


- 1Department of Clinical Immunology and Internal medicine, Graduate School of Medicine, Yokohama City University, Yokohama, Japan
- 2Department of Pathology, Graduate School of Medicine, Yokohama City University, Yokohama, Japan
Asthma is a serious health and socioeconomic issue all over the world, affecting more than 300 million individuals. The disease is considered as an inflammatory disease in the airway, leading to airway hyperresponsiveness, obstruction, mucus hyper-production and airway wall remodeling. The presence of airway inflammation in asthmatic patients has been found in the nineteenth century. As the information in patients with asthma increase, paradigm change in immunology and molecular biology have resulted in an extensive evaluation of inflammatory cells and mediators involved in the pathophysiology of asthma. Moreover, it is recognized that airway remodeling into detail, characterized by thickening of the airway wall, can be profound consequences on the mechanics of airway narrowing and contribute to the chronic progression of the disease. Epithelial to mesenchymal transition plays an important role in airway remodeling. These epithelial and mesenchymal cells cause persistence of the inflammatory infiltration and induce histological changes in the airway wall, increasing thickness of the basement membrane, collagen deposition and smooth muscle hypertrophy and hyperplasia. Resulting of airway inflammation, airway remodeling leads to the airway wall thickening and induces increased airway smooth muscle mass, which generate asthmatic symptoms. Asthma is classically recognized as the typical Th2 disease, with increased IgE levels and eosinophilic inflammation in the airway. Emerging Th2 cytokines modulates the airway inflammation, which induces airway remodeling. Biological agents, which have specific molecular targets for these Th2 cytokines, are available and clinical trials for asthma are ongoing. However, the relatively simple paradigm has been doubted because of the realization that strategies designed to suppress Th2 function are not effective enough for all patients in the clinical trials. In the future, it is required to understand more details for phenotypes of asthma.
Introduction
Asthma is characterized by the action of airway leading to reversible airflow obstruction in association with airway hyperresponsiveness (AHR) and airway inflammation (Holgate, 2012). The disease is affecting more than 300 million persons all over the world, with approximately 250,000 annual deaths (Bousquet et al., 2007). In the last couple of decades, as the inhaled corticosteroid has become the major treatment agent for asthma, the mortality of asthma has decreased (Wijesinghe et al., 2009). Meanwhile, allergic diseases, such as asthma, have markedly increased in the past half centuries associated with urbanization (Alfvén et al., 2006). Children have the greatest percentage of asthma compared with other generation groups (Centers for Disease Control and Prevention, 2011). Then, it is expected that the number of the patients will increase by more than 100 million by 2025 (Masoli et al., 2004).
Generally, most asthma starts from childhood in relation to sensitization to common inhaled allergens, such as house dust mites, cockroaches, animal dander, fungi, and pollens. These inhaled allergens stimulate T helper type 2 (Th2) cell proliferation, subsequently Th2 cytokines, interleukin (IL)-4, IL-5 and IL-13 production and release. Many basic and clinical studies suggested that airway inflammation was a central key to the disease pathophysiology. The existence of chronic airway inflammation in asthma has been recognized for over a century. The inflammation is induced by the release of potent chemical mediators from inflammatory cells. Resulted of chronic airway inflammation, airway remodeling, characterized by thickening of all compartments of the airway wall, is occurred and may have profound consequences on the mechanics of airway narrowing in asthma and contribute to the chronicity and progression of the disease.
As allergic sensitization, allergen can be taken up by dendritic cells (DCs), which process antigenic molecules and present them to naїve T helper cells. Consequently the activation of allergen-specific Th2 cells is occurred, the cells play an important role in developing the asthma. Nowadays, it is known that Th17 cells and Th9 cells also modulate the disease. Th17 cells produce IL-17A, IL-17F, and IL-22. These cytokines induce airway inflammation and IL-17A enhance smooth muscle contractility.
Allergic diseases are caused by inappropriate immunological responses to allergens without pathogenesis driven by a Th2-mediated immune response. The hygiene hypothesis has been used to explain the increase in allergic diseases since industrialization and urbanization, and the higher incidence of allergic diseases in more developed countries. The hypothesis has now expanded to include exposure to symbiotic bacteria and parasites as important modulators of immune system development, along with infectious agents (Grammatikos, 2008). Recently, asthma has not been recognized as a simple Th2 disease, which is characterized by IgE elevation and relatively eosinophilia. Th17 and Th9 cell subtype are known to contribute the inflammation or enhancing smooth muscle contraction or stimulating mast cells.
Histopathology of Asthmatic Airway
Asthma is considered in terms of its hallmarks of reversible airflow obstruction, non-specific bronchial hyperreactivity and chronic airway inflammation (American Thoracic Society, 1987). Osler (1892) mentioned in the classic textbook, the inflammatory process, affecting the conducting airways with relative sparing of the lung parenchyma. Huber and Koesser (1922) provided a comprehensive perspective of the histopathological features of asthma. That is, the lungs are usually hyperinflated as a consequence of extensive mucous plugging in segmental, subsegmental bronchus and peripheral airways, but the lung parenchyma in general, remains relatively intact in subjects who die in exacerbation, so-called status asthmatics. The composition of mucous includes cellular debris from necrotic airway epithelial cells, an inflammatory cells including lymphocytes, eosinophils, and neutrophils, plasma protein exudate, and mucin that is produced by goblet cells (Unger, 1945; Bullen, 1952; Dunnill, 1960; Messer et al., 1960). The airway epithelium typically shows sloughing of ciliated columnar cells, with goblet cell and squamous cell metaplasia as a sign of airway epithelial repair. There is increased thickness of the subepithelial basement membrane, however, some studies have established that the true basal lamina is of normal thickness, and the apparent increase in thickness is related to accumulation of other extracellular matrix components beneath the basal lamina (Roche et al., 1989). The asthmatic airway showed a thickness with inflammatory cell infiltration consisting of an admixture of T lymphocytes and eosinophils, mast cells (Carroll et al., 1997; Hamid et al., 1997). Interestingly, prominent neutrophil infiltrates have been reported to be a specific feature of the clinical entity of sudden onset fatal asthma (Sur et al., 1993).
Nowadays investigators can easily obtain lung tissue and bronchoalveolar lavage (BAL) specimens from the patients with asthma (Salvato, 1968; Djukanovic et al., 1991). Results of studies of BAL (Robinson et al., 1992) and lung tissue specimens (Minshall et al., 1998) have strongly implicated a role for cytokines produced by the Th2 subset of CD4+ T cells in the pathogenesis of asthma. For example, IL-13 plays an important role in regulating the airway inflammation in asthma (Wills-Karp et al., 1998; Zhu et al., 1999).
In recent years, there has been increasing interest in the mechanism of airway wall remodeling in asthma, owing to the increasing realization that airway inflammation alone is not enough to explain the chronicity or progression of asthma (Holgate et al., 1999). The nature of airway remodeling may be considered in terms of extracellular matrix deposition. It is postulated that the injured airway epithelium acts as a continuous stimulus for airway remodeling (Holgate et al., 1999), and this is supported by results of recent cell culture experiments examining interactions of bronchial epithelial cells with myofibroblasts in response to injurious stimuli (Zhang et al., 1999). The remodeling is predicted to have little effect on baseline respiratory mechanics, the physiological effects of extracellular matrix accumulation are predicted to result in an exaggerated degree of narrowing for a given amount of airway smooth muscle (ASM) contraction.
Airway wall thickening is greater in the asthmatic patients than normal subjects, and severe patients have greater (Awadh et al., 1998). This thickness is due to an increase in ASM mass and mucous glands (Johns et al., 2000). The airflow limitation is also compounded by the presence of increased mucous secretion and inflammatory exudate (Chiappara et al., 2001). Thus, the results from many studies have supported that airway remodeling related to airway inflammation. Surprisingly, physical force generated by ASM in bronchoconstriction without additional inflammation induces airway remodeling in patients with asthma (Grainge et al., 2011). Despite these recent advances, further work is necessary to establish a causal relationship between airway remodeling and the severity of asthma (Bento and Hershenson, 1998).
Airway Epithelium
The structural changes in the asthmatic airway result from interdependent inflammatory and remodeling processes (Chiappara et al., 2001). In the processes, inflammation occurs common features, vascular congestion, exudaution, and inflammatory cell recruitment to the interstitial tissue. Furthermore mucus secretion and desquamation of epithelial cells are increased. The chronic inflammatory changes develop epithelium-mesenchymal interactions (Holgate et al., 2000). The number of myofibroblasts, which deposit collagens, increases in the understructure of epithelium, the proximity of the smooth muscle layer and the lamina reticularis in the patients. Subepithelial collagens cause thickening and increasing density of the basement membrane.
The airway inflammation gives damage to the epithelium and damaged epithelial cells will be repaired in the injury-repair cycle. Some studies showed that epithelial cells of untreated asthmatic patients had low level expression of proliferating markers, despite extensive damage, revealing a potential failure in the epithelial injury-repair cycle in response to local inflammation and inhaled agents (Bousquet et al., 2000). Injury to the epithelium results in a localized and persistent increase in epidermal growth factor (EGF) receptor, a mechanism that may cause the epithelium to be locked in a repair phenotype (Puddicombe et al., 2000). Epithelial cells which are in repair phase produced some profibrotic mediators, including transforming growth factor-β (TGF-β), fibroblast growth factor and endothelin, which regulate fibroblast and myofibroblast to release collagen, elastic fiber, proteoglycan, and glycoprotein and these substances induce airway wall thickening (Holgate et al., 2000). Myofibroblast is a rich source of collagen types I, II, and V, fibronectin and tenascin that also accumulate in the airway wall and induce thickening lamina reticularis (Roche et al., 1989; Brewster et al., 1990). This process may contribute phenomena by augmentation of airway narrowing because the inner airway wall volume increases.
Eosinophils seem to contribute to airway remodeling in several ways, including through release of eosinophil-derived TGF-β, cationic proteins, and cytokines, as well as through interactions with mast cell and epithelial cells. Many of these factors can directly activate epithelium and mesenchymal cells, deeply related to the development of airway remodeling (Kariyawasam and Robinson, 2007; Aceves and Broide, 2008; Venge, 2010). Eosinophil-derived cytokines are in the modulation of Th2 responses that trigger macrophage production of TGF-β1, which serves as a stimulus for extracellular matrix production (Fanta et al., 1999; Holgate, 2001). TGF-β1 induced epithelial to mesenchymal transition (EMT) in alveolar epithelial cells and could contribute to enhance fibrosis in idiopathic lung fibrosis (Wilson and Wynn, 2009). TGF-β1 might also contribute to enhance airway remodeling through EMT. Indeed, anti-TGF-β1 treatment inhibits EMT in airway epithelial cells (Yasukawa et al., 2013).
Airway epithelium is a barrier in the frontline against stimuli from the environment, but in asthmatic epithelium is defective in barrier function with incomplete formation of tight junctions, that prevent allergen from penetrating into the airway tissue (Xiao et al., 2011). The defect would induce that a proportion of the asthma-related had biological properties to infiltrate the epithelial barrier and trigger a danger signal to DCs. Components of house dust mite, cockroach, animal, and fungal can disrupt epithelial tight junctions and activate protease-activated receptors (Jacquet, 2011). The defective epithelial barrier function has also been described in the pathophysiology of other allergic disease. Therefore, healthy barrier function is important to avoid sensitization and development in allergic disease.
Airway Smooth Muscle
Abnormalities of asthmatic ASM structure and morphology have been described by Huber and Koesser (1922) in the first quarter of twentieth century when they reported that smooth muscle from the patients who died by acute exacerbation was increase much greater than in those who died from another disease. Airflow limitation mainly due to reversible smooth muscle contraction is a most important symptom of the disease. Therefore, ASM plays a material role in asthma. Abnormal accumulation of smooth muscle cells is another mechanism of airway remodeling. Some in vivo animal studies confirmed that prolonged allergen exposure increase smooth muscle thickness in the airway (Salmon et al., 1999). It is still unknown whether the phenomenon is occurred by fundamental changes in the phenotype of the smooth muscle cells, is caused by structural or mechanical changes in the non-contractile elements of the airway wall. There are two different ways by which cyclic generation of length and force could influence ASM contracting and airway narrowing. The processes, which are myosin binding and plasticity, have different biochemical and physical mechanisms and consequences. They have the potential to interact and to have a fundamental effect on the contractual capacity of smooth muscle and its potential to cause excessive airway narrowing (King et al., 1999).
Like other muscles, ASM is also provoked to contract with intracellular calcium ions (Ca2+), which comes from the extracellular environment through voltage-dependent calcium channel or from the sarcoplasmic reticulum stores (Figure 1). The source of Ca2+ surge in ASM is mainly from intracellular sarcoplasmic reticulum stores rather than from the extracellular Ca2+ seen in cardiac, skeletal, and vascular muscle cells. Ligands to G-ptotein coupled receptor (GPCR), such as acetylcholine and methacholine, induce the activation of phospholipase C (PLC), which in turn leads to the formation of the inositol triphosphate (IP3; Chen et al., 2012). Then, IP3 occurs to release Ca2+ from sarcoplasmic reticulum (SR) stores, then Ca2+ forms a calcium-calmodulin comlex, activates MLC kinase (MLCK) which phosphorylates regulatory MLCs (rMLCs) forming phosphorylated-MLC (p-MLC; Berridge, 2009). Finally, this mechanism occurs to the activation of actin and myosin crossbridges resulting in shortening and contraction (Gunst and Tang, 2000).
