


- Julius Kühn-Institut, Federal Research Centre for Cultivated Plants, Braunschweig, Germany
Considering their key role for ecosystem processes, it is important to understand the response of microbial communities in unpolluted soils to pollution with polycyclic aromatic hydrocarbons (PAH). Phenanthrene, a model compound for PAH, was spiked to a Cambisol and a Luvisol soil. Total community DNA from phenanthrene-spiked and control soils collected on days 0, 21, and 63 were analyzed based on PCR-amplified 16S rRNA gene fragments. Denaturing gradient gel electrophoresis (DGGE) fingerprints of bacterial communities increasingly deviated with time between spiked and control soils. In taxon specific DGGE, significant responses of Alphaproteobacteria and Actinobacteria became only detectable after 63 days, while significant effects on Betaproteobacteria were detectable in both soils after 21 days. Comparison of the taxonomic distribution of bacteria in spiked and control soils on day 63 as revealed by pyrosequencing indicated soil type specific negative effects of phenanthrene on several taxa, many of them belonging to the Gamma-, Beta-, or Deltaproteobacteria. Bacterial richness and evenness decreased in spiked soils. Despite the significant differences in the bacterial community structure between both soils on day 0, similar genera increased in relative abundance after PAH spiking, especially Sphingomonas and Polaromonas. However, this did not result in an increased overall similarity of the bacterial communities in both soils.
Introduction
Anthropogenic activities such as combustion of wood, coal, and petroleum or mining accidents frequently result in the pollution of soils with polycyclic aromatic hydrocarbons (PAH) which have several potential adverse effects on environments and human health due to their toxicity, persistence, and carcinogenicity (Johnsen et al., 2005; Pumphrey and Madsen, 2007; Schafer et al., 2009). Microbes play an important role in the bioremediation of PAH-polluted sites. Microbial populations in polluted soils or sediments are very well studied (Gomes et al., 2005, 2007; Leys et al., 2005; Singleton et al., 2005; Ni Chadhain et al., 2006; Zhou et al., 2006; Flocco et al., 2009; Liang et al., 2009). Degradative populations were frequently affiliated to the genera Sphingomonas, Polaromonas, Burkholderia, Pseudomonas, Mycobacterium, Nocardia, and Rhodococcus. The type of PAH substrates (Singleton et al., 2005; Ni Chadhain et al., 2006; Gray et al., 2011) together with several biotic and abiotic factors, e.g., plant exudates (Sipila et al., 2008; Cebron et al., 2011) or particle sizes of soil minerals (Uyttebroek et al., 2006), were shown to influence the composition and abundance of total or degradative bacterial populations in polluted soils. However, the dynamics of microbial communities in unpolluted soils which are under the threat of PAH pollutions were only rarely explored.
Polycyclic aromatic hydrocarbons or their metabolites may have toxic effects on microbes (Eom et al., 2007). Previously unpolluted soils which were reported to have a low abundance of PAH-degrading populations (Johnsen et al., 2007; Flocco et al., 2009; Ding et al., 2010) and thus detoxification of PAH are assumed to be retarded. As a consequence, PAH pollution of non-adapted soils might reduce soil microbial diversity and cause more severe effects on soil microbial function as a high microbial diversity is assumed to be important for the proper functioning of soil ecosystems (Giovannoni, 2004; Bell et al., 2005). Toxic effects of PAH on plants (Li et al., 2011) and animals (Brown et al., 2004) were reported, but so far microbial ecology studies mainly focused on microbes which are able to degrade the pollutants or their metabolites. In a study by Sipila et al. (2008) the relative abundance of Acidobacteria subgroup GP1 was lower in the PAH-polluted bulk soil and rhizosphere than in the non-polluted soil.
In a previously described experiment, a Cambisol and a Luvisol soil contrasting in their texture were either spiked with phenanthrene or not and incubated in soil microcosms for 63 days (Ding et al., 2010). The diversity and abundance of enriched populations carrying PAH-hydroxylating dioxygenase (PAH-RHDα) genes depended on the soil type (Ding et al., 2010). Here we investigated for the same experiment how the bacterial communities change after spiking of the Cambisol and Luvisol with phenanthrene, in comparison to the unpolluted controls, by analyzing 16S rRNA gene fragments amplified from total community (TC) DNA by denaturing gradient gel electrophoresis (DGGE). An integrated data processing pipeline was developed for the analysis of data from pyrosequencing of 16S rRNA gene amplicons from the spiked soils and unpolluted controls after 63 days of incubation, in order to systematically identify taxa with increased or decreased abundance, to compare community structure and diversity of total and important groups between different samples.
Materials and Methods
Experimental Design
Details of the experimental setup were described previously (Ding et al., 2010). Briefly, Cambisol and Luvisol soil samples taken from the long-term observation sites in Ultuna (Sweden) and Scheyern (Germany), respectively, were contaminated with phenanthrene to reach a final concentration of 2 mg g−1 soil. Four replicates of each soil, phenanthrene-spiked or not, were incubated at room temperature (23°C) in the dark. Samples were taken on days 0, 21, and 63 after phenanthrene spiking and kept at −20°C before DNA extraction with Bio-101 DNA spin kit for soil (QBiogene, Heidelberg, Germany).
DGGE Analysis of 16S rRNA Gene Fragments
Bacterial 16S rRNA gene fragments from the soil samples were amplified with the primers F984-GC and R1378 as described by Heuer et al. (1997). A semi-nested or nested PCR approach was applied for amplification of 16S rRNA genes of Actinobacteria, Alphaproteobacteria, Betaproteobacteria, or Pseudomonas as previously described (Heuer et al., 1997; Gomes et al., 2001; Costa et al., 2007). All primers used in this study are provided in Table A1 in Appendix. DGGE of the 16S rRNA gene amplicons was performed according to Gomes et al. (2005), using a double gradient gel composed of 46.5–65% denaturants (100% denaturant was defined as 7 M urea and 40% formamide and 6.2–9% of mixture of bis-acrylamide and acrylamide (1:37.5). The following electrophoresis was performed at a constant voltage of 140 V for 17 h at 58°C in 1× Tris-acetate-EDTA buffer with a PhorU2 apparatus (Ingeny, Goes, Netherlands). The DGGE gels were silver stained according to Heuer et al. (2001). GelCompar II 4.5 was used for pairwise comparisons of microbial DGGE profiles. Dendrograms were constructed based on pairwise Pearson correlation indices by means of unweighted pair group method using arithmetic averages (UPGMA). The pairwise Pearson correlation indices were used to test for significant treatment effects by application of the previously described PERMTEST software (Kropf et al., 2004). Cloning and sequencing of the selected bands from DGGE gels were done as previously described (Gomes et al., 2008). The partial 16S rRNA gene sequences excluding the primers were classified by the Naïve Bayesian rRNA Classifier of the Ribosomal Database Project1 and BLASTN in the GenBank database.2
Pyrosequencing
Three replicate samples per treatment and control from day 63 were studied more in depth by barcoded pyrosequencing. PCR amplification and sequencing were done at Biotechnology Innovation Center (BIOCANT, Cantanhede, Portugal). Briefly, the hypervariable V3–V4 regions of 16S rRNA genes were amplified with bacterial primers 338F and 802R (RDP’s Pyrosequencing Pipeline3) which were fused to the 454 A and B adaptors, respectively. Standard PCR reaction conditions were employed for reactions with Fast Start polymerase (Roche, Pensberg, Germany), 3 mM MgCl2, 6% DMSO, 200 nM each primer and 200 mM dNTP. The PCR conditions were 94°C for 3 min, followed by 35 cycles of 94°C for 30 s, 44°C for 45 s, and 72°C for 60 s, and a final elongation step at 72°C for 2 min. Sequencing was performed on a 454 Genome Sequencer FLX platform according to standard 454 protocols (Roche – 454 Life Sciences, Branford, CT, USA).
Sequence and Statistical Analyses
A semi-automatic pipeline for analyzing 16S rRNA gene sequences was integrated by Perl4 (5.12). The pipeline consists of two major parts: an operational taxonomic unit (OTU) report generator and an OTU report analyzer (supplement).
Denoizing, multiple alignments, OTU assignment, classification, and generation of OTU reports were linked according to the following steps and criteria: the unpaired region for each sequence was truncated based on standalone BLASTN analysis against a SILVA 16S rRNA gene database (Pruesse et al., 2007). Only sequences of more than 200 bp were analyzed. The software package Mothur (v1.14.0; Schloss et al., 2009; Schloss, 2010) was used for multiple alignments and OTU assignment. Classification of sequences was done by using the Naïve Bayesian classifier at >80% confidence (Wang et al., 2007). Aligned sequences, their corresponding taxonomy, as well as OTU assignment were stored in a local MYSQL database. A Perl script including the package database interface (DBI) was used to construct an OTU level report with each row representing one OTU containing taxonomic position (domain, phylum, class, order, family, and genus) and number of sequences for each sample.
Analyses based on the OTU (>97%) report were done with R5 (2.12). To compare the community structure between samples, cluster analyses were performed, including all taxonomic groups with more than 50 OTUs, based on the pairwise Pearson correlation which is suitable to compare samples with a different number of sequence reads. The reliability of clusters was tested by 500 times bootstrap analyses. The difference in community structure between treatments was measured using the average of Pearson correlation within treatment minus average of Pearson correlation between treatments. Rarefaction analysis for different treatments and different taxonomic groups was also performed with R to compare the detected diversity between treatments. Diversity indices such as Chao1, Pielous’s evenness, and Shannon were also analyzed in a similar manner as the rarefaction analysis to alleviate the bias caused by the different numbers of sequence reads per sample.
Discriminative taxa between treatments or soil types were identified by Tukey’s honest significance tests under a generalized linear model via a logistic function for binomial data with the package multcomp (Hothorn et al., 2008). The analyses were performed systematically from phylum to species level (OTU > 97% similarity). Representative sequences for those discriminative OTUs (>97% sequences similarity) as well as their closest related 16S rRNA gene sequences were aligned with Mothur. A neighbor-joining phylogenetic tree was constructed and tested using software package SeaView4 (Gouy et al., 2010). Compression of the branches was performed to reduce the complexity of the phylogenetic tree generated. Nodes in the phylogenetic tree were marked for compression at defined distances using a local Perl package (gardener) including TreeIO (Bioperl) and DBI. Phylogenetic trees were edited with software Archaeopteryx (Han and Zmasek, 2009).
