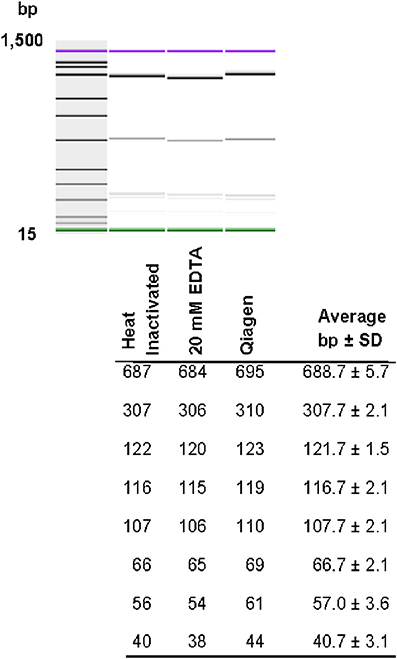
Figure 3. Effect of different restriction enzyme inactivation methods in the number and sizes of restriction fragments. The fliC gene of S. Typhimurium was PCR-amplified and cut with HhaI restriction enzyme as described in the Materials and Methods. Restriction enzyme activity was stop by heat inactivation, addition of 20 mM EDTA or the reaction was cleaned with a commercially available kit.
Differentiation of Salmonellae by RFLP Cluster Analysis
The relationship among restriction patterns was analyzed by cluster analysis. A total of possible 63 individual and multiple combinations were analyzed. Dendrograms were drawn using BioNumerics (Applied Maths). S. enterica is comprised of six subspecies: enterica (I), salamae (II), arizonae (IIIa), diarizonae (IIIb), houtenae (IV), and indica (VI). For simplicity, S. bongori is still commonly referred to as subsp. V (Tindall et al., 2005). Some derivatives of S. enterica subsp. houtenae (IV) have been reported and identified as subgroup VII (Boyd et al., 1996). Based on biotype these are very similar to subsp. IV but can be distinguished by multilocus enzyme electrophoresis (Boyd et al., 1996). To establish whether PCR-RFLP has the potential to differentiate among Salmonella subspecies we conducted cluster analysis as described in the Material and Methods Section. Based on the distribution of subspecies and the number of members in each subspecies group (Figure 1A), we expected that the best clustering would consist of seven to eight clusters depending on whether the derivatives of subsp. IV could be separated in two distinct clusters. These theoretical subspecies clustering show a discriminatory power (DP) equal to 0.167 (Hunter and Gaston, 1988; Hunter, 1990). The current RFLP cluster analysis showed that restriction patterns obtained cutting the mutS gene with the restriction enzyme AciI was indeed sufficient to differentiate the different subspecies of Salmonella (Figure 4). S. enterica subsp. II, IIIa, IIIb, IV, V, and VI were grouped in single homogeneous clusters (Figure 4). S. enterica subsp. I was grouped into six homogeneous clusters consisting of 109, 2, 15, 3, 5, and 12 members, respectively. This clustering corresponds to a DP of 0.5219. This suggests that Salmonella subspecies can be differentiated by mutS-AciI RFLP cluster analysis.
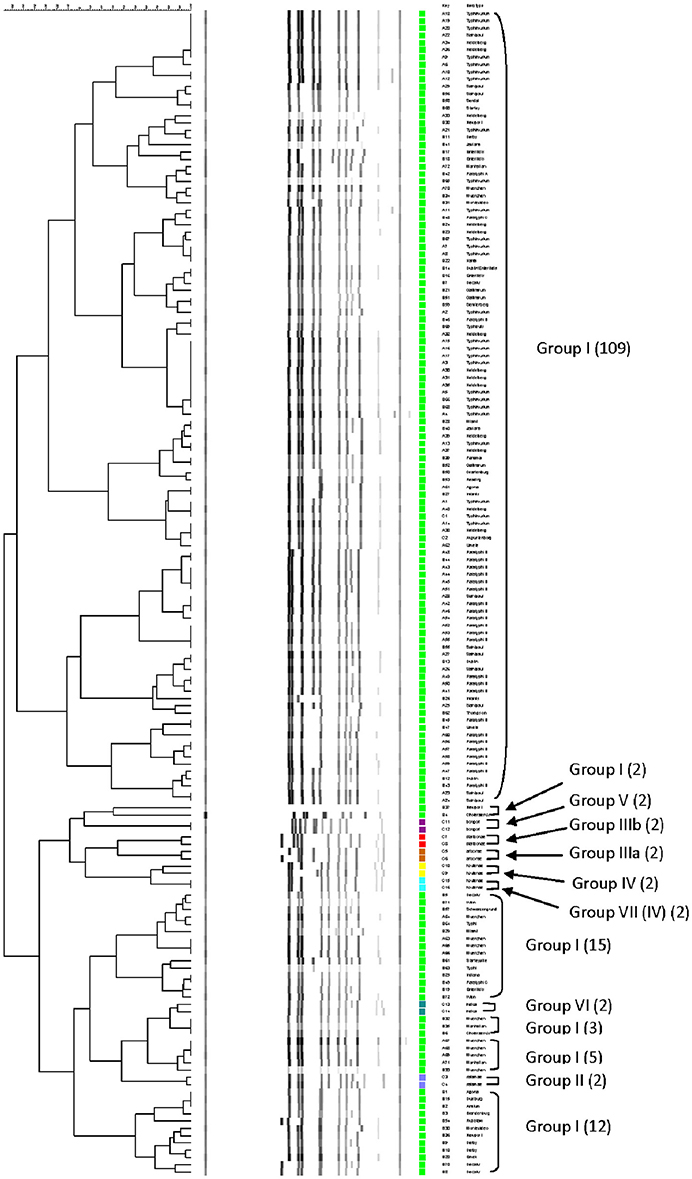
Figure 4. Differentiation of Salmonella species and subspecies by PCR-RFLP and cluster analysis. The mutS gene was PCR-amplified on the 160 Salmonella strains. The PCR product was cut with AciI. Restriction patterns were analyzed as described in the Materials and Methods. The relationship among restriction patterns was analyzed by cluster analysis using BioNumerics. The mutS-AciI banding pattern is shown. Homogeneous clusters consisting of Salmonella subspecies are indicated.
There are a total of 2,579 serovars in the genus Salmonella distributed between the two species and six subspecies, the bulk of which (1531 serovars) are in S. enterica subsp. enterica (Grimont and Weill, 2007). The current study represents 41 serovars of S. enterica subsp. enterica (Figure 1B). To establish whether PCR-RFLP has the potential to differentiate among Salmonella species, subspecies and serovars, we conducted cluster analysis as described in the Material and Methods Section. Based on the distribution of S. enterica subsp. enterica serovars and the five subspecies we expect that the best clustering for our subset should consist of the following: 28 homogeneous clusters containing at least two representatives of a selected serovar or subspecies, and 19 individual serovars (due to their representation with 1 member), for a total of 47 different types among the 160 strains. This clustering distribution corresponds to a DP of 0.9331 (Hunter and Gaston, 1988; Hunter, 1990).
The six restrictions patterns obtained by the digestion of the fliC, gnd, and mutS genes were analyzed in BioNumerics. We obtained best differential clustering using the combination of the fliC gene cut with HhaI and Sau3AI; gnd gene cut with AciI and AluI; the mutS gene cut with HaeII. Forty-three different clusters and eleven single serovars were identified for a total of 54 different types (Figure 5). This cluster distribution corresponds to a DP of 0.9725. The 43 clusters and the relationship among strains on each cluster are described in Table 2. Twenty-six out of 43 (60.5%) clusters consisted of different homogeneous serovar groups. Nineteen out of the 28 (68%) serovars and/or subspecies represented by more than one strain were grouped into homogeneous clusters (Table 2). In twelve out of these 19 (63.2%) homogeneous clusters representing serovars and subspecies containing more than one strain, 100% of the representing strains were grouped together. Seventeen out of 43 (39.5%) clusters were defined as Type I clusters consisting of S. enterica subsp. enterica strains. Five (29.4%), five (29.4%) and eight (47%) of the 17 Type I clusters did not share, shared one or two elements in their antigenic formula, respectively. Four out of five (80%) clusters sharing one element shared either H1 or H2 antigen. Five out of eight (62.5%) clusters sharing two elements shared the O and the H2 antigen. Thirty-seven % (3/8) shared both H1 and H2 antigens.