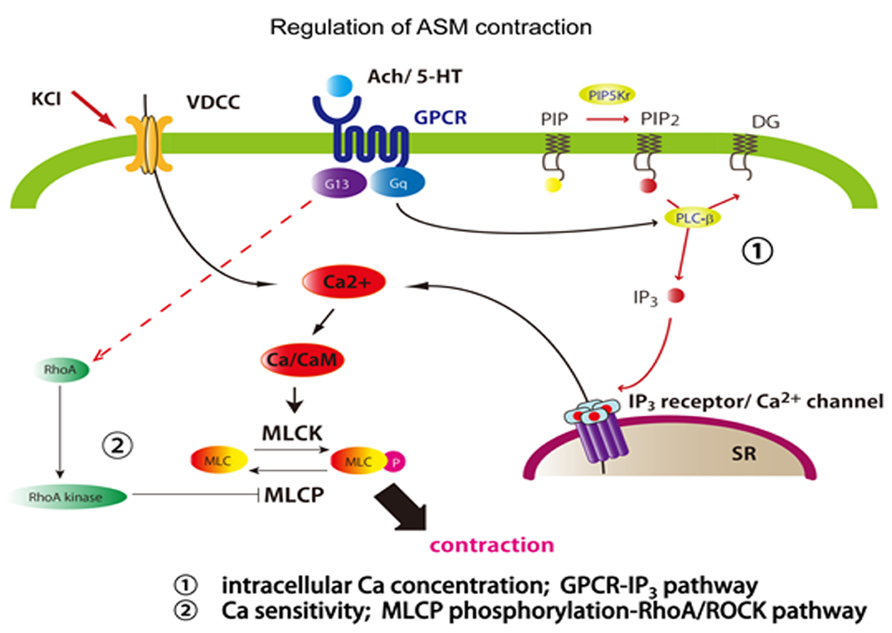
FIGURE 1. Regulation of ASM contractility. ASM contraction is induced by calcium, regulated two different pathways. First, ASM is evoked by intracellular calcium influx from SR depending on GPCR stimulation or from the extracellular environment through voltage-dependent calcium channel. Second, smooth muscle can be induced calcium sensitivity by RhoA/Rho kinase pathway. RhoA activates Rho-kinase which phosphorylates MLCP. pMLC phosphatase fails to dephosphorylate MLC. KCl, potassium chloride; Ach, acetylcholine; 5-HT, 5-hydroxytryptamine (serotonin); PIP, phosphatidylinositol 4-phosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP5K, 1-phosphatidylinositol-4-phosphate 5-kinase; DG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate.
And the contraction is also regulated by calcium sensitivity of myosin light chain (MLC; Kudo et al., 2012). The p-MLC is regulated by MLC phosphatase (MLCP) which converts p-MLC back to inactive MLC. MLCP is negatively controlled by Ras homolog gene family, member A (RhoA) and its target Rho Kinase such as Rho-associated, coiled-coil containing protein kinase (ROCK) which phosphorylates myosin phosphatase target subunit 1 (MYPT-1). Upregulation of the RhoA/Rho kinase signaling pathway inducing to inhibition of MLCP would result in increased levels of p-MLC and subsequently increased ASM contraction force. Increased levels of RhoA protein and mRNA were found in airway hyperresponsive animal models and this is probably medicated through inflammatory cytokines, such as IL-13 and IL-17A that themselves directly enhance the contractility of ASM (Chiba et al., 2009; Kudo et al., 2012). For IL-17A, sensitized mouse conditional lacking integrin αvβ8 on DCs shows attenuated reactivity against IL-17A-induce antigen challenge. This is induced by that IL-17A itself enhances the contractile force of ASM, through RhoA/Rho kinase signaling change.
Airway smooth muscle cells also contribute to the inflammatory mechanisms and airway remodeling of asthma. The proactivating signals, including viruses and immunoglobulin E could convert ASM cells into a proliferative and secretory cell in asthma. Naureckas et al. (1999) demonstrated the presence of smooth muscle mitogens in the BAL fluids from asthmatic individuals who underwent allergen challenge. Smooth muscle proliferation is also caused by the production of matrix metalloproteinase (MMP)-2, which has been demonstrated to be an important autocrine factor that is required for proliferation (Johnson and Knox, 1999). Production of MMP-2 from smooth muscle cells suggests that ASM contributes to the extracellular matrix turnover and airway remodeling. These cells may also participate in chronic airway inflammation by interacting with both Th1- and Th2-derived cytokines to modulate chemoattractant activity for eosinophils, activated T lymphocytes, and monocytes/macrophages (Teran et al., 1999).
In addition, recent studies demonstrated that eosinophils can also contribute to airway remodeling during an asthma by enhancing ASM cell proliferation. Halwani et al. (2013) verified that preventing eosinophil contact with ASM cells using specific antibodies or blocking cysteinyl leukotrienes derived from eosinophils was associated with inhibition of ASM proliferation. Moreover, ASM-synthesized cytokines seem to direct the eosinophil differentiation and maturation from progenitor cells, which can promote perpetuation of eosinophilic inflammation and consequently the tissue remodeling in asthma (Fanat et al., 2009). It was also reported that TGF-β alone induces only weak mitogenic effect on ASM cells, however, it synergistically stimulates ASM proliferation with methacholine which is agonist for the muscarinic receptor (Oenema et al., 2013). These smooth muscle cell proliferations related to airway remodeling can be the target to treat asthma.
Epithelial to Mesenchymal Transition on Asthma
As airway remodeling on asthma attracts investigators interested in airway remodeling on asthma, EMTs are recognized to be more important in asthma than before. EMTs are biological processes that epithelial cells lose their polarity and cell adhesion resulted in fragility of tight junction and gain migratory and invasive properties to change their cell formation to mesenchymal cells (Kalluri and Neilson, 2003). It is essential for processes including mesoderm formation and neural tube formation in the development and recently has also been reported to involve in wound healing, in organ fibrosis and in cancer metastasis. First, EMTs were found in the embryogenesis. Epithelial cells are different from mesenchymal cells in their phenotype. Epithelial cells connect each other, forming tight junction. These cells have polarity in cytoskeleton and bound to basal lamina. For mesenchymal cells, the polarity is lost and shaped in spindle. Lately, EMTs are divided into three subtypes, developmental (Type I), fibrosis, tissue regeneration and wound healing (Type II), and cancer progression and metastasis (Type III; Kalluri and Weinberg, 2009).
Type II EMT involves in wound healing, resulted that it contributes airway remodeling in asthma after airway epithelial injury induced by inflammation. Type II EMT indicates that epithelial tissue can be expressed plasticity (Thiery and Sleeman, 2006). It is initiated by extracellular signals, such as connection with extracellular matrix; collagen or hyaluronic acids and by growth factors; TGF-β and EGF. Among those signals, TGF-β is established how it plays important role in airway remodeling and EMT (Phipps et al., 2004; Boxall et al., 2006; Hackett et al., 2009). TGF-β induces the expression of α-smooth muscle actin and vimentin and the downregulation of E-cadherin expression, inducing the dissolution of polarity of the epithelial cell and intercellular adhesion. The such physiological effects of TGF-β signaling in the system have been shown to depend on microenvironment. Bone morphogenesis protein (BMP)-7 fails to attenuate TGF-β-induced EMT, however, one of the family member BMP-4 plays the role of EMT in the airway (Molloy et al., 2008; Hackett et al., 2009). This TGF-β-induced attenuation of intercellular adhesion and wound repair in EMT can be enhanced by the proinflammatory cytokines tumor necrosis factor (TNF)-α (Camara and Jarai, 2010). Furthermore, it was showed that house dust mite, through EGF receptor enhanced TGF-β-induce downregulation of E-cadherin in the bronchial epithelial cells (Heijink et al., 2010). And house dust mite and TGF-β synergistically induced expression of mesenchymal markers vimentin and fibronectin. In chronic house dust mite-exposure model, the airway epithelial cells were shown to elevate TGF-β expression and nuclear phosphorylated Smad3. And in these cells, the tight-junction protein was dissolved, occluding and expressed α-smooth muscle actin and collagen (Johnson et al., 2011). Inhaled allergens might modify EMT, cooperating with cytokines which also promote asthma.
Mast Cells and Eosinophils
Mact cells can induce the activation of mesenchymal cells (Holgate, 2000). The serine protease, tryptase which is released from degranulating mast cells is a potent stimulant of fibroblast and smooth muscle cell proliferation, and is capable of stimulating synthesis of type I collagen by human fibroblasts. A major mechanism involved in the regulation of fibroblast proliferation appears to be cleavage and activation of protease activated receptor-2 on fibroblasts (Akers et al., 2000). Mast cells may also influence the development of airway remodeling in asthma by releasing large amounts of plasminogen activator inhibitor type1. Moreover, Sugimoto et al. (2012) have shown that other mast cell proteases regulate airway hyperreactivity. Mice lacking αvβ6 integrin are protected from exaggerated airway narrowing. Mast cell proteases are differentially expressed, in mouse mast cell protease 1 (mMCP-1) induced by allergen challenge in wild-type (WT) mice and mMCP-4 increased at baseline in β6-deficient mice. MCPs from intraepithelial mast cell and their proteolytic substrates could be regulate airway hyperreactivity.
Eosinophils are circulating granulocytes and at relatively low levels in the bloodstream, upto 3% of white blood cells. These are the major cell types that can be recruited to sites of inflammatory responses (Huang et al., 2009; Isobe et al., 2012; Uhm et al., 2012). The function of eosinophils in asthma is related to their release of toxic granule proteins, reactive oxygen species (ROS), cytokines, and lipid mediators (Liu et al., 2006). The recruit of eosinophils into the epithelium and eosinophilic inflammation is involved in the pathogenesis of asthma. The proinflammatory mediators derived by eosinophil are major contributors to inflammation in asthma, including airway epithelial cell damage and desquamation, airway dysfunction of cholinergic nerve receptors, AHR, mucus hypersecretion, and airway remodeling, characterized by fibrosis and collagen deposition (Kay, 2005; Watt et al., 2005; Kanda et al., 2009; Walsh, 2010). Eosinophils are likely to contribute to airway remodeling with release of eosinophil-derived mediators such as TGF-β, secretion of cationic proteins, and cytokines, as well as having interactions with mast cell and epithelial cells. Those factors can directly activate epithelium and mesenchymal cells (Venge, 2010). Moreover, recent data demonstrated that eosinophils can also contribute to airway remodeling with ASM cell proliferation.
Extracellular Matrix
The airways of asthmatic patients showed excess accumulation of extracellular matrix components, particularly collagen, in the subepithelial connective tissue and adventitia of the airway wall (Kuwano et al., 1993; Gillis and Lutchen, 1999). The cellular interactions in mast cells and fibroblasts through protease activated receptor-2 may contribute an abnormal mesenchymal cell proliferation, and may account for the increased number of fibroblasts and myofibroblasts that are found in the airways of asthmatic subjects. Fibroblasts retain the capacity for growth and regeneration, and may evolve into various cell types, including smooth muscle cells that subsequently become myofibroblasts. Myofibroblasts can contribute to tissue remodeling by releasing extracellular matrix components such as elastin, fibronectin and laminin (Vignola et al., 2000). It was seen that the numbers of myofibroblasts in the airway of asthmatic subjects increased and their number appeared to correlate with the size of the basement reticular membrane (Holgate et al., 2000). Smooth muscle cells also have the potential to alter the composition of the extracellular matrix environment. The reticular basement membrane thickening is a characteristic typical feature of the asthmatic airways. It appears to consist of a plexiform deposition of immunoglobulins, collagen types I and III, tenascin and fibronectin (Jeffery et al., 2000), but not of laminin.
Remodeling processes of the extracellular matrix are less known than the thickening of the lamina reticularis. Most asthmatic subjects present with an abnormal superficial elastic fiber network, with fragmented fibers (Bousquet et al., 2000). In the deeper layer of elastic fibers is also abnormal, the fibers often being often patchy, tangled, and thickened. Some studies using transmission electron microscopy have shown that an elastolytic process occurs in asthmatic patients, and in some patients disruption of fibers has been observed. In the case of fatal asthma, fragmentation of elastic fiber has also been found in central airways, and was associated with marked elastolysis (Mauad et al., 1999). These bundles are seen to be hypertrophied as a result of an increased amount of collagen and myofibroblast matrix deposition occurring during exaggerated elastic fiber deposition (Carroll et al., 1997). Loss of lung elastic recoil force has been shown in adults with persistent asthma and irreversible expiratory airflow obstruction. Persistent asthmatic patients have severe abnormal flow-volume curves in expiration at both high and low lung volumes, and hyperinflation can be seen by residual volume, at forced residual capacity and total lung capacity (Gelb and Zamel, 2000). The increased elastolysis is part of a more complex process that regulates the size of a submucosal network formed by elastic fibers dispersed in a collagen and myofibroblast matrix (Chiappara et al., 2001). These features induce changes in airway, as demonstrated by airway compliance, particularly in those patients who are suffering from asthma for long period, supporting the concept that chronic inflammation and remodeling of the airway wall may result in stiffer dynamic elastic properties of the asthmatic airway (Brackel et al., 2000). Furthermore, disruption of elastic fibers may contribute to a reduction in the preload and afterload for smooth muscle contraction. Though it is difficult to associate aspects of remodeling with disease severity or degree of airways obstruction and hyperresponsiveness (Mauad et al., 2007), some investigators indicated that smooth muscle remodeling is related to the severity of asthma (James et al., 2009). It has shown that the clinical expression of asthma (Brightling et al., 2002), AHR (Siddiqui et al., 2008) and impaired airway relaxation (Slats et al., 2007) are associated with mast cell counts in the ASM layer in asthma. The deposition of extracellular matrix inside and outside the smooth muscle layer in asthma also seems to be related to its clinical severity and is altered as compared to healthy controls (Araujo et al., 2008; Klagas et al., 2009). Yick et al. (2012) have shown that extracellular matrix in ASM was related to the dynamics of airway function in asthma.
