 Cheng-Yao Chen
Cheng-Yao Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 24 June 2014
Sec. Evolutionary and Genomic Microbiology
Volume 5 - 2014 | https://doi.org/10.3389/fmicb.2014.00305
This article is part of the Research Topic DNA polymerases in Biotechnology View all 14 articles
Next-generation sequencing (NGS) technologies have revolutionized modern biological and biomedical research. The engines responsible for this innovation are DNA polymerases; they catalyze the biochemical reaction for deriving template sequence information. In fact, DNA polymerase has been a cornerstone of DNA sequencing from the very beginning. Escherichia coli DNA polymerase I proteolytic (Klenow) fragment was originally utilized in Sanger’s dideoxy chain-terminating DNA sequencing chemistry. From these humble beginnings followed an explosion of organism-specific, genome sequence information accessible via public database. Family A/B DNA polymerases from mesophilic/thermophilic bacteria/archaea were modified and tested in today’s standard capillary electrophoresis (CE) and NGS sequencing platforms. These enzymes were selected for their efficient incorporation of bulky dye-terminator and reversible dye-terminator nucleotides respectively. Third generation, real-time single molecule sequencing platform requires slightly different enzyme properties. Enterobacterial phage ϕ29 DNA polymerase copies long stretches of DNA and possesses a unique capability to efficiently incorporate terminal phosphate-labeled nucleoside polyphosphates. Furthermore, ϕ29 enzyme has also been utilized in emerging DNA sequencing technologies including nanopore-, and protein-transistor-based sequencing. DNA polymerase is, and will continue to be, a crucial component of sequencing technologies.
Since the advent of enzymatic dideoxy-DNA sequencing by Frederic Sanger (Sanger et al., 1977), sequencing DNA/RNA has become standard practice in most molecular biology research. The proliferation of next-generation sequencing (NGS) technologies has further transformed modern biological and biomedical research. Today, large-scale whole genome sequencing has become routine in life science research. Although technical advances in current NGS technologies have dramatically changed the way nucleic acids are sequenced, the engine ultimately responsible for these modern innovations remains unchanged. Like Sanger sequencing, today’s NGS technologies, with the exception of oligonucleotide-based ligation sequencing (Drmanac et al., 2010), still require a DNA polymerase to carry out the necessary biochemical reaction for replicating template sequence information. This unique, polymerase-dependent sequencing approach is generally referred to as DNA sequencing-by-synthesis (SBS), because the consecutive sequencing reaction concurrently generates a newly synthesized DNA strand as a result.
However, unlike Sanger sequencing, DNA polymerases utilized in NGS technologies are more diverse and tailor-made. The Klenow enzyme, a proteolytic fragment of Escherichia coli DNA polymerase I. was originally utilized in Sanger’s dideoxy chain-terminating DNA sequencing chemistry (Sanger et al., 1977). This enzyme was chosen for its efficient incorporation of 2′, 3′-dideoxynucleotides (ddNTPs) that leads to chain termination of DNA synthesis (Atkinson et al., 1969). From this humble beginning, followed by a robust sequencing chemistry improvement, the nucleotide substrates used for DNA sequencing became larger and bulkier. First, four fluorescent dyes with distinct, non-overlapping optical spectra were attached to either purine or pyrimidine bases, respectively, and even the terminal gamma phosphate of four (A, T, C, and G) nucleotides for the ease of signal detection (Smith et al., 1986; Ju et al., 2006; Guo et al., 2008, 2010; Eid et al., 2009; Korlach et al., 2010). Second, the 3′ hydroxyl group on deoxyribose of four nucleotides was replaced with a larger, cleavable chemical group used to reversibly terminate DNA synthesis (Ju et al., 2006; Guo et al., 2008, 2010). As a result, the original Klenow enzyme no longer efficiently incorporated these newly modified nucleotides. DNA polymerases with different enzymatic properties were required for improving the nucleotide incorporation reactions. Fortunately, the adoption of NGS sequencing in life science research allowed a rapid expansion of organism-specific, genome sequence information accessible via public database. Various DNA polymerases from mesophilic/thermophilic viruses, bacteria, and archaea were discovered and later screened for efficient incorporation of modified nucleotides in new DNA sequencing methods. A pool of new, advantageous DNA polymerases from a wide variety of microorganisms were selected and served as protein backbones for further improvement via protein engineering or directed enzyme evolution (Patel and Loeb, 2001). Evolved DNA polymerases with improved biochemical performances were ultimately utilized for each, unique sequencing technology.
This article briefly covers (1) the progression of decades’ enzymatic DNA sequencing methods reliant on functions of DNA polymerase for synthesizing new DNA strands; (2) the novel properties of DNA polymerase that are required for high-precision DNA sequencing; (3) the influence of nucleotide modifications on DNA polymerase research that ultimately lead to improved sequencing performance; (4) the application of DNA polymerases in emerging DNA sequencing methods. Readers interest in learning more about other sequencing methods can refer to these literatures (Landegren et al., 1988; Ding et al., 2012) for more information.
Since the discovery of DNA polymerase I in E. coli by Arthur Kornberg’s group in the late 1950s (Lehman et al., 1958a,b), multiple DNA polymerases have been discovered, purified and characterized from bacteria, eukaryotes, archaea, and their viruses. The expansion of organism-specific, genome sequence information accessible via public database, together with advanced search-algorithms based on DNA polymerase structure–function relationships, have reduced the time necessary for identification of additional, putative DNA polymerases from a variety of sources (Burgers et al., 2001). Based on the phylogenetic relationships of E. coli and human DNA polymerases, DNA polymerases are generally classified into seven main families: A (E. coli Pol I), B (E. coli Pol II), C (E. coli Pol III), D, X (human Pol β-like), Y (E. coli Pol IV and V and TLS polymerases), and RT (reverse transcriptase) (Burgers et al., 2001; Langhorst et al., 2012). All living organisms, except viruses, harbor multiple types of DNA polymerases for cellular functions. Interestingly, neither bacteria, eukaryotes nor archaea contain all families of DNA polymerases. As summarized in Table 1, the family C DNA polymerases are unique to bacteria, and have not been found in either eukaryotes or archaea (Hübscher et al., 2010; Langhorst et al., 2012). Likewise, the family D polymerases are restricted to archaea (Euryarchaeota), and do not exist in bacteria or eukaryotes (Hübscher et al., 2010; Langhorst et al., 2012). Another characteristic exclusive to archaeal DNA polymerases is the presence of intervening sequences (inteins) within the polymerase coding genes (Perler et al., 1992). These inteins cause in-frame insertions in archaeal DNA pols and must be spliced out in order to form mature enzymes (Hodges et al., 1992).
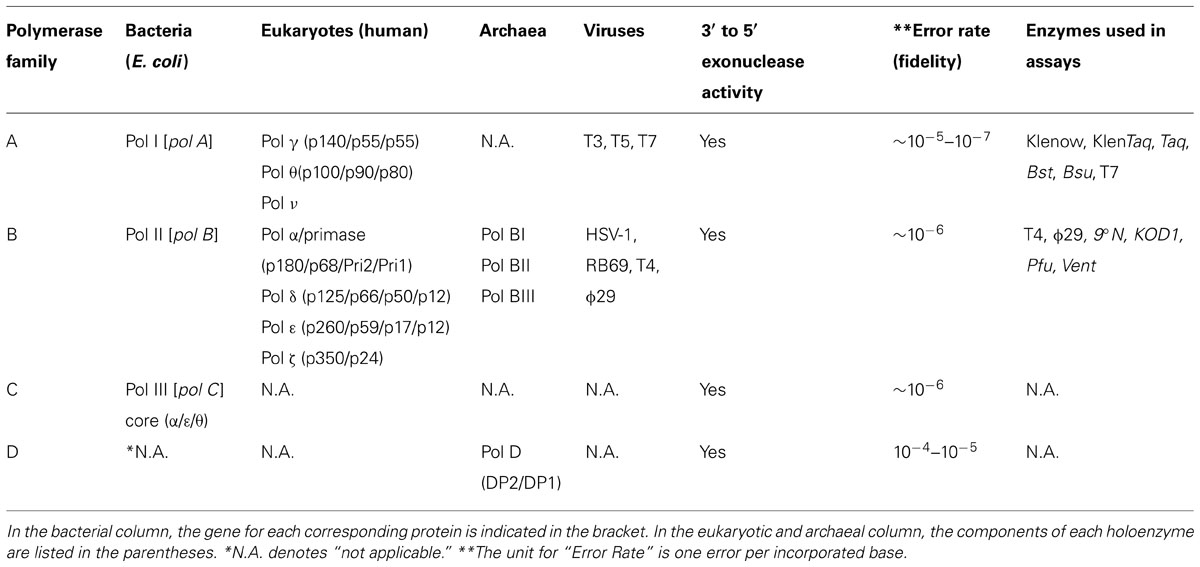
TABLE 1. Families and properties of cellular DNA replicases (Kunkel, 2004; Hübscher et al., 2010; Greenough et al., 2014).