Results
Responses of Soil Bacterial Communities to Phenanthrene Depending on Soil Type, Exposure Time, and Taxonomic Group as Revealed by PCR-DGGE
Community fingerprints of Bacteria, Pseudomonas, Actinobacteria, Beta-, and Alphaproteobacteria were clustered by UPGMA based on Pearson correlation indices. Phenanthrene significantly affected the bacterial community structure in both soils (Table 1).

Table 1. Percent dissimilarity between microbial DGGE fingerprints of different taxa from phenanthrene-spiked and control soils.
The bacterial taxonomic groups analyzed responded differently to phenanthrene spiking (Table 1). Despite the relatively high variability of the Pseudomonas-specific gacA fingerprints some differentiating bands were detected in the patterns of phenanthrene-polluted Luvisol and Cambisol on day 21 (Figures A1A,B in Appendix). A pronounced difference (d = 23%) of the Pseudomonas community structure between phenanthrene-spiked Cambisol and the control was observed (Table 1). But on day 63, no differentiating bands were identified in response to the phenanthrene contamination in the Pseudomonas fingerprints (Figures A1C,D in Appendix), and the dissimilarity of Pseudomonas microbial DGGE fingerprints between phenanthrene-polluted soil and the corresponding control was small (Table 1). In contrast to Pseudomonas communities, a difference of the actinobacterial communities of phenanthrene-polluted and control soils was observed only on day 63 but not on day 21 (Table 1). Responses of betaproteobacterial communities to phenanthrene spiking were observed for both soils on days 21 and 63. The populations (accession numbers: JF810414-JF810419) behind the bands with strongly increased intensity (most dominant responder) in both soils shared the highest sequence similarity with Polaromonas sp. (AM492164; Figures A2A–C in Appendix). The alphaproteobacterial community patterns for soil samples taken on day 21 displayed a high variability among replicates. But some differentiating bands were identified when comparing polluted and unpolluted Luvisol (data not shown). On day 63, significant differences were observed between polluted and non-polluted treatments for both soils (Table 1 and Figures A3A,B in Appendix). As for the total bacterial community, differences of the alphaproteobacterial, betaproteobacterial, and actinobacterial community between the polluted soils and the corresponding controls strongly increased with time in both soils, while the community of Pseudomonas became more similar between phenanthrene-treated and non-treated soils (Table 1). Changes in the bacterial community structure of the untreated control soils during the experiment were minor for Cambisol and not detectable for Luvisol, indicating that microbial community changes were directly or indirectly induced by the phenanthrene spiking (Figure A4 in Appendix). Based on the d-values (Table 1), differences in the bacterial community structure after phenanthrene spiking were more pronounced in Luvisol than in Cambisol at both samplings.
Soil Type and Taxonomic Group Dependent Effects of Phenanthrene Spiking on Bacterial Community Structure and Diversity as Revealed by 16S rRNA Gene Amplicon Sequencing
Three replicate samples per treatment from day 63 were analyzed by barcoded pyrosequencing of 16S rRNA genes. Altogether 34,054 sequences for all 12 samples were examined, of which 31,201 sequences could be classified to 21 phyla. The remaining 2,853 sequences were only classified as Bacteria. More than 40% could not be reliably classified at genus level indicating novel diversity not yet described (Table A2 in Appendix). The most abundant phyla in both soils belonged to the Proteobacteria, Actinobacteria, Acidobacteria, Firmicutes, and Gemmatimonadetes (Figure A5 in Appendix).
A taxonomic report containing 8,889 OTUs at species level (>97% sequences similarity) was obtained for comparing community structures. Phenanthrene spiking strongly influenced the total bacterial community structure in both soils. The differences between polluted soils and their corresponding control were comparable for both soils (37% for Cambisol and 30% for Luvisol; Figure 1A). Bootstrap analysis confirmed the high reliability of the formed clusters for each treatment (bootstrap value > 90), suggesting that the total bacterial community differed between treated and control soils. The difference (d = 30%) between two polluted soils was similar to the difference (d = 34%) between the control soils (Figure 1A), indicating that bacterial communities in both soils did not converge or diverge after pollution. To compare DGGE and pyrosequencing in analyzing the community structure of bacterial taxonomic groups, Alphaproteobacteria, Betaproteobacteria, and Actinobacteria were also analyzed using OTUs belonging to these taxa, respectively. Like DGGE, clear soil type and treatment clusters were formed for alphaproteobacterial communities (Figure 1B). In contrast to the DGGE results the differences between polluted soils and the corresponding controls were comparable for both soil types (Figure 1B). Pyrosequencing analysis additionally showed that the topology of the UPGMA clusters for Alphaproteobacteria was highly similar to that observed for the total bacterial community, suggesting that the dominant members of the bacterial communities and responders belonged to Alphaproteobacteria. Both DGGE and pyrosequencing analyses suggested that differences between betaproteobacterial communities of polluted and unpolluted Luvisol (d = 66%) were stronger than for Cambisol (d = 43%; Table 2 and Figure 1C). The polluted soils shared more similarity to each other than to their corresponding control soil. Additionally, pyrosequencing analysis showed that replicates were more similar for the polluted Luvisol (>74%) than the control (55–59%), indicating convergence of the betaproteobacterial communities in the Luvisol after phenanthrene pollution. DGGE analysis revealed a significant effect of the phenanthrene spiking on actinobacterial communities in both soils. The similarities shared by replicates within each type of polluted soil were high. Pyrosequencing analysis confirmed the significant difference between polluted soils and the corresponding control except for one replicate of the control Cambisol (Figure 1D). Unlike the DGGE analysis, the similarity among replicates of dominant actinobacterial communities in both polluted soils was low, probably due to the high diversity of Actinobacteria and the rather limited amount of sequences acquired for some of the phenanthrene-spiked soils.
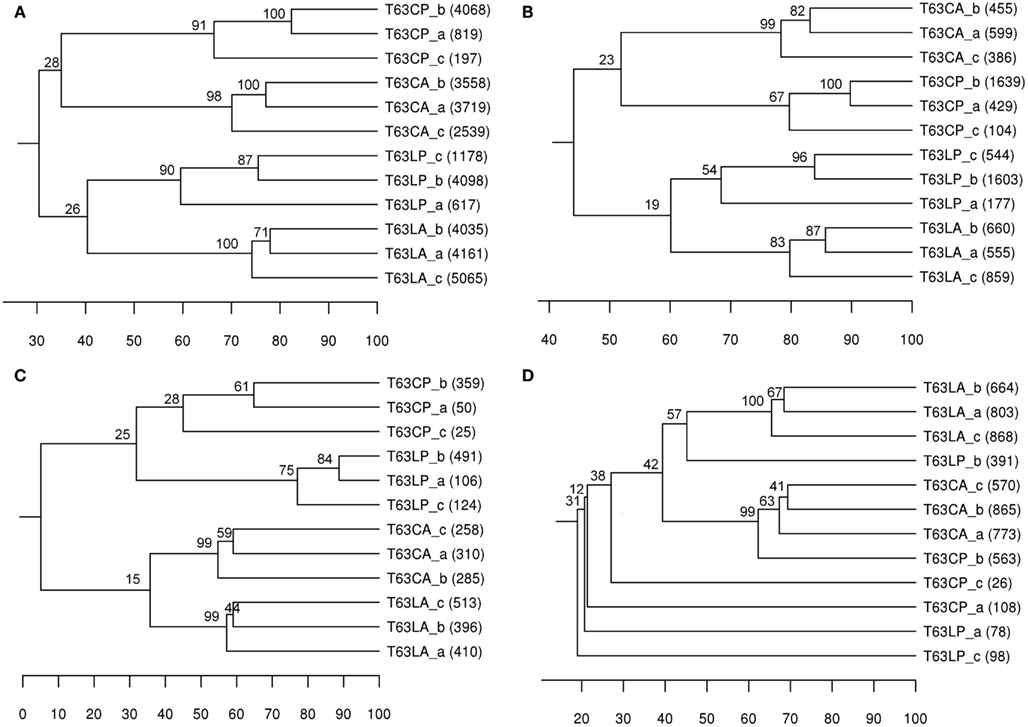
Figure 1. UPGMA cluster analysis of bacterial communities’ structure based on Pearson correlation using OTUs (> 97% sequence similarity) as the species data. Number in brackets: number of sequences obtained for the sample. Numbers on node: Bootstrapping value for each node means the percent frequency of all samples under the node grouping exclusively together. (A) total bacterial community; (B) Alphaproteobacteria; (C) Betaproteobacteria; (D) Actinobacteria. T63, 63 days after phenanthrene spiking; CA: Cambisol control, CP: phenanthrene-spiked Cambisol, LA: Luvisol control, LP: phenanthrene-spiked Luvisol.