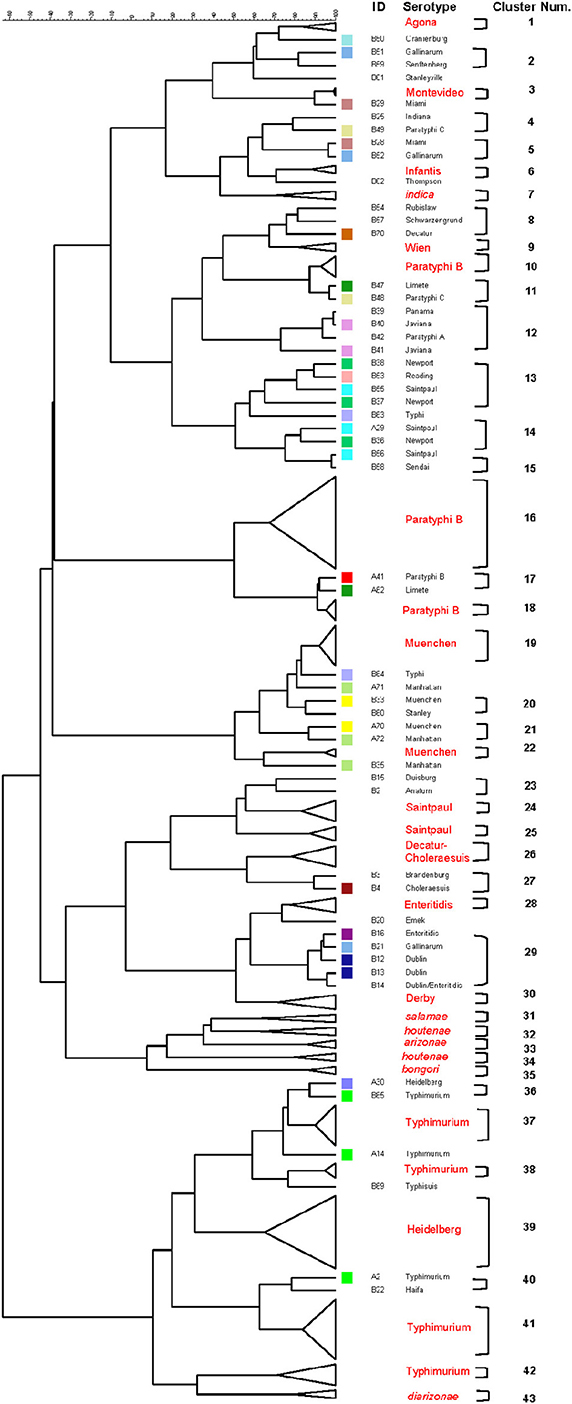
Figure 5. Differentiation of Salmonella species, subspecies and serovars by PCR-RFLP and cluster analysis. The fliC, gnd, mutS genes were PCR-amplified on the 160 Salmonella strains. The PCR products were cut with specific restriction enzymes and the relationship among restriction patterns were analyzed as described in the Material and Methods. Homogeneous clusters consisting of one Salmonella species, subspecies, and serovars are identified in red.
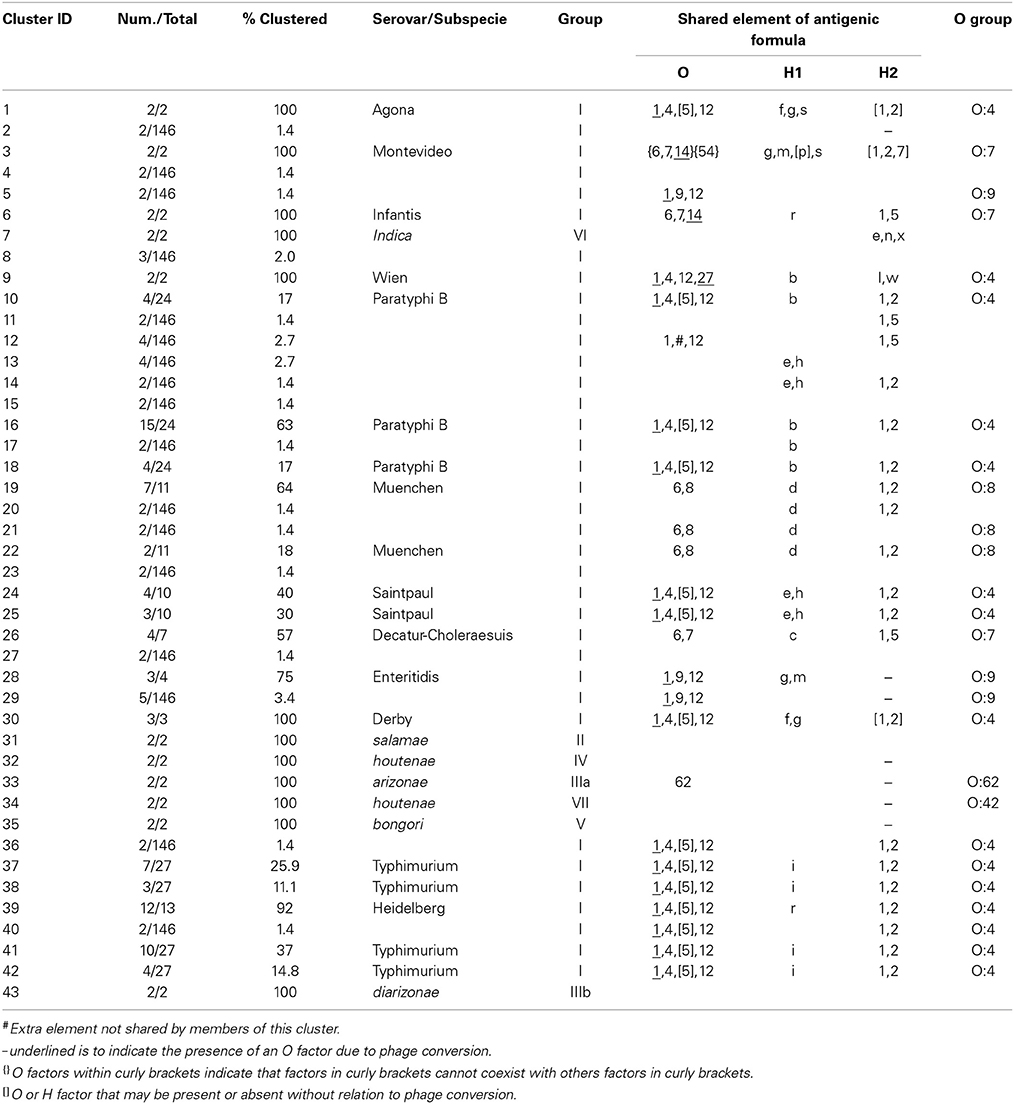
Table 2. Three-genes PCR-RFLP clusters.
Detection of Salmonella in Artificially Inoculated Produce
To test the applicability of the PCR-RFLP in the detection of S. enterica subsp. enterica in contaminated produce after 24 h of pre-enrichment, we artificially inoculated food commodities with Salmonella serovars known to have been responsible for past outbreaks associated with the those food commodities: alfalfa sprouts with S. Newport, jalapeno peppers with S. Saintpaul, and tomatoes with S. Typhimurium (CDC, 2006, 2008, 2010). Food commodities were inoculated as described before the Materials and Methods Section. Four 1 ml aliquots were collected after 24 h pre-enrichment and different DNA extraction methods were tested to assess the effect of these different extraction methods on the amplification of the fliC, gnd, and mutS genes. We used a Salmonella-specific qPCR as a comparator (Deer et al., 2010). Conventional PCRs of fliC, gnd and mutS genes were affected by the DNA extraction and the food commodity (data not shown). Salmonella spp. was detected by qPCR in all DNA extraction methods in all food commodities tested. However, a positive amplification of the three RFLP genes by conventional PCR was obtained using the commercially available DNA extraction kit in jalapeño peppers and tomatoes. In tomatoes, pelleting bacteria from pre-enrichment followed by resuspension in water and boiling was also a good source for DNA template for conventional PCR. Although amplification of the three RFLP genes was observed using DNA extracted with the commercially available kit from alfalfa sprouts pre-enrichment, the yield of PCR product was too low for further manipulation (data not shown). Restriction patterns obtained from pre-enrichment samples exhibited identical patterns when compared to pure culture controls.
We next compared our PCR-RFLP method with the BAM standard method (Andrews et al., 2007). After artificial inoculation Salmonella strains were detected by the BAM standard method, confirmed by biochemical fingerprint (Biolog, Inc.) and serotyped using the Salmonella standard molecular serotyping method (Table 3). Using our previously established cluster analysis, PCR-RFLP identified and serotyped S. Saintpaul and S. Typhimurium after 24 h pre-enrichment (Table 3). However, low amplification yield for S. Newport precluded further manipulation of three RFLP genes. Following the BAM Method, all three Salmonella strains used for the artificially inoculation were isolated, identified and serotyped from all food commodities.
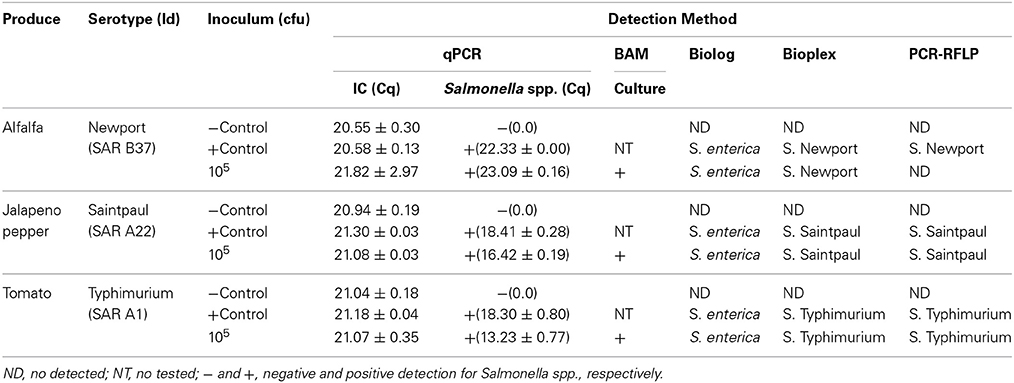
Table 3. Summary of artificially inoculated food commodities.