The basic function of DNA polymerases (cellular DNA replicases) are to faithfully replicate the organism’s whole genome and pass down the correct genetic information to future generations. In bacteria, family C DNA polymerases, such as Pol III holoenzyme in E. coli or Bacillus subtilis, are the key element for driving chromosomal replication and thus absolutely mandatory for cell viability (Gefter et al., 1971; Nusslein et al., 1971; Gass and Cozzarelli, 1973, 1974). Besides the Pol III holoenzyme, the A-family Pol I also participates in bacterial DNA replication (Olivera and Bonhoeffer, 1974). Pol I contains a separate 5′ to 3′ exonuclease, independent of the DNA polymerase domain, that can remove RNA primers and concurrently fill in the nucleotide gaps between Okazaki fragments during lagging strand DNA synthesis (Okazaki et al., 1971; Konrad and Lehman, 1974; Xu et al., 1997). Unlike bacterial cells, eukaryotic B-family DNA polymerases, such as Pol δ and ε in human and yeast, are responsible for nuclear chromosomal replication (Miyabe et al., 2011). Recent studies in yeast by Thomas Kunkel’s group suggest that Pol δ and ε divide their roles during DNA replication and are responsible for lagging and leading strand DNA synthesis, respectively (Pursell et al., 2007; Kunkel and Burgers, 2008; Nick McElhinny et al., 2008; Miyabe et al., 2011). In archaea, both B- and D-family pols are involved in genomic replication. However, the role of each Pol in vivo remains controversial. From biochemical studies, both Pol B and D enzymes from hyperthermophilic Pyrococcus abyssi are proposed to function together in DNA replication (Henneke et al., 2005). In contrast, a recent genetic study in Thermococcus kodakarensis showed that Pol D alone is sufficient for cell viability and genomic replication which argues that Pol D, rather than Pol B, is the main replicative DNA polymerase in this archaeon (Cubonova et al., 2013). It is possible that the requirements for Pol B and D enzymes in DNA replication are different in separate phyla of Archaea.
In summary, all DNA polymerases engaged in cellular genome replication, regardless of origin, have the following common features (See Table 1): (1) they appear to form a multi-subunit enzyme complex (holoenzyme); (2) they all possess an intrinsic 3′ to 5′ exonuclease, or proofreading activity, that removes misincorporated nucleotides immediately after nucleotide incorporation to ensure high-fidelity of DNA synthesis (Figure 3A). In contrast to the major cellular DNA polymerases, functions of X, Y, and RT families of Pols are more diverse and specialized in many DNA processes, such as DNA repair, translesion synthesis, and eukaryotic telomere maintenance (Hübscher et al., 2010). None of these Pols have any intrinsic 3′ to 5′ proofreading exonuclease activity and are thus more error-prone during DNA synthesis (Kunkel and Bebenek, 2000; Kunkel, 2004, 2009).
Growing numbers of DNA polymerases, each with distinct functions, provide abundant enzymatic resources for improving current and emerging DNA sequencing techniques. However, not all families of DNA polymerases are suitable for high-precision DNA sequencing reactions. To be considered, and ultimately applied for a particular method of sequencing, the DNA polymerase should possess the following properties:
(1) The pol has to be a DNA-dependent DNA polymerase. Some X and RT-family enzymes do not require a DNA template for replication and are thus not suitable for DNA sequencing; for instance, X-family terminal deoxynucleotidyl transferases (Tdt) are template-independent DNA polymerases which catalyze the addition of deoxynucleotides (dNTPs) to the 3′-OH ends of DNA in the absence of a DNA template (Kato et al., 1967; Coleman et al., 1974). Similarly, RT-family eukaryotic telomerases are ribonucleoproteins which utilize their own, endogenous RNA template for elongation at the telomeric DNA ends (Morin, 1989; Blackburn et al., 2006). These enzymes bypass the requirement of a DNA template to function and cannot be used for DNA sequencing.
(2) The pol should rapidly incorporate nucleotides. Despite the diverse functions among DNA polymerases, the basic mechanism of nucleotide incorporation remains relatively fixed. All replicative DNA pols require a duplex primer-template DNA with a free 3′-OH group at the primer terminus, all four nucleoside triphosphates (dATP, dTTP, dCTP, and dGTP), and catalytic, divalent cations (Mg2+ or Mn2+, etc.) for the sequencing reaction. Addition of nucleotides to the 3’ end of a primer by DNA pols proceeds through a highly ordered, temporal mechanism. The minimal catalytic mechanism of single-nucleotide incorporation by DNA pol has been proposed (Donlin et al., 1991; Johnson, 1993) and is illustrated in Figure 1. A brief description for each reaction step can be found in the figure legend. As shown in Figure 1, the nucleotide incorporation efficiency (specificity) of a DNA polymerase (kpol/kd,dNTP) is determined by the rate of phosphodiester bond formation (kpol) and the binding constant for the cognate nucleotide (kd,dNTP; Wong et al., 1991; Johnson, 1993). DNA pols with a faster nucleotide incorporation rate and lower kd,dNTP (large kpol and small kd,dNTP) can catalyze DNA synthesis much more efficiently. In this aspect, none of the X and Y-family pols can meet this requirement. Both X and Y-family Pols have much lower nucleotide incorporation efficiency (Brown et al., 2011a,b) compared to cellular DNA replicases from A, B, C, or D-family enzymes (Patel et al., 1991; Wong et al., 1991; Bloom et al., 1997; Zhang et al., 2009). Therefore, they are not ideal for DNA sequencing.
(3) The pol must have high replicative fidelity to minimize systematic sequencing errors. In order to accurately read DNA template sequence information, the DNA pol must faithfully incorporate the correct, matched nucleotides along the DNA template. The fidelity of nucleotide incorporation by X, Y, and RT Pols range from ~10-1 to 10-4 error per base incorporated, two to three orders of magnitude lower than high-fidelity cellular DNA polymerases from A, B, or C-family enzymes (Kunkel, 2004). These repair pols generally make errors during DNA synthesis (Kunkel and Bebenek, 2000; Kunkel, 2004, 2009) and are not appropriate for high-precision DNA sequencing applications.
(4) The pol should possess long, intrinsic, replicative processivity. The processivity of DNA polymerase is defined as the number of dNTPs incorporated during complex formation with a primer/template (P/T) DNA. As illustrated in Figure 1, the processivity of DNA pol is directly related to two parameters: (1) the rate of dNTP incorporation by the enzyme (kpol of step 5); (2) the enzyme’s dissociation rate from the enzyme–DNA binary complex (koff,DNA of step 1). Under these parameters, the enzyme remains associated with the template DNA, it carries out sequential rounds of nucleotide incorporation until it dissociates from the binary complex (Figure 1, steps 2–7). Theoretically, processivity of the DNA polymerase can be estimated by calculating the ratio of kpol/koff, DNA. Amongst DNA polymerases, only viral DNA polymerases have unusually intrinsic, long processivity. For instance, the enterobacterial phage ϕ29 DNA polymerase (a B-family enzyme) possesses intrinsic, long, replicative processivity and can replicate its own genomic DNA (~18,000 base pairs) in vitro without any accessory cofactors (Blanco and Salas, 1985). In contrast, most cellular DNA replicases from A, B, C, and D families are distributive, and limited to only a few nucleotide incorporations. These DNA replicases must physically interact with their processivity factors, including β-sliding clamp in bacteria, and PCNA in eukaryotes and archaea, in order to achieve a long processivity during DNA replication (Johnson and O’Donnell, 2005). No X, Y, or RT-family enzymes are processive.
(5) The pol should function as a monomer for ease of protein production and further modification. As illustrated in Table 1, most A, B, C, and D-family DNA replicases form a multi-subunit enzyme complex (holoenzyme). Components of these replicative holoezymes are difficult to purify, and whole enzyme complexes are very challenging to reconstitute. Therefore, these types of enzyme complexes are seldom used in any DNA sequencing chemistry.
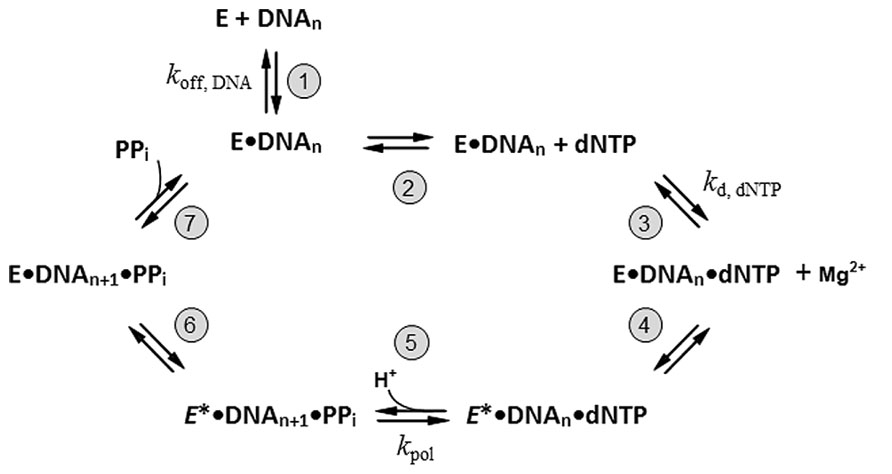
FIGURE 1. The minimal catalytic steps required for single-nucleotide incorporation by DNA polymerase. The addition of nucleotide to the 3′ end of a primer by DNA polymerase passes through a temporally ordered mechanism. The reaction begins with the binding of free DNA polymerase (E) to a duplex primer/template DNA complex (DNAn) resulting in a binary enzyme-DNA complex (E•DNAn; step 1). The koff, DNA represents the rate of enzyme dissociation from the E•DNAn complex. Addition of the correct nucleotide (dNTP) in the presence of divalent cations, such as Mg2+, promotes the enzyme-DNA-dNTP ternary complex formation (E•DNAn•dNTP; step 2 and 3). The kd, dNTP denotes the nucleotide binding constant of the enzyme. The binding of the dNTP induces the first conformational change of the enzyme in the ternary complex (E*•DNAn•dNTP; step 4; Wong et al., 1991). The actual chemistry happens (step 5). The phosphodiester bond is formed between the α-phosphate of the incoming dNTP and 3′-OH of the primer terminus and produces an added nucleotide base to the primer terminus (DNAn+1). The chemical reaction generates a pyrophosphate (PPi) and proton molecule (H+). This is followed by a second conformational change of the enzyme (step 6), which allows the final release of the PPi leaving group (step 7). The nucleotide incorporation cycle is complete after PPi release. If the enzyme remains associated with DNA, a new round of nucleotide addition will continue until the enzyme dissociates from the DNA (processive synthesis).