Immune Response
Allergic Sensitization
Regarding to the immune system against allergy, it seems that hygiene hypothesis would provide the reason why the number of the patients with asthma is increasing, in relation with urbanization. The hypothesis is that the Th1 cells polarized response is not induced early in life leaving the body more susceptible to developing Th2 induced disease (Strachan, 2000). First, Strachan (1989) mentioned that the hypothesis was proposed to explain the observation that hay fever and eczema were less common in children from larger families, which were presumably exposed to more infectious agents through their siblings, than in children from small families, especially without siblings. Many bacteria and viruses derive a Th1-mediated immune response, which down-regulates Th2 responses. The urban-rural gradient in prevalence has been demonstrated most strongly in children who grew up in environments with a wide range of microbial exposures, who are protected from childhood asthma and atopy (the predisposition to develop IgE against common environmental allergens) in proportion to their level of exposure to bacterial and fungal microbes (Ege et al., 2011).
In association with the airway epithelium and underling mucosa is a specialized population of antigen-presenting cells (APCs) called DCs (Holgate, 2012). As allergen sensitization, DCs take up the allergens and present small peptide from them. DCs express receptors of the innate immune system and process allergens into small peptides and then present them through the major histocompatibility complexes, MHC class I and MHC class II for recognition by T cell receptors. In allergic individuals, it is promoted by interaction of the allergen with IgE attached to FcεRI, the high-affinity receptor for IgE (Sallmann et al., 2011). When individual is born, there is no DCs in the airway. Damage to and activation of the respiratory epithelium are the major stimuli that initiate the ingression of immature DCs from the bone marrow (McWilliam et al., 1994) and cause the release of C–C chemokines which direct DCs migration toward the epithelium and underlying mucosa (Hammad et al., 2010). GM-CSF, which is released from epithelial cells and immune cells in the presence of IL-4 and TNF-α, leads to DCs maturation to a fully competent as APCs. During initial allergen entering to airways to sensitize, Th2 lymphocyte differentiation from naїve T cells requires IL-4 release. The cellular source of the IL-4 is still unclear. There are some hypotheses to explain that (Holgate, 2012). Polarization to Th2 cells subtype is also under epigenetic regulation. From the study with mouse, microRNA-21 has been shown to exert a pivotal role in setting a balance between Th1 and Th2 responses. It works through binding the promoter of the gene encoding IL-12 p35 and inhibiting its activation in favor of a Th2 profile. Conversely, reduced microRNA levels lead DCs to produce more IL-12, and allergen-stimulated T cells to produce more interferon-γ (IFN-γ ) and less IL-4, enhancing Th1 delayed-type hypersensitivity (Lu et al., 2011).
Dendritic Cell Activation
As described above, DCs present small peptide from antigens through MHC class I and II/ T cell receptors. Once sensitized, T cells drive the allergic response in progress through interactions with DCs (Veres et al., 2011). DCs spread their processes into the lumen between airway epithelial cells and can detect allergen by forming tight junctions, keeping the epithelial barrier (Blank et al., 2011). In mouse, two distinct DC subsets have been described in accordance with their expression of the CD11c as myeloid [conventional DCs (cDCs), CD11c+] or plasmacytoid DCs (pDCs, CD11c-; Lambrecht and Hammad, 2009). Similarly, human DCs are subdivided into CD11c- pDCs and CD11c+ myeloid DCs. Induced sputum from asthmatic airways and peripheral blood contain increased numbers of both pDCs and cDCs, which further increase in number upon allergen challenge (Dua et al., 2010). Proteolytic activities of allergens initiate to mature DCs. In a few hours after contact with allergen, pattern-recognition receptors activation, such as Toll-like receptors (TLRs) on DCs augments their homing capacity by upregulating chemokine receptors. It is cDC subtypes that are predominantly responsible for antigen presentation. Mature DCs shape an immunological synapse with the allergen-specific T lymphocytes to initiate a Th response (Holgate, 2012). Whereas some of the Th cells make their way to the B-cell follicle to facilitate immunoglobulin class switching from IgM to IgE, others move back to the airway mucosa to elicit the classical Th2 response through the secretion of the proallergic cytokines. Pattern-recognition receptors have a crucial adjuvant role in directing allergen sensitization. TLRs are key components of the innate immune system that mediate recognition and response to pathogen-associated molecular patterns (PAMPs) in the form of microbial, fungal and viral products and their ligands, including endotoxin which is recognized by TLR4, lipoproteins (TLR2 and TLR6), viral double- and single-stranded RNA (TLR3 and TLR7/8) and bacterial CpG-containing DNA (TLR9) (Akira et al., 2006). Other pattern recognition receptors respond to endogenously generated damage-associated molecular pattern molecules (DAMPs) produced during tissue damage. Inflammatory DCs have been suggested to be necessary and sufficient for the development of Th2 immunity to house dust mite allergen when the first exposure occurs by inhalation. For inhaled allergens, it is proposed that DCs amplify the Th2 immunity through basophiles and, in part, influenced by innate signaling through TLR4 and C-type lectin signaling on epithelial cells and DCs (Trompette et al., 2009). A cooperation of airway epithelium and DCs controls asthma development Th2 activation requires DCs-mediated antigen-presentation. Then, allergic sensitization fails to develop in the absence of DCs (Hammad et al., 2010), while DCs remain inactive in the absence of TLR ligation (Perros et al., 2009). That is, TLRs activation on epithelial cells enhances DCs motility and antigen sampling through the production of Th2-promoting chemokines and cytokines (IL-25, IL-33, GM-CSF).
Viral Infection to Predisposition
The fact that early-in-life sensitization to multiple allergens carries the greatest risk for developing asthma (Simpson et al., 2010) brings the question of what factors result in a predisposition to this phenotype. Although infection with rhinovirus is the major cause of acute exacerbation, in those genetically at risk of asthma, rhinovirus-induced wheezing in the first three years in the life is also the greatest risk factor for developing asthma at 6 years of age (Jackson et al., 2008). Impaired TLR3-mediated IFN-β and -λ production by asthmatic epithelial cells would make susceptible to both viral infection and allergic sensitization (Wark et al., 2005; Contoli et al., 2006; Bosco et al., 2010; Jartti and Korppi, 2011). Reduced primary IFN production by lower-airway epithelial cells enables some viruses to replicate, leading to cytotoxic cell, release of inflammatory products and enhanced viral shedding. Such events provide a strong stimulus for recruitment of immature DCs and their priming for allergen sensitization (McWilliam et al., 1994, 1996). When asthmatic epithelial cells are received to damage by rhinovirus infection, the cells generate increased amounts of the pro-Th2 cytokine thymic stromal lympoietin (Uller et al., 2010), which stimulates DCs and increases allergic inflammation, whereas exogenous IFN-b applied to asthmatic epithelium exerts anti-Th2 as well as antiviral properties (Cakebread et al., 2011).
Cellular Immunity
Asthma is classically considered Th2 disease, with increased IgE and eosinophilic inflammation caused by increased levels of Th2-type cytokines. However, this paradigm has been challenged because of the realization that strategies designed to suppress Th2 function are not effective for all patients. The clinical phenotype of asthma is notoriously heterogeneous. It is shown that cellular immune process in the asthmatic airways in Figure 2. Th2 cells activation requires antigen-presentation by DCs. DCs play a role both in the initiation and maintenance of allergic airway inflammation and asthma, and control many aspects of the disease, including bronchial hyperresponsiveness and goblet cell metaplasia, by controlling the recruitment and activation of Th2 cells (Lambrecht and Hammad, 2009; Schuijs et al., 2013). Researches in both mouse and human, mentioned the expression of Th2-type cytokines, such as IL-4, IL-5, and IL-13, in the allergic lung. Experimental asthma models indicate that these cytokines, IL-13 in particular, are critical in driving key pathologic features of the allergic response. Moreover, Th2 blockade is very effective in suppressing these features of allergic disease in mice (Finkelman et al., 2010). The classical asthmatic phenotype is one of eosinophilia concomitant with high IgE levels. However, a proportion of patients are not atopic and do not have eosinophilic inflammation. In fact, it is estimated that as many as 50% of adult patients are encompassed by this non-atopic, non-eosinophilic, non-IgE-dependent subgroup (Lloyd and Saglani, 2013). Molecular therapy data support an overall Th2 association with phenotypes, such that they might satisfy a definition of Th2-associated asthma. However, even these distinctions are too simple, especially when disease severity is considered. Although children with severe asthma have eosinophilic inflammation, high-dose steroids effectively suppress Th2-type cytokines, such as IL-13 and IL-5, but symptoms remain with persistent eosinophilia (Bossley et al., 2012), thus raising the importance of identifying other less steroidsensitive, non-Th2 mediators driving disease. Then, it is apparent that asthma can no longer be considered simply a Th2-mediated disease.
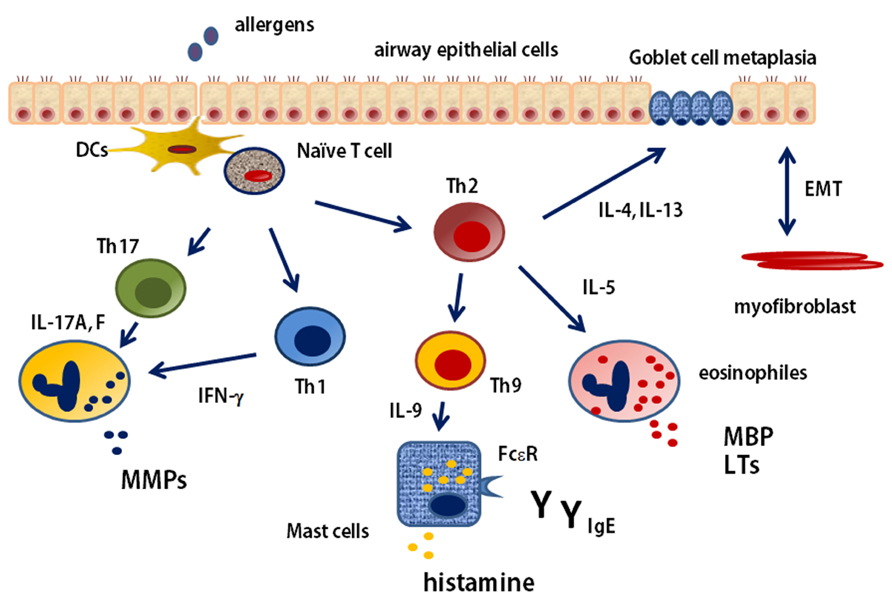
FIGURE 2. T cell immune response in the asthmatic airways. naїve T cell is received allergen presentation by DCs. The pathway begins with the development of Th2 cells and their production of the cytokines IL-4, IL-5, and IL-13. These cytokines stimulate allergic and eosinophilic inflammation as well as epithelial and smooth-muscle changes that contribute to asthma pathobiology. Th9 cell can be induced and stimulate mast cells by IL-9. naїve T cell is also differentiated to Th1 or Th17 cells depending on the existence of cytokines in the microenvironment. Th1 cell and Th17 cell stimulate and induce neutrophilic inflammation. EMT, epithelial-mesenchymal-myofibroblast transition; MMP, matrix metalloproteinase; MBP, major basic protein; LT, leukotriene.
Effector CD4 cells expressing IL-17A, IL-17F were first described in 2005 (Harrington et al., 2005; Park et al., 2005) and were thought to represent a distinct T-cell lineage that promoted the first revision of the Th1/Th2 paradigm of immunity. Differentiation of naive effector T cells in the presence of IL-6 and TGF-β, leading to the expression of the transcription factor RORγt, results in IL-17 expression through the transcription factors Smad 2/3, signal transducer and activation of transcription (STAT) 3, and nuclear factor κB. naїve T cells can differentiate several cell types and have specific immune response through the release of cell-type specific cytokines (Figure 3). Th17 cells have a role in regulating both neutrophilic and macrophage inflammation in autoimmune disease, and more recently they have been suggested to be involved in asthma and corticosteroid insensitivity (Nembrini et al., 2009). Conversely, their differentiation is restricted by both Th1 and Th2 cytokines including IFN-γ, IL-4, and IL-13 (Park et al., 2005). Specifically, the induction of CXCL8, a potent neutrophil chemokine whose expression is elevated in airway secretions in severe asthma, has directly implicated Th17 cells in neutrophilic airway inflammation. IL-17A itself, but not IL-17F or IL-22, enhances the contractile force of ASM. Sensitized mice lacking the integrin αvβ8 on DCs show reduced activation of this IL-17A-linked pathway with antigen challenge. This reduction in smooth muscle contraction in the airways is reversible by IL-17A, indicating involvement of this cytokine on allergen-induced AHR by acting directly on ASM (Kudo et al., 2012). Allergic induces a strong Th17 response in association with airway neutrophilia and hyperresponsiveness, and this response is abrogated in IL-17F knockout mice (Yang et al., 2008). However, although a good case can be made for IL-17A and IL-17F in mouse models of neutrophilic and corticosteroid-refractory lung responses to allergens, evidence for IL-17 involvement in human asthma is less robust, despite some emerging genetic evidence and a potential role for IL-17A and IL-17F in moderate-to-severe disease (Chakir et al., 2003; Doe et al., 2010). In humans, a subset of Th2 memory and effector cells has been recognized expressing both GATA3 and RORγ t and, as a consequence, producing both Th17 and Th2 cytokines (Cosmi et al., 2010). Studies have reported that the number of circulating Th17 cells as well as plasma concentrations of IL-17 and IL-22 increase in proportion to disease severity. In a bronchial biopsy in asthma vs. normal controls, there was no correlation between IL-17A or IL-17F expression and the extent of neutrophilia, nor any link to asthma severity (Doe et al., 2010). The contribution of Th17 cells in human asthma has not been established enough. It is required to clear association of Th17 cells and subphenotype in human asthma.