Differentiation of Salmonellae by Concatenated Sequence Analyses of MLST Housekeeping, fliC, gnd, and mutS Genes
The length of the seven concatenated housekeeping genes was 3,138 bp. The number of variable nucleotides was 59.4% among the 160 Salmonella species, subspecies, and serovars. Evolutionary analyses were conducted in MEGA5 (Tamura et al., 2011). The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei, 1987). The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., 2004) and are in the units of the number of base substitutions per site (Figure 6). Forty-one different clusters and 21 single serovars were identified for a total of 62 different types (Figure 6). This cluster distribution corresponds to a DP of 0.9652. The 41 clusters and the relationship among strains on each cluster are described in Table 4. Twenty-six out of 41 (63.4%) clusters consisted of different homogeneous serovar groups. Twenty out of the 28 (71%) serovars and/or subspecies represented by more than one strain were grouped into homogeneous clusters (Table 4). In eleven out of these 20 (52.4%) homogeneous clusters representing serovars and subspecies containing more than one strain, 100% of the representing strains were grouped together. Fourteen out of 41 (34.1%) clusters were defined as Type I clusters consisting of S. enterica subsp. enterica strains. Five (35.7%), one (7.1%) and eight (57.1%) of the 14 Type I clusters did not share, shared one or two elements in their antigenic formula, respectively. Seven out of eight (87.5%) clusters sharing two elements shared O and the H2 antigen. The remaining cluster sharing two elements of the antigenic formula was composed of strains sharing the O and the H1 (1/8; 12.5%). Only one cluster out of 41 (2.4%) was composed of strains from two different subspecies (Table 4).
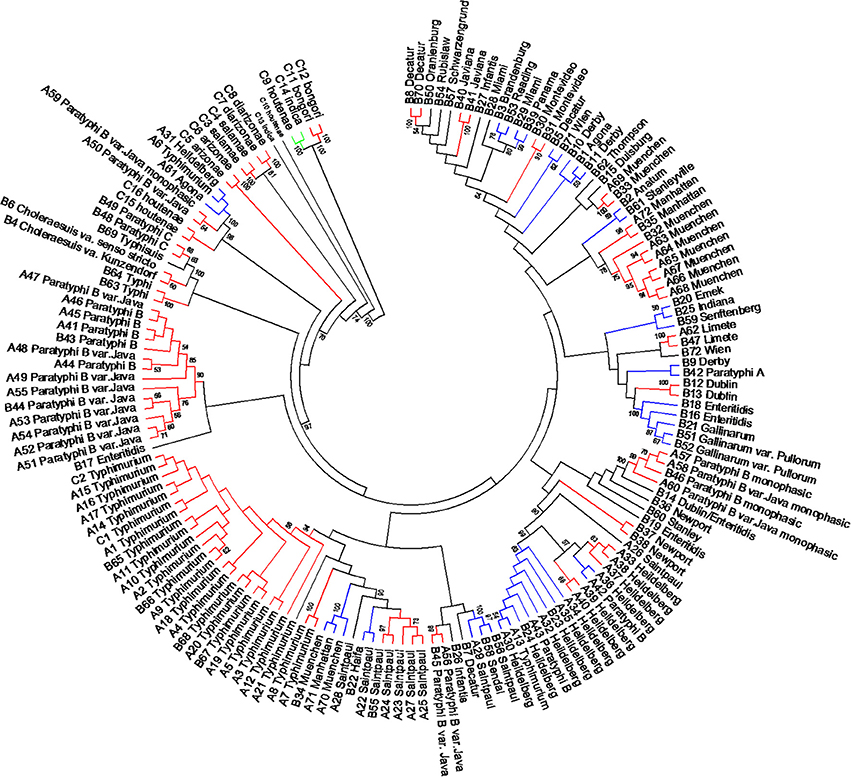
Figure 6. Concatenated seven housekeeping genes sequences evolutionary relationships of taxa. The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei, 1987). The bootstrap consensus tree inferred from 1000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., 2004) and are in the units of the number of base substitutions per site. The analysis involved 160 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 3010 positions in the final dataset. Evolutionary analyses were conducted in MEGA5 (Tamura et al., 2011). Specific, Type I and mix clusters are represented in red, blue, and green, respectively.
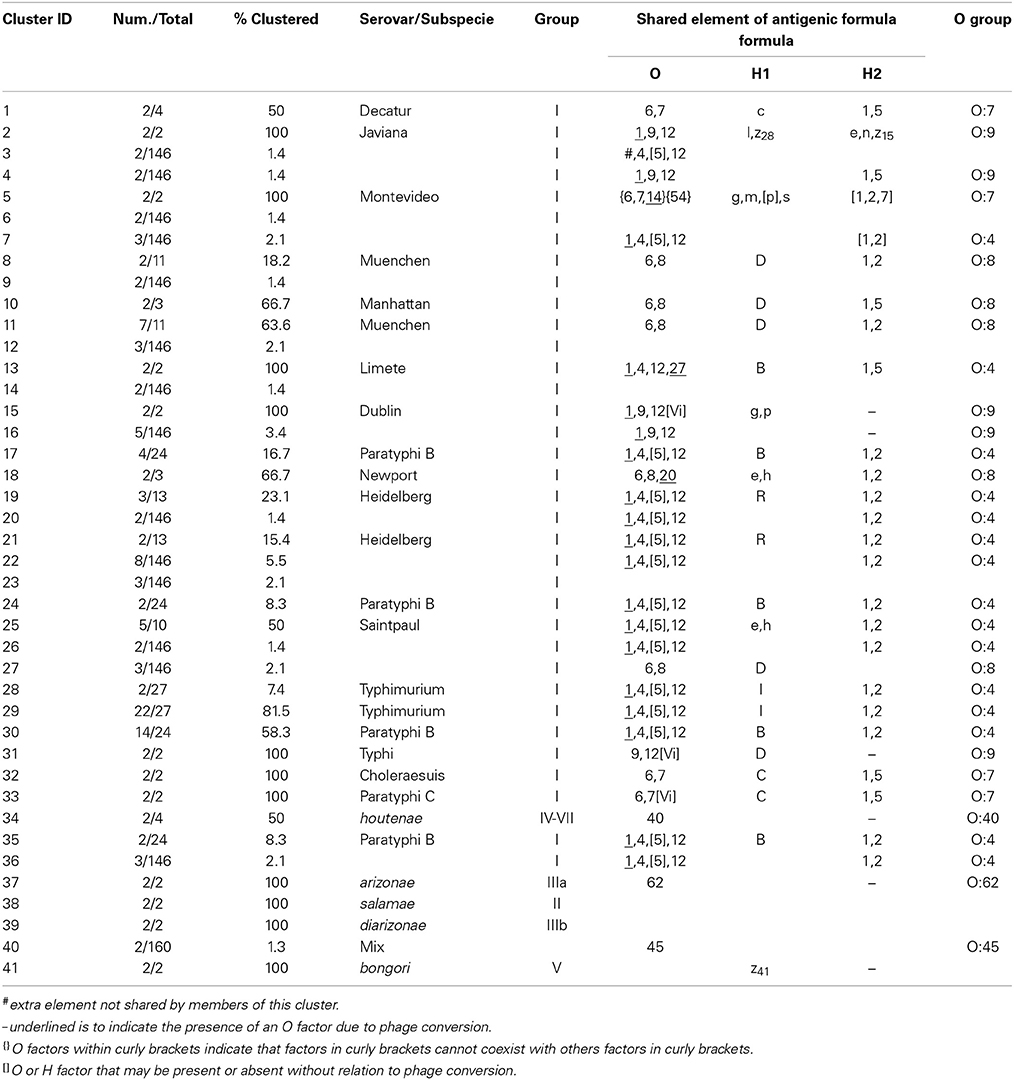
Table 4. Concatenated sequence analysis of the seven housekeeping genes clusters.
Concatenated analysis of three RFLP genes showed the following. The length of the concatenated fliC, gnd, and mutS genes was 3,907 bp. The number of variable nucleotides was 73.5% among the 160 Salmonella species, subspecies, and serovars. Evolutionary analysis, history and distances were determined as for the housekeeping genes in MEGA5 (Saitou and Nei, 1987; Tamura et al., 2004, 2011) (Figure 7). Forty-four different clusters and 34 single serovars were identified for a total of 78 different types (Figure 7). This cluster distribution corresponds to a DP of 0.988. The 44 clusters and the relationship among strains on each cluster are described in Table 5. Twenty-two out of 44 (50.0%) clusters consisted of different homogeneous serovar groups. Thirteen out of the 28 (46.4%) serovars and/or subspecies represented by more than one strain were grouped in homogeneous clusters (Table 5). In six out of these third-teen (46.1%) homogeneous clusters representing serovars and subspecies containing more than one strain, 100% of the representing strains were grouped together. Twenty-one out of 44 (47.7 %) clusters were defined as Type I clusters consisting of S. enterica subsp. enterica strains. Six (28.6%), four (19.0%) and ten (47.6%) of the 21 Type I clusters did not share, shared one or two elements in their antigenic formula, respectively. Three out of four (75%) clusters shared only the O antigen. Nine out of 10 (90.0%) clusters sharing 2 elements, shared the O and the H2 antigen. Cluster 20 (4.8%) is a Type I cluster composed of two members that shared the H1 and H2 flagellar antigens and differed in one element of the O antigen. Only one cluster out of 44 (2.7%) was composed of strains from two different subspecies but shared the H1 antigen (Table 5).