In summary, to fulfill the above requirements for high-precision DNA sequencing, only A-family enzymes from bacteria and phage viruses (such as T5 and T7 phages), and B-family pols from bacterial viruses (such as T4, Rb69, and ϕ29 phages), bacteria, and archaea (Vent, 9°N, Pfu, and KOD1) have been evaluated for sequencing chemistry development (See Table 1). All family A and B enzymes have an associated, intrinsic 3′ to 5′ exonuclease proofreading activity. When these enzymes incorporate an incorrect nucleotide at the primer terminus, the enzymes’ ability to extend the primer terminus diminishes, and allows the nascent DNA strand to migrate to the 3′ exonuclease site for excision (See Figure 3A; Donlin et al., 1991; Joyce and Steitz, 1994; Patel and Loeb, 2001). This unique partitioning mechanism of the 3′ exonuclease proofreading domain among A and B-family polymerases is disfavored for DNA sequencing. It causes asynchronous DNA sequencing reactions and generates systematic sequencing errors (Figures 3B,C). Therefore, the majority of A and B-family pols used for DNA sequencing are either lacking, or have an attenuated, 3′ exonuclease proofreading activity.
Generations of DNA polymerase-based sequencing methods and their corresponding commercial platforms are summarized in Table 2. As shown in Table 2, all methods require a DNA polymerase to catalyze the necessary biochemical reaction for extracting DNA sequence information. The fundamental difference amongst these technologies is the type of nucleotide substrate incorporated. The structures of these nucleotides are illustrated in Figure 2. More in-depth information regarding these nucleotides can be found in the following articles (Metzker et al., 1996; Lee et al., 1997; Kumar et al., 2005; Metzker, 2010; Chen et al., 2013a). From classical Sanger sequencing to modern NGS technologies, the nucleotide substrates used for sequencing have changed over time. In the original Sanger sequencing method, four 2′, 3′-ddNTPs (Figure 2B) are utilized (Sanger et al., 1977). Unlike normal dNTPs (Figure 2A), the ddNTPs lack the 3′-hydroxyl group (3′-OH), which is required for the phosphodiester bond formation between the incorporating nucleotide and primer terminus. Once ddNTPs are incorporated by the DNA polymerase, they terminate further addition of nucleotides from the primer terminus, and cease elongation of the DNA chain (Atkinson et al., 1969). Besides the utilization of ddNTPs, Sanger’s protocol requires a set of radioisotope-labeled primers in four, separate (A, T, C, and G) reactions. The resulting dideoxy-terminated DNA fragments must be analyzed side-by-side using slab gel electrophoresis while sequence information is deduced via autoradiography (Sanger et al., 1977). The procedure itself is extremely time consuming and further compounded by low data output. This makes such an approach insufficient at meeting the growing demand for high-throughput DNA sequencing.
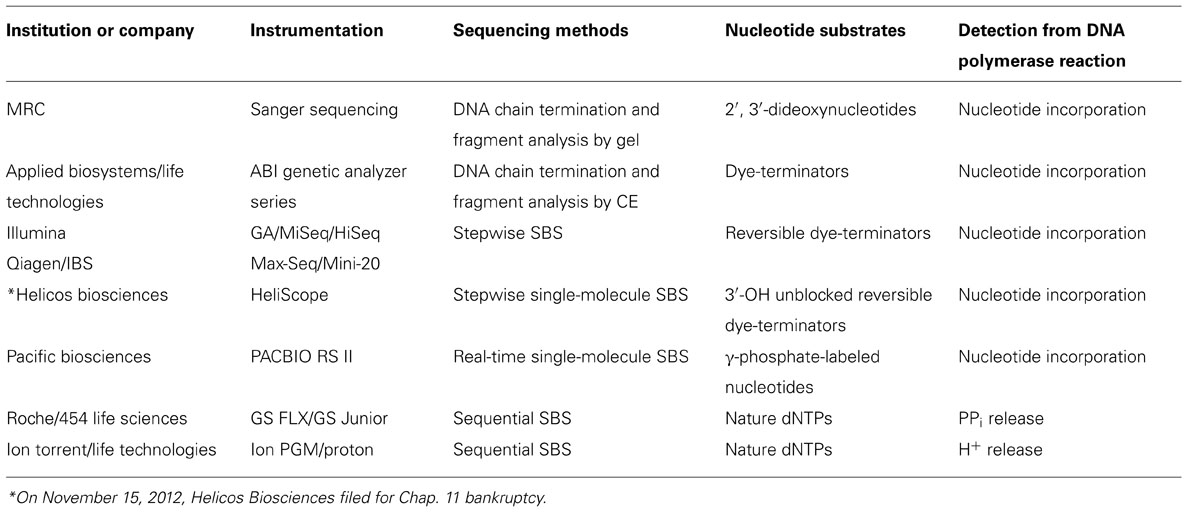
TABLE 2. Generations of DNA polymerase-based DNA sequencing technologies.
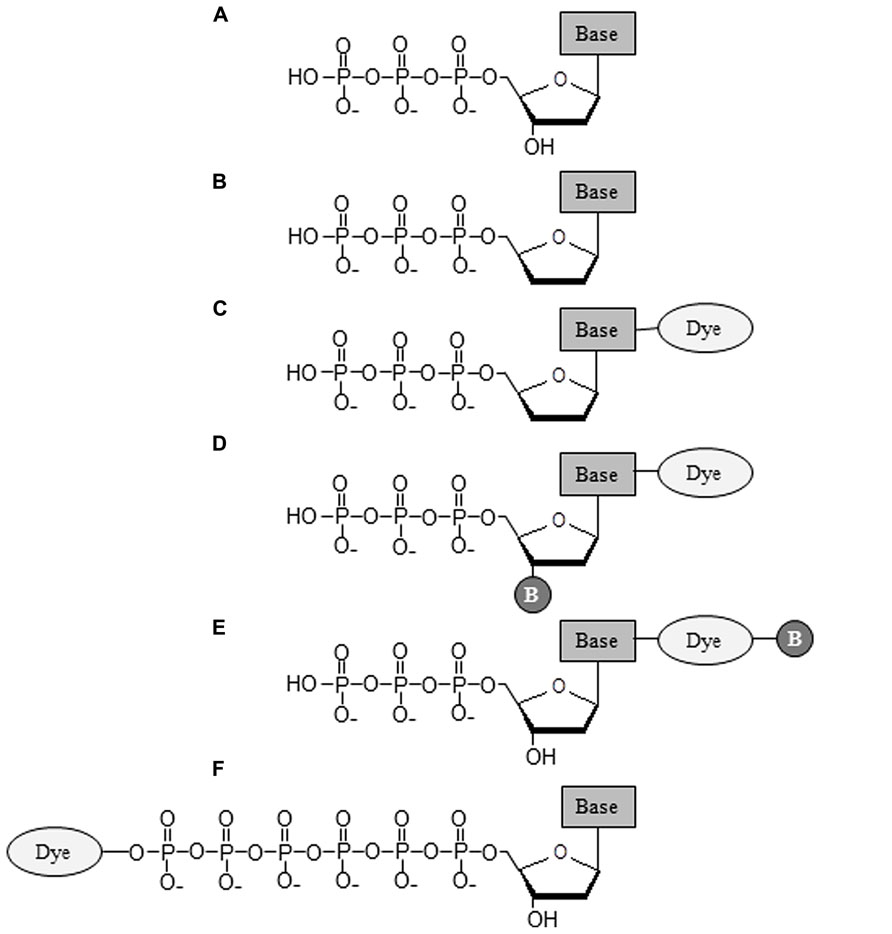
FIGURE 2. Structures of nucleotides utilized in the generations of DNA polymerase-based sequencing methods.(A) Deoxynucleotides (dNTPs); (B) 2′, 3′-dideoxynucleotides (ddNTPs); (C) Dye-terminators; (D) Reversible dye-terminators; (E) 3′-OH unblocked reversible dye-terminators; (F) Dye-labeled hexaphosphate nucleotides. The “Base” in the diagram represents an A, T, C or G base, and “B” indicates a cleavable chemical blockage group.