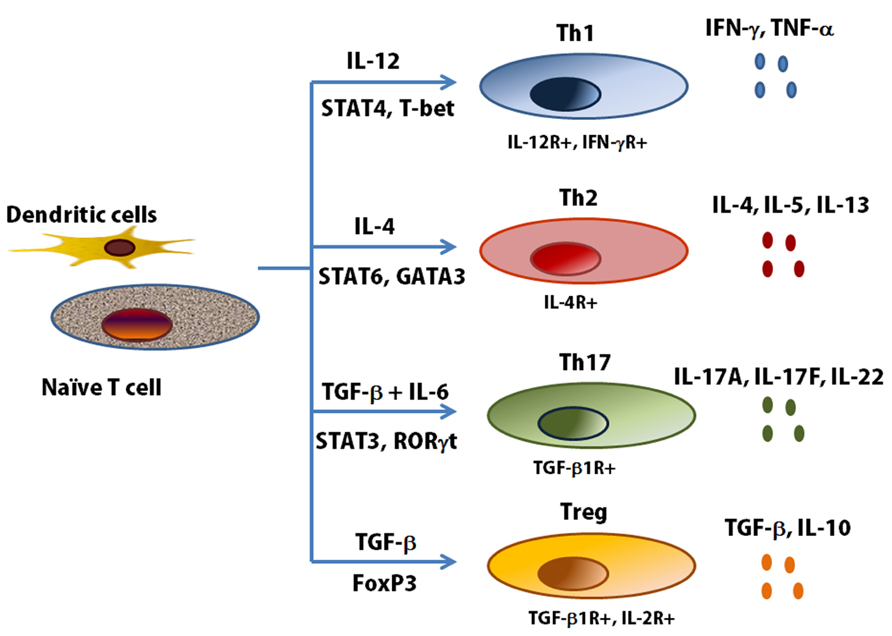
FIGURE 3. T helper cell subsets and cytokine profiles. Th1, Th2 and Th17 cells are a separate lineage of CD4+ T cells, distinct from other T cell subsets. Every specific T helper cells produce its specific cytokines (Lazarevic and Glimcher, 2011). T-bet, T-box expressed in T cells; FoxP3, forkhead box P3; ROR, retinoid-related orphan receptor.
Cytokine Targets
IL-4/IL-13
The key cytokines involved in Th2-type immunoreaction are those encoded in the IL-4 gene cluster on chromosome 5q31, containing the genes encoding IL-3, IL-4, IL-5, IL-9, IL-13, and GM-CSF (Bowen et al., 2008). The fact that the Th2 pathway is crucial to asthma pathophysiology has been the driving force for a range of biologics targeting the specific cytokines. The signals of Th2-cell-associated cytokines, IL-4 and IL-13, transmit through the IL-4Ra/IL-13Ra1 complex. IL-4 promotes B-cell isotype switching, the upregulation of adhesion molecules, eotaxin production, and the development of AHR and goblet cell metaplasia. In animal model, IL-4 deficient mice were shown to be protected from developing asthma (Brusselle et al., 1994). IL-13 can have most of these functions (Wills-Karp et al., 1998; Webb et al., 2000). Furthermore, those cytokines have the potential to induce TSLP, GM-CSF, and CCL20 production by the airway epithelium (Reibman et al., 2003; Kato et al., 2007). Furthermore, IL-13 was shown to have direct effect to enhance ASM, upregulating RhoA protein which stimulates Rho-kinase inducing calcium sensitivity (Chiba et al., 2009). Therefore, a good example is the IL-4 and IL-13 pathway for anti cytokine treatment against asthma.
Given the clear evidence for IL-4 and/or IL-13 in mouse models of disease were launched and a humanized anti-IL-4 neutralizing antibody (pascolizumab) was introduced and showed promising results in human-derived cell lines and monkeys (Hart et al., 2002). However, IL-4-specific antagonists used in clinical trials have failed (Wenzel et al., 2007). More recently, a human monoclonal anti-IL-4Ra antibody (AMG317) has been developed but did not show clinical efficacy (Corren et al., 2010). For IL-13, several neutralizing antibodies have been developed, but trials are still in their infancy. The latter IL-13-antibody (CAT-354) has recently been shown to be safe for use in humans in a phase I clinical trial but its real clinical efficacy remains to be proven (Singh et al., 2010). Attempts to validate importance of IL-13 in human asthma revealed that only 50% of individuals with asthma had elevated IL-13 levels in sputum, irrespective of the severity of the disease (Berry et al., 2004). And Woodruff et al. (2009) have also shown that only 50% of patients express IL-13-responsive genes in the airway epithelial cells, and this is linked to a strong Th2 response in bronchial biopsies, as opposed to in other asthmatics, whose IL-13-responsive gene expression was almost same level from that of normal subjects. Th2-high subjects had greater expression of IL-13 in bronchial biopsies along with greater AHR and higher serum IgE, blood and airway eosinophilia. It was suggested that one IL-13 biomarker was periostin (Woodruff et al., 2009). In a recently published trial, the monoclonal antibody (mAb) to IL-13, lebrikizumab, when administered to patients with chronic moderate-to-severe asthma for 12 weeks, significantly increased baseline spirometry (5.5%). This result was enhanced in those with elevated serum periostin (high periostin 8.2% vs. low periostin 1.2%; Corren et al., 2011).
IL-5
IL-5 is a key cytokine crucial to eosinophil growth, maturation, activation, and survival whose blockade in various animal models has a strong effect on acute and more sustained pulmonary eosinophilia and attendant changes in lung function. It is mainly produced by Th2-lymphocytes, mast cells and eosinophils. Interestingly, IL-5 regulates its own receptor expression during eosinophil ontogeny consisting of an IL-5- specific receptor α-chain, and common β-chain. Because of its restriction to the eosinophil/basophil lineage in humans, IL-5 therapy may attenuate key characteristics of allergic airway inflammation, such as airway eosinophilia, airway remodeling, and AHR, without affecting the function of other immune cells (Trifilieff et al., 2001; Flood-Page et al., 2003; Humbles et al., 2004). It has also been implicated in the induction of AHR, as IL-5 inhalation by asthmatic patients induces eosinophil influx and AHR (Leckie et al., 2000). However, despite markedly reducing both circulating and sputum eosinophilia, two humanized mAbs, mepolizumab and reslizumab, when administered to patients with moderate-to-severe asthma, had no overall effect on any asthma outcome measures. Nonetheless, the studies of mepolizumab for patients with severe asthma requiring oral corticosteroids and persistent sputum eosinophilia showed a good clinical response (Haldar et al., 2009; Nair et al., 2009), as also found in Churg-Strauss and other hypereosinophilic syndromes (Abonia and Putnam, 2011). Similar results have also been obtained with reslizumab (Castro et al., 2011; Spergel et al., 2012). Efficacy of mepolizumab has also been described in severe eosinophilic nasal polyposis in proportion to nasal lavage IL-5 levels (Gevaert et al., 2006). A further development of this approach has been the introduction of a highly active mAb targeting IL-5Rα (benralizumab), which has been defucosylated to enhance its antibody-dependent cell-mediated cytotoxicity potential (Kolbeck et al., 2010). The studies demonstrate that anti-IL-5 therapy is effective in reducing exacerbation frequency in severe asthma, with highest efficacy in subgroups of patients where eosinophils have a pathogenic role. A phase 1 study in mild asthma has shown a strong dose-related reduction of circulating eosinophils lasting 8–12 weeks after a single injection (Busse et al., 2010). It seems, however, that for the majority of asthmatic patients the anti-IL-5 treatment will need to be administered in combination with other therapies that suppress asthma features through other mechanisms. Results of clinical trials targeting the IL5Rα subunit to obtain long-term depletion of eosinophils and basophils are eagerly awaited.
IL-17/IL-22
The rapid emergence and characterization of the Th17 lineage (CD4 T cells producing IL-17 family; IL-17A, IL-17F, IL-22) refines the existing model and provides a more unified perspective of allergic inflammation by CD4+ T cell subsets. Interestingly, some asthmatic individuals, especially those poorly responding to steroid treatment, show airway infiltrations primarily composed of neutrophils. These cells are probably recruited to the airways by IL-17-producing cells that also produce IL-4 (Wang et al., 2010a). In mice, allergic sensitization followed by challenge of the airways induces a strong Th17 response and IL-17 controls bronchial hyperresponsiveness and airway remodeling, and some of these effects are mediated directly on bronchial smooth muscle cells (Pichavant et al., 2008; Wang et al., 2010b; Bellini et al., 2012; Kudo et al., 2012). Moreover, IL-17 can also induce steroid insensitivity in bronchial epithelial cells (Zijlstra et al., 2012). IL-22 can also be produced by Th17 cells. In mouse asthma models, IL-22 seems to exert a dual role. Indeed, IL-22 blockade in Th2 sensitization dramatically reduced eosinophil recruitment, Th2 cytokine and chemokine production, AHR, and mucus production. In contrast, IL-22 inhibition in allergen challenge induced lung inflammation and increased Th2 cytokine production. On epithelial cells, IL-22 has the potential to induce the production of antimicrobial peptides and to promote epithelial repair as well as suppressing the production of proinflammatory chemokines and cytokines (Pennino et al., 2012). Despite these studies, our knowledge of IL-22 in asthma pathophysiology is still limited.
IL-17A has been considered as one of most important player in asthma, however, clinical attempts for anti-IL-17A therapy to asthma has just begun (Table 1). Any data in anti-IL-17A trials for asthma are not available so far. Some clinical trials targeted at IL-17A have conducted and substantiated importance of IL-17A in autoimmune disorders. Phase II data on secukinumab, ixekizumab, and brodalumab in psoriasis indicate rapid and pronounced effects on measures of disease activity (Hueber et al., 2010). Early clinical trials in psoriatic arthritis, rheumatoid arthritis, and ankylosing spondylitis also support the therapeutic utility of IL-17A inhibition.
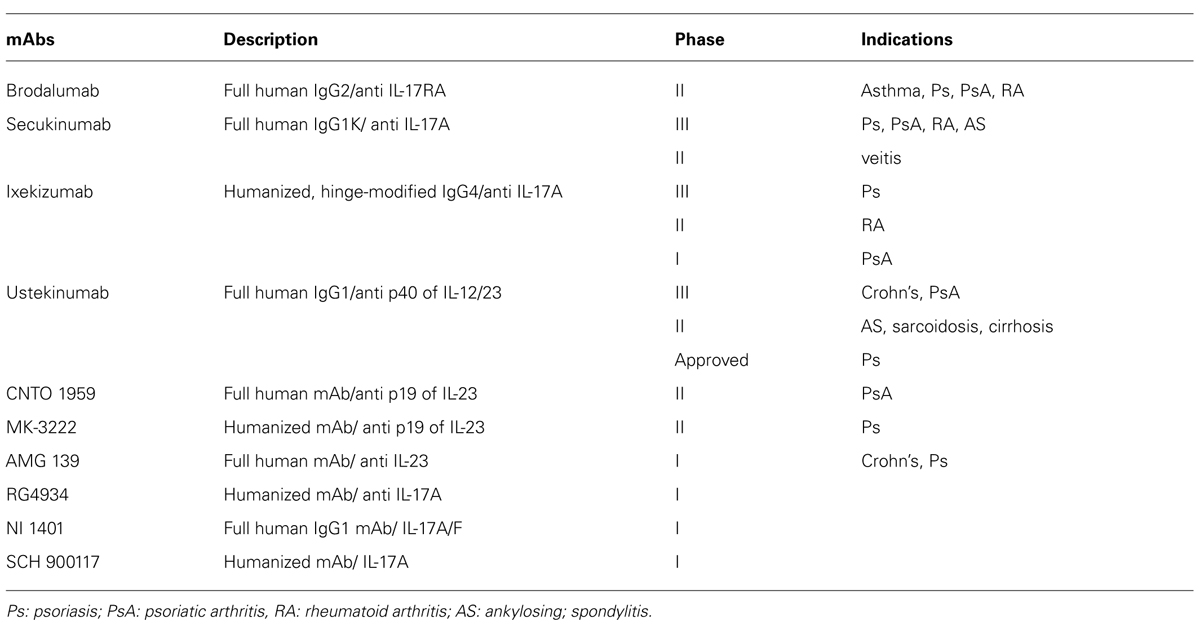
TABLE 1. Monoclonal antibodies against IL-17 pathway clinical trials.
In addition, whereas secukinumab and ixekizumab selectively target and neutralize IL-17A, brodalumab binds the IL-17RA chain of the heteromeric IL-17 receptor, which is shared with multiple members of the IL-17 cytokine family and is therefore expected to inhibit the biological activity of IL-17A and IL-17F as well as IL-17C (Ramirez-Carrozzi et al., 2011), IL-17E (IL-25) and potentially other not yet discovered IL-17 family members that utilize IL-17RA (Papp et al., 2012). Considering with these data from clinical trials for autoimmune disease, this hypothetical advantage for IL-17A inhibitors against asthma can be expected to have clinical benefits. We have to wait that data from asthma studies becomes available.