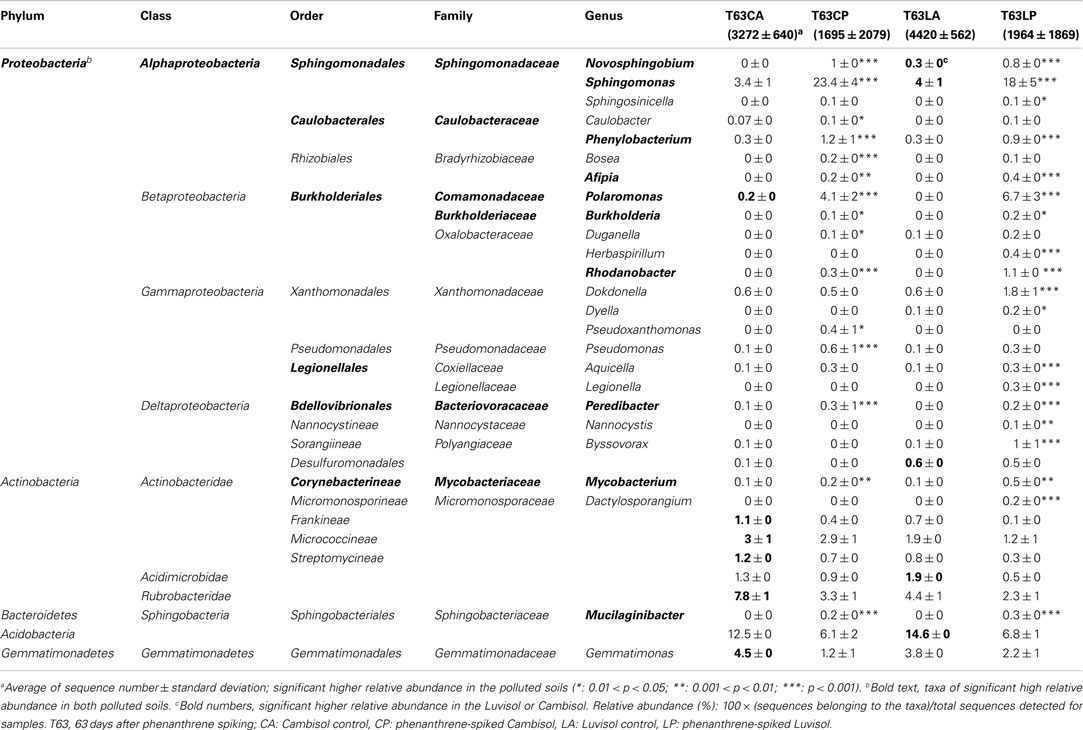
Table 2. Taxa enriched in the polluted soils or with significant (p < 0.05) different relative abundance (%) between two soils.
To compare the diversity of detected sequences from different treatments, rarefaction analyses were performed based on OTUs defined at four similarity levels (97% and 90% = Figure 2A, 80% and 70% = Figure A6 in Appendix). The bacterial diversity was lower in the polluted soils than in the unpolluted control at all similarity levels (Figure 2A and Table A3 in Appendix). The lowest diversity was found for the polluted Cambisol (Figure 2A). A lower bacterial diversity (richness and evenness) in the polluted soils was also confirmed by Chao1 (Figure A7A in Appendix), Pielous’s evenness (Figure A7B in Appendix), and Shannon index (Figure A7C in Appendix). For Alphaproteobacteria (Figure 2B), Betaproteobacteria (Figure 2C), and Actinobacteria (Figure 2D), the detected diversities were also lower for polluted soils than for the controls. The average of observed OTUs for 400 sequences was used to compare the diversity in phenanthrene-spiked and control soils (Alphaproteobacteria, Betaproteobacteria, and Actinobacteria could be analyzed with this amount of sequences). The percentage of reduction in diversity after pollution depended on the taxonomic group and the soil type. For the polluted Luvisol, the highest decrease in diversity was observed for Betaproteobacteria (27%), followed by Bacteria (15%), Alphaproteobacteria (13%), and Actinobacteria (9%). For the polluted Cambisol, the decrease in betaproteobacterial diversity was only 9%, while a similar decrease in diversity was found for Bacteria, Alphaproteobacteria, and Actinobacteria in both polluted soils.
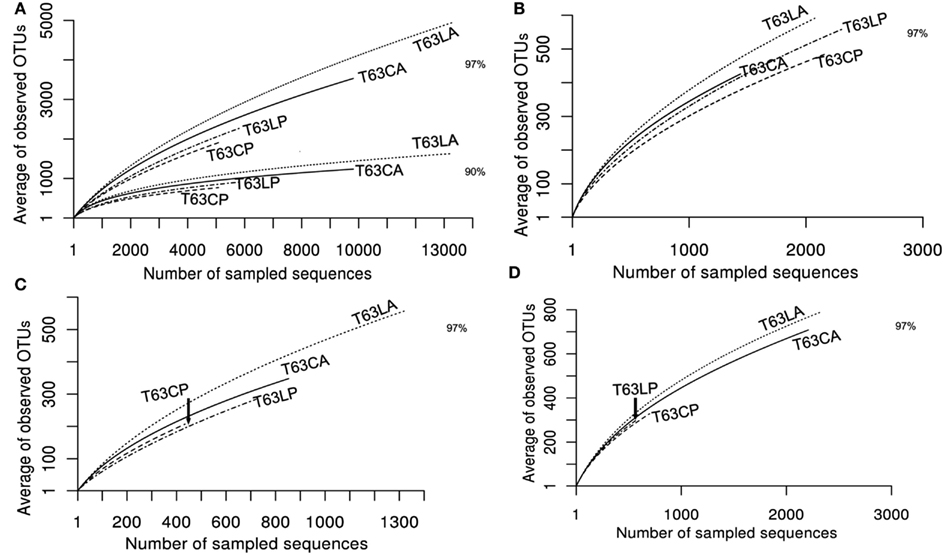
Figure 2. Plot of rarefaction curves for soils spiked with phenanthrene (LP, CP) or not (LA, CA) on day 63. (A) Total bacteria; (B) Alphaproteobacteria; (C) Betaproteobacteria; (D) Actinobacteria. T63, 63 days after phenanthrene spiking; CA: Cambisol control, CP: phenanthrene-spiked Cambisol, LA: Luvisol control, LP: phenanthrene-spiked Luvisol.
Numerous Common and Soil Type Specific Taxa Enriched in Phenanthrene-Polluted Soils
To identify those taxa which were enriched in the polluted soils, multiple Tukey’s tests were applied (unadjusted p < 0.05). Phenanthrene spiking selectively enriched diverse bacterial taxonomic groups (Table 2). About 10% of the genera detected were significantly enriched in one or both polluted soils. Most genera belonged to Proteobacteria (23 genera), and few to Actinobacteria (2 genera) or Bacteroidetes (1 genus; Table 2). Many genera were never reported being involved in PAH degradation, e.g., Sphingosinicella, Afipia, Duganella, Herbaspirillum, Rhodanobacter, Dokdonella, Dyella, Aquicella, Peredibacter, Legionella, Nannocystis, Byssovorax, Dactylosporangium, and Mucilaginibacter. The relative abundance of most enriched genera was still low (<0.4%) in both polluted soils. Sphingomonas, Polaromonas, Novosphingobium, Phenylobacterium, Burkholderia, Mycobacterium, and Mucilaginibacter were enriched in both polluted soils. Among them, Sphingomonas (>18%) and Polaromonas (>4%) became the most dominant genera in both soils. In contrast to these genera commonly enriched in both polluted soils, Herbaspirillum, Dokdonella, Dyella, Legionella, and Dactylosporangium were enriched in the polluted Luvisol, while Pseudoxanthomonas showed an increased abundance only in the polluted Cambisol. Interestingly, the genus Dokdonella had a similar relative abundance (0.6%) in both control soils, but an increased abundance of this genus was only observed in the polluted Luvisol (Table 2). Further analysis at the species level confirmed that the most abundant Dokdonella OTUs were present in both control soils but a significantly increased abundance was observed only in the polluted Luvisol. Other examples of soil type specific responses were observed for Novosphingobium, Sphingomonas, and Polaromonas (Table 2).
To pinpoint those species which significantly differed between phenanthrene-polluted and non-polluted soils, all OTUs at the species level were subjected to Tukey’s tests. A total of 88 OTUs were identified as significantly enriched in the polluted soils, and the majority of them were affiliated to Proteobacteria and Actinobacteria. The diversity of enriched OTUs was higher in the polluted Luvisol (69 OTUs belonging to Proteobacteria or Actinobacteria) than in the polluted Cambisol (49 OTUs belonging to Proteobacteria, Actinobacteria, Acidobacteria, or Bacteroidetes) at species level. Interestingly, 30 out of 88 OTUs (Table A4 in Appendix) were commonly enriched in both polluted soils. Some enriched OTUs (Sphingomonadaceae) were also abundant in the control soils. Eight out of the 10 most abundant and significantly enriched OTUs were more frequently detected than average in the control soils. Three of them even ranked among the most detected seven OTU in the control Luvisol. In contrast, the most dominant OTUs detected for both polluted soils were not detected from their corresponding control at all. Their relative abundance was extremely high, reaching 3.9% in the polluted Cambisol and 2.5% in the polluted Luvisol. Interestingly, they were affiliated to Sphingomonas (Sphingomonadaceae) and Polaromonas (Burkholderiales), respectively, corresponding to the results in the previous study on PAH-RHDα genes.
Diverse Soil Type Specific Taxa with Decreased Abundance in the Polluted Soils
To identify those taxa numerically reduced (termed in the following as diminished OTUs) in the polluted soils, stringent criteria were applied. Only those OTUs were regarded as diminished OTUs which were five times more often detected than average in the control soil but were never detected in the corresponding polluted soils, or which were at least four times more often detected than average and significantly higher (p < 0.05) in relative abundance in control soil than in the corresponding polluted soil.
A total of 30 OTUs were identified, belonging to five phyla (Proteobacteria, Actinobacteria, Bacteroidetes, Acidobacteria, and Gemmatimonadetes). Compared to those enriched OTUs, the OTUs with decreased abundance seemed more soil type specific. Only one OTU affiliated to Massilia sp. (D84590) was found to be reduced in both polluted soils. Most diminished OTUs belonged to the Proteobacteria, but no one was affiliated with Alphaproteobacteria to which most enriched OTUs belonged. The diminished OTUs belonging to the Proteobacteria were affiliated to Gammaproteobacteria (eight OTUs), Deltaproteobacteria (four OTUs), or Betaproteobacteria (four OTUs). To compare the diminished OTUs and enriched OTUs phylogenetically, representative sequences for each OTU as well as their closest related reference sequences were retrieved from the SILVA 16S rRNA gene database (Pruesse et al., 2007) and subjected to phylogenetic analysis (Figure 3). Interestingly, the diminished OTUs and those enriched OTUs belong to different phylogenetic branches (Figure 3). For example, diminished OTUs belonging to Actinobacteria were affiliated with Conexibacter, Solirubrobacter, and Streptomyces while sequences of enriched OTUs were affiliated to Mycobacterium and Dactylosporangium (Figure 3). Other examples were also observed for Acidobacteria and Bacteroidetes (Figure 3).