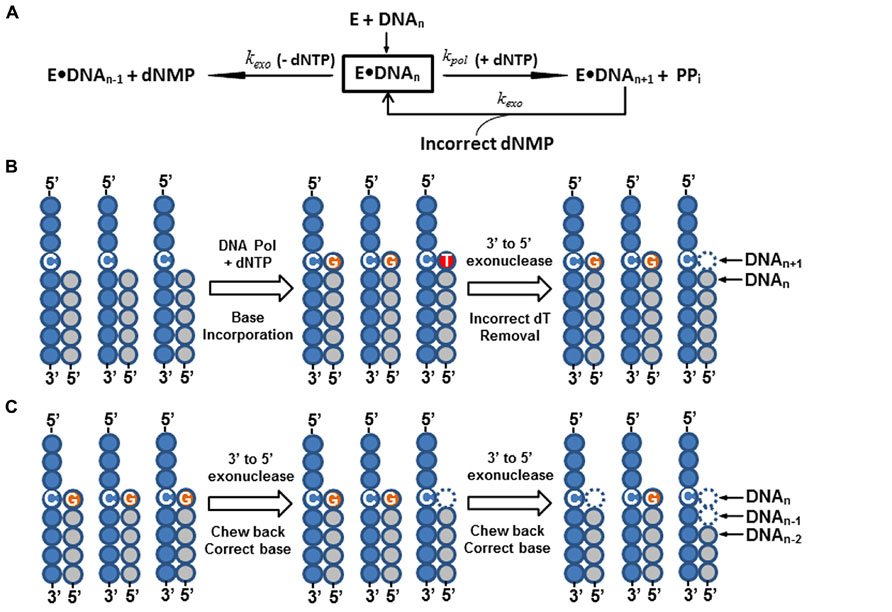
FIGURE 3. Intrinsic 3′ to 5′ exonuclease activity of DNA polymerase and its impact on DNA sequencing reactions. (A) A simplified kinetic model illustrating the proofreading function and nucleotide excision activity of 3′ to 5′ exonuclease of DNA polymerase (Donlin et al., 1991; Johnson, 1993). As shown in the figure, when a free DNA polymerase (E) is mixed with a duplex primer/template DNA complex (DNAn), they form a stable, binary enzyme-DNA complex (E•DNAn). In the presence of nucleotide (+dNTP) and divalent cations (Mg2+, Mn2+, etc.), the enzyme rapidly incorporates (kpol) a single-nucleotide base to the primer terminus (DNAn+1) and concurrently drives release of free pyrophosphate (PPi). However, when an incorrect nucleotide is misincorporated by the enzyme, it causes a base-pair mismatch at the primer terminus (DNAn+1; Panel B, middle cartoon, a dC:dT mismatch). This mismatched nucleotide base at the primer terminus greatly impedes the DNA polymerase’s capability to incorporate the next nucleotide base (greatly reduced kpol value) and triggers a rapid transfer of DNA primer strand to the intrinsic 3′ to 5′ exonuclease domain. The mismatched nucleotide base is then removed (incorrect deoxynucleoside monophosphate, dNMP) by the 3′ to 5′ exonuclease (kexo). Once the mismatched nucleotide base is excised by the 3′ to 5′ exonuclease, the corrected primer strand is shifted back to the DNA polymerase catalytic domain (E•DNAn). As a result, the misincorporated nucleotide is removed and the enzyme is ready to incorporate the correct nucleotide (see Panel B, left to right cartoons). In addition to the base-mismatched proofreading function of the 3′ to 5′ exonuclease domain, it will also gradually chew back the primer strand (DNAn-1, DNAn-2, etc.) and release dNMPs in the absence of nucleotide (-dNTP; see Panel C, left to right cartoons). An asynchronous DNA sequencing reaction occurs when the sequencing DNA polymerase misincorporates a nucleotide base (Panel B), or the DNA sequencing primer is chewed back by the enzyme’s 3′ to 5′ exonuclease (Panel C). The outcome of both reactions produces a non-uniform duplex primer-template DNA for DNA sequencing (Panels B,C, the right cartoons), and causes systematic DNA sequencing errors. In the panels B,C, each filled circle indicates a nucleotide base. A string of filled-gray circles represents the primer strand, and a string of filled-blue circles is the template DNA strand. Specific bases (dC, dG, and dT) are indicated inside the circles.
To simplify and subsequently automate Sanger’s method, Leroy Hood’s group, then at California Institute of Technology, invented the first fluorescent sequencing (dye-primer) method based on Sanger’s approach (Smith et al., 1986). In Hood’s revised protocol, the primers used for sequencing reactions are covalently attached to four distinct colors of fluorophores at the 5′-end, corresponding to each of the A, T, C, and G reactions in Sanger sequencing. The advantages to this approach are (1) the four reaction mixtures can be combined and analyzed in a single sequencing lane; (2) the results can be directly monitored by a computer-aided fluorescence detection system, specifically matched to the emission spectra of the four dyes. These advantages allow DNA sequence information to be analyzed automatically by the computer.
Hood’s dye-primer method simplifies traditional Sanger sequencing processes but it is not, however, completely ideal for fully automated DNA sequencing, mainly due to the four, separate reactions still required. To solve this problem, the fluorescently labeled chain-terminating ddNTPs (dye-terminators) were soon introduced by Prober et al. (1987) from DuPont. Similar to the dye-primers, a set of fluorescently distinguished fluorophores are covalently attached to each of four ddNTPs (See Figure 2C). Adaptation of dye-terminators for Sanger sequencing workflow makes the four, base-specific chain termination reactions happen in one, single reaction tube. DNA polymerase is able to simultaneously incorporate four dye-terminators and generate the terminated DNA pieces for sequence analysis (Rosenthal and Charnock-Jones, 1992, 1993). The speed and throughput of dye-terminator sequencing was drastically improved when the automated capillary-array electrophoresis (CAE) was adopted for DNA analysis (Drossman et al., 1990; Luckey et al., 1990; Zagursky and McCormick, 1990; Dovichi and Zhang, 2000).
The dye-terminator-CE method has greatly improved sequencing performance and has become the laboratory standard for DNA sequencing over the past few decades. However, the technique itself is still very limited, especially for large-scale, whole genome sequencing. Increasing the sequencing throughput of dye-terminator-CE chemistry requires additional capillary tubes to be implemented. This becomes impractical for the application of high-throughput, multiplexing sequencing that is capable of sequencing millions of different DNA strands concurrently. To alleviate this limitation, reversible dye-terminators were introduced to the modified, dye-terminating sequencing scheme. Similar to dye-terminators (Figure 2C), reversible dye-terminators (Figure 2D) are also missing the 3′-OH group needed for DNA polymerase extension of the primer terminus. Incorporation of these modified nucleotides by DNA polymerase terminates DNA chain elongation (Bentley et al., 2008; Guo et al., 2008; Hutter et al., 2010). When these reversible dye-terminators are used in parallel with immobilization of DNA molecules on a solid-state surface, the individual DNA sequence can be directly ascertained from the base-specific, terminated DNA molecules recognized by the fluorescent imaging system (Bentley et al., 2008; Guo et al., 2008, 2010). As a result, the requirements for capillary electrophoresis (CE) analysis in a typical dye-terminator approach are no longer necessary, and millions of different DNA molecules can be sequenced simultaneously. Differentiating themselves from dye-terminators, reversible dye-terminators contain cleavable chemical groups at the 3′ position of the pentose and linker region, located between the base and attached fluorophore (Figure 2D; Bentley et al., 2008; Guo et al., 2008; Hutter et al., 2010). These cleavable chemical groups can be removed in order to restore the normal 3′-OH group of deoxyribose and maintain the integrity of bases attached with dye. DNA chains can thus be further extended by the DNA polymerase and incorporation can resume once more in the next reaction cycle (Bentley et al., 2008; Guo et al., 2008, 2010). A similar sequencing scheme was also carried out using another class of reversible dye-terminators with normal 3′-OH groups (Wu et al., 2007; Pushkarev et al., 2009; Litosh et al., 2011; Gardner et al., 2012). These 3′ unblocked, reversible terminators possess both chemical blockage group and fluorescent dye attached to the same base (Figure 2E), and can be removed by either chemical cleavage or UV light (Pushkarev et al., 2009; Litosh et al., 2011).
In both classes of reversible dye-terminators, cleavage of the linker group carrying the fluorescent dye leaves extra chemical molecules on the normal purine and pyrimidine bases. These molecular remnants may perturb the protein–DNA interaction and eventually impact the sequencing performance of the DNA polymerase (Metzker, 2010; Chen et al., 2013a). To circumvent this concern, terminal γ-phosphate, fluorescently labeled nucleoside polyphosphates (Figure 2F) were developed for the more advanced, third-generation DNA sequencing technique (Kumar et al., 2005; Korlach et al., 2010). There are two major advantages of performing DNA sequencing with γ-phosphate-labeled nucleotides over conventional chain terminators. First, the nucleotides, once incorporated, don’t generate a molecular scar on the newly synthesized DNA, and second, they enable real-time, single-molecule SBS (Korlach et al., 2010). Because the phosphoryl transfer reaction only occurs between the 3′-OH group of the primer terminus and α-phosphate of the incoming nucleotide, the conclusion of each enzymatic reaction results in one nucleotide addition to the primer terminus plus a pyrophosphate (PPi) leaving group (Figure 1, steps 5–7; Steitz, 1997, 1999). Hence, any fluorophore covalently attached to the PPi leaving group will be released after nucleotide addition to the primer terminus, and thus leave no molecular vestige in the DNA. Since the added nucleotide possesses no blockage group to hinder DNA elongation from the primer terminus, the sequencing reaction can continue uninterrupted.