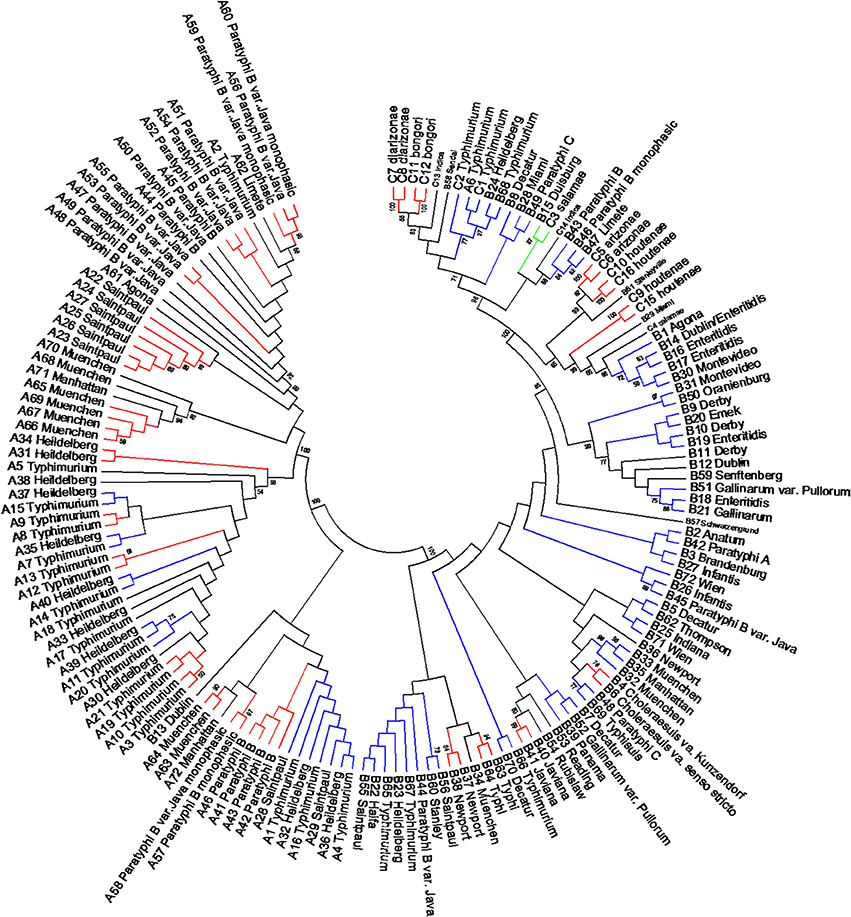
Figure 7. Concatenated fliC, gnd and mutS genes partial sequences evolutionary relationships of taxa. The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei, 1987). The bootstrap consensus tree inferred from 1000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., 2004) and are in the units of the number of base substitutions per site. The analysis involved 160 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 1047 positions in the final dataset. Evolutionary analyses were conducted in MEGA5 (Tamura et al., 2011). Specific, Type I and mix clusters are represented in red, blue and green, respectively.
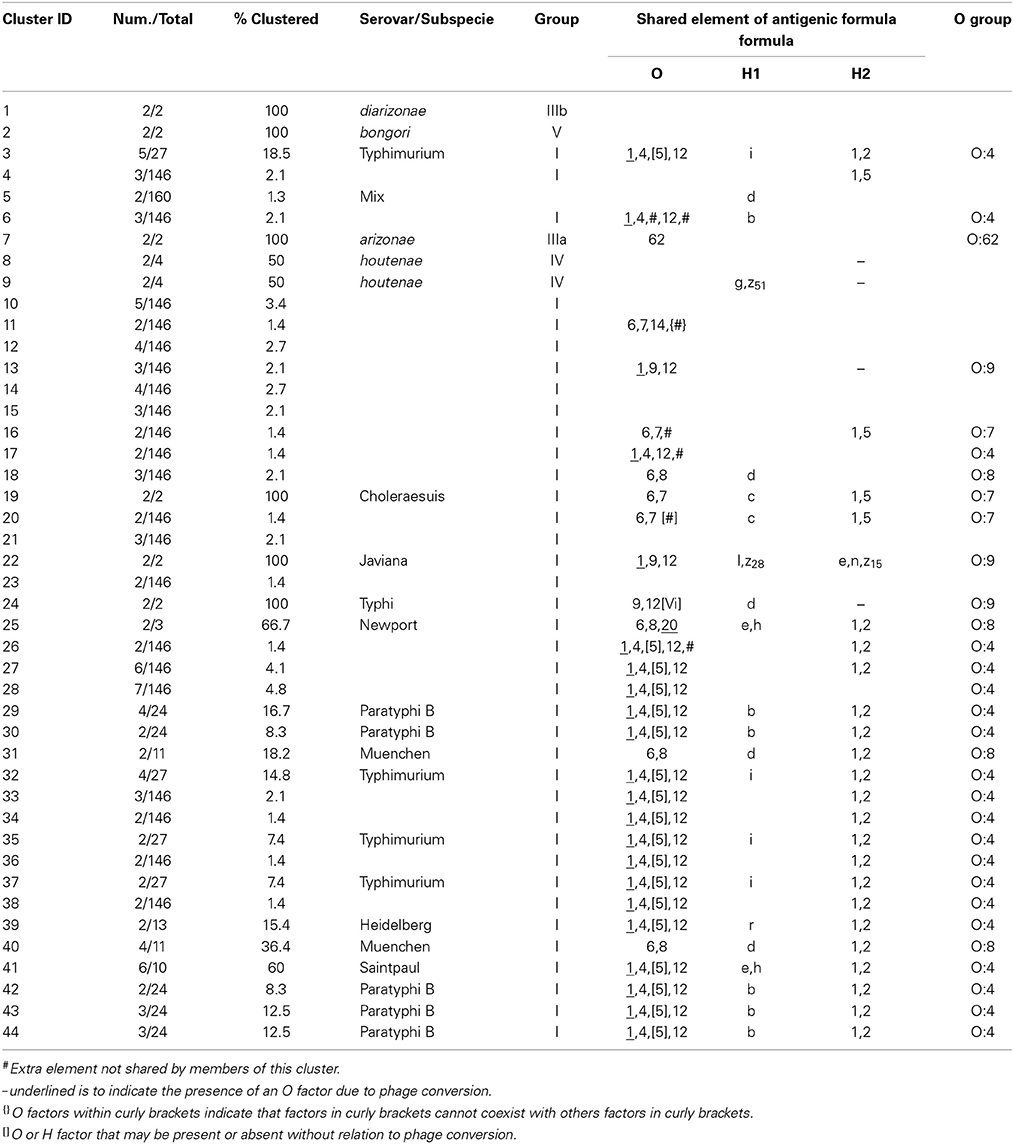
Table 5. Concatenated sequence analysis of fliC, gnd and mutS genes clusters.
Multilocus Sequence Type (st) vs. PCR-RFLP Restriction Type (RT)
MLST assigns an independent allele number based on sequence differences to each of the seven housekeeping genes. The combination of alleles defines an individual strain multilocus sequence type (ST) (Maiden et al., 1998). To test a different approach in the analysis of PCR-RFLP data collected we assigned a numerical identifier to each of the 930 restriction patterns generated by the restriction digestion of fliC, gnd, and mutS genes PCR products and investigated the relatedness of the 160 Salmonella strains by assigning a restriction type (RT). To assign the restriction patterns numbers we took in consideration the following: number of bands, differences in fragments sizes and presence and/or absence of a fragment (Van Belkum et al., 2007). Based on that, we identified 71 and 39 different restriction patterns by cutting the fliC gene with HhaI and Sau3AI, respectively. In the case of the gnd gene, we found 39 and 23 different restriction patterns by cutting with AciI and AluI, respectively. For the mutS gene, 41 and 40 different restriction patterns were assigned after digesting the PCR product with AciI and HaeII, respectively.