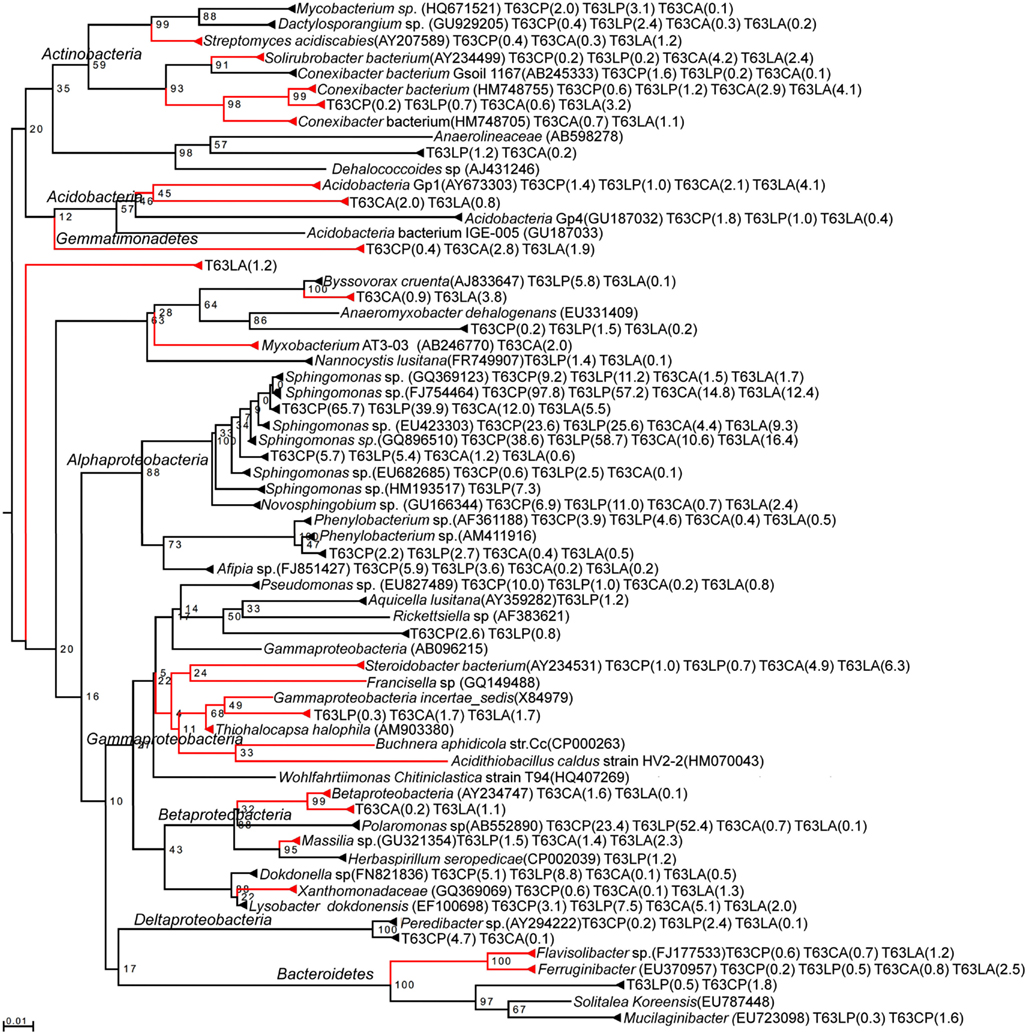
Figure 3. Neighbor-joining rooted phylogenetic tree based on multiple alignments of representative sequences for discriminative OTUs. Red branch: OTUs numerically reduced in the polluted soils. Black branch: OTUs enriched in the polluted soils. Values at each node = (bootstrap value/100) × 100. Number in brackets: relative abundance in %. T63, 63 days after phenanthrene spiking; CA: Cambisol control, CP: phenanthrene-spiked Cambisol, LA: Luvisol control, LP: phenanthrene-spiked Luvisol.
Discussion
The response to phenanthrene spiking of bacterial communities in two previously unpolluted soils was explored by DGGE and pyrosequencing analysis of 16S rRNA gene fragments amplified from TC DNA. A novel semi-automatic pipeline for analyzing 16S rRNA gene sequences consisting of an OTU report generator and OTU report analyzer allowed us to determine the community structure but also to reveal taxonomic affiliation of the responders to phenanthrene spiking. The deviation of the bacterial communities between the spiked and non-spiked control increased over time, suggesting that specific bacteria grew on the added substrate or its metabolites. The concentration of phenanthrene in both soils continually decreased after spiking (Ding et al., 2010). Many enriched ribotypes belonged to genera that were never reported as responders to PAH pollution. However, the bacterial populations which showed the strongest enrichment in both soils belonged to genera such as Sphingomonas, Polaromonas, which were previously found to be involved in PAH degradation in polluted soils or sediments (Jeon et al., 2003; Singleton et al., 2006; Sun et al., 2010; Jones et al., 2011). Several bacterial isolates affiliated to the two genera were shown to grow on media with one of PAH substrates as sole carbon source (Jeon et al., 2006; Kobayashi et al., 2009; Schuler et al., 2009), suggesting they could be the dominant populations utilizing added PAH substrates. However, microorganisms were reported degrading organic pollutants cooperatively (Sorensen et al., 2002; Dejonghe et al., 2003; Guo et al., 2010) and the populations controlling the flux of metabolic intermediates were assumed to be important for accelerating bioremediation of polluted environments (Head et al., 2006).
In the present study, only a small proportion of enriched Sphingomonas populations seemed to carry RHDα-like genes (Ding et al., 2010) that are essential for the first step of the aerobic phenanthrene degradation, and that the majority was benefiting from the initial degraders by syntrophic interactions. This finding is supported by the GeoChip analysis (Ding et al., 2012) as most enriched organic pollutant remediation genes were linked with the degradation of one-ring aromatic hydrocarbons and chloro-hydrocarbons. Some of the enriched OTUs belonging to Sphingomonas were also highly abundant in the control soils from which no PAH-RHDα gene amplicons were previously detected (Ding et al., 2010), suggesting that dominant Sphingomonas probably had a better chance to utilize metabolic intermediates of phenanthrene.
Bacterial consortia consisting of functional complementary groups might facilitate synergistic degradation of organic pollutant by decreasing the possible inhibition effects of metabolic intermediates (Keck et al., 1989). A fast degradation of PAH substrates by co-culture of Sphingomonas and Mycobacterium was indeed observed by Guo et al. (2010).
Enrichment of Polaromonas (Burkholderiales) in both phenanthrene-spiked soils was evident by both DGGE and pyrosequencing analysis. Polaromonas is an important genus responsible for aromatic hydrocarbon degradation in soil or sediments (Jeon et al., 2003; Sun et al., 2010). NagA genes encode the key enzyme for naphthalene (PAH) degradation in Polaromonas naphthalenivorans CJ2 (Jeon et al., 2006). However, the enriched Polaromonas populations probably differed between polluted soils in this experiment, as phnAc was the dominant degradative gene in phenanthrene-spiked Luvisol but was not detected in the polluted Cambisol and both control soils (Ding et al., 2010). The gene phnAc was reported in strains of the genus Burkholderia (Laurie and Lloyd-Jones, 1999; Vacca et al., 2005), Acidovorax (Singleton et al., 2009), and Delftia (Hickey et al., 2012). All host bacteria of the phnAc gene reported so far belong to the order Burkholderiales (Betaproteobacteria). Based on the complete genome analysis of P. naphthalenivorans CJ2, Yagi et al. (2009) suggested that extensive horizontal gene transfer contributed to the evolution and metabolic versatility of this strain. Pyrosequencing analysis revealed that the most dominant OTU (2.5%) for phenanthrene-spiked Luvisol was also affiliated to Polaromonas, while this OTU was not detected for both control soils and was less abundant (0.3%) in polluted Cambisol. The relative abundance of this OTU (0.3%) was much lower than all OTUs affiliated to Polaromonas in the polluted Cambisol (4.1%). These results support that the majority of enriched Polaromonas populations in the polluted Cambisol likely also benefit from the initial degraders. The study of Yagi et al. (2009) also predicted that P. naphthalenivorans CJ2 can utilize aromatic hydrocarbons such as benzoate and biphenyl which share metabolic pathways with intermediates in the degradation of phenanthrene or other PAH.
In the present study, pyrosequencing data revealed that the relative abundance of Pseudomonas was significantly higher in the polluted Cambisol than in the corresponding control (Table 2), suggesting that some Pseudomonas bacteria grew in the polluted Cambisol. Pseudomonas was reported to be enriched in PAH-polluted sites (Edlund and Jansson, 2008; Jones et al., 2011). Unlike the pyrosequencing data, the DGGE analysis did not indicate an effect of phenanthrene on Pseudomonas which may be explained by the inefficient amplification of some Pseudomonas populations by the gacA primers (Costa et al., 2007). The role of the enriched Pseudomonas in the polluted Cambisol remains to be elucidated as no nahAc-like genes were found in the previous study on PAH-RHDα genes (Ding et al., 2010). NahAc-like genes are often located on IncP-9 plasmids (Dennis and Zylstra, 2004; Sota et al., 2006; Heinaru et al., 2009). The host range of IncP-9 plasmids is confined mainly to Pseudomonas. In our experiment, oriV-rep regions of IncP-9 plasmids were detected for most soil samples but no enhanced abundance of IncP-9 plasmids was found for the polluted soils (data not shown).