Finally, there are no DNA scar issues for both pyrosequencing technology (Ronaghi et al., 1996, 1998), which detects the release of PPi after nucleotide addition by DNA polymerase, and semiconductor-based proton sequencing technique (Rothberg et al., 2011), which monitors the proton (H+) release during phosphodiester bond formation between the 3′-OH and α-phosphate of incoming nucleotide. Both technologies utilize natural nucleoside triphosphates (dNTPs) for their sequencing reactions (Table 2 and Figure 2A).
A series of nucleotide modifications, created for rapidly changing DNA polymerase-based sequencing technologies has created a daunting task for DNA polymerase researchers to look for, design or evolve compatible enzymes for ever-changing DNA sequencing chemistries. From the beginning, A-family E. coli DNA polymerase I (Pol I) or its proteolytic (Klenow) fragment was chosen by Dr. Sanger for his dideoxy-sequencing chemistry (Sanger et al., 1977). This was the only DNA polymerase available at the time and, quite fortunately, tolerated incorporation of 2′, 3′-ddNTPs (Atkinson et al., 1969). However, Pol I effectively discriminates between a deoxy- and dideoxyribose in the nucleoside triphosphate, and does not incorporate ddNTPs very well (Atkinson et al., 1969). In fact, the incorporation rate of ddNTP by Pol I is several hundred-fold slower than that of normal dNTPs and is also sequence context-dependent (Tabor and Richardson, 1989). This sequence-specific ddNTP incorporation by Pol I creates non-uniform band intensities on the sequencing gel. This phenomenon becomes increasingly problematic, especially in the dye-primer/terminator sequencing, because the method of sequence information retrieval relies on the interpretation of fluorescent intensity of each dideoxy-terminated DNA band from the gel or capillary tubes. Similar results were reported with thermostable, Family A, Thermus aquaticus (Taq) DNA polymerase I (Innis et al., 1988).
In contrast, phage T7 DNA polymerase does not distinguish ddNTPs from dNTPs, and incorporates both types of nucleotides at nearly equal efficiencies (Tabor and Richardson, 1987; Brandis et al., 1996). Thus, the intensities of dideoxy-terminated bands are significantly more uniform with T7 pol in Sanger sequencing. To understand the molecular basis for this discrepancy, sequence analysis and biochemical studies were conducted among these three, A-family enzymes. The results indicate that a single phenylalanine to tyrosine residue change (Y526) on T7 pol, homologous position (F672), of a highly conserved finger motif (motif B) in A-family pols greatly reduces the enzyme’s ability to select against ddNTPs (Tabor and Richardson, 1995). Biochemical studies further confirm that mutant Pol I, or Taq, carrying a F672Y or F667Y mutation, respectively, loses its discriminatory ability for ddNTPs, and thus incorporates ddNTPs very efficiently (Patel and Loeb, 2001). Additionally, these two mutant proteins were demonstrated to incorporate fluorescein- and rhodamine-labeled dye-terminators, three orders of magnitude more efficiently than their wild-type parent enzymes (Tabor and Richardson, 1995). Subsequently, T7, F672Y Pol I, and F667Y Taq pols were all used for manual and automated Sanger sequencing (Tabor and Richardson, 1987, 1989; Rosenthal and Charnock-Jones, 1992; Tabor and Richardson, 1995). However, Taq pol has become preferred for dye-terminator sequencing, because the enzyme has several advantages over Pol I or T7. The enzyme is more readily purified and modifiable for further improvement. It also has no intrinsic, 3′ to 5′ exonuclease proofreading activity, and is active over a broad range of temperatures (Innis et al., 1988). The thermostablility of Taq pol became essential for sequencing after the PCR-based “cycle sequencing” approach was introduced (Rosenthal and Charnock-Jones, 1993).
The Phe to Tyr mutation at position 667 on conserved motif B of Taq pol only addresses the deoxy- and dideoxyribose selectivity problem in dye-terminator sequencing. The enzyme, like Pol I, possesses bias. Uneven ddNTP incorporation results in variable DNA band intensities, and unequal peak heights in CE analysis, creating unwanted sequencing errors (Parker et al., 1995; Li et al., 1999). Kinetic analysis reveals that Taq pol favors ddGTP incorporation over other ddNTPs, with a much more robust nucleotide incorporation rate (kpol; Brandis et al., 1996). To investigate the cause of ddGTP bias, structural analysis of all four, ddNTP-trapped ternary complexes of the large fragment of Taq pol (Klentaq1) was implemented. The data reveals a selective interaction between the guanidinium side chain of arginine residue 660 (R660) and the O6/N7 atoms of the guanine base of the incoming ddGTP. Substitution of the Arg660 residue with a negatively charged aspartic acid completely eliminates preference for ddGTP incorporation. The R660D/F667Y double mutant of Taq pol greatly improves dye-terminator sequencing quality and accuracy (Li et al., 1999).
Although the F667Y mutation on Taq pol greatly improves the enzyme’s incorporation efficiency for dideoxy-dye-terminators, the improvement becomes marginal for the reversible dye-terminators, which carry larger chemical blocking groups than the normal 3′-OH at the 3′ position of deoxyribose (Bentley et al., 2008; Guo et al., 2008; Chen et al., 2010, 2013a; Hutter et al., 2010). The 3′ reversible terminating group is normally linked to the deoxyribose of the nucleotide through the oxygen atom of 3′-OH. A series of 3′-O-blocking groups have been developed including 3′-O-allyl (Ruparel et al., 2005; Wu et al., 2007), 3′-O-(2-nitrobenzyl) (Wu et al., 2007), and 3′-O-azidomethylene (Bentley et al., 2008). Serendipitously, reversible dye-terminators bearing either blockage group were found to be incorporated well by a variant of archaeal 9°N DNA polymerase (a B-family Pol) of hyperthermophilic Thermococcus sp. 9°N-7 (Southworth et al., 1996; Ruparel et al., 2005; Ju et al., 2006; Bentley et al., 2008). The enzyme variant bearing A485L and Y409V double mutations on conserved motifs A and B, respectively, of the DNA polymerase shows enhanced preference for incorporating both acyclic and dideoxy dye-terminators over the parent enzyme (Gardner and Jack, 2002). The same mutational effects were also found in enzyme mutants possessing homologous mutations in other archaeal, B-family DNA polymerase species (Gardner and Jack, 1999; Gardner et al., 2004). Similarly, the analogous combination of mutations (P410L/A485T) at the same conserved protein regions of closely related, B-family DNA polymerase Thermococcus sp. JDF-3 also shows an additive effect on improving dye-terminator incorporation (Arezi et al., 2002). Furthermore, an A485L variant of 9°N DNA pol, termed Therminator DNA polymerase commercially, was recently demonstrated to efficiently incorporate 3′-OH unblocked dye-terminators with a terminating 2-nitrobenzyl moiety attached to hydroxymethylated nucleobases (Gardner et al., 2012). Thus, mutations at these two conserved protein motifs of archaeal, B-family DNA polymerase might affect the enzyme’s selectivity and tolerance for modifications and substitutions on the deoxyribose and nucleobase.
Recently, a more rational approach was taken to search for variants of Taq pol that can accept new types of reversible terminators possessing a 3′-ONH2 blocking group (dNTP-ONH2; Chen et al., 2010). Using the structure-guided reconstruction of ancestral DNA sequence analysis on Taq pol, a library of 93 protein variants carrying different combinations of mutations were designed and screened for the ability to incorporate dNTP-ONH2 in primer-extension assays. One beneficial mutation (L616A) on Taq pol was identified. The L616A Taq enzyme variants incorporated both dNTP-ONH2 and ddNTPs faithfully and efficiently.
The path toward acquisition of a compatible DNA polymerase for incorporation of fluorescent, terminal polyphosphate-labeled nucleotides has not been so straightforward. Historically, the specificities of DNA polymerases toward γ-phosphate modified dNTPs are found to be very different, due to the various degrees of steric effects of substituted chemical groups on each enzyme’s dNTP binding pocket (Arzumanov et al., 1996; Martynov et al., 1997). For instance, a bulky 2, 4-dinitrophenyl group substitution at the γ-phosphate of dNTP is a good substrate for the RT-family AMV RT, but is not acceptable for A or B-family DNA polymerases (Alexandrova et al., 1998). Similar findings were reported with the bis-(2′-deoxynucleoside) 5′, 5′-triphosphates (Victorova et al., 1999). HIV-RT utilizes this type of γ-phosphate modified nucleotide very effectively, while E. coli Pol I and Taq pol do not. Interestingly, in the same study, both Pol I and Taq pol were found to incorporate the bis-(2′-deoxynucleoside) 5′, 5′-tetraphosphates more efficiently than the triphosphate analog (Victorova et al., 1999). Thus, the addition of an extra-phosphate moiety to the terminal γ-phosphate of dNTP seems to attenuate the steric effects on the enzyme. Alternatively stated, the extra phosphate spacer, linked to the terminal γ-phosphate of dNTP, makes the modified nucleotide better tolerated by the enzyme. Indeed, when nucleotide incorporation rates were evaluated with fluorescent, terminal phosphate-labeled nucleoside polyphosphates containing 3, or more, phosphates at the 5′-position of the nucleoside, the nucleotides possessing greater than three phosphates were more effective substrates for A and B-family DNA polymerases (Kumar et al., 2005). Later studies proved both dye-labeled nucleoside penta/hexaphosphates (dN5Ps and dN6P) alone can be used by enterobacterial phage ϕ29 DNA polymerase for incorporating thousands of bases in length, approaching natural dNTP rates (Korlach et al., 2008, 2010). This unique, long, replicative processivity of ϕ29 DNA pol, together with intrinsic, superior capability of incorporating dye-labeled, terminal polyphosphate nucleotides plays a key role in real-time, single-molecule SBS (Korlach et al., 2010).