To assign the numerical RT we used the same restriction patterns that best clustered the different species, subspecies, and serovars (Figure 5). We assigned 128 different RTs vs. 87 different STs among the 160 strains studied (Tables S4, S5). Among the species, subspecies and serovars with more than one representative (28 possible clusters), we identified in eleven out of 28 (39.3%) the same number of RTs and STs. In four out of the 28 possible homogeneous clusters (14.3%) no STs were identified in the database. These strains belong to S. bongori, and S. enterica subspecies arizonae, houtenae and salamae. In third-teen out of 28 (46.4%) a higher number of RTs than STs were assigned. Higher diversity in the number of RTs increased with the number of representative in a specific Salmonella serovar. In the case of S. Typhimurium, Paratyphi B, Heidelberg, Muenchen and Saintpaul, 15, 12, 8, 7, and 10 different RTs were assigned vs. 4,7,2,5, and 4 STs, respectively (Tables S4, S5). To compare and illustrate the clonal structure of S. Typhimurium, Paratyphi B, Heidelberg, Muenchen and Saintpaul derived from STs and RTs we used eBURST program (Feil et al., 2004). Results are summarized in Table 6. Given the fact that more RTs were assigned as compared to STs, a more complex clonal structure is observed in all five serotypes using the RTs (Table 6). S. Typhimurium clonal structure using the STs consisted of a founder ST 19 containing the majority of the strains (20 out 26, 77%), and connecting two single locus variable STs, 98 and 99, and a singleton ST 36. The S. Typhimurium clonal structure based on RTs is composed of one founder RT 6 connecting two subgroup founders RTs 9 and 38; and 3 singletons RTs 33, 92 and 93. Subgroup founder RT 9 is diversified by a third subgroup founder 69 (Table 6). It is interesting to mention that based on ST only singletons STs were observed in S. Muenchen. However, using RTs three different clonal groups were identified (Table 6). The typeability of the RT approach when compared to cluster and concatenated sequence analysis of fliC, gnd and mutS was superior (Table S6). We tried to determine epidemiology concordance in respect to the source of isolation. Even though some RTs were unique to the source of isolation for some Salmonella strains, the strain specific information was limited to reach a conclusion (Table S7).
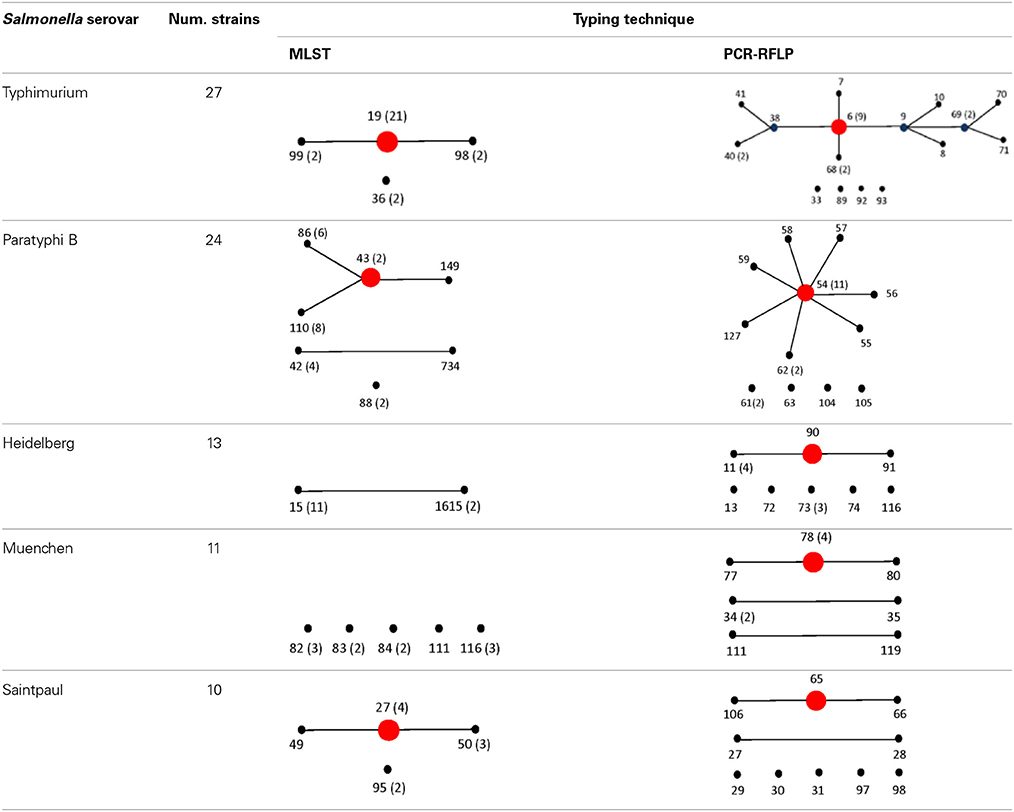
Table 6. Comparison of eBURST diagrams of selected Salmonella enterica subsp. enterica serovars.
Discussion
In order to accelerate the process of conventional serotyping researchers have developed and evaluated various molecular methods and combination techniques such as microarrays, sequencing of housekeeping and antimicrobial resistance genes, and whole genome sequence in an attempt to improve the ability to differentiate not only between Salmonella serovars, but also between different strains of the same serovar (Porwollik et al., 2004; Grimont and Weill, 2007; Franklin et al., 2011; Braun et al., 2012; Allard et al., 2013; Ranieri et al., 2013). Several methods have been applied for epidemiological studies. Phage typing and MLST have been used for epidemiological studies with limitations (Zheng et al., 2014). MLST can be used for epidemiological studies of any bacterial pathogen that exhibits variability in its housekeeping gene sequences. As an example, in a global collection of S. Typhi isolates, only three polymorphic sites were identified among the seven housekeeping genes, partitioning the isolates into four STs (Kidgell et al., 2002).
Here we describe the development and applicability of a typing method for differentiation of Salmonella species, subspecies and serovars based on a three genes PCR-RFLP using the fliC, gnd, and mutS genes as targets and the incorporation of the 2100 Agilent Bioanalyzer to facilitate data collection for further analysis. One caveat worth noting in our study was the selection of several genes which are known to have been subject to substantial reticulate evolutionary change in the form of horizontal gene transfer (Nelson and Selander, 1994; Thampapillai et al., 1994; Brown et al., 2002, 2003). Even though that the gnd is located in highly variable region, the rate of recombination due to horizontally transferred gnd sequences is only moderately higher than the rates for other chromosomal housekeeping genes. This is in contrast with E. coli in which several studies of nucleotide sequence variation in gnd have identified interstrain transfer and recombination as a factor contributing to an unusually high level of alleles (Nelson and Selander, 1994; Thampapillai et al., 1994). Previous studies have suggested that the Salmonella mutS gene has gone intragenic recombination. These recombination events have been however restricted among members of Salmonella enterica subspecies I and more limited beyond the other subspecies (Brown et al., 2002, 2003).
While numerous studies have noted extensive recombination among mutS and gnd across subspecies I Salmonellae, it is important to recall the context in which evidence for this allelic shuffling was documented. Evidence for lateral transfer was noted across disparate serovars and, in several cases, across Salmonella subspecies (Brown et al., 2002, 2003). However, these changes likely accrued across evolution time during the radiation of S. enterica. For molecular epidemiologic utility in real time, the polymorphism itself is what is useful in delimiting the relatedness of outbreak strains and tracing back to an outbreak's source, thus more rapidly evolving markers can often be useful in this regard. Albeit, for evolutionary divergence over a longer time, changes that occur too often could easily obscure phylogenetic relationships. Restated, in this particular instance, the conserved nature of S. enterica genomes among closely related serovars confounded an effective differentiation of these serovars, making it difficult to find phylogenetic characters that have undergone change. mutS and gnd both retained sufficient genetic changes for molecular epidemiological purposes and these changes are not beholden to a rigid evolutionary model for their purposes here (Zheng et al., 2014).
First, we were able to differentiate the two species of Salmonella by conventional PCR based on the lack of the PCR amplification of the gnd gene. All (8 out of 8 samples) of the S. bongori strains tested were PCR negative for the gnd gene and positive for the fliC and the mutS genes under our PCR conditions (Figure 2). We know that the gnd gene is present in S. bongori, however we determined that the lack of gnd amplification was due to a nucleotide mismatch at the 3′-end of the gnd F-1 primer (Figure S1) (Beard et al., 2004; Johnson and Beese, 2004; Stadhouders et al., 2010). This mismatch provided the opportunity to differentiate S. enterica from S. bongori, and as far as we know this is a first report of a simple test to differentiate the two Salmonella species. In contrast, with the exception of a S. Newport strain (SAR B37) that was negative for the mutS gene, all the remaining strains tested were PCR positive for the three RFLP target genes.
Salmonellae consist of six subspecies. A multiplex PCR assay for Salmonella subspecies identification has been published before (Lee et al., 2009). This assay consisted in the PCR amplification of six different target genes and the differentiation was based on the patterns generated by the positive or negative PCR amplification of the selected markers. Fifty of the 53 Salmonella strains (94.3%) shown a unique band pattern. Although PCR is a simple test, multiplex PCR can be tricky to apply. By cluster analysis we found that the restriction patterns generated cutting the mutS gene PCR product with the AciI restriction enzyme has the sufficient potential sufficient to differentiate the six Salmonella subspecies into homogeneous clusters (Figure 4). One hundred and forty-six out 160 strains tested in this study belonged to S. enterica subsp. enterica (Group I) and as a consequence six different homogeneous clusters were identified.