Pyrosequencing analysis revealed that the bacterial diversity [richness (Chao1, rarefaction analysis) and evenness (Pielous’s evenness indices)] was lower in the phenanthrene-spiked soils than the non-spiked control, probably due to the remarkable increase in relative abundance of few taxa. The influence of PAH pollution on bacterial diversities seemed inconsistent as Dos Santos et al. (2011) observed an increase of bacterial richness for sediments after adding oil which is a complex mixture containing different PAH compounds. However, bacterial species diversity in soil or sediments is often extremely high (Curtis et al., 2002; Gans et al., 2005; Hong et al., 2006), only the relatively dominant populations are likely to be detected and the estimation of the bacterial richness is frequently biased by the amount of sequences (see Figure A7 in Appendix) or the algorithm. Populations that decreased in abundance after PAH pollution might be useful indicators of adverse effects of PAH pollutants. In the present study, bacteria belonging to Conexibacteria, Acidobacteria Gp1, Myxobacterium were numerically reduced in the phenanthrene-polluted soils. Among these taxa, only Acidobacteria Gp1 was previously reported to be affected by phenanthrene in soil (Sipila et al., 2008). Bacteria belonging to Solirubrobacter, Conexibacter, Myxobacterium, and Acidobacteria GP1 prevail in soils but are difficult to be cultivated under standard lab conditions (Davis et al., 2011).
Several studies suggested an important role of fungi in PAH metabolization by direct degradation, by transport of bacterial degraders to the source of PAH (Kohlmeier et al., 2005; Mollea et al., 2005; Furuno et al., 2010; Hong et al., 2010). However, DGGE analysis of PCR-amplified ITS fragments (Weinert et al., 2009) revealed that the fungal communities were less affected by phenanthrene in the present study, except for Cambisol on day 63 (Table 1). Compared to the Luvisol, the Cambisol has a much higher clay content. High clay contents were found to impact bacterial motility or the rate of substrate diffusion in soil (Raynaud and Leadley, 2004; Long and Or, 2009). The transport of bacterial degraders to phenanthrene (Kohlmeier et al., 2005; Furuno et al., 2010) or vice versa on fungal hyphae, might be more important in the Cambisol than in the Luvisol. However, the different response of fungal community to phenanthrene spiking might be also related to the differences in fungal community compositions of both soils (data not shown).
In summary, the responses of soil indigenous bacteria to phenanthrene depended on soil type, exposure time and taxonomic group. In relation to the abundance and diversity of enriched PAH-RHDα genes and the relative abundance of enriched taxa in the corresponding control soil, possible synergistic interactions of bacteria belonging to Sphingomonadaceae in degrading phenanthrene were suggested. In response to phenanthrene pollution of previously unpolluted soils, numerous OTUs with decreased abundance were identified.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by DFG SPP1315 (SM59/8–1) and the BMBF project MÄQNU.
Footnotes
References
Bell, T., Newman, J. A., Silverman, B. W., Turner, S. L., and Lilley, A. K. (2005). The contribution of species richness and composition to bacterial services. Nature 436, 1157–1160.
Brown, P. J., Long, S. M., Spurgeon, D. J., Svendsen, C., and Hankard, P. K. (2004). Toxicological and biochemical responses of the earthworm Lumbricus rubellus to pyrene, a non-carcinogenic polycyclic aromatic hydrocarbon. Chemosphere 57, 1675–1681.
Cebron, A., Louvel, B., Faure, P., France-Lanord, C., Chen, Y., Murrell, J. C., and Leyval, C. (2011). Root exudates modify bacterial diversity of phenanthrene degraders in PAH-polluted soil but not phenanthrene degradation rates. Environ. Microbiol. 13, 722–736.
Costa, R., Gomes, N. C. M., Krögerrecklenfort, E., Opelt, K., Berg, G., and Smalla, K. (2007). Pseudomonas community structure and antagonistic potential in the rhizosphere: insights gained by combining phylogenetic and functional gene-based analyses. Environ. Microbiol. 9, 2260–2273.
Curtis, T. P., Sloan, W. T., and Scannell, J. W. (2002). Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. U.S.A. 99, 10494–10499.
Davis, K. E., Sangwan, P., and Janssen, P. H. (2011). Acidobacteria, Rubrobacteridae and Chloroflexi are abundant among very slow-growing and mini-colony-forming soil bacteria. Environ. Microbiol. 13, 798–805.
Dejonghe, W., Berteloot, E., Goris, J., Boon, N., Crul, K., Maertens, S., Hofte, M., De Vos, P., Verstraete, W., and Top, E. M. (2003). Synergistic degradation of linuron by a bacterial consortium and isolation of a single linuron-degrading Variovorax strain. Appl. Environ. Microbiol. 69, 1532–1541.
Dennis, J. J., and Zylstra, G. J. (2004). Complete sequence and genetic organization of pDTG1, the 83 kilobase naphthalene degradation plasmid from Pseudomonas putida strain NCIB 9816-4. J. Mol. Biol. 341, 753–768.
Ding, G. C., Heuer, H., He, Z. L., Xie, J. P., Zhou, J. Z., and Smalla, K. (2012). More functional genes and convergent overall functional patterns detected by GeoChip in phenanthrene spiked soils. FEMS Microbiol. Ecol. doi:10.1111/j.1574-6941.2012.01413.x
Ding, G. C., Heuer, H., Zühlke, S., Spiteller, M., Pronk, G. J., Heister, K., Kögel-Knabner, I., and Smalla, K. (2010). Soil type-dependent responses to phenanthrene as revealed by determining the diversity and abundance of polycyclic aromatic hydrocarbon ring-hydroxylating dioxygenase genes by using a novel PCR detection system. Appl. Environ. Microbiol. 76, 4765–4771.
Dos Santos, H. F., Cury, J. C., Do Carmo, F. L., Dos Santos, A. L., Tiedje, J., Van Elsas, J. D., Rosado, A. S., and Peixoto, R. S. (2011). Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution. PLoS ONE 6, e16943. doi:10.1371/journal.pone.0016943
Edlund, A., and Jansson, J. K. (2008). Use of bromodeoxyuridine immunocapture to identify psychrotolerant phenanthrene-degrading bacteria in phenanthrene-enriched polluted Baltic Sea sediments. FEMS Microbiol. Ecol. 65, 513–525.
Eom, I. C., Rast, C., Veber, A. M., and Vasseur, P. (2007). Ecotoxicity of a polycyclic aromatic hydrocarbon (PAH)-contaminated soil. Ecotoxicol. Environ. Saf. 67, 190–205.
Flocco, C. G., Gomes, N. C. M., Mac, C. W., and Smalla, K. (2009). Occurrence and diversity of naphthalene dioxygenase genes in soil microbial communities from the Maritime Antarctic. Environ. Microbiol. 11, 700–714.
Furuno, S., Pazolt, K., Rabe, C., Neu, T. R., Harms, H., and Wick, L. Y. (2010). Fungal mycelia allow chemotactic dispersal of polycyclic aromatic hydrocarbon-degrading bacteria in water-unsaturated systems. Environ. Microbiol. 12, 1391–1398.
Gans, J., Wolinsky, M., and Dunbar, J. (2005). Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309, 1387–1390.
Gomes, N. C. M., Borges, L. R., Paranhos, R., Pinto, F. N., Krögerrecklenfort, E., Mendonca-Hagler, L. C., and Smalla, K. (2007). Diversity of ndo genes in mangrove sediments exposed to different sources of polycyclic aromatic hydrocarbon pollution. Appl. Environ. Microbiol. 73, 7392–7399.
Gomes, N. C. M., Borges, L. R., Paranhos, R., Pinto, F. N., Mendonca-Hagler, L. C. S., and Smalla, K. (2008). Exploring the diversity of bacterial communities in sediments of urban mangrove forests. FEMS Microbiol. Ecol. 66, 96–109.
Gomes, N. C. M., Heuer, H., Schönfeld, J., Costa, R., Mendonca-Hagler, L., and Smalla, K. (2001). Bacterial diversity of the rhizosphere of maize (Zea mays) grown in tropical soil studied by temperature gradient gel electrophoresis. Plant Soil 232, 167–180.
Gomes, N. C. M., Kosheleva, I. A., Abraham, W. R., and Smalla, K. (2005). Effects of the inoculant strain Pseudomonas putida KT2442 (pNF142) and of naphthalene contamination on the soil bacterial community. FEMS Microbiol. Ecol. 54, 21–33.
Gouy, M., Guindon, S., and Gascuel, O. (2010). Seaview version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224.
Gray, N. D., Sherry, A., Grant, R. J., Rowan, A. K., Hubert, C. R., Callbeck, C. M., Aitken, C. M., Jones, D. M., Adams, J. J., Larter, S. R., and Head, I. M. (2011). The quantitative significance of Syntrophaceae and syntrophic partnerships in methanogenic degradation of crude oil alkanes. Environ. Microbiol. 13, 2957–2975.
Guo, C. L., Dang, Z., Wong, Y. S., and Tam, N. F. (2010). Biodegradation ability and dioxgenase genes of PAH-degrading Sphingomonas and Mycobacterium strains isolated from mangrove sediments. Int. Biodeterior. Biodegradation 64, 419–426.
Han, M. V., and Zmasek, C. M. (2009). phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics 10, 356. doi:10.1186/1471-2105-10-356
Head, I. M., Jones, D. M., and Roling, W. F. (2006). Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 4, 173–182.
Heinaru, E., Vedler, E., Jutkina, J., Aava, M., and Heinaru, A. (2009). Conjugal transfer and mobilization capacity of the completely sequenced naphthalene plasmid pNAH20 from multiplasmid strain Pseudomonas fluorescens PC20. FEMS Microbiol. Ecol. 70, 563–574.
Heuer, H., Krsek, M., Baker, P., Smalla, K., and Wellington, E. M. (1997). Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 63, 3233–3241.
Heuer, H., Wieland, J., Schönwälder, A., Gomes, N. C. M., and Smalla, K. (2001). “Bacterial community profiling using DGGE or TGGE analysis,” in Environmental Molecular Microbiology: Protocols and Applications, ed. I. P. Rouchelle (Wymondham: Horizon Scientific Press), 177–190.
Hickey, W. J., Chen, S., and Zhao, J. (2012). The phn island: a new genomic island encoding catabolism of polynuclear aromatic hydrocarbons. Front. Microbiol. 3:125. doi:10.3389/fmicb.2012.00125
Hong, J. W., Park, J. Y., and Gadd, G. M. (2010). Pyrene degradation and copper and zinc uptake by Fusarium solani and Hypocrea lixii isolated from petrol station soil. J. Appl. Microbiol. 108, 2030–2040.