IL-9
Interleukin-9 produced from CD4+ T cell (Th9) has been identified as a subset definite from the classical Th2 cells, requiring the transcription factors IRF4, PU1, STAT6, Smad3, and Notch signaling for development. The cells differentiate in response to IL-4 and TGF-β and are described to promote T cell proliferation, IgE and IgG production by B-cells, survival and maturation of eosinophils, increasing the number of mast cell (Veldhoen et al., 2008; Staudt et al., 2010; Kearley et al., 2011; Elyaman et al., 2012; Goswami et al., 2012). Studies in human have also shown that IL-9 expression increased markedly in response to allergen challenge (Erpenbeck et al., 2003) and IL-9 is highly expressed and localized to tissue lymphocytes during intestinal parasite infection (Faulkner et al., 1998) and to CD3+ cells in bronchial submucosa and BAL (Shimbara et al., 2000). In studies using IL-9 transgenic and knockout mice, direct IL-9 instillation into the lungs and blocking mAbs, it has been shown that IL-9 drives mucus production, both by a direct effect on airway epithelia (Bryce, 2011) and also by interacting with IL-13 (Steenwinckel et al., 2007). Mice with IL-9 overexpression in lung have increased airway inflammation and AHR (Bisgaard et al., 2007; Gern, 2011). IL-9 is also made by ILC2s and boosts production of IL-5 and IL-13 (Rabinovitch et al., 2005). Along with IL-4 and stem cell factor, IL-9 is also a potent stimulus for mast-cell development (Kearley et al., 2011). As IL-9 has been implicated in both inflammatory and remodeling components in mouse models of allergic airway disease, it seems an attractive therapeutic target. Currently, clinical data on anti-IL-9 therapeutics are modest and larger clinical trials are eagerly awaited to conclude whether this form of therapy can be used in the treatment of asthma (Shalev et al., 2011). Two first-in-human, open-label dose-escalation trials of a monoclonal antibody against IL-9, MEDI-528, in normal subjects and subjects with mild asthma have been successfully completed, showing some evidence of efficacy (Parker et al., 2011).
TNF-α
Tumor necrosis factor α, a multifunctional cytokine that exerts a variety of effects, such as growth promotion, apoptosis, angiogenesis, cytotoxicity, inflammation, and immunomodulation, has been implicated in several inflammatory conditions. This cytokine is not only produced predominantly by activated macrophages but also by other immune (lymphocytes, natural killer cells, mast cells) as well as stromal (endothelial cells, fibroblasts, microglial cells) cells and presents in increased concentrations in bronchoalveolar fluid from the airways of patients with asthma (Broide et al., 1992). Some studies mentioned a relationship between TNF-α and severity of asthma.
The rates of death and complications are high among patients with refractory asthma and account for a disproportionate amount of the health resource burden attributed to asthma (Serra-Batlles et al., 1998). The airway abnormality in severe asthma is different from that in more mild asthma in having a more heterogeneous pattern of inflammatory response (Wenzel et al., 1999), with greater involvement of neutrophilic inflammation and the distal lung (Berry et al., 2005) and increased airway remodeling (Busse et al., 1999). Interest in the role of TNF-α in refractory asthma has been increased by a study showing increased concentrations of TNF-α in BAL from patients with more severe asthma and by an uncontrolled study showing that treatment with the recombinant soluble TNF-α receptor etanercept markedly improved AHR in patients with refractory asthma (Howarth et al., 2005). On the other hand, targeting TNF-α in severe asthma with golimumab yields responders and non-responders (Wenzel et al., 2009). And administration with infliximab for severe asthma also does responders and non-responders (Taillé et al., 2013). Therefore, controlled studies have shown controversial results and the risk-benefit profile of TNF-blocking agents is still debated (Cox, 2009).
The studies suggest that anti-TNF-α agents might improve the condition of a subgroup of patients severe steroid-dependent asthma, who have life-threatening exacerbations and complications of long-term steroid therapy. In the studies, the identification of more neutrophilic asthma that is less dependent upon Th2 mechanisms and, as a consequence, less responsive to corticosteroids might help identify a responsive target subpopulation. Such patients have been shown to have high circulating TNF-α and CXCL-8 as biomarkers (Silvestri et al., 2006). A transcriptomic analysis applied to induced sputum has identified a unique signature with prominence of TNF-α and nuclear factor-κB pathways (Baines et al., 2010). This stratification of asthma into pathway-selective phenotypes is likely to be a key driver for future drug development, as is proving so successful for cancer treatments (Holgate, 2012).
Conclusion
Bronchial asthma is a world-wide common disease and characterized by reversible airflow limitation, with non-specific AHR related to airway inflammation. Airway inflammation induces not only asthmatic symptoms which are the reversible airway obstruction and ASM contraction but also airway remodeling. Lately, the information for airway remodeling is increasing, the number of myofibroblasts increases in the understructure of epithelium, the proximity of the smooth muscle layer and the lamina reticularis. And it is more understood what EMT is. EMT can play a important role in airway remodeling. These epithelial and mesenchymal cells cause persistence of the inflammatory infiltrate and induce histological changes in the airway wall, increasing thickness of the basement membrane, collagen deposition and smooth muscle hypertrophy and hyperplasia. Subepithelial collagens cause thickening and increasing density of the basement membrane.
Classically, asthma is considered as Th2 disease, relating to increased IgE and eosinophilic inflammation in the airway. Recent results have shown that not only Th2, but Th17 and Th9 cells subset also contributed the disease, releasing their specific cytokines. These different cytokine give different biological effect. These can be targeted as an anti-cytokine treatment in asthma and some monoclonal antibodies against specific cytokines or their receptors are available. The results of those clinical trials have said that trials failed to control disease, despite it was clearly confirmed that those cytokines contributed the disease in animal model studies. It is required that more information for subphenotype of human asthma and its mechanism in more detail.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abonia, J. P., and Putnam, P. E. (2011). Mepolizumab in eosinophilic disorders. Expert Rev. Clin. Immunol. 7, 411–417. doi: 10.1586/eci.11.27
Aceves, S. S., and Broide, D. H. (2008). Airway fibrosis and angiogenesis due to eosinophil trafficking in chronic asthma. Curr. Mol. Med. 8, 350–358. doi: 10.2174/156652408785161023
Akers, I. A., Parsons, M., Hill, M. R., Hollenberg, M. D., Sanjar, S., Laurent, G. J., et al. (2000). Mast cell tryptase stimulates human lung fibroblast proliferation via protease-activated receptor-2. Am. J. Physiol. Lung Cell. Mol. Physiol. 278, L193–L201.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Alfvén, T., Braun-Fahrländer, C., Brunekreef, B., von Mutius, E., Riedler, J., Scheynius, A., et al. (2006). Allergic diseases and atopic sensitization in children related to farming and anthroposophic lifestyle-the PARSIFAL study. Allergy 61, 414–421. doi: 10.1111/j.1398-9995.2005.00939.x
American Thoracic Society. (1987). Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. Am. J. Respir. Crit. Care Med. 136, 225–244. doi: 10.1164/ajrccm/136.1.225
Araujo, B. B., Dolhnikoff, M., Silva, L. F., Elliot, J., Lindeman, J. H., Ferreira, D. S., et al. (2008). Extracellular matrix components and regulators in the airway smooth muscle in asthma. Eur. Respir. J. 32, 61–69. doi: 10.1183/09031936.00147807
Awadh, N., Muller, N. L., Park, C. S., Abboud, R. T., and FitzGerald, J. M. (1998). Airway wall thickness in patients with near fatal asthma and control groups: assessment with high resolution computed tomographic scanning. Thorax 53, 248–253. doi: 10.1136/thx.53.4.248
Baines, K. J., Simpson, J. L., Bowden, N. A., Scott, R. J., and Gibson, P. G. (2010). Differential gene expression and cytokine production from neutrophils in asthma phenotypes. Eur. Respir. J. 35, 522–531. doi: 10.1183/09031936.00027409
Bellini, A., Marini, M. A., Bianchetti, L., Barczyk, M., Schmidt, M., and Mattoli, S. (2012). Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. 5, 140–149. doi: 10.1038/mi.2011.60
Bento, A. M., and Hershenson, M. B. (1998). Airway remodeling: potential contributions of subepithelial fibrosis and airway smooth muscle hypertrophy/hyperplasia to airway narrowing in asthma. Allergy Asthma Proc. 19, 353–358. doi: 10.2500/108854198778612672
Berridge, M. J. (2009). Inositol trisphosphate and calcium signaling mechanisms. Biochim. Biophys. Acta 1793, 933–940. doi: 10.1016/j.bbamcr.2008.10.005
Berry, M. A., Parker, D., Neale, N., Woodman, L., Morgan, A., Monk, P., et al. (2004). Sputum and bronchial submucosal IL-13 expression in asthma and eosinophilic bronchitis. J. Allergy Clin. Immunol. 114, 1106–1109. doi: 10.1016/j.jaci.2004.08.032
Berry, M. A., Hargadon, B., Morgan, A., Shelley, M., Richter, J., Shaw, D., et al. (2005). Alveolar nitric oxide in adults with asthma: evidence of distal lung inflammation in refractory asthma. Eur. Respir. J. 25, 986–991. doi: 10.1183/09031936.05.00132404
Bisgaard, H., Hermansen, M. N., Buchvald, F., Loland, L., Halkjaer, L. B., Bønnelykke, K., et al. (2007). Childhood asthma after bacterial colonization of the airway in neonates. N. Engl. J. Med. 357, 1487–1495. doi: 10.1056/NEJMoa052632
Blank, F., Wehrli, M., Lehmann, A., Baum, O., Gehr, P., von Garnier, C., et al. (2011). Macrophages and dendritic cells express tight junction proteins and exchange particles in an in vitro model of the human airway wall. Immunobiology 216, 86–95. doi: 10.1016/j.imbio.2010.02.006
Bosco, A., Ehteshami, S., Stern, D. A., and Martinez, F. D. (2010). Decreased activation of inflammatory networks during acute asthma exacerbations is associated with chronic airflow obstruction. Mucosal Immunol. 3, 399–409 doi: 10.1038/mi.2010.13
Bossley, C. J., Fleming, L., Gupta, A., Regamey, N., Frith, J., Oates, T., et al. (2012). Pediatric severe asthma is characterized by eosinophilia and remodeling without TH2 cytokines. J. Allergy Clin. Immunol. 129, 974–982. doi: 10.1016/j.jaci.2012.01.059
Bousquet, J., Clark, T. J., Hurd, S., Khaltaev, N., Lenfant, C., O’Byrne, P., et al. (2007). GINA guidelines on asthma and beyond. Allergy 62, 102–112. doi: 10.1111/j.1398-9995.2006.01305.x
Bousquet, J., Jeffery, P. K., Busse, W. W., Johnson, M., and Vignola, A. M. (2000). Asthma. From bronchoconstriction to airways inflammation and remodeling. Am. J. Respir. Crit. Care Med. 161, 1720–1745. doi: 10.1164/ajrccm.161.5.9903102
Bowen, H., Kelly, A., Lee, T., and Lavender, P. (2008). Control of cytokine gene transcription in Th1 and Th2 cells. Clin. Exp. Allergy 38, 1422–1431. doi: 10.1111/j.1365-2222.2008.03067.x
Boxall, C., Holgate, S. T., and Davies, D. E. (2006). The contribution of transforming growth factor-beta and epidermal growth factor signalling to airway remodelling in chronic asthma. Eur. Respir. J. 27, 208–229. doi: 10.1183/09031936.06.00130004
Brackel, H. J., Pedersen, O. F., Mulder, P. G., Overbeek, S. E., Kerrebjin, K. F., and Bogaard, J. M. (2000). Central airways behave more stiffly during forced expiration in patients with asthma. Am. J. Respir. Crit. Care Med. 162, 896–904. doi: 10.1164/ajrccm.162.3.9905034
Brewster, C. E., Howarth, P. H., Djukanovic, R., Wilson, J., Holgate, S. T., and Roche, W. R. (1990). Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 3, 507–511. doi: 10.1165/ajrcmb/3.5.507
Brightling, C. E., Bradding, P., Symon, F. A., Holgate, S. T., Wardlaw, A. J., and Pavord, I. D. (2002). Mast-cell infiltration of airway smooth muscle in asthma. N. Engl. J. Med. 346, 1699–1705. doi: 10.1056/NEJMoa012705
Broide, D. H., Lotz, M., Cuomo, A. J., Coburn, D. A., Federman, E. C., and Wasserman, S. I. (1992). Cytokines in symptomatic asthma airways. J. Allergy Clin. Immunol. 89, 958–967. doi: 10.1016/0091-6749(92)90218-Q
Brusselle, G. G., Kips, J. C., Tavernier, J., van der Heyden, J. G., Cuvelier, C. A., Pauwels, R. A., et al. (1994). Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin. Exp. Allergy 24, 73–80. doi: 10.1111/j.1365-2222.1994.tb00920.x
Bryce, P. J. (2011). Revolution 9: the backwards and forwards evidence surrounding interleukin-9. Am. J. Respir. Crit. Care Med. 183, 834–835. doi: 10.1164/rccm.201009-1464ED
Bullen, S. S. (1952). Correlation of clinical and autopsy findings in 176 cases of asthma. J. Allergy Clin. Immunol. 23, 193–203.