Forty-one different S. enterica subsp. enterica serovars forming subsp. I and the five remaining subspecies were represented among the 160 strains tested. The best cluster differentiation among S. enterica subsp. enterica serovars and the five remaining subspecies was achieved by combining restriction patterns obtained from the fliC gene cut with HhaI and Sau3AI; gnd gene cut with AciI and AluI; and the mutS gene cut HaeII. This enabled us to group 19 out of 28 different S. enterica subsp. enterica serovars and subspecies into homogeneous clusters (Figure 5 and Table 2). While validating these results will require a larger number of different serovars and strains, we have demonstrated the potential of this technique in the identification of Salmonella by its ability to identify S. Saintpaul and S. Typhimurium in artificially inoculated jalapeño peppers and tomatoes, respectively. Amplification of the fliC, gnd, and mutS genes from DNA extracted from alfalfa sprouts pre-enrichment was not successful. Low yields of PCR product were observed, however S. Newport, the serovar used during artificial inoculation of alfalfa sprouts, was identified using the Salmonella standard molecular serotyping method (Fitzgerald et al., 2007; Mcquiston et al., 2011). PCR-RFLP has been attempted before for the identification of Salmonella strains in cantaloupe and chile peppers production systems in Mexico (Gallegos-Robles et al., 2008) and in shellfish (Albarnaz et al., 2007). Contrary to our study, in which the identification and serotyping was done directly from the 24 h pre-enrichments of jalapeño peppers and tomatoes, these previous studies used pure culture isolates from collected samples and compared to reference Salmonella strains (Albarnaz et al., 2007; Gallegos-Robles et al., 2008).
In the present study, we compared the three genes PCR-RFLP cluster analysis, concatenated sequence analysis of the MLST housekeeping genes (Figure 6) and partial sequences of fliC, gnd, and mutS genes (Figure 7) of the 160 strains representing the 2, 6, and 41 Salmonella species, subspecies, and serovars, respectively. Discriminatory power was higher in the two sequence based type of analyses (Table S6). However, the simplicity of our PCR-RFLP and its direct application to food samples increases its future potential use. MLST has the advantage of providing unambiguous results because DNA sequences, rather than banding patterns, are analyzed (Maiden et al., 1998). Sequence types are easy to compare between laboratories. Unlike serotyping, MLST recognizes evolutionary groupings and recently has been recommended that Salmonella classification by serotyping should be replaced by this technique or its equivalents (Achtman et al., 2012). However, having the same multilocus sequence type does not mean that the strains are genetically identical, because given the fact that only a tiny fraction of the genome is sequenced. For every strain analyzed, seven gene fragments must be sequenced in both directions for a total of 14 sequences. Although sequencing services are becoming more available, the analysis of the sequences can be time-consuming. Simpler and more economical phylogenetic schemes with high discriminative power that are free of recombination bias are preferable. As an example, the complete sequence of the rpoB genes were used for the serotyping of 100 Salmonella strains representing 40 serovars (Seong et al., 2012). In addition, Seong et al. (2012) introduced the concept of 60 rpoB sequence type (RSTs) identifier based on nucleotide differences among test strains gene sequences when compared with an rpoB consensus sequence. Phylogenetic analysis showed 60 different RSTs. MLST in the same group of strains identified 49 different ST.
We explored a different approach to analyze the collected RFLP data. Here we introduce the concept of restriction type (RT). A similar concept known as RFLP type was described by Hathaway et al. (2007) for Streptcoccus pneumoniae. In their case, numeric IDs were assigned to each one of multiple restriction patterns. However, all the restriction patterns generated came from only one region in the genome of S. pneumonie. Our RT concept can be considered a hybrid among RFLP and MLST. In our case we assigned numeric IDs to each one of the different restriction patterns as described before (Hathaway et al., 2007) however we used three different regions of the Salmonella genome, the fliC, gnd, and mutS genes. The combination of five out the six restriction patterns among the three genes formed a unique strain ID or RT. Based on that we were able to assigned a total of 128 unique RTs (Tables S4, S6). When compared to MLST, 81 STs were identified among the same strains. All the RTs identified were specific for a given serotype demonstrating a higher typeability when compared to the any other of the clusters or concatenated sequence analysis tested (Tables S4, S6).
Our current study is the first research adapting PCR-RFLP for Salmonella molecular typing using the 2100 Agilent 2100 Bioanalyzer. The Agilent 2100 Bioanalyzer is relatively inexpensive (~$24,000.00) and simple to operate, compared to other commercially available capillary electrophoresis devices. Analysis with the Agilent 2100 Bioanalyzer yields several important advantages compared to traditional separation, imaging, and analysis techniques. Due to its sensitivity 1 μl of sample is required for the analysis of nucleic acids in real time. Results of the resolved nucleic acids are delivered within 30 min. Prepackaged kits, standardized sample preparation and automated analysis yield more accurate and reproducible data due to decreased manual intervention. These characteristics allows the comparative analysis of data obtained at different dates. Overall, the Agilent 2100 Bioanalyzer allows analysis of DNA fragments, including chip preparation, separation, detection and data analysis to be done in a shorter period of time when compared to other serotyping methods.
The cost of subgrouping and serotyping Salmonella using the three genes PCR-RFLP per sample ranges from $12.25 (using NEB restriction enzymes) to $19.28 (using Fermentas restriction enzymes). The use of NEB restriction enzymes will decrease the cost per sample but will increase the time of processing due to the additional hour required for restriction digestion of the DNA. In contrast, typing techniques such as MLST has been estimated to cost closer to $35.00 per sample, and between $35.00 and $135.00 per isolate for traditional serotyping (Achtman et al., 2012; Guard et al., 2012). Given the frequency of Salmonella outbreaks, these cost savings could become significant over time.
Conclusions
Previous Salmonella RFLP studies and those using the Agilent 2100 Bioanalyzer have been concentrated in one area of the genome. Our method increases the discriminatory power of RFLP by using three genes (fliC, gnd, and mutS) and digesting each gene with two enzymes. The combination of five out the six restriction patterns generated digesting the fliC, gnd, and mutS genes showed a good discriminatory power by cluster analysis but it was superior using the RTs. While PCR amplification of the fliC, gnd, and mutS genes appears to be affected by the type of food commodity tested, we believe our method of PCR-RFLP may be a cost-effective tool for narrowing down the number of possible Salmonella serovars in 24-hour pre-enrichment samples. Contrary to conventional agarose gels, the sensitivity of the bioanalyzer can be adjusted to increasing its capacity of detection of the fragments. Co-migrating bands are reported by this device facilitating the determination of complete restriction digestion. The report of the molar concentration of the restriction fragments allows the use stoichiometric distribution as an indicator of complete digestion under the conditions tested. In addition, different runs from different dates can be compared facilitating the normalization and identification of different restriction patterns. The incorporation of automation in PCR-RFLP will facilitate the creation of databases that can be compared between laboratories following a standard procedure describing the preparation, processing, and analysis of the samples.
Author Contributions
Ángel A. Soler-García and Eric W. Brown contributed with the conception and design of the study. Ángel A. Soler-García, Antonio J. De Jesús, and Kishana Taylor were involved in the collection and assembly of data. Ángel A. Soler-García, Antonio J. De Jesús, Kishana Taylor, and Eric W. Brown were involved in the analysis and interpretation of data; and drafting of the article, critical revision of the article for important intellectual content and final approval of the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Ángel A. Soler-García was supported by an appointment with the Research Participation Program at the US FDA-Center for Food Safety and Applied Nutrition administered by the Oak Ridge Institute for Science and Education. We thank Lili Velez, Technical writer at US FDA-CFSAN for critical review of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00417/abstract
References
Achtman, M., Hale, J., Murphy, R. A., Boyd, E. F., and Porwollik, S. (2013). Population structures in the SARA and SARB reference collections of Salmonella enterica according to MLST, MLEE and microarray hybridization. Infect. Genet. Evol. 16, 314–325. doi: 10.1016/j.meegid.2013.03.003
Achtman, M., Wain, J., Weill, F. X., Nair, S., Zhou, Z., Sangal, V., et al. (2012). Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 8:e1002776. doi: 10.1371/journal.ppat.1002776
Albarnaz, J. D., Toso, J., Correa, A. A., Simoes, C. M., and Barardi, C. R. (2007). Relationship between the contamination of gulls (Larus dominicanus) and oysters (Crassostrea gigas) with Salmonella serovar Typhimurium by PCR-RFLP. Int. J. Environ. Health Res. 17, 133–140. doi: 10.1080/09603120701219816
Allard, M. W., Luo, Y., Strain, E., Pettengill, J., Timme, R., Wang, C., et al. (2013). On the evolutionary history, population genetics and diversity among isolates of Salmonella Enteritidis PFGE pattern JEGX01.0004. PLoS ONE 8:e55254. doi: 10.1371/journal.pone.0055254
Andrews, W. H., Jacobson, A., and Hammack, T. (2007). “Salmonella,” in Bacteriological Analytical Manual online, eds G. J. E. A. Jackson (Gaithersburg, MD: Center for Food Safety and Applied Nutrition, U.S. Food and Drug Administration). Available online at: http://www.cfsan.fda.gov/~ebam/bam-5.html
Beard, W. A., Shock, D. D., and Wilson, S. H. (2004). Influence of DNA structure on DNA polymerase beta active site function: extension of mutagenic DNA intermediates. J. Biol. Chem. 279, 31921–31929. doi: 10.1074/jbc.M404016200
Bell, R. L., Gonzalez-Escalona, N., Stones, R., and Brown, E. W. (2011). Phylogenetic evaluation of the ‘Typhimurium’ complex of Salmonella strains using a seven-gene multi-locus sequence analysis. Infect. Genet. Evol. 11, 83–91. doi: 10.1016/j.meegid.2010.10.005
Beltran, P., Plock, S. A., Smith, N. H., Whittam, T. S., Old, D. C., and Selander, R. K. (1991). Reference collection of strains of the Salmonella typhimurium complex from natural populations. J. Gen. Microbiol. 137, 601–606. doi: 10.1099/00221287-137-3-601
Bertrand, S., Dierick, K., Heylen, K., De, B. T., Pochet, B., Robesyn, E., et al. (2010). Lessons learned from the management of a national outbreak of Salmonella ohio linked to pork meat processing and distribution. J. Food Prot. 73, 529–534.