Hong, S. H., Bunge, J., Jeon, S. O., and Epstein, S. S. (2006). Predicting microbial species richness. Proc. Natl. Acad. Sci. U.S.A. 103, 117–122.
Hothorn, T., Bretz, F., and Westfall, P. (2008). Simultaneous inference in general parametric models. Biom. J. 50, 346–363.
Jeon, C. O., Park, M., Ro, H. S., Park, W., and Madsen, E. L. (2006). The naphthalene catabolic (nag) genes of Polaromonas naphthalenivorans CJ2: evolutionary implications for two gene clusters and novel regulatory control. Appl. Environ. Microbiol. 72, 1086–1095.
Jeon, C. O., Park, W., Padmanabhan, P., Derito, C., Snape, J. R., and Madsen, E. L. (2003). Discovery of a bacterium, with distinctive dioxygenase, that is responsible for in situ biodegradation in contaminated sediment. Proc. Natl. Acad. Sci. U.S.A. 100, 13591–13596.
Johnsen, A. R., Schmidt, S., Hybholt, T. K., Henriksen, S., Jacobsen, C. S., and Andersen, O. (2007). Strong impact on the polycyclic aromatic hydrocarbon (PAH)-degrading community of a PAH-polluted soil but marginal effect on PAH degradation when priming with bioremediated soil dominated by mycobacteria. Appl. Environ. Microbiol. 73, 1474–1480.
Johnsen, A. R., Wick, L. Y., and Harms, H. (2005). Principles of microbial PAH-degradation in soil. Environ. Pollut. 133, 71–84.
Jones, M. D., Crandell, D. W., Singleton, D. R., and Aitken, M. D. (2011). Stable-isotope probing of the polycyclic aromatic hydrocarbon-degrading bacterial guild in a contaminated soil. Environ. Microbiol. 13, 2623–2632.
Keck, J., Sims, R. C., Coover, M., Park, K., and Symons, B. (1989). Evidence for cooxidation of polynuclear aromatic-hydrocarbons in soil. Water Res. 23, 1467–1476.
Kobayashi, T., Murai, Y., Tatsumi, K., and Iimura, Y. (2009). Biodegradation of polycyclic aromatic hydrocarbons by Sphingomonas sp. enhanced by water-extractable organic matter from manure compost. Sci. Total Environ. 407, 5805–5810.
Kohlmeier, S., Smits, T. H., Ford, R. M., Keel, C., Harms, H., and Wick, L. Y. (2005). Taking the fungal highway: mobilization of pollutant-degrading bacteria by fungi. Environ. Sci. Technol. 39, 4640–4646.
Kropf, S., Heuer, H., Grüning, M., and Smalla, K. (2004). Significance test for comparing complex microbial community fingerprints using pairwise similarity measures. J. Microbiol. Methods 57, 187–195.
Laurie, A. D., and Lloyd-Jones, G. (1999). The phn genes of Burkholderia sp. strain RP007 constitute a divergent gene cluster for polycyclic aromatic hydrocarbon catabolism. J. Bacteriol. 181, 531–540.
Leys, N. M., Ryngaert, A., Bastiaens, L., Wattiau, P., Top, E. M., Verstraete, W., and Springael, D. (2005). Occurrence and community composition of fast-growing Mycobacterium in soils contaminated with polycyclic aromatic hydrocarbons. FEMS Microbiol. Ecol. 51, 375–388.
Li, J. H., Yu, X. Z., Wu, S. C., Wang, X. R., Wang, S. H., Tam, N. F., and Wong, M. H. (2011). Responses of bioaugmented ryegrass to PAH soil contamination. Int. J. Phytoremediation 13, 441–455.
Liang, Y., Li, G., Van Nostrand, J. D., He, Z., Wu, L., Deng, Y., Zhang, X., and Zhou, J. (2009). Microarray-based analysis of microbial functional diversity along an oil contamination gradient in oil field. FEMS Microbiol. Ecol. 70, 324–333.
Long, T., and Or, D. (2009). Dynamics of microbial growth and coexistence on variably saturated rough surfaces. Microb. Ecol. 58, 262–275.
Mollea, C., Bosco, F., and Ruggeri, B. (2005). Fungal biodegradation of naphthalene: microcosms studies. Chemosphere 60, 636–643.
Ni Chadhain, S. M., Norman, R. S., Pesce, K. V., Kukor, J. J., and Zylstra, G. J. (2006). Microbial dioxygenase gene population shifts during polycyclic aromatic hydrocarbon biodegradation. Appl. Environ. Microbiol. 72, 4078–4087.
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., and Glöckner, F. O. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196.
Pumphrey, G. M., and Madsen, E. L. (2007). Naphthalene metabolism and growth inhibition by naphthalene in Polaromonas naphthalenivorans strain CJ2. Microbiology 153, 3730–3738.
Raynaud, X., and Leadley, P. W. (2004). Soil characteristics play a key role in modeling nutrient competition in plant communities. Ecology 85, 2200–2214.
Schafer, A. N., Snape, I., and Siciliano, S. D. (2009). Influence of liquid water and soil temperature on petroleum hydrocarbon toxicity in Antarctic soil. Environ. Toxicol. Chem. 28, 1409–1415.
Schloss, P. D. (2010). The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput. Biol. 6, e1000844. doi:10.1371/journal.pcbi.1000844
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. J., and Weber, C. F. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541.
Schuler, L., Jouanneau, Y., Chadhain, S. M., Meyer, C., Pouli, M., Zylstra, G. J., Hols, P., and Agathos, S. N. (2009). Characterization of a ring-hydroxylating dioxygenase from phenanthrene-degrading Sphingomonas sp. strain LH128 able to oxidize benz[a]anthracene. Appl. Microbiol. Biotechnol. 83, 465–475.
Singleton, D. R., Powell, S. N., Sangaiah, R., Gold, A., Ball, L. M., and Aitken, M. D. (2005). Stable-isotope probing of bacteria capable of degrading salicylate, naphthalene, or phenanthrene in a bioreactor treating contaminated soil. Appl. Environ. Microbiol. 71, 1202–1209.
Singleton, D. R., Ramirez, L. G., and Aitken, M. D. (2009). Characterization of a polycyclic aromatic hydrocarbon degradation gene cluster in a phenanthrene-degrading Acidovorax strain. Appl. Environ. Microbiol. 75, 2613–2620.
Singleton, D. R., Sangaiah, R., Gold, A., Ball, L. M., and Aitken, M. D. (2006). Identification and quantification of uncultivated Proteobacteria associated with pyrene degradation in a bioreactor treating PAH-contaminated soil. Environ. Microbiol. 8, 1736–1745.
Sipila, T. P., Keskinen, A. K., Akerman, M. L., Fortelius, C., Haahtela, K., and Yrjala, K. (2008). High aromatic ring-cleavage diversity in birch rhizosphere: PAH treatment-specific changes of I.E.3 group extradiol dioxygenases and 16S rRNA bacterial communities in soil. ISME J. 2, 968–981.
Sorensen, S. R., Ronen, Z., and Aamand, J. (2002). Growth in coculture stimulates metabolism of the phenylurea herbicide isoproturon by Sphingomonas sp strain SRS2. Appl. Environ. Microbiol. 68, 3478–3485.
Sota, M., Yano, H., Ono, A., Miyazaki, R., Ishii, H., Genka, H., Top, E. M., and Tsuda, M. (2006). Genomic and functional analysis of the IncP-9 naphthalene-catabolic plasmid NAH7 and its transposon Tn4655 suggests catabolic gene spread by a tyrosine recombinase. J. Bacteriol. 188, 4057–4067.
Sun, W., Xie, S., Luo, C., and Cupples, A. M. (2010). Direct link between toluene degradation in contaminated-site microcosms and a Polaromonas strain. Appl. Environ. Microbiol. 76, 956–959.
Uyttebroek, M., Breugelmans, P., Janssen, M., Wattiau, P., Joffe, B., Karlson, U., Ortega-Calvo, J. J., Bastiaens, L., Ryngaert, A., Hausner, M., and Springael, D. (2006). Distribution of the Mycobacterium community and polycyclic aromatic hydrocarbons (PAHs) among different size fractions of a long-term PAH-contaminated soil. Environ. Microbiol. 8, 836–847.
Vacca, D. J., Bleam, W. F., and Hickey, W. J. (2005). Isolation of soil bacteria adapted to degrade humic acid-sorbed phenanthrene. Appl. Environ. Microbiol. 71, 3797–3805.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267.
Weinert, N., Meincke, R., Gottwald, C., Heuer, H., Gomes, N. C. M., Schloter, M., Berg, G., and Smalla, K. (2009). Rhizosphere communities of genetically modified zeaxanthin-accumulating potato plants and their parent cultivar differ less than those of different potato cultivars. Appl. Environ. Microbiol. 75, 3859–3865.
Yagi, J. M., Sims, D., Brettin, T., Bruce, D., and Madsen, E. L. (2009). The genome of Polaromonas naphthalenivorans strain CJ2, isolated from coal tar-contaminated sediment, reveals physiological and metabolic versatility and evolution through extensive horizontal gene transfer. Environ. Microbiol. 11, 2253–2270.
Zhou, H. W., Guo, C. L., Wong, Y. S., and Tam, N. F. (2006). Genetic diversity of dioxygenase genes in polycyclic aromatic hydrocarbon-degrading bacteria isolated from mangrove sediments. FEMS Microbiol. Lett. 262, 148–157.