Busse, W., Elias, J., Sheppard, D., and Banks-Schlegel, S. (1999). Airway remodeling and repair. Am. J. Respir. Crit. Care Med. 160, 1035–1042. doi: 10.1164/ajrccm.160.3.9902064
Busse, W. W., Katial, R., Gossage, D., Sari, S., Wang, B., Kolbeck, R., et al. (2010). Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor α antibody, in a phase I study of subjects with mild asthma. J. Allergy Clin. Immunol. 125, 1237–1244. doi: 10.1016/j.jaci.2010.04.005
Cakebread, J. A., Xu, Y., Grainge, C., Kehagia, V., Howarth, P. H., Holgate, S. T., et al. (2011). Exogenous IFN-β has antiviral and anti-inflammatory properties in primary bronchial epithelial cells from asthmatic subjects exposed to rhinovirus. J. Allergy Clin. Immunol. 127, 1148–1154. doi: 10.1016/j.jaci.2011.01.023
Camara, J., and Jarai, G. (2010). Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair 3, 2. doi: 10.1186/1755-1536-3-2
Carroll, N., Cooke, C., and James, A. (1997). The distribution of eosinophils and lymphocytes in the large and small airways of asthmatics. Eur. Respir. J. 10, 292–300. doi: 10.1183/09031936.97.10020292
Castro, M., Mathur, S., Hargreave, F., Boulet, L. P., Xie, F., Young, J., et al. (2011). Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am. J. Respir. Crit. Care Med. 184, 1125–1132. doi: 10.1164/rccm.201103-0396OC
Centers for Disease Control and Prevention. (2011). Vital signs: asthma prevalence, disease characteristics, and self-management education: United States, 2001–2009. Morb. Mortal. Wkly. Rep. 60, 547–552.
Chakir, J., Shannon, J., Molet, S., Fukakusa, M., Elias, J., Laviolette, M., et al. (2003). Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-β, IL-11, IL-17, and type I and type III collagen expression. J. Allergy Clin. Immunol. 111, 1293–1298. doi: 10.1067/mai.2003.1557
Chen, C., Kudo, M., Rutaganira, F., Takano, H., Lee, C., Atakilit, A., et al. (2012). Integrin α 9β1 in airway smooth muscle suppresses exaggerated airway narrowing. J. Clin. Invest. 122, 2916–2927. doi: 10.1172/JCI60387
Chiappara, G., Gagliardo, R., Siena, A., Bonsignore, M. R., Bousquet, J., Bonsignore, G., et al. (2001). Airway remodelling in the pathogenesis of asthma. Curr. Opin. Allergy Clin. Immunol. 1, 85–93.
Chiba, Y., Nakazawa, S., Todoroki, M., Shinozaki, K., Sakai, H., and Misawa, M. (2009). Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am. J. Respir. Cell Mol. Biol. 40, 159–167. doi: 10.1165/rcmb.2008-0162OC
Contoli, M., Message, S. D., Laza-Stanca, V., Edwards, M. R., Wark, P. A., Bartlett, N. W., et al. (2006). Role of deficient type III interferon-lambda production in asthma exacerbations. Nat. Med. 12, 1023–1026. doi: 10.1038/nm1462
Corren, J., Busse, W., Meltzer, E. O., Mansfield, L., Bensch, G., Fahrenholz, J., et al. (2010). A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am. J. Respir. Crit. Care Med. 181, 788–796. doi: 10.1164/rccm.200909-1448OC
Corren, J., Lemanske, R. F., Hanania, N. A., Korenblat, P. E., Parsey, M. V., Arron, J. R., et al. (2011). Lebrikizumab treatment in adults with asthma. N. Engl. J. Med. 365, 1088–1098. doi: 10.1056/NEJMoa1106469
Cosmi, L., Maggi, L., Santariasci, V., Capone, M., Cardilicchia, E., Frosali, F., et al. (2010). Identification of a novel subset of human circulating memory CD4+ T cells that produce both IL-17A and IL-4. J. Allergy Clin. Immunol. 125, 222–230. doi: 10.1016/j.jaci.2009.10.012
Cox, L. S. (2009). How safe are the biologicals in treating asthma and rhinitis? Allergy Asthma Clin. Immunol. 5, 4. doi: 10.1186/1710-1492-5-4
Djukanovic, R., Wilson, J. W., Lai, C. K. W., Holgate, S. T., and Howarth, P. H. (1991). The safety aspects of fiberoptic bronchoscopy, bronchoalveolar lavage, and endobronchial biopsy in asthma. Am. J. Respir. Crit. Care Med. 143, 772–777. doi: 10.1164/ajrccm/143.4_Pt_1.772
Doe, C., Bafadhel, M., Siddiqui, S., Desai, D., Mistry, V., Rugman, P., et al. (2010). Expression of the T helper 17–associated cytokines IL-17A and IL-17F in asthma and COPD. Chest 138, 1140–1147. doi: 10.1378/chest.09-3058
Dua, B., Watson, R. M., Gauvreau, G. M., and O’Byrne, P. M. (2010). Myeloid and plasmacytoid dendritic cells in induced sputum after allergen inhalation in subjects with asthma. J. Allergy Clin. Immunol. 126, 133–139. doi: 10.1016/j.jaci.2010.04.006
Dunnill, M. S. (1960). The pathology of asthma with special reference to changes in the bronchial mucosa. J. Clin. Pathol. 13, 27–33. doi: 10.1136/jcp.13.1.27
Ege, M. J., Mayer, M., Normand, A. C., Genuneit, J., Cookson, W. O., Braun-Fahrländer, C., et al. (2011). Exposure to environmental microorganisms and childhood asthma. N. Engl. J. Med. 364, 701–709. doi: 10.1056/NEJMoa1007302
Elyaman, W., Bassil, R., Bradshaw, E. M., Orent, W., Lahoud, Y., Zhu, B., et al. (2012). Notch receptors and Smad3 signaling cooperate in the induction of interleukin-9- producing T cells. Immunity 36, 623–634. doi: 10.1016/j.immuni.2012.01.020
Erpenbeck, V. J., Hohlfeld, J. M., Volkmann, B., Hagenberg, A., Geldmacher, H., Braun, A., et al. (2003). Segmental allergen challenge in patients with atopic asthma leads to increased IL-9 expression in bronchoalveolar lavage fluid lymphocytes. J. Allergy Clin. Immunol. 111, 1319–1327. doi: 10.1067/mai.2003.1485
Fanat, A. I., Thomson, J. V., Radford, K., Nair, P., and Sehmi, R. (2009). Human airway smooth muscle promotes eosinophil differentiation. Clin. Exp. Allergy 39, 1009–1017. doi: 10.1111/j.1365-2222.2009.03246.x
Fanta, C., Bohle, B., Hirt, W., Siemann, U., Horak, F., Kraft, D., et al. (1999). Systemic immunological changes induced by administration of grass pollen allergens via the oral mucosa during sublingual immunotherapy. Int. Arch. Allergy Immunol. 120, 218–224. doi: 10.1159/000024270
Faulkner, H., Renauld, J. C., Van Snick, J., and Grencis, R. K. (1998). Interleukin-9 enhances resistance to the intestinal nematode Trichuris muris. Infect. Immun. 66, 3832–3840.
Finkelman, F. D., Hogan, S. P., Hershey, G. K. K., Rothenberg, M. E., and Wills-Karp, M. (2010). Importance of cytokines in murine allergic airway disease and human asthma. J. Immunol. 184, 1663–1674. doi: 10.4049/jimmunol.0902185
Flood-Page, P., Menzies-Gow, A., Phipps, S., Ying, S., Wangoo, A., Ludwig, M. S., et al. (2003). Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J. Clin. Invest. 112, 1029–1036.
Gelb, A. F., and Zamel, N. (2000). Unsuspected pseudophysiologic emphysema in chronic persistent asthma. Am. J. Respir. Crit. Care. Med. 162, 1778–1782. doi: 10.1164/ajrccm.162.5.2001037
Gern, J. E. (2011). Barnyard microbes and childhood asthma. N. Engl. J. Med. 364, 769–770. doi: 10.1056/NEJMe1013713
Gevaert, P., Lang-Loidolt, D., Lackner, A., Stammberger, H., Staudinger, H., Van Zele, T., et al. (2006). Nasal IL-5 levels determine the response to anti–IL-5 treatment in patients with nasal polyps. J. Allergy Clin. Immunol. 118, 1133–1141. doi: 10.1016/j.jaci.2006.05.031
Gillis, H. L., and Lutchen, K. R. (1999). Airway remodeling in asthma amplifies heterogeneities in smooth muscle shortening causing hyperresponsiveness. J. Appl. Physiol. 86, 2001–2012.
Goswami, R., Jabeen, R., Yagi, R., Pham, D., Zhu, J., Goenka, S., et al. (2012). STAT6-dependent regulation of Th9 development. J. Immunol. 188, 968–975. doi: 10.4049/jimmunol.1102840
Grainge, C. L., Lau, L. C., Ward, J. A., Dulay, V., Lahiff, G., Wilson, S., et al. (2011). Effect of bronchoconstriction on airway remodeling in asthma. N. Engl. J. Med. 364, 2006–2015. doi: 10.1056/NEJMoa1014350
Grammatikos, A. P. (2008). The genetic and environmental basis of atopic diseases. Ann. Med. 40, 482–495 doi: 10.1080/07853890802082096
Gunst, S. J., and Tang, D. D. (2000). The contractile apparatus and mechanical properties of airway smooth muscle. Eur. Respir. J. 15, 600–616. doi: 10.1034/j.1399-3003.2000.15.29.x
Hackett, T. L., Warner, S. M., Stefanowicz, D., Shaheen, F., Pechkovsky, D. V., Murray, L. A., et al. (2009). Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am. J. Respir. Crit. Care Med. 180, 122–133. doi: 10.1164/rccm.200811-1730OC
Haldar, P., Brightling, C. E., Hargadon, B., Gupta, S., Monteiro, W., Sousa, A., et al. (2009). Mepolizumab and exacerbations of refractory eosinophilic asthma. N. Engl. J. Med. 360, 973–984. doi: 10.1056/NEJMoa0808991
Halwani, R., Vazquez-Tello, A., Sumi, Y., Pureza, M. A., Bahammam, A., Al-Jahdali, H., et al. (2013). Eosinophils induce airway smooth muscle cell proliferation. J. Clin. Immunol. 33, 595–604. doi: 10.1007/s10875-012-9836-3
Hamid, Q., Song, Y., Kotsimbos, T. C., Minshall, E., Bai, T. R., Hegele, R. G., et al. (1997). Inflammation of small airways in asthma. J. Allergy Clin. Immunol. 100, 44–51. doi: 10.1016/S0091-6749(97)70193-3
Hammad, H., Plantinga, M., Deswarte, K., Pouliot, P., Willart, M. A., Kool, M., et al. (2010). Inflammatory dendritic cells – not basophils – are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J. Exp. Med. 207, 2097–2111. doi: 10.1084/jem.20101563
Harrington, L. E., Hatton, R. D., Mangan, P. R., Turner, H., Murphy, T. L., Murphy, K. M., et al. (2005). Interleukin 17-producing CD41 effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages 325. Nat. Immunol. 6, 1123–1132. doi: 10.1038/ni1254
Hart, T. K., Blackburn, M. N., Brigham-Burke, M., Dede, K., Al-Mahdi, N., Zia-Amirhosseini, P., et al. (2002). Preclinical efficacy and safety of pascolizumab (SB 240683): a humanized anti-interleukin-4 antibody with therapeutic potential in asthma. Clin. Exp. Immunol. 130, 93–100. doi: 10.1046/j.1365-2249.2002.01973.x
Heijink, I. H., Postma, D. S., Noordhoek, J. A., Broekema, M., and Kapus, A. (2010). House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am. J. Respir. Cell Mol. Biol. 42, 69–79. doi: 10.1165/rcmb.2008-0449OC
Holgate, S. (2012). Innate and adaptive immune responses in asthma. Nat. Med. 18, 673–83. doi: 10.1038/nm.2731
Holgate, S. T. (2000). The role of mast cells and basophils in inflammation. Clin. Exp. Allergy 1, 28–32. doi: 10.1046/j.1365-2222.2000.00093.x
Holgate, S. T. (2001). Mechanisms of allergy and adult asthma. Curr. Opin. Allergy Clin. Immunol. 1, 47–50.
Holgate, S. T., Davies, D. E., Lackie, P. M., Wilson, S. J., Puddicombe, S. M., and Lordan, J. L. (2000). Epithelial-mesenchymal interactions in the pathogenesis of asthma. J. Allergy Clin. Immunol. 105, 193–204. doi: 10.1016/S0091-6749(00)90066-6
Holgate, S. T., Lackie, P. M., Davies, D. E., Roche, W. R., and Walls, A. F. (1999). The bronchial epithelium as a key regulator of airway inflammation and remodelling in asthma. Clin. Exp. Allergy 29(Suppl. 2), 90–95. doi: 10.1046/j.1365-2222.1999.00016.x
Howarth, P. H., Babu, K. S., Arshad, H. S., Lau, L., Buckley, M., McConnell, W., et al. (2005). Tumour necrosis factor (TNF-α ) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax 60, 1012–1018. doi: 10.1136/thx.2005.045260
Huang, F. Y., Wang, C. C., Zhou, S. L., Huang, Y. H., Wang, H., Chen, F., et al. (2009). Antisense interleukin-5 reduces eosinophil infiltration and hyper-responsiveness in an allergic asthma model. Asian Pac. J. Allergy Immunol. 27, 35–41.