Bikandi, J., San, M. R., Rementeria, A., and Garaizar, J. (2004). In silico analysis of complete bacterial genomes: PCR, AFLP-PCR and endonuclease restriction. Bioinformatics 20, 798–799. doi: 10.1093/bioinformatics/btg491
Boyd, E. F., Wang, F. S., Beltran, P., Plock, S. A., Nelson, K., and Selander, R. K. (1993). Salmonella reference collection B (SARB): strains of 37 serovars of subspecies I. J. Gen. Microbiol. 139(Pt 6), 1125–1121132. doi: 10.1099/00221287-139-6-1125
Boyd, E. F., Wang, F. S., Whittam, T. S., and Selander, R. K. (1996). Molecular genetic relationships of the salmonellae. Appl. Environ. Microbiol. 62, 804–808.
Braun, S. D., Ziegler, A., Methner, U., Slickers, P., Keiling, S., Monecke, S., et al. (2012). Fast DNA serotyping and antimicrobial resistance gene determination of salmonella enterica with an oligonucleotide microarray-based assay. PLoS ONE 7:e46489. doi: 10.1371/journal.pone.0046489
Brown, E. W., Kotewicz, M. L., and Cebula, T. A. (2002). Detection of recombination among Salmonella enterica strains using the incongruence length difference test. Mol. Phylogenet. Evol. 24, 102–120. doi: 10.1016/S1055-7903(02)00222-1
Brown, E. W., Mammel, M. K., Leclerc, J. E., and Cebula, T. A. (2003). Limited boundaries for extensive horizontal gene transfer among Salmonella pathogens. Proc. Natl. Acad. Sci. U.S.A. 100, 15676–15681. doi: 10.1073/pnas.2634406100
Buchholz, U., Brodhun, B., Brockmann, S. O., Dreweck, C. M., Prager, R., Tschape, H., et al. (2005). An outbreak of Salmonella Munchen in Germany associated with raw pork meat. J. Food Prot. 68, 273–276.
Bustin, S. A., Beaulieu, J. F., Huggett, J., Jaggi, R., Kibenge, F. S., Olsvik, P. A., et al. (2010). MIQE precis: practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Mol. Biol. 11:74. doi: 10.1186/1471-2199-11-74
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
CDC. (2006). Salmonellosis-outbreak Investigation, October 2006, as of November 3. [Online]. Available online at: http://www.cdc.goc/ncidod/dbmd/diseaseinfo/salmonellosis_2006/110306_outbreak_notice.htm
CDC. (2008). Investigation of Outbreak of Infections Caused by Salmonella Saintpaul, as of August 28 9 PM EDT [Online]. Available online at: http://www.cdc.goc/salmonella/saintpaul/jalapeno
CDC. (2010). Investigation Update: Multistate Outbreak of Human Salmonella Newport Infections Linked to Raw Alfalfa Sprouts, as of June 29 [Online]. Available online at: http://www.cdc.goc/salmonella/newport
CDC. (2011a). CDC 2011 Estimates: Findings [Online]. Available online at: http://www.cdc.gov
CDC. (2011b). Vital signs: incidence and trends of infection with pathogens transmitted commonly through food-Foodborne Diseases Active Surveillance Network, 10 U.S. Sites, 1996-2010. Morb. Mortal. Weekly Rep. 60, 749–755.
Dauga, C., Zabrovskaia, A., and Grimont, P. A. (1998). Restriction fragment length polymorphism analysis of some flagellin genes of Salmonella enterica. J. Clin. Microbiol. 36, 2835–2843.
Deer, D. M., Lampel, K. A., and Gonzalez-Escalona, N. (2010). A versatile internal control for use as DNA in real-time PCR and as RNA in real-time reverse transcription PCR assays. Lett. Appl. Microbiol. 50, 366–372. doi: 10.1111/j.1472-765X.2010.02804.x
Enright, M. C., and Spratt, B. G. (1999). Multilocus sequence typing. Trends Microbiol. 7, 482–487. doi: 10.1016/S0966-842x(99)01609-1
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.2307/2408678
Fitzgerald, C., Collins, M., Van, D. S., Mikoleit, M., Brown, T., and Fields, P. (2007). Multiplex, bead-based suspension array for molecular determination of common Salmonella serogroups. J. Clin. Microbiol. 45, 3323–3334. doi: 10.1128/JCM.00025-07
Foley, S. L., Lynne, A. M., and Nayak, R. (2009). Molecular typing methodologies for microbial source tracking and epidemiological investigations of Gram-negative bacterial foodborne pathogens. Infect. Genet. Evol. 9, 430–440. doi: 10.1016/j.meegid.2009.03.004
Foley, S. L., Zhao, S., and Walker, R. D. (2007). Comparison of molecular typing methods for the differentiation of Salmonella foodborne pathogens. Foodborne Pathog. Dis. 4, 253–276. doi: 10.1089/fpd.2007.0085
Franklin, K., Lingohr, E. J., Yoshida, C., Anjum, M., Bodrossy, L., Clark, C. G., et al. (2011). Rapid genoserotyping tool for classification of Salmonella serovars. J. Clin. Microbiol. 49, 2954–2965. doi: 10.1128/JCM.02347-10
Gallegos-Robles, M. A., Morales-Loredo, A., Alvarez-Ojeda, G., Vega, P., Chew, M., Velarde, S., et al. (2008). Identification of Salmonella serotypes isolated from cantaloupe and chile pepper production systems in Mexico by PCR-restriction fragment length polymorphism. J. Food Prot. 71, 2217–2222.
Grimont, P. A. D., and Weill, F. X. (2007). Antigenic Formulae of the Salmonella Serovars. Paris: Pasteur Institute; World Health Organization Collaborating Centre for Reference and Research on Salmonella.
Guard, J., Sanchez-Ingunza, R., Morales, C., Stewart, T., Liljebjelke, K., Van, K. J., et al. (2012). Comparison of dkgB-linked intergenic sequence ribotyping to DNA microarray hybridization for assigning serotype to Salmonella enterica. FEMS Microbiol. Lett. 337, 61–72. doi: 10.1111/1574-6968.12010
Hathaway, L. J., Brugger, S., Martynova, A., Aebi, S., and Muhlemann, K. (2007). Use of the Agilent 2100 bioanalyzer for rapid and reproducible molecular typing of Streptococcus pneumoniae. J. Clin. Microbiol. 45, 803–809. doi: 10.1128/JCM.02169-06
Herikstad, H., Motarjemi, Y., and Tauxe, R. V. (2002). Salmonella surveillance: a global survey of public health serotyping. Epidemiol. Infect. 129, 1–8. doi: 10.1017/S0950268802006842
Hong, Y., Liu, T., Hofacre, C., Maier, M., White, D. G., Ayers, S., et al. (2003). A restriction fragment length polymorphism-based polymerase chain reaction as an alternative to serotyping for identifying Salmonella serotypes. Avian Dis. 47, 387–395. doi: 10.1637/0005-2086(2003)047[0387:ARFLPP]2.0.CO;2
Hu, Y. S., Liu, J. H., Pang, X. L., Chen, S. Y., and Chen, X. G. (2009). Phylogenetic analysis and PCR-restriction fragment length polymophism identification of Salmonella based on grOEL gene sequence. Nan Fang Yi Ke Da Xue Xue Bao 29, 2037–2039, 2043.
Hunter, P. R. (1990). Reproducibility and indices of discriminatory power of microbial typing methods. J. Clin. Microbiol. 28, 1903–1905.
Hunter, P. R., and Gaston, M. A. (1988). Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J. Clin. Microbiol. 26, 2465–2466.
Johnson, S. J., and Beese, L. S. (2004). Structures of mismatch replication errors observed in a DNA polymerase. Cell 116, 803–816. doi: 10.1016/S0092-8674(04)00252-1
Kidgell, C., Reichard, U., Wain, J., Linz, B., Torpdahl, M., Dougan, G., et al. (2002). Salmonella typhi, the causative agent of typhoid fever, is approximately 50,000 years old. Infect. Genet. Evol. 2, 39–45. doi: 10.1016/S1567-1348(02)00089-8
Kilger, G., and Grimont, P. A. (1993). Differentiation of Salmonella phase 1 flagellar antigen types by restriction of the amplified fliC gene. J. Clin. Microbiol. 31, 1108–1110.