In contrast to current, SBS approaches, emergent DNA sequencing methods rely on unconventional applications of DNA polymerase. These techniques utilize DNA polymerase as a traditional incorporating enzyme, and alternatively as a molecular motor, responsible for controlled DNA translocation across the protein nanopore. Traditional, nanopore-based, SBS uses commercial Therminator γ DNA polymerase, a variant9°N DNA pol, to incorporate terminal, γ-phosphate-labeled nucleoside tetraphosphates. These modified nucleotides are coupled with four, different-length PEG-coumarin tags corresponding to base A, T, C, and G (Kumar et al., 2012). DNA sequence information can be ascertained by measuring current (amp) fluctuations of the orderly, released PEG-coumarin tags through the α-hemolysin nanopore following DNA polymerase incorporation. A related, but fundamentally different approach involves mutant Mycobacterium smegmatis porin A (MspA) nanopore, ϕ29 DNA polymerase, and natural dNTPs (Manrao et al., 2012). In this approach, the enzyme functions as both DNA replicative enzyme, and molecular motor, which control the speed of DNA translocation through the MspA nanopore.
Besides the nanopore-based sequencing approach, a protein, transistor-based sequencing method, leveraging electrical conductance measurement of ϕ29 DNA polymerase reactions has been reported (Chen et al., 2013b). Unfortunately, this study is currently called into question, and the merits of this particular method must be reevaluated (Chen et al., 2013b).
Since the introduction of the first enzymatic DNA sequencing by Frederic Sanger in the mid-1970s, decades of scientific research on various DNA polymerases, starting with Arthur Kornberg’s enzyme discovery in the mid-1950s, have provided the basic understanding of how these enzymes function and replicate DNAs, further cementing the foundation for improving enzyme properties and applications in current, and future, DNA polymerase-based sequencing technologies. The large-scale of organism-specific, genome research reveals the intrinsic diversity and unique characteristics of DNA polymerases present in all kingdoms of life, including their viruses. Diverse DNA polymerases with distinct functions and properties provide a large pool of natural protein variants that can be tested, and later utilized, for continuously evolving sequencing-chemistries. Tailor-made protein variants designed via protein engineering or directed-enzyme evolution have created powerful protein-engines that have propelled the progression of DNA sequencing technologies over the past few decades. Without a doubt, DNA polymerase has been, and will continue to remain, a crucial component of future sequencing technologies.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author thanks Ali Nikoomanzar for editing the manuscript, and Dr. Lawrence A. Loeb, Eddie Fox, and Thomas Lie for critical reading the manuscript.
Alexandrova, L. A., Skoblov, A. Y., Jasko, M. V., Victorova, L. S., and Krayevsky, A. A. (1998). 2′-Deoxynucleoside 5′-triphosphates modified at alpha-, beta- and gamma-phosphates as substrates for DNA polymerases. Nucleic Acids Res. 26, 778–786. doi: 10.1093/nar/26.3.778
Arezi, B., Hansen, C. J., and Hogrefe, H. H. (2002). Efficient and high fidelity incorporation of dye-terminators by a novel archaeal DNA polymerase mutant. J. Mol. Biol. 322, 719–729. doi: 10.1016/S0022-2836(02)00843-4
Arzumanov, A. A., Semizarov, D. G., Victorova, L. S., Dyatkina, N. B., and Krayevsky, A. A. (1996). Gamma-phosphate-substituted 2′-deoxynucleoside 5′-triphosphates as substrates for DNA polymerases. J. Biol. Chem. 271, 24389–24394. doi: 10.1074/jbc.271.40.24389
Atkinson, M. R., Deutscher, M. P., Kornberg, A., Russell, A. F., and Moffatt, J. G. (1969). Enzymatic synthesis of deoxyribonucleic acid. XXXIV. Termination of chain growth by a 2′,3′-dideoxyribonucleotide. Biochemistry 8, 4897–4904. doi: 10.1021/bi00840a037
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G., et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59. doi: 10.1038/nature07517
Blackburn, E. H., Greider, C. W., and Szostak, J. W. (2006). Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 12, 1133–1138. doi: 10.1038/nm1006-1133
Blanco, L., and Salas, M. (1985). Replication of phage phi 29 DNA with purified terminal protein and DNA polymerase: synthesis of full-length phi 29 DNA. Proc. Natl. Acad. Sci. U.S.A. 82, 6404–6408. doi: 10.1073/pnas.82.19.6404
Bloom, L. B., Chen, X., Fygenson, D. K., Turner, J., O’Donnell, M., and Goodman, M. F. (1997). Fidelity of Escherichia coli DNA polymerase III holoenzyme. The effects of beta, gamma complex processivity proteins and epsilon proofreading exonuclease on nucleotide misincorporation efficiencies. J. Biol. Chem. 272, 27919–27930. doi: 10.1074/jbc.272.44.27919
Brandis, J. W., Edwards, S. G., and Johnson, K. A. (1996). Slow rate of phosphodiester bond formation accounts for the strong bias that Taq DNA polymerase shows against 2′,3′-dideoxynucleotide terminators. Biochemistry 35, 2189–2200. doi: 10.1021/bi951682j
Brown, J. A., Pack, L. R., Fowler, J. D., and Suo, Z. (2011a). Pre-steady-state kinetic analysis of the incorporation of anti-HIV nucleotide analogs catalyzed by human X- and Y-family DNA polymerases. Antimicrob. Agents Chemother. 55, 276–283. doi: 10.1128/AAC.01229-10
Brown, J. A., Pack, L. R., Sanman, L. E., and Suo, Z. (2011b). Efficiency and fidelity of human DNA polymerases lambda and beta during gap-filling DNA synthesis. DNA Repair (Amst.) 10, 24–33. doi: 10.1016/j.dnarep.2010.09.005
Burgers, P. M., Koonin, E. V., Bruford, E., Blanco, L., Burtis, K. C., Christman, M. F., et al. (2001). Eukaryotic DNA polymerases: proposal for a revised nomenclature. J. Biol. Chem. 276, 43487–43490. doi: 10.1074/jbc.R100056200
Chen, F., Dong, M., Ge, M., Zhu, L., Ren, L., Liu, G., et al. (2013a). The history and advances of reversible terminators used in new generations of sequencing technology. Genomics Proteomics Bioinformatics 11, 34–40. doi: 10.1016/j.gpb.2013.01.003
Chen, Y. S., Lee, C. H., Hung, M. Y., Pan, H. A., Chiou, J. C., and Huang, G. S. (2013b). DNA sequencing using electrical conductance measurements of a DNA polymerase. Nat. Nanotechnol. 8, 452–458. doi: 10.1038/nnano.2013.71
Chen, F., Gaucher, E. A., Leal, N. A., Hutter, D., Havemann, S. A., Govindarajan, S., et al. (2010). Reconstructed evolutionary adaptive paths give polymerases accepting reversible terminators for sequencing and SNP detection. Proc. Natl. Acad. Sci. U.S.A. 107, 1948–1953. doi: 10.1073/pnas.0908463107
Coleman, M. S., Hutton, J. J., and Bollum, F. J. (1974). Terminal deoxynucleotidyl transferase and DNA polymerase in classes of cells from rat thymus. Biochem. Biophys. Res. Commun. 58, 1104–1109. doi: 10.1016/S0006-291X(74)80257-3
Cubonova, L., Richardson, T., Burkhart, B. W., Kelman, Z., Connolly, B. A., Reeve, J. N., et al. (2013). Archaeal DNA polymerase D but not DNA polymerase B is required for genome replication in Thermococcus kodakarensis. J. Bacteriol. 195, 2322–2328. doi: 10.1128/JB.02037-12
Ding, F., Manosas, M., Spiering, M. M., Benkovic, S. J., Bensimon, D., Allemand, J. F., et al. (2012). Single-molecule mechanical identification and sequencing. Nat. Methods 9, 367–372. doi: 10.1038/nmeth.1925
Donlin, M. J., Patel, S. S., and Johnson, K. A. (1991). Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry 30, 538–546. doi: 10.1021/bi00216a031
Dovichi, N. J., and Zhang, J. (2000). How Capillary Electrophoresis Sequenced the Human Genome This Essay is based on a lecture given at the Analytica 2000 conference in Munich (Germany) on the occasion of the Heinrich-Emanuel-Merck Prize presentation. Angew. Chem. Int. Ed. Engl. 39, 4463–4468. doi: 10.1002/1521-3773(20001215)39:24<4463::AID-ANIE4463>3.0.CO;2-8
Drmanac, R., Sparks, A. B., Callow, M. J., Halpern, A. L., Burns, N. L., Kermani, B. G., et al. (2010). Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81. doi: 10.1126/science.1181498
Drossman, H., Luckey, J. A., Kostichka, A. J., D’Cunha, J., and Smith, L. M. (1990). High-speed separations of DNA sequencing reactions by capillary electrophoresis. Anal. Chem. 62, 900–903. doi: 10.1021/ac00208a003
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Gardner, A. F., and Jack, W. E. (1999). Determinants of nucleotide sugar recognition in an archaeon DNA polymerase. Nucleic Acids Res. 27, 2545–2553. doi: 10.1093/nar/27.12.2545
Gardner, A. F., and Jack, W. E. (2002). Acyclic and dideoxy terminator preferences denote divergent sugar recognition by archaeon and Taq DNA polymerases. Nucleic Acids Res. 30, 605–613. doi: 10.1093/nar/30.2.605
Gardner, A. F., Joyce, C. M., and Jack, W. E. (2004). Comparative kinetics of nucleotide analog incorporation by vent DNA polymerase. J. Biol. Chem. 279, 11834–11842. doi: 10.1074/jbc.M308286200
Gardner, A. F., Wang, J., Wu, W., Karouby, J., Li, H., Stupi, B. P., et al. (2012). Rapid incorporation kinetics and improved fidelity of a novel class of 3′-OH unblocked reversible terminators. Nucleic Acids Res. 40, 7404–7415. doi: 10.1093/nar/gks330
Gass, K. B., and Cozzarelli, N. R. (1973). Further genetic and enzymological characterization of the three Bacillus subtilis deoxyribonucleic acid polymerases. J. Biol. Chem. 248, 7688–7700.