Appendix
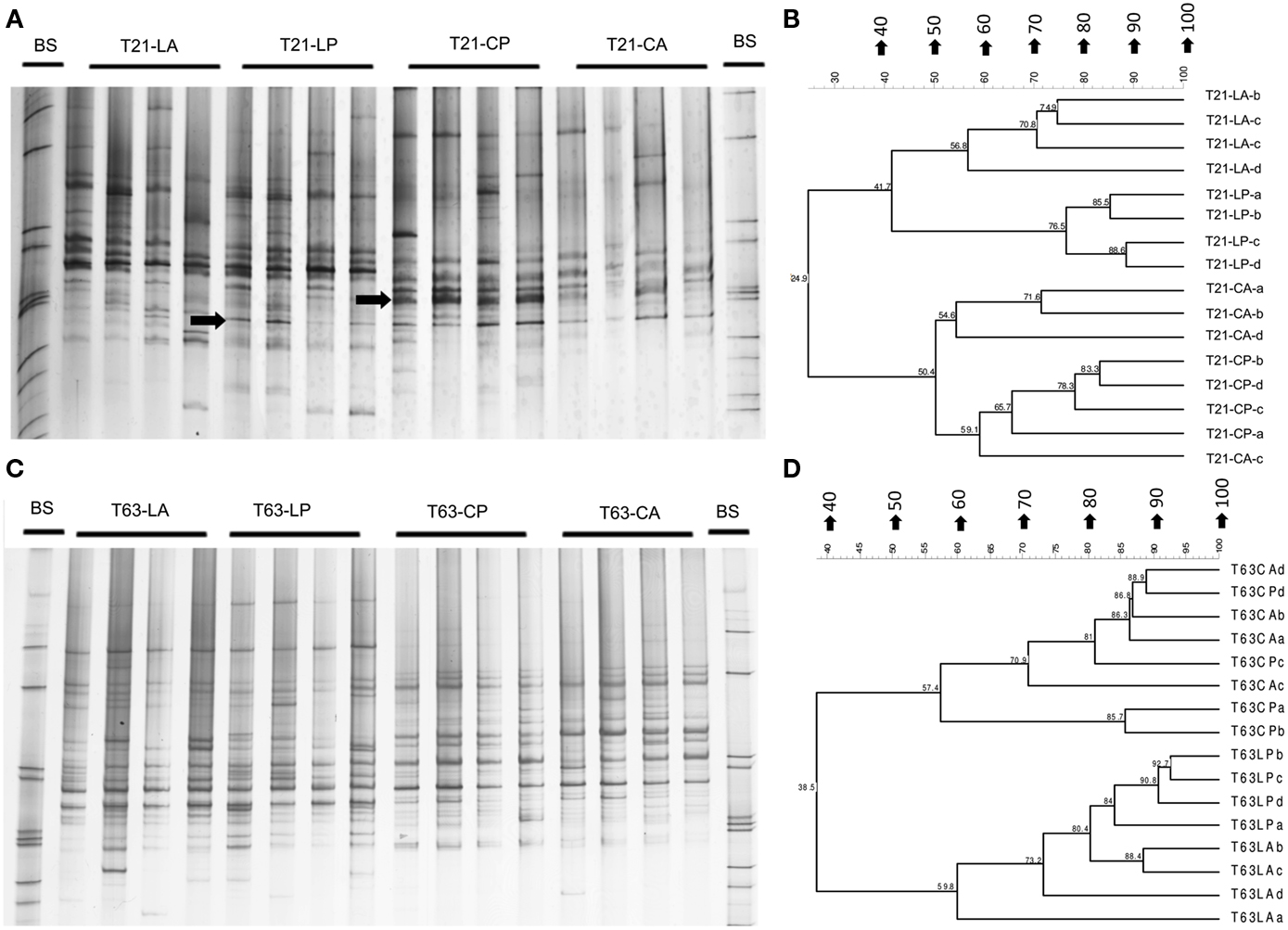
Figure A1. Pseudomonas-specific gacA DGGE fingerprint (A,C) and the corresponding UPGMA clusters (B,D) using pairwise Pearson correlation indices for soil spiked with phenanthrene (LP: phenanthrene-spiked Luvisol, CP: phenanthrene-spiked Cambisol) or not (LA: Luvisol control, CA: Cambisol control) on days 21 and 63. Arrows showing bands with enhanced intensities in phenanthrene spiked soils. BS: bacterial standards.
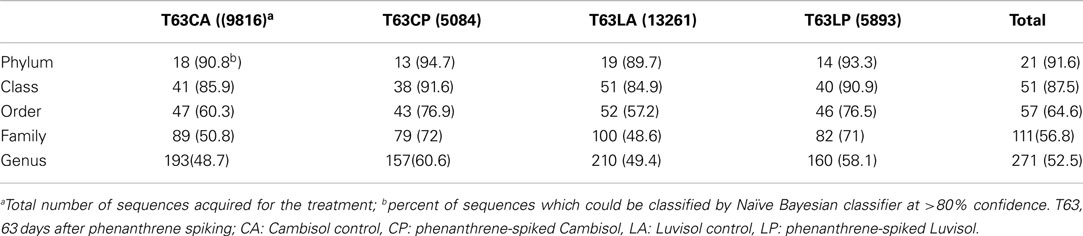
Table A1. Primers used in this study.
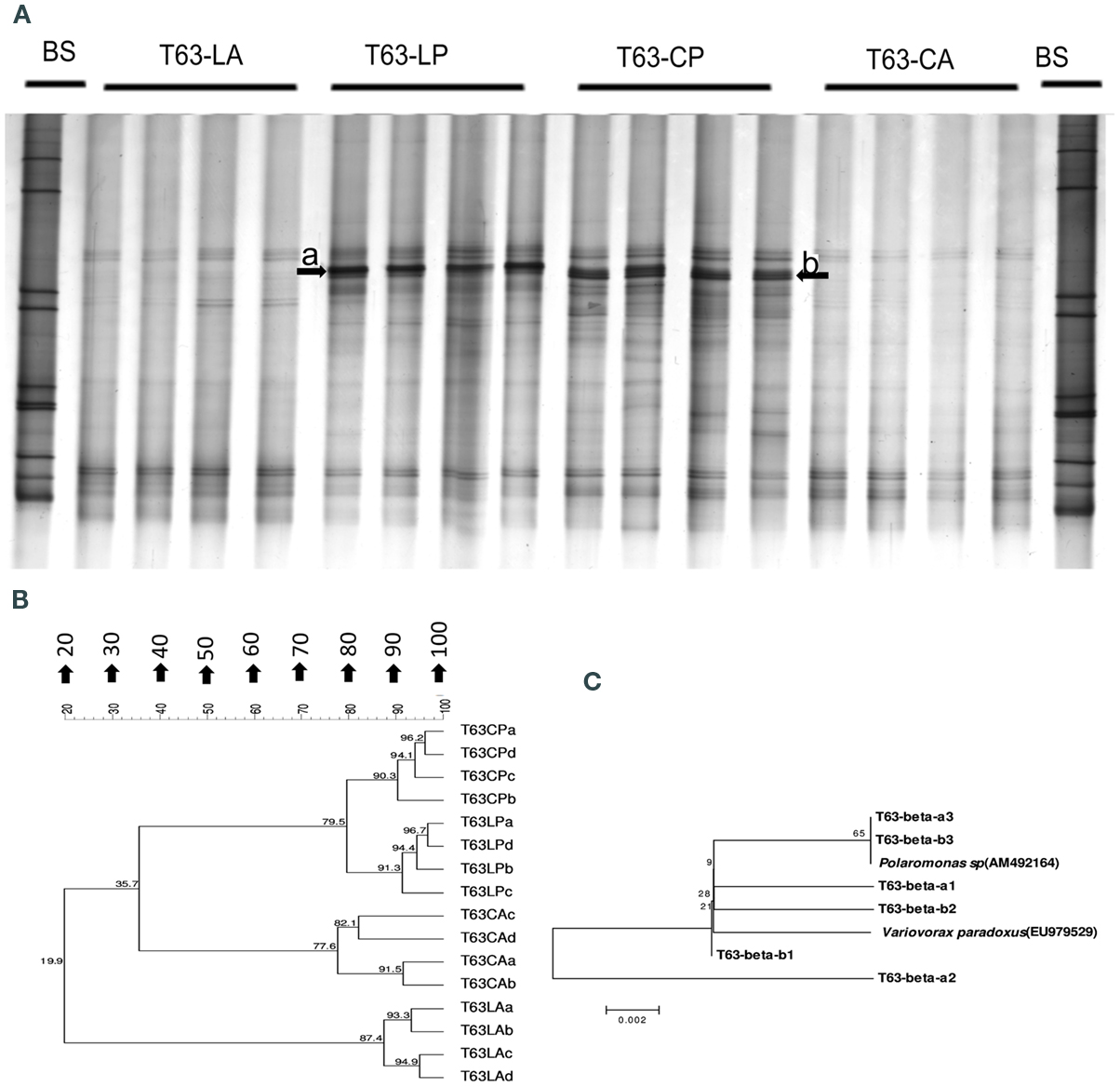
Figure A2. Betaproteobacteria-specific DGGE fingerprint (A), corresponding UPGMA clusters (B) for soils spiked with phenanthrene (LP: phenanthrene-spiked Luvisol, CP: phenanthrene-spiked Cambisol) or not (LA: Luvisol control, CA: Cambisol control) and neighbor-joining rooted phylogenetic tree (C) based on sequences (three clones were sequenced for each band) behind dominant enriched bands (band a and band b) in both polluted soils on day 63 (T63).
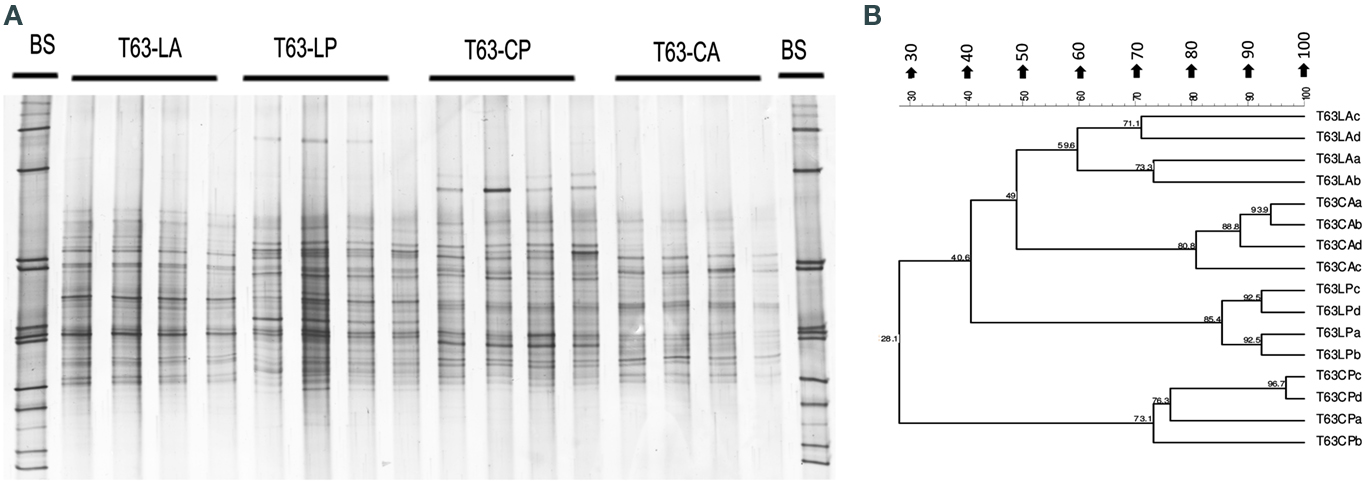
Figure A3. Alphaproteobacteria-specific DGGE fingerprints (A) and the corresponding UPGMA clusters (B) for soil spiked with phenanthrene (LP: phenanthrene-spiked Luvisol, CP: phenanthrene-spiked Cambisol) or not (LA: Luvisol control, CA: Cambisol control) on day 63 (T63).