Kisiela, D., Kuczkowski, M., Kiczak, L., Wieliczko, A., and Ugorski, M. (2005). Differentiation of Salmonella Gallinarum biovar Gallinarum from Salmonella Gallinarum biovar Pullorum by PCR-RFLP of the fimH gene. J. Vet. Med. B Infect. Dis. Vet. Public Health 52, 214–218. doi: 10.1111/j.1439-0450.2005.00846.x
Kwon, H. J., Park, K. Y., Yoo, H. S., Park, J. Y., Park, Y. H., and Kim, S. J. (2000). Differentiation of Salmonella enterica serotype gallinarum biotype pullorum from biotype gallinarum by analysis of phase 1 flagellin C gene (fliC). J. Microbiol. Methods 40, 33–38. doi: 10.1016/S0167-7012(99)00129-3
Lee, K., Iwata, T., Shimizu, M., Taniguchi, T., Nakadai, A., Hirota, Y., et al. (2009). A novel multiplex PCR assay for Salmonella subspecies identification. J. Appl. Microbiol. 107, 805–811. doi: 10.1111/j.1365-2672.2009.04263.x
Li, J., Nelson, K., Mcwhorter, A. C., Whittam, T. S., and Selander, R. K. (1994). Recombinational basis of serovar diversity in Salmonella enterica. Proc. Natl. Acad. Sci. U.S.A. 91, 2552–2556. doi: 10.1073/pnas.91.7.2552
Lu, C. Y., Tso, D. J., Yang, T., Jong, Y. J., and Wei, Y. H. (2002). Detection of DNA mutations associated with mitochondrial diseases by Agilent 2100 bioanalyzer. Clin. Chim. Acta. 318, 97–105. doi: 10.1016/S0009-8981(01)00809-9
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Matsui, T., Matsuda, M., Murayama, O., Millar, B. C., and Moore, J. E. (2001). recA genotyping of Salmonella enteritidis phage type 4 isolates by restriction fragment length polymorphism analysis. Lett. Appl. Microbiol. 32, 424–427. doi: 10.1046/j.1472-765X.2001.00935.x
Mcquiston, J. R., Herrera-Leon, S., Wertheim, B. C., Doyle, J., Fields, P. I., Tauxe, R. V., et al. (2008). Molecular phylogeny of the salmonellae: relationships among Salmonella species and subspecies determined from four housekeeping genes and evidence of lateral gene transfer events. J. Bacteriol. 190, 7060–7067. doi: 10.1128/JB.01552-07
Mcquiston, J. R., Parrenas, R., Ortiz-Rivera, M., Gheesling, L., Brenner, F., and Fields, P. I. (2004). Sequencing and comparative analysis of flagellin genes fliC, fljB, and flpA from Salmonella. J. Clin. Microbiol. 42, 1923–1932. doi: 10.1128/JCM.42.5.1923-1932.2004
Mcquiston, J. R., Waters, R. J., Dinsmore, B. A., Mikoleit, M. L., and Fields, P. I. (2011). Molecular determination of H antigens of Salmonella by use of a microsphere-based liquid array. J. Clin. Microbiol. 49, 565–573. doi: 10.1128/JCM.01323-10
Nachamkin, I., Panaro, N. J., Li, M., Ung, H., Yuen, P. K., Kricka, L. J., et al. (2001). Agilent 2100 bioanalyzer for restriction fragment length polymorphism analysis of the Campylobacter jejuni flagellin gene. J. Clin. Microbiol. 39, 754–757. doi: 10.1128/JCM.39.2.754-757.2001
Nelson, K., and Selander, R. K. (1994). Intergeneric transfer and recombination of the 6-phosphogluconate dehydrogenase gene (gnd) in enteric bacteria. Proc. Natl. Acad. Sci. U.S.A. 91, 10227–10231. doi: 10.1073/pnas.91.21.10227
Owen, R. J., and Leeton, S. (1999). Restriction fragment length polymorphism analysis of the flaA gene of Campylobacter jejuni for subtyping human, animal and poultry isolates. FEMS Microbiol. Lett. 176, 345–350. doi: 10.1111/j.1574-6968.1999.tb13682.x
Panaro, N. J., Yuen, P. K., Sakazume, T., Fortina, P., Kricka, L. J., and Wilding, P. (2000). Evaluation of DNA fragment sizing and quantification by the agilent 2100 bioanalyzer. Clin. Chem. 46, 1851–1853.
Porwollik, S., Boyd, E. F., Choy, C., Cheng, P., Florea, L., Proctor, E., et al. (2004). Characterization of Salmonella enterica subspecies I genovars by use of microarrays. J. Bacteriol. 186, 5883–5898. doi: 10.1128/JB.186.17.5883-5898.2004
Ranieri, M. L., Shi, C., Moreno Switt, A. I., Den Bakker, H. C., and Wiedmann, M. (2013). Comparison of typing methods with a new procedure based on sequence characterization for Salmonella serovar prediction. J. Clin. Microbiol. 51, 1786–1797. doi: 10.1128/JCM.03201-12
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2007). REBASE–enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 35, D269–D270. doi: 10.1093/nar/gkl891
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Scallan, E., Griffin, P. M., Angulo, F. J., Tauxe, R. V., and Hoekstra, R. M. (2011a). Foodborne illness acquired in the United States–unspecified agents. Emerging. Infect. Dis. 17, 16–22. doi: 10.3201/eid1701.091101p2.
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M. A., Roy, S. L., et al. (2011b). Foodborne illness acquired in the United States–major pathogens. Emerging Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.091101p1.
Schwartz, D. C., and Cantor, C. R. (1984). Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell 37, 67–75. doi: 10.1016/0092-8674(84)90301-5
Seong, W. J., Kwon, H. J., Kim, T. E., Lee, D. Y., Park, M. S., and Kim, J. H. (2012). Molecular serotyping of Salmonella enterica by complete rpoB gene sequencing. J. Microbiol. 50, 962–969. doi: 10.1007/s12275-012-2547-x
Shah, S. A., and Romick, T. L. (1997). Subspecies differentiation of Salmonella by PCR-RFLP of the ribosomal operon using universal primers. Lett. Appl. Microbiol. 25, 54–57. doi: 10.1046/j.1472-765X.1997.00174.x
Sirinavin, S., Jayanetra, P., and Thakkinstian, A. (1999). Clinical and prognostic categorization of extraintestinal nontyphoidal Salmonella infections in infants and children. Clin. Infect. Dis. 29, 1151–1156. doi: 10.1086/313469
Stadhouders, R., Pas, S. D., Anber, J., Voermans, J., Mes, T. H., and Schutten, M. (2010). The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J. Mol. Diagn. 12, 109–117. doi: 10.2353/jmoldx.2010.090035
Tamura, K., Nei, M., and Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A. 101, 11030–11035. doi: 10.1073/pnas.0404206101
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thampapillai, G., Lan, R., and Reeves, P. R. (1994). Molecular evolution in the gnd locus of Salmonella enterica. Mol. Biol. Evol. 11, 813–828.
Tindall, B. J., Grimont, P. A., Garrity, G. M., and Euzeby, J. P. (2005). Nomenclature and taxonomy of the genus Salmonella. Int. J. Syst. Evol. Microbiol. 55, 521–524. doi: 10.1099/ijs.0.63580-0
Van Belkum, A., Tassios, P. T., Dijkshoorn, L., Haeggman, S., Cookson, B., Fry, N. K., et al. (2007). Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin. Microbiol. Infect. 13(Suppl. 3), 1–46. doi: 10.1111/j.1469-0691.2007.01786.x
Wattiau, P., Boland, C., and Bertrand, S. (2011). Methodologies for Salmonella enterica subsp. enterica subtyping: gold standards and alternatives. Appl. Environ. Microbiol. 77, 7877–7885. doi: 10.1128/AEM.05527-11
Zhang, G., Brown, E. W., and Gonzalez-Escalona, N. (2011). Comparison of real-time PCR, reverse transcriptase real-time PCR, loop-mediated isothermal amplification, and the FDA conventional microbiological method for the detection of Salmonella spp. in produce. Appl. Environ. Microbiol. 77, 6495–6501. doi: 10.1128/AEM.00520-11
Keywords: Salmonella enterica, Bioanalyzer, PCR-RFLP, restriction type, reference collection
Citation: Soler-García ÁA, De Jesús AJ, Taylor K and Brown EW (2014) Differentiation of Salmonella strains from the SARA, SARB and SARC reference collections by using three genes PCR-RFLP and the 2100 Agilent Bioanalyzer. Front. Microbiol. 5:417. doi: 10.3389/fmicb.2014.00417
Received: 31 March 2014; Accepted: 22 July 2014;
Published online: 11 August 2014.
Edited by:
Michael Gänzle, Alberta Veterinary Research Institute, CanadaReviewed by:
Scott H. Harrison, North Carolina A&T State University, USALuca Settanni, Università degli Studi di Palermo, Italy
Copyright © 2014 Soler-García, De Jesús, Taylor and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ángel A. Soler-García, US Food and Drug Administration, Urology and Lithotripsy Devices Branch, Division of Reproductive, Gastro-Renal, and Urological Devices, Center for Devices and Radiological Health, WO66 RmG223, 10903 New Hampshire Ave., Silver Spring, MD 20993, USA e-mail: angel.soler-garcia@fda.hhs.gov
†Present address: Kishana Taylor, Department of Population Health, Biomedical and Health Science Institute, University of Georgia, Athens, GA, USA