Gass, K. B., and Cozzarelli, N. R. (1974). Bacillus subtilis DNA polymerases. Methods Enzymol. 29, 27–38. doi: 10.1016/0076-6879(74)29006-2
Gefter, M. L., Hirota, Y., Kornberg, T., Wechsler, J. A., and Barnoux, C. (1971). Analysis of DNA polymerases II and 3 in mutants of Escherichia coli thermosensitive for DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 68, 3150–3153. doi: 10.1073/pnas.68.12.3150
Greenough, L., Menin, J. F., Desai, N. S., Kelman, Z., and Gardner, A. F. (2014). Characterization of Family D DNA polymerase from Thermococcus sp. 9 degrees N. Extremophiles doi: 10.1007/s00792-014-0646-9 [Epub ahead of print].
Guo, J., Xu, N., Li, Z., Zhang, S., Wu, J., Kim, D. H., et al. (2008). Four-color DNA sequencing with 3′-O-modified nucleotide reversible terminators and chemically cleavable fluorescent dideoxynucleotides. Proc. Natl. Acad. Sci. U.S.A. 105, 9145–9150. doi: 10.1073/pnas.0804023105
Guo, J., Yu, L., Turro, N. J., and Ju, J. (2010). An integrated system for DNA sequencing by synthesis using novel nucleotide analogues. Acc. Chem. Res. 43, 551–563. doi: 10.1021/ar900255c
Henneke, G., Flament, D., Hubscher, U., Querellou, J., and Raffin, J. P. (2005). The hyperthermophilic euryarchaeota Pyrococcus abyssi likely requires the two DNA polymerases D and B for DNA replication. J. Mol. Biol. 350, 53–64. doi: 10.1016/j.jmb.2005.04.042
Hodges, R. A., Perler, F. B., Noren, C. J., and Jack, W. E. (1992). Protein splicing removes intervening sequences in an archaea DNA polymerase. Nucleic Acids Res. 20, 6153–6157. doi: 10.1093/nar/20.23.6153
Hübscher, U., Spadari, S., Villani, G., and Giovanni, M. (2010). DNA Polymerases: Discovery, Characterization and Functions in Cellular DNA Transactions. Singapore: World Scientific Publishing Co. Pte. Ltd.
Hutter, D., Kim, M. J., Karalkar, N., Leal, N. A., Chen, F., Guggenheim, E., et al. (2010). Labeled nucleoside triphosphates with reversibly terminating aminoalkoxyl groups. Nucleosides Nucleotides Nucleic Acids 29, 879–895. doi: 10.1080/15257770.2010.536191
Innis, M. A., Myambo, K. B., Gelfand, D. H., and Brow, M. A. (1988). DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA. Proc. Natl. Acad. Sci. U.S.A. 85, 9436–9440. doi: 10.1073/pnas.85.24.9436
Johnson, A., and O’Donnell, M. (2005). Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 74, 283–315. doi: 10.1146/annurev.biochem.73.011303.073859
Johnson, K. A. (1993). Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 62, 685–713. doi: 10.1146/annurev.bi.62.070193.003345
Joyce, C. M., and Steitz, T. A. (1994). Function and structure relationships in DNA polymerases. Annu. Rev. Biochem. 63, 777–822. doi: 10.1146/annurev.bi.63.070194.004021
Ju, J., Kim, D. H., Bi, L., Meng, Q., Bai, X., Li, Z., et al. (2006). Four-color DNA sequencing by synthesis using cleavable fluorescent nucleotide reversible terminators. Proc. Natl. Acad. Sci. U.S.A. 103, 19635–19640. doi: 10.1073/pnas.0609513103
Kato, K. I., Goncalves, J. M., Houts, G. E., and Bollum, F. J. (1967). Deoxynucleotide-polymerizing enzymes of calf thymus gland. II. Properties of the terminal deoxynucleotidyltransferase. J. Biol. Chem. 242, 2780–2789.
Konrad, E. B., and Lehman, I. R. (1974). A conditional lethal mutant of Escherichia coli K12 defective in the 5′ leads to 3′ exonuclease associated with DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 71, 2048–2051. doi: 10.1073/pnas.71.5.2048
Korlach, J., Bibillo, A., Wegener, J., Peluso, P., Pham, T. T., Park, I., et al. (2008). Long, processive enzymatic DNA synthesis using 100% dye-labeled terminal phosphate-linked nucleotides. Nucleosides Nucleotides Nucleic Acids 27, 1072–1083. doi: 10.1080/15257770802260741
Korlach, J., Bjornson, K. P., Chaudhuri, B. P., Cicero, R. L., Flusberg, B. A., Gray, J. J., et al. (2010). Real-time DNA sequencing from single polymerase molecules. Methods Enzymol. 472, 431–455. doi: 10.1016/S0076-6879(10)72001-2
Kumar, S., Sood, A., Wegener, J., Finn, P. J., Nampalli, S., Nelson, J. R., et al. (2005). Terminal phosphate labeled nucleotides: synthesis, applications, and linker effect on incorporation by DNA polymerases. Nucleosides Nucleotides Nucleic Acids 24, 401–408. doi: 10.1081/NCN-200059823
Kumar, S., Tao, C., Chien, M., Hellner, B., Balijepalli, A., Robertson, J. W., et al. (2012). PEG-labeled nucleotides and nanopore detection for single molecule DNA sequencing by synthesis. Sci. Rep. 2:684. doi: 10.1038/srep00684
Kunkel, T. A. (2004). DNA replication fidelity. J. Biol. Chem. 279, 16895–16898. doi: 10.1074/jbc.R400006200
Kunkel, T. A. (2009). Evolving views of DNA replication (in)fidelity. Cold Spring Harb. Symp. Quant. Biol. 74, 91–101. doi: 10.1101/sqb.2009.74.027
Kunkel, T. A., and Bebenek, K. (2000). DNA replication fidelity. Annu. Rev. Biochem. 69, 497–529. doi: 10.1146/annurev.biochem.69.1.497
Kunkel, T. A., and Burgers, P. M. (2008). Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 18, 521–527. doi: 10.1016/j.tcb.2008.08.005
Landegren, U., Kaiser, R., Sanders, J., and Hood, L. (1988). A ligase-mediated gene detection technique. Science 241, 1077–1080. doi: 10.1126/science.3413476
Langhorst, B. W., Jack, W. E., Reha-Krantz, L., and Nichols, N. M. (2012). Polbase: a repository of biochemical, genetic and structural information about DNA polymerases. Nucleic Acids Res. 40, D381–D387. doi: 10.1093/nar/gkr847
Lee, L. G., Spurgeon, S. L., Heiner, C. R., Benson, S. C., Rosenblum, B. B., Menchen, S. M., et al. (1997). New energy transfer dyes for DNA sequencing. Nucleic Acids Res. 25, 2816–2822. doi: 10.1093/nar/25.14.2816
Lehman, I. R., Bessman, M. J., Simms, E. S., and Kornberg, A. (1958a). Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli. J. Biol. Chem. 233, 163–170.