Huber, H., and Koesser, K. (1922). The pathology of bronchial asthma. Arch. Int. Med. Exp. 30, 689–760. doi: 10.1001/archinte.1922.00110120002001
Hueber, W., Patel, D. D., Dryja, T., Wright, A. M., Koroleva, I., Bruin, G., et al. (2010). Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2, 52ra72. doi: 10.1126/scitranslmed.3001107
Humbles, A. A., Lloyd, C. M., McMillan, S. J., Friend, D. S., Xanthou, G., McKenna, E. E., et al. (2004). A critical role for eosinophils in allergic airways remodeling. Science 305, 1776–1779. doi: 10.1126/science.1100283
Isobe, Y., Kato, T., and Arita, M. (2012). Emerging roles of eosinophils and eosinophil-derived lipid mediators in the resolution of inflammation. Front. Immunol. 3:270. doi: 10.3389/fimmu.2012.00270
Jackson, D. J., Gangnon, R. E., Evans, M. D., Roberg, K. A., Anderson, E. L., Pappas, T. E., et al. (2008). Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 178, 667–672 doi: 10.1164/rccm.200802-309OC
Jacquet, A. (2011). Interactions of airway epithelium with protease allergens in the allergic response. Clin. Exp. Allergy 41, 305–311. doi: 10.1111/j.1365-2222.2010.03661.x
James, A. L., Bai, T. R., Mauad, T., Abramson, M. J., Dolhnikoff, M., McKay, K. O., et al. (2009). Airway smooth muscle thickness in asthma is related to severity but not duration of asthma. Eur. Respir. J. 34, 1040–1045. doi: 10.1183/09031936.00181608
Jartti, T., and Korppi, M. (2011). Rhinovirus-induced bronchiolitis and asthma development. Pediatr. Allergy Immunol. 22, 350–355 doi: 10.1111/j.1399-3038.2011.01170.x
Jeffery, P. K., Laitinen, A., and Venge, P. (2000). Biopsy markers of airway inflammation and remodelling. Respir. Med. 94(Suppl. 6), S9–S15. doi: 10.1016/S0954-6111(00)90127-6
Johns, D. P., Wilson, J., Harding, R., and Walters, E. H. (2000). Airway distensibility in healthy and asthmatic subjects: effect of lung volume history. J. Appl. Physiol. 88, 1413–1420.
Johnson, J. R., Roos, A., Berg, T., Nord, M., and Fuxe, J. (2011). Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS ONE 6, e16175. doi: 10.1371/journal.pone.0016175
Johnson, S., and Knox, A. (1999). Autocrine production of matrix metalloproteinase-2 is required for human airway smooth muscle proliferation. Am. J. Physiol. 277, L1110–L1117.
Kalluri, R., and Neilson, E. G. (2003). Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 112, 1776–1784.
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi: 10.1172/JCI39104
Kanda, A., Driss, V., Hornez, N., Abdallah, M., Roumier, T., Abboud, G., et al. (2009). Eosinophil-derived IFN-gamma induces airway hyper- responsiveness and lung inflammation in the absence of lymphocytes. J. Allergy Clin. Immunol. 124, 573–582. doi: 10.1016/j.jaci.2009.04.031
Kariyawasam, H. H., and Robinson, D. S. (2007). The role of eosinophils in airway tissue remodelling in asthma. Curr. Opin. Immunol. 19, 681–686. doi: 10.1016/j.coi.2007.07.021
Kato, A., Favoreto, S. Jr., Avila, P. C., and Schleimer, R. P. (2007). TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 179, 1080–1087.
Kay, A. B. (2005). The role of eosinophils in the pathogenesis of asthma. Trends Mol. Med. 11, 148–152. doi: 10.1016/j.molmed.2005.02.002
Kearley, J., Erjefalt, J. S., Andersson, C., Benjamin, E., Jones, C. P., Robichaud, A., et al. (2011). IL-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. Am. J. Respir. Crit. Care Med. 183, 865–875. doi: 10.1164/rccm.200909-1462OC
King, G. G., Paré, P. D., and Seow, C. Y. (1999). The mechanics of exaggerated airway narrowing in asthma: the role of smooth muscle. Respir. Physiol. 118, 1-13 doi: 10.1016/S0034-5687(99)00076-6
Klagas, I., Goulet, S., Karakiulakis, G., Zhong, J., Baraket, M., Black, J. L., et al. (2009). Decreased hyaluronan in airway smooth muscle cells from patients with asthma and COPD. Eur. Respir. J. 34, 616–628. doi: 10.1183/09031936.00070808
Kolbeck, R., Kozhich, A., Koike, M., Peng, L., Andersson, C. K., Damschroder, M. M., et al. (2010). MEDI-563, a humanized anti-IL-5 receptor alpha mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J. Allergy Clin. Immunol. 125, 1344–1353. doi: 10.1016/j.jaci.2010.04.004
Kudo, M., Melton, A. C., Chen, C., Engler, M. B., Huang, K. E., Ren, X., et al. (2012). IL-17A produced by α β T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Med. 18, 547–554. doi: 10.1038/nm.2684
Kuwano, K., Bosken, C. H., Pare, P. D., Bai, T. R., Wiggs, B. R., and Hogg, J. C. (1993). Small airways dimensions in asthma and in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 148, 1220–1225. doi: 10.1164/ajrccm/148.5.1220
Lambrecht, B. N., and Hammad, H. (2009). Biology of lung dendritic cells at the origin of asthma. Immunity 31, 412–424. doi: 10.1016/j.immuni.2009.08.008
Lazarevic, V., and Glimcher, L. H. (2011). T-bet in disease. Nat. Immunol. 12, 597–606. doi: 10.1038/ni.2059
Leckie, M. J., ten Brinke, A., Khan, J., Diamant, Z., O’Connor, B. J., Walls, C. M., et al. (2000). Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 356, 2144–2148. doi: 10.1016/S0140-6736(00)03496-6
Liu, L. Y., Mathur, S. K., Sedgwick, J. B., Jarjour, N. N., Busse, W. W., and Kelly, E. (2006). Human airway and peripheral blood eosinophils enhance Th1 and Th2 cytokine secretion. Allergy 61, 589–597. doi: 10.1111/j.1398-9995.2006.01060.x
Lloyd, C. M., and Saglani, S. (2013). T cells in asthma: influences of genetics, environment, and T-cell plasticity. J. Allergy Clin. Immunol. 131, 1267–1274. doi: 10.1016/j.jaci.2013.02.016
Lu, T. X., Hartner, J., Lim, E. J., Fabry, V., Mingler, M. K., Cole, E. T., et al. (2011). MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-γ pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J. Immunol. 187, 3362–3373. doi: 10.4049/jimmunol.1101235
Masoli, M., Fabian, D., Holt, S., Beasley, R., and Global Initiative for Asthma (GINA) Program. (2004). The global burden of asthma: executive summary of the GINA dissemination committee report. Allergy 59, 469–478. doi: 10.1111/j.1398-9995.2004.00526.x
Mauad, T., Bel, E. H., and Sterk, P. J. (2007). Asthma therapy and airway remodeling. J. Allergy Clin. Immunol. 120, 997–1009. doi: 10.1016/j.jaci.2007.06.031
Mauad, T., Xavier, A. C., Saldiva, P. H., and Dolhnikoff, M. (1999). Elastosis and fragmentation of fibers of the elastic system in fatal asthma. Am. J. Respir. Crit. Care Med. 160, 968–975. doi: 10.1164/ajrccm.160.3.9809088
McWilliam, A. S., Napoli, S., Marsh, A. M., Pemper, F. L., Nelson, D. J., Pimm, C. L., et al. (1996). Dendritic cells are recruited into the airway epithelium during the inflammatory response to a broad spectrum of stimuli. J. Exp. Med. 184, 2429–2432. doi: 10.1084/jem.184.6.2429
McWilliam, A. S., Nelson, D., Thomas, J. A., and Holt, P. G. (1994). Rapid dendritic cell recruitment is a hallmark of the acute inflammatory response at mucosal surfaces. J. Exp. Med. 179, 1331–1336. doi: 10.1084/jem.179.4.1331
Messer, J., Peters, G. A., and Bennet, W. A. (1960). Cause of death and pathological findings in 304 cases of bronchial asthma. Dis. Chest 38, 616–624. doi: 10.1378/chest.38.6.616
Minshall, E., Hogg, J., and Hamid, Q. (1998). Cytokine mRNA expression in asthma is not restricted to the large airways. J. Allergy Clin. Immunol. 101, 386–390. doi: 10.1016/S0091-6749(98)70252-0
Molloy, E. L., Adams, A., Moore, J. B., Masterson, J. C., Madrigal-Estebas, L., Mahon, B. P., et al. (2008). BMP4 induces an epithelial-mesenchymal transition-like response in adult airway epithelial cells. Growth Factors 26, 12–22. doi: 10.1080/08977190801987166
Nair, P., Pizzichini, M. M., Kjarsgaard, M., Inman, M. D., Efthimiadis, A., Pizzichini, E., et al. (2009). Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N. Engl. J. Med. 360, 985–993. doi: 10.1056/NEJMoa0805435
Naureckas, E. T., Ndukwu, I. M., Halayko, A. J., Maxwell, C., Hershenson, M. B., and Solway, J. (1999). Bronchoalveolar lavage fluid from asthmatic subjects is mitogenic for human airway smooth muscle. Am. J. Respir. Crit. Care Med. 160, 2062–2066. doi: 10.1164/ajrccm.160.6.9903131
Nembrini, C., Marsland, B. J., and Kopf, M. (2009). IL-17–producing T cells in lung immunity and inflammation. J. Allergy Clin. Immunol. 123, 986–994. doi: 10.1016/j.jaci.2009.03.033
Oenema, T. A., Mensink, G., Smedinga, L., Halayko, A. J., Zaagsma, J., Meurs, H., et al. (2013). Cross-talk between transforming growth factor-β1 and muscarinic M2 receptors augments airway smooth muscle proliferation. Am. J. Respir. Cell Mol. Biol. 49, 8–27. doi: 10.1165/rcmb.2012-0261OC
Osler, W. (1892). “Bronchial asthma,” in The Principles and Practice of Medicine, 1st Edn. New York, NY: Appleton and Company, 497–501.