Figure A4. Bacterial DGGE fingerprint (A,C) and the corresponding UPGMA clusters (B,D) for soil spiked with phenanthrene (LP: phenanthrene-spiked Luvisol, CP: phenanthrene-spiked Cambisol) or not (LA: Luvisol control, CA: Cambisol control) on day 21 (T21) and 63 (T63) and the initial soils (L: Luvisol and C: Cambisol) on day 0 (T0).
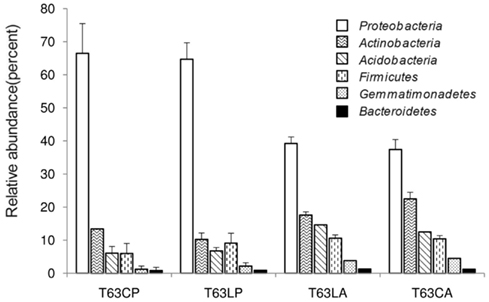
Figure A5. Relative abundance of dominant phyla detected for soils spiked with phenanthrene (LP: phenanthrene-spiked Luvisol, CP: phenanthrene-spiked Cambisol) or not (LA: Luvisol control, CA: Cambisol control) on day 63 (T63).
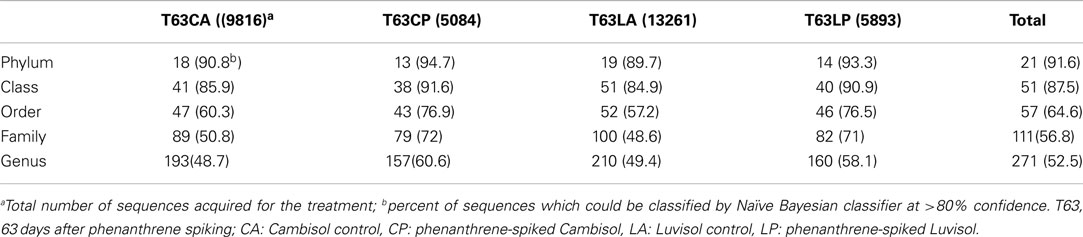
Table A2. Number of taxa detected.
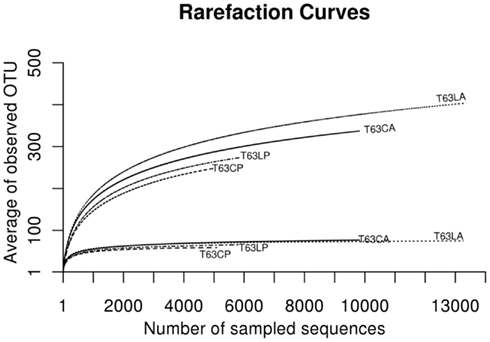
Figure A6. Plot of rarefaction curves at 80 and 70% similarities for soils spiked with phenanthrene (LP: phenanthrene-spiked Luvisol, CP: phenanthrene-spiked Cambisol) or not (LA: Luvisol control, CA: Cambisol control) on day 63 (T63).
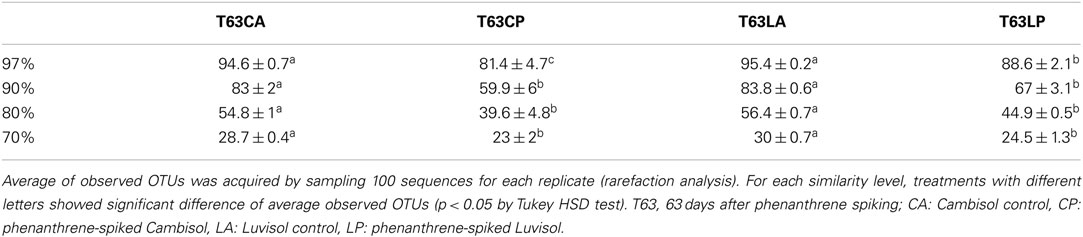
Table A3. Average of observed OTUs at four different sequences similarities.
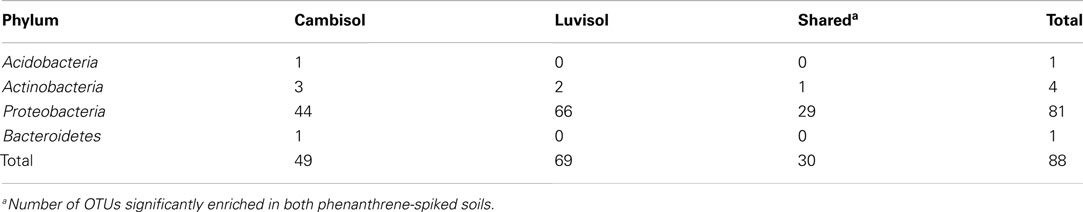
Table A4. Number of OTUs (>97% similarity) significantly (unadjusted p < 0.05) enriched in phenanthrene-spiked soils.
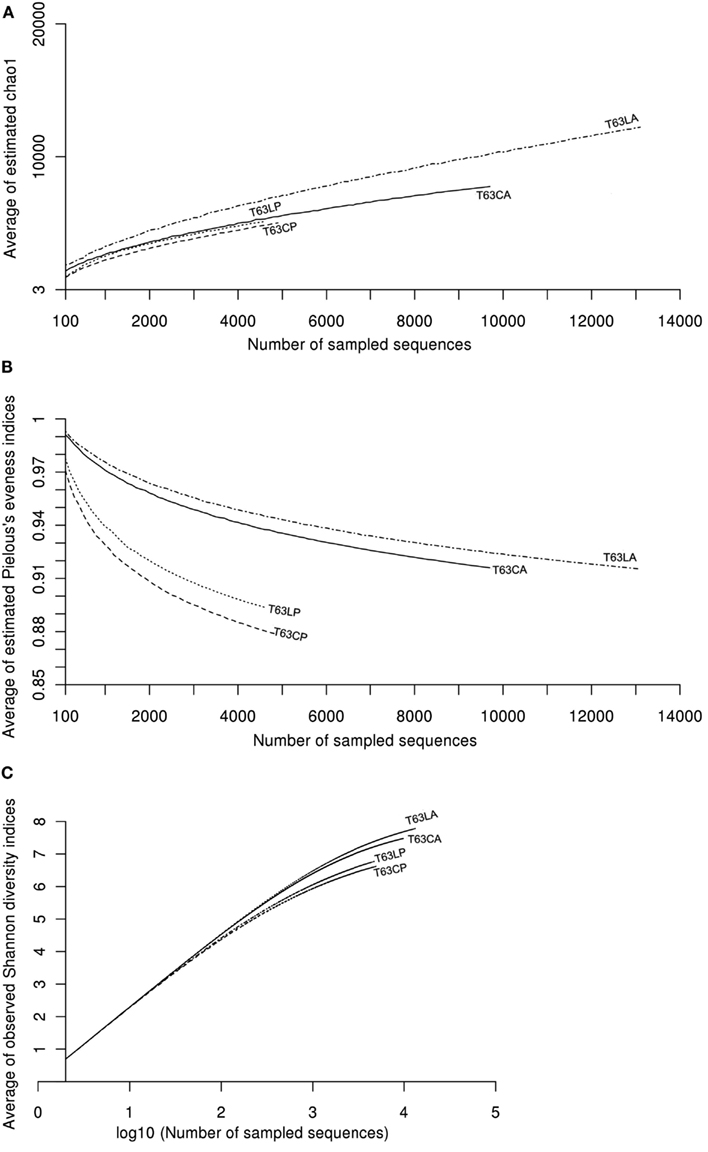
Figure A7. Plot of curves of average diversity indices [(A): CHAO1, (B): Pielous’s evenness, and (C): Shannon] for soils spiked with phenanthrene (LP: phenanthrene-spiked Luvisol, CP: phenanthrene-spiked Cambisol) or not (LA: Luvisol control, CA: Cambisol control) on day 63 (T63). Average diversity indices were calculated in a similar way to rarefaction analysis with 100 times repeating sampling.
Keywords: unpolluted soil, phenanthrene, bacterial communities, DGGE, pyrosequencing
Citation: Ding G-C, Heuer H and Smalla K (2012) Dynamics of bacterial communities in two unpolluted soils after spiking with phenanthrene: soil type specific and common responders. Front. Microbio. 3:290. doi: 10.3389/fmicb.2012.00290
Received: 29 May 2012; Accepted: 23 July 2012;
Published online: 21 August 2012.
Edited by:
Matthew Fields, Montana State University, USAReviewed by:
Uwe Strotmann, Fachhochschule Gelsenkirchen, GermanyYingying Wang, Nankai University, China
Copyright: © 2012 Ding, Heuer and Smalla. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Kornelia Smalla, Julius Kühn-Institut, Federal Research Centre for Cultivated Plants, Messeweg 11-12, D-38104 Braunschweig, Germany. e-mail:a29ybmVsaWEuc21hbGxhQGpraS5idW5kLmRl