Lehman, I. R., Zimmerman, S. B., Adler, J., Bessman, M. J., Simms, E. S., and Kornberg, A. (1958b). Enzymatic Synthesis of deoxyribonucleic acid. V. Chemical composition of enzymatically synthesized deoxyribonucleic acid. Proc. Natl. Acad. Sci. U.S.A. 44, 1191–1196. doi: 10.1073/pnas.44.12.1191
Li, Y., Mitaxov, V., and Waksman, G. (1999). Structure-based design of Taq DNA polymerases with improved properties of dideoxynucleotide incorporation. Proc. Natl. Acad. Sci. U.S.A. 96, 9491–9496. doi: 10.1073/pnas.96.17.9491
Litosh, V. A., Wu, W., Stupi, B. P., Wang, J., Morris, S. E., Hersh, M. N., et al. (2011). Improved nucleotide selectivity and termination of 3′-OH unblocked reversible terminators by molecular tuning of 2-nitrobenzyl alkylated HOMedU triphosphates. Nucleic Acids Res. 39:e39. doi: 10.1093/nar/gkq1293
Luckey, J. A., Drossman, H., Kostichka, A. J., Mead, D. A., D’Cunha, J., Norris, T. B., et al. (1990). High speed DNA sequencing by capillary electrophoresis. Nucleic Acids Res. 18, 4417–4421. doi: 10.1093/nar/18.15.4417
Manrao, E. A., Derrington, I. M., Laszlo, A. H., Langford, K. W., Hopper, M. K., Gillgren, N., et al. (2012). Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotechnol. 30, 349–353. doi: 10.1038/nbt.2171
Martynov, B. I., Shirokova, E. A., Jasko, M. V., Victorova, L. S., and Krayevsky, A. A. (1997). Effect of triphosphate modifications in 2′-deoxynucleoside 5′-triphosphates on their specificity towards various DNA polymerases. FEBS Lett. 410, 423–427. doi: 10.1016/S0014-5793(97)00577-2
Metzker, M. L. (2010). Sequencing technologies – the next generation. Nat. Rev. Genet. 11, 31–46. doi: 10.1038/nrg2626
Metzker, M. L., Lu, J., and Gibbs, R. A. (1996). Electrophoretically uniform fluorescent dyes for automated DNA sequencing. Science 271, 1420–1422. doi: 10.1126/science.271.5254.1420
Miyabe, I., Kunkel, T. A., and Carr, A. M. (2011). The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS Genet. 7:e1002407. doi: 10.1371/journal.pgen.1002407
Morin, G. B. (1989). The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59, 521–529. doi: 10.1016/0092-8674(89)90035-4
Nick McElhinny, S. A., Gordenin, D. A., Stith, C. M., Burgers, P. M., and Kunkel, T. A. (2008). Division of labor at the eukaryotic replication fork. Mol. Cell 30, 137–144. doi: 10.1016/j.molcel.2008.02.022
Nusslein, V., Otto, B., Bonhoeffer, F., and Schaller, H. (1971). Function of DNA polymerase 3 in DNA replication. Nat. New Biol. 234, 285–286. doi: 10.1038/newbio234285a0
Okazaki, R., Arisawa, M., and Sugino, A. (1971). Slow joining of newly replicated DNA chains in DNA polymerase I-deficient Escherichia coli mutants. Proc. Natl. Acad. Sci. U.S.A. 68, 2954–2957. doi: 10.1073/pnas.68.12.2954
Olivera, R. M., and Bonhoeffer, E. (1974). Replication of Escherichia coli requires DNA polymerase I. Nature 250, 513–514. doi: 10.1038/250513a0
Parker, L. T., Deng, Q., Zakeri, H., Carlson, C., Nickerson, D. A., and Kwok, P. Y. (1995). Peak height variations in automated sequencing of PCR products using Taq dye-terminator chemistry. Biotechniques 19, 116–121.
Patel, P. H., and Loeb, L. A. (2001). Getting a grip on how DNA polymerases function. Nat. Struct. Biol. 8, 656–659. doi: 10.1038/90344
Patel, S. S., Wong, I., and Johnson, K. A. (1991). Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525. doi: 10.1021/bi00216a029
Perler, F. B., Comb, D. G., Jack, W. E., Moran, L. S., Qiang, B., Kucera, R. B., et al. (1992). Intervening sequences in an Archaea DNA polymerase gene. Proc. Natl. Acad. Sci. U.S.A. 89, 5577–5581. doi: 10.1073/pnas.89.12.5577
Prober, J. M., Trainor, G. L., Dam, R. J., Hobbs, F. W., Robertson, C. W., Zagursky, R. J., et al. (1987). A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science 238, 336–341. doi: 10.1126/science.2443975
Pursell, Z. F., Isoz, I., Lundstrom, E. B., Johansson, E., and Kunkel, T. A. (2007). Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 317, 127–130. doi: 10.1126/science.1144067
Pushkarev, D., Neff, N. F., and Quake, S. R. (2009). Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 27, 847–850. doi: 10.1038/nbt.1561
Ronaghi, M., Karamohamed, S., Pettersson, B., Uhlen, M., and Nyren, P. (1996). Real-time DNA sequencing using detection of pyrophosphate release. Anal. Biochem. 242, 84–89. doi: 10.1006/abio.1996.0432
Ronaghi, M., Uhlen, M., and Nyren, P. (1998). A sequencing method based on real-time pyrophosphate. Science 281, 363, 365. doi: 10.1126/science.281.5375.363
Rosenthal, A., and Charnock-Jones, D. S. (1992). New protocols for DNA sequencing with dye terminators. DNA Seq. 3, 61–64.
Rosenthal, A., and Charnock-Jones, D. S. (1993). Linear amplification sequencing with dye terminators. Methods Mol. Biol. 23, 281–296. doi: 10.1385/0-89603-248-5:281
Rothberg, J. M., Hinz, W., Rearick, T. M., Schultz, J., Mileski, W., Davey, M., et al. (2011). An integrated semiconductor device enabling non-optical genome sequencing. Nature 475, 348–352. doi: 10.1038/nature10242
Ruparel, H., Bi, L., Li, Z., Bai, X., Kim, D. H., Turro, N. J., et al. (2005). Design and synthesis of a 3′-O-allyl photocleavable fluorescent nucleotide as a reversible terminator for DNA sequencing by synthesis. Proc. Natl. Acad. Sci. U.S.A. 102, 5932–5937. doi: 10.1073/pnas.0501962102
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 74, 5463–5467. doi: 10.1073/pnas.74.12.5463
Smith, L. M., Sanders, J. Z., Kaiser, R. J., Hughes, P., Dodd, C., Connell, C. R., et al. (1986). Fluorescence detection in automated DNA sequence analysis. Nature 321, 674–679. doi: 10.1038/321674a0
Southworth, M. W., Kong, H., Kucera, R. B., Ware, J., Jannasch, H. W., and Perler, F. B. (1996). Cloning of thermostable DNA polymerases from hyperthermophilic marine Archaea with emphasis on Thermococcus sp. 9 degrees N-7 and mutations affecting 3′-5′ exonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 93, 5281–5285. doi: 10.1073/pnas.93.11.5281
Steitz, T. A. (1997). DNA and RNA polymerases: structural diversity and common mechanisms. Harvey Lect. 93, 75–93.
Steitz, T. A. (1999). DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 274, 17395–17398. doi: 10.1074/jbc.274.25.17395
Tabor, S., and Richardson, C. C. (1987). DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 84, 4767–4771. doi: 10.1073/pnas.84.14.4767
Tabor, S., and Richardson, C. C. (1989). Effect of manganese ions on the incorporation of dideoxynucleotides by bacteriophage T7 DNA polymerase and Escherichia coli DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 86, 4076–4080. doi: 10.1073/pnas.86.11.4076
Tabor, S., and Richardson, C. C. (1995). A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc. Natl. Acad. Sci. U.S.A. 92, 6339–6343. doi: 10.1073/pnas.92.14.6339
Victorova, L., Sosunov, V., Skoblov, A., Shipytsin, A., and Krayevsky, A. (1999). New substrates of DNA polymerases. FEBS Lett. 453, 6–10. doi: 10.1016/S0014-5793(99)00615-8
Wong, I., Patel, S. S., and Johnson, K. A. (1991). An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry 30, 526–537. doi: 10.1021/bi00216a030
Wu, J., Zhang, S., Meng, Q., Cao, H., Li, Z., Li, X., et al. (2007). 3′-O-modified nucleotides as reversible terminators for pyrosequencing. Proc. Natl. Acad. Sci. U.S.A. 104, 16462–16467. doi: 10.1073/pnas.0707495104
Xu, Y., Derbyshire, V., Ng, K., Sun, X. C., Grindley, N. D., and Joyce, C. M. (1997). Biochemical and mutational studies of the 5′-3′ exonuclease of DNA polymerase I of Escherichia coli. J. Mol. Biol. 268, 284–302. doi: 10.1006/jmbi.1997.0967
Zagursky, R. J., and McCormick, R. M. (1990). DNA sequencing separations in capillary gels on a modified commercial DNA sequencing instrument. Biotechniques 9, 74–79.
Keywords: Sanger sequencing, chain terminators, reversible terminators, sequencing-by-synthesis, DNA polymerase, next-generation sequencing, protein engineering
Citation: Chen C-Y (2014) DNA polymerases drive DNA sequencing-by-synthesis technologies: both past and present. Front. Microbiol. 5:305. doi: 10.3389/fmicb.2014.00305
Received: 07 April 2014; Paper pending published: 08 May 2014;
Accepted: 03 June 2014; Published online: 24 June 2014.
Edited by:
Andrew F. Gardner, New England Biolabs, USAReviewed by:
Suleyman Yildirim, Walter Reed Army Institute of Research, USACopyright © 2014 Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng-Yao Chen, Protein Engineering Group, Illumina, 5200 Illumina Way, San Diego, CA 92122, USA e-mail:Y2NoZW4yQGlsbHVtaW5hLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.