Papp, K. A., Leonardi, C., Menter, A., Ortonne, J. P., Krueger, J. G., Kricorian, G., et al. (2012). Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 366, 1181–1189. doi: 10.1056/NEJMoa1109017
Park, H., Li, Z., Yang, X. O., Chang, S. H., Nurieva, R., Wang, Y. H., et al. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6, 1133–1141. doi: 10.1038/ni1261
Parker, J. M., Oh, C. K., LaForce, C., Miller, S. D., Pearlman, D. S., Le, C., et al. (2011). Safety profile and clinical activity of multiple subcutaneous doses of MEDI-528, a humanized anti–interleukin-9 monoclonal antibody, in two randomized phase 2a studies in subjects with asthma. BMC Pulm. Med. 11:14. doi: 10.1186/1471-2466-11-14
Pennino, D., Bhavsar, P. K., Effner, R., Avitabile, S., Venn, P., Quaranta, M., et al. (2012). IL-22 suppresses IFN-gamma-mediated lung inflammation in asthmatic patients. J. Allergy Clin. Immunol. 131, 562–570. doi: 10.1016/j.jaci.2012.09.036
Perros, F., Hoogsteden, H. C., Coyle, A. J., Lambrecht, B. N., and Hammad, H. (2009). Blockade of CCR4 in a humanized model of asthma reveals a critical role for DC-derived CCL17 and CCL22 in attracting TH2 cells and inducing airway inflammation. Allergy 64, 995–1002. doi: 10.1111/j.1398-9995.2009.02095.x
Phipps, S., Benyahia, F., Ou, T. T., Barkans, J., Robinson, D. S., and Kay, A. B. (2004). Acute allergen-induced airway remodeling in atopic asthma. Am. J. Respir. Cell Mol. Biol. 31, 626–632. doi: 10.1165/rcmb.2004-0193OC
Pichavant, M., Goya, S., Meyer, E. H., Johnston, R. A., Kim, H. Y., Matangkasombut, P., et al. (2008). Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J. Exp. Med. 205, 385–393. doi: 10.1084/jem.20071507
Puddicombe, S. M., Polosa, R., Richter, A., Krishna, M. T., Howarth, P. H., Holgate, S. T., et al. (2000). Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 14, 1362–1374. doi: 10.1096/fj.14.10.1362
Rabinovitch, N., Liu, A. H., Zhang, L., Rodes, C. E., Foarde, K., Dutton, S. J., et al. (2005). Importance of the personal endotoxin cloud in school-age children with asthma. J. Allergy Clin. Immunol. 116, 1053–1057. doi: 10.1016/j.jaci.2005.08.045
Ramirez-Carrozzi, V., Sambandam, A., Luis, E., Lin, Z., Jeet, S., Lesch, J., et al. (2011). IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 12, 1159–1166. doi: 10.1038/ni.2156
Reibman, J., Hsu, Y., Chen, L. C., Bleck, B., and Gordon, T. (2003). Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. Am. J. Respir. Cell Mol. Biol. 28, 648–654. doi: 10.1165/rcmb.2002-0095OC
Robinson, D. S., Hamid, Q., Ying, S., Tsicopoulos, A., Barkans, J., Bentley, A. M., et al. (1992). Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N. Engl. J. Med. 326, 298–304. doi: 10.1056/NEJM199201303260504
Roche, W. R., Beasley, R., Williams, J. H., and Holgate, S. T. (1989). Subepithelial fibrosis in the bronchi of asthmatics. Lancet 1, 520–524. doi: 10.1016/S0140-6736(89)90067-6
Sallmann, E., Reininger, B., Brandt, S., Duschek, N., Hoflehner, E., Gomer-Spitzer, E., et al. (2011). High-affinity IgE receptors on dendritic cells exacerbate Th2-dependent inflammation. J. Immunol. 187, 164–171. doi: 10.4049/jimmunol.1003392
Salmon, M., Walsh, D. A., Koto, H., Barnes, P. J., and Chung, K. F. (1999). Repeated allergen exposure of sensitized Brown-Norway rats induce airway cell DNA synthesis and remodelling. Eur. Respir. J. 14, 633–641. doi: 10.1034/j.1399-3003.1999.14c25.x
Salvato, G. (1968). Some histological changes in chronic bronchitis and asthma. Thorax 23, 168–172. doi: 10.1136/thx.23.2.168
Schuijs, M. J., Willart, M. A., Hammad, H., and Lambrecht, B. N. (2013). Cytokine targets in airway inflammation. Curr. Opin. Pharmacol. 13, 351–361. doi: 10.1016/j.coph.2013.03.013
Serra-Batlles, J., Plaza, V., Morejon, E., Comella, A., and Brugues, J. (1998). Costs of asthma according to the degree of severity. Eur. Respir. J. 12, 1322–1326. doi: 10.1183/09031936.98.12061322
Shalev, I., Schmelzle, M., Robson, S. C., and Levy, G. (2011). Making sense of regulatory T cell suppressive function. Semin. Immunol. 23, 282–292. doi: 10.1016/j.smim.2011.04.003
Shimbara, A., Christodoulopoulos, P., Soussi-Gounni, A., Olivenstein, R., Nakamura, Y., Levitt, R. C., et al. (2000). IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J. Allergy Clin. Immunol. 105, 108–115. doi: 10.1016/S0091-6749(00)90185-4
Siddiqui, S., Mistry, V., Doe, C., Roach, K., Morgan, A., Wardlaw, A., et al. (2008). Airway hyperresponsiveness is dissociated from airway wall structural remodeling. J. Allergy Clin. Immunol. 122, 335–41 doi: 10.1016/j.jaci.2008.05.020
Silvestri, M., Serpero, L., Petecchia, L., Sabatini, F., Cerasoli, F. Jr., and Rossi, G. A. (2006). High serum levels of tumour necrosis factor-α and interleukin-8 in severe asthma: markers of systemic inflammation? Clin. Exp. Allergy 36, 1373–1381. doi: 10.1111/j.1365-2222.2006.02502.x
Simpson, A., Tan, V. Y., Winn, J., Svensén, M., Bishop, C. M., Heckerman, D. E., et al. (2010). Beyond atopy: multiple patterns of sensitization in relation to asthma in a birth cohort study. Am. J. Respir. Crit. Care Med. 181, 1200–1206. doi: 10.1164/rccm.200907-1101OC
Singh, D., Kane, B., Molfino, N. A., Faggioni, R., Roskos, L., and Woodcock, A. (2010). A phase 1 study evaluating the pharmacokinetics, safety and tolerability of repeat dosing with a human IL-13 antibody (CAT-354) in subjects with asthma. BMC Pulm. Med. 10:3. doi: 10.1186/1471-2466-10-3
Slats, A. M., Janssen, K., van Schadewijk, A., van der Plas, D. T., Schot, R., van den Aardweg, J. G., et al. (2007). Bronchial inflammation and airway responses to deep inspiration in asthma and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 176, 121–128. doi: 10.1164/rccm.200612-1814OC
Spergel, J. M., Rothenberg, M. E., Collins, M. H., Furuta, G. T., Markowitz, J. E., Fuchs, G. 3rd., et al. (2012). Reslizumab in children and adolescents with eosinophilic esophagitis: results of a double-blind, randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 129, 456–463. doi: 10.1016/j.jaci.2011.11.044
Staudt, V., Bothur, E., Klein, M., Lingnau, K., Reuter, S., Grebe, N., et al. (2010). Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 33, 192–202. doi: 10.1016/j.immuni.2010.07.014
Steenwinckel, V., Louahed, J., Orabona, C., Huaux, F., Warnier, G., McKenzie, A., et al. (2007). IL-13 mediates in vivo IL-9 activities on lung epithelial cells but not on hematopoietic cells. J. Immunol. 178, 3244–3251.
Strachan, D. P. (1989). Hay fever, hygiene, and household size. Br. Med. J. 299, 1259–1260. doi: 10.1136/bmj.299.6710.1259
Strachan, D. P. (2000). Family size, infection and atopy: the first decade of the “hygiene hypothesis”. Thorax 55, S2–S10. doi: 10.1136/thorax.55.suppl_1.S2
Sugimoto, K., Kudo, M., Sundaram, A., Ren, X., Huang, K., Bernstein, X., et al. (2012). The αvβ6 integrin modulates airway hyperresponsiveness in mice by regulating intraepithelial mast cells. J. Clin. Invest. 122, 748–758. doi: 10.1172/JCI58815
Sur, S., Crotty, T. B., Kephart, G. M., Hyma, B. A., Colby, T. V., Reed, C. E., et al. (1993). Sudden onset fatal asthma: a distinct entity with few eosinophils and relatively more neutrophils in the airway submucosa? Am. J. Respir. Crit. Care Med. 148, 713–719. doi: 10.1164/ajrccm/148.3.713
Taillé, C., Poulet, C., Marchand-Adam, S., Borie, R., Dombret, M. C., Crestani, B., et al. (2013). Monoclonal anti-TNF-α antibodies for severe steroid-dependent asthma: a case series. Open Respir. Med. J. 7, 21–25. doi: 10.2174/1874306401307010021
Teran, L. M., Mochizuki, M., Bartels, J., Valencia, E. L., Nakajima, T., Hirai, K., et al. (1999). Th1- and Th2-type cytokines regulate the expression and production of eotaxin and RANTES by human lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 20, 777–786. doi: 10.1165/ajrcmb.20.4.3508
Thiery, J. P., and Sleeman, J. P. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142. doi: 10.1038/nrm1835
Trifilieff, A., Fujitani, Y., Coyle, A. J., Kopf, M., and Bertrand, C. (2001). IL-5 deficiency abolishes aspects of airway remodelling in a murine model of lung inflammation. Clin. Exp. Allergy 31, 934–942. doi: 10.1046/j.1365-2222.2001.01084.x
Trompette, A., Divanovic, S., Visintin, A., Blanchard, C., Hegde, R. S., Madan, R., et al. (2009). Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature 457, 585–588. doi: 10.1038/nature07548
Uhm, T. G., Kim, B. S., and Chung, I. Y. (2012). Eosinophil development, regulation of eosinophil-specific genes, and role of eosinophils in the pathogenesis of asthma. Allergy Asthma Immunol. Res. 4, 68–79. doi: 10.4168/aair.2012.4.2.68
Uller, L., Leino, M., Bedke, N., Sammut, D., Green, B., Lau, L., et al. (2010). Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-β in bronchial epithelial cells from donors with asthma. Thorax 65, 626–632. doi: 10.1136/thx.2009.125930
Unger, L. (1945). The pathology of bronchial asthma. South. Med. J. 38, 513–522. doi: 10.1097/00007611-194508000-00004
Veldhoen, M., Uyttenhove, C., van Snick, J., Helmby, H., Westendorf, A., Buer, J., et al. (2008). Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 9, 1341–1346. doi: 10.1038/ni.1659
Venge, P. (2010). The eosinophil and airway remodeling in asthma. Clin. Respir. J. 4, 15–19. doi: 10.1111/j.1752-699X.2010.00192.x
Veres, T. Z., Voedisch, S., Spies, E., Tschernig, T., and Braun, A. (2011). Spatiotemporal and functional behavior of airway dendritic cells visualized by two-photon microscopy. Am. J. Pathol. 179, 603–609. doi: 10.1016/j.ajpath.2011.04.039
Vignola, A. M., Kips, J., and Bousquet, J. (2000). Tissue remodeling as a feature of persistent asthma. J. Allergy Clin. Immunol. 105, 1041–1053. doi: 10.1067/mai.2000.107195
Walsh, G. M. (2010). Targeting eosinophils in asthma: current and future state of cytokine-and chemokine-directed monoclonal therapy. Expert Rev. Clin. Immunol. 6, 701–704. doi: 10.1586/eci.10.58
Wang, Q., Li, H., Yao, Y., Xia, D., and Zhou, J. (2010a). The overexpression of heparin-binding epidermal growth factor is responsible for Th17-induced airway remodeling in an experimental asthma model. J. Immunol. 185, 834–841. doi: 10.4049/jimmunol.0901490
Wang, Y. H., Voo, K. S., Liu, B., Chen, C. Y., Uygungil, B., Spoede, W., et al. (2010b). A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J. Exp. Med. 207, 2479–2491. doi: 10.1084/jem.20101376
Wark, P. A., Johnston, S. L., Bucchieri, F., Powell, R., Puddicombe, S., Laza-Stanca, V., et al. (2005). Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 201, 937–947. doi: 10.1084/jem.20041901
Watt, A. P., Schock, B. C., and Ennis, M. (2005). Neutrophils and eosinophils: clinical implications of their appearance, presence and disappearance in asthma and COPD. Curr. Drug Targets Inflamm. Allergy 4, 415–423. doi: 10.2174/1568010054526313
Webb, D. C., McKenzie, A. N., Koskinen, A. M., Yang, M., Mattes, J., and Foster, P. S. (2000). Integrated signals between IL-13, IL-4, and IL-5 regulate airways hyperreactivity. J. Immunol. 165, 108–113.
Wenzel, S., Wilbraham, D., Fuller, R., Getz, E. B., and Longphre, M. (2007). Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet 370, 1422–1431. doi: 10.1016/S0140-6736(07)61600-6
Wenzel, S. E., Barnes, P. J., Bleecker, E. R., Bousquet, J., Busse, W., Dahlén, S. E., et al. (2009). A randomized, double-blind, placebo-controlled study of tumor necrosis factor-α blockade in severe persistent asthma. Am. J. Respir. Crit. Care Med. 179, 549–558. doi: 10.1164/rccm.200809-1512OC
Wenzel, S. E., Schwartz, L. B., Langmack, E. L., Halliday, J. L., Trudeau, J. B., Gibbs, R. L., et al. (1999). Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am. J. Respir. Crit. Care Med. 160, 1001–1008. doi: 10.1164/ajrccm.160.3.9812110
Wijesinghe, M., Weatherall, M., Perrin, K., Crane, J., and Beasley, R. (2009). International trends in asthma mortality rates in the 5- to 34-year age group: a call for closer surveillance. Chest 135, 1045–1049. doi: 10.1378/chest.08-2082
Wills-Karp, M., Luyimbazi, J., Xu, X., Schofield, B., Neben, T. Y., Karp, C. L., et al. (1998). Interleukin-13: central mediator of allergic asthma. Science 282, 2258–2261. doi: 10.1126/science.282.5397.2258
Wilson, M. S., and Wynn, T. A. (2009). Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2, 103–121. doi: 10.1038/mi.2008.85
Woodruff, P. G., Modrek, B., Choy, D. F., Jia, G., Abbas, A. R., Ellwanger, A., et al. (2009). T-helper type 2–driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 180, 388–395. doi: 10.1164/rccm.200903-0392OC
Xiao, C., Puddicombe, S. M., Field, S., Haywood, J., Broughton-Head, V., Puxeddu, I., et al. (2011). Detective epithelial barrier function in asthma. J. Allergy Clin. Immunol. 128, 549–556. doi: 10.1016/j.jaci.2011.05.038
Yang, X. O., Chang, S. H., Park, H., Nurieva, R., Shah, B., Acero, L., et al. (2008). Regulation of inflammatory responses by IL-17F. J. Exp. Med. 205, 1063–1075. doi: 10.1084/jem.20071978
Yasukawa, A., Hosoki, K., Toda, M., Miyake, Y., Matsushima, Y., Matsumoto, T., et al. (2013). Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS ONE 8:e64281. doi: 10.1371/journal.pone.0064281
Yick, C. Y., Ferreira, D. S., Annoni, R., von der Thusen, J. H., Kunst, P. W., Bel, E. H., et al. (2012). Extracellular matrix in airway smooth muscle is associated with dynamics of airway function in asthma. Allergy 67, 552–559. doi: 10.1111/j.1398-9995.2011.02773.x
Zhang, S., Smartt, H., Holgate, S. T., and Roche, W. R. (1999). Growth factors secreted by bronchial epithelial cells control myofibroblast proliferation: an in vitro co-culture model of airway remodeling in asthma. Lab. Invest. 79, 395–405.
Zhu, Z., Homer, R. J., Wang, Z., Chen, Q., Geba, G. P., Wang, J., et al. (1999). Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalties, and eotaxin production. J. Clin. Invest. 103, 779–788. doi: 10.1172/JCI5909
Keywords: asthma, remodeling, epithelial to mesenchymal transition, Th2 cells, cytokines, Th17 cells, Th9 cell
Citation: Kudo M, Ishigatsubo Y and Aoki I (2013) Pathology of asthma. Front. Microbiol. 4:263. doi: 10.3389/fmicb.2013.00263
Received: 30 July 2013; Accepted: 16 August 2013;
Published online: 10 September 2013.
Edited by:
Akihide Ryo, Yokohama City University, JapanReviewed by:
Masatoshi Nakazawa, Yokohama City University, JapanHiroyuki Tsukagoshi, Gunma Prefectural Institute of Public Health and Environmental Sciences, Japan
Copyright © 2013 Kudo, Ishigatsubo and Aoki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ichiro Aoki, Department of Pathology, Graduate School of Medicine, Yokohama City University, 3-9 Fuku-ura, Kanazawa-ku Yokohama 236-0004, Japan e-mail:aWFva2lAbWVkLnlva29oYW1hLWN1LmFjLmpw