
 Mun Lok Chew1
Mun Lok Chew1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 22 June 2012
Sec. Virology
Volume 3 - 2012 | https://doi.org/10.3389/fmicb.2012.00227
This article is part of the Research Topic Retroviruses, retroelements and their restrictions View all 13 articles
In retroviral infections, a copy of the viral DNA is first synthesized from genomic RNA by reverse transcription and subsequently integrated into host chromatin. This integration step, executed by the viral enzyme integrase (IN), is one of the hallmarks of retroviral infection. Although an obligate role for IN in retroviral integration has been clearly defined by numerous biochemical analysis of its recombinant protein and genetic analysis of the viral IN gene, several host cellular proteins have also been implicated as key factors involved in the integration step during viral replication. Although studies on integration cofactors have mostly emphasized factors that aid the integration process either through direct or indirect association with IN, it has become apparent that host cells may also harbor proteins that act as inhibitors of retroviral integration. Intriguingly, some of these inhibitory proteins appear to hamper the integration process via posttranslational modifications of the components of the preintegration complex including IN. A better understanding of the molecular mechanisms leading to the inhibition of integration will provide us with clues for the development of new strategies for treating retroviral infections. In this review, we draw attention to recent insights regarding potential host cellular factors that restrict integration, and illustrate how these inhibitory effects are achieved.
After entry into a target host cell, retroviruses carry out reverse transcription for the synthesis of double-stranded DNA from their RNA genome in a complex derived from the viral core. Subsequently, the newly synthesized viral DNA remains associated with viral and cellular proteins and forms a high-ordered nucleoprotein complex called the preintegration complex (PIC). The PIC guides nuclear entry and the subsequent integration process of the viral DNA genome (Suzuki and Craigie, 2007). The viral DNA that is integrated into chromosomal DNA, termed the provirus, acts as a template for the efficient transcription of viral RNA and ensures the equal segregation of genetic material to daughter cells upon division. Once established, proviruses cannot be distinguished or excised from the cellular genomic DNA of infected cells. Therefore, integration occupies an important place not only in viral replication but also in the treatment of retroviral infectious diseases (Lewinski and Bushman, 2005).
The viral enzyme catalyzing the integration is integrase (IN). As shall be shown, the chemical mechanism of the integration reaction has been elucidated by biochemical studies using oligonucleotide DNA substrates and purified IN protein produced by the Escherichia coli system (Bushman et al., 1990; Craigie et al., 1990; Katz et al., 1990; Sherman and Fyfe, 1990). These studies indicate that IN alone can carry out the DNA breakage and joining reactions during integration (Engelman, 2003; Turlure et al., 2004). However, the reaction by recombinant IN lacks the full fidelity of DNA integration observed in virus-infected cells. In contrast, PICs extracted from infected cells efficiently insert both viral DNA ends into a target DNA in a concerted manner in vitro, a hallmark of the integration reaction in vivo, indicating that the PIC harbors additional factors essential for the authentic integration in cells (Fujiwara and Mizuuchi, 1988; Bowerman et al., 1989; Brown et al., 1989; Farnet and Haseltine, 1990; Turlure et al., 2004). Indeed, a number of cellular proteins have since been identified as co-factors of retroviral IN and PICs (Turlure et al., 2004; Al-Mawsawi and Neamati, 2007).
As seen with the integration step, retroviruses need to hijack host biological processes to execute their efficient replication in the cell (Goff, 2007). Under the selection pressure of such viral virulence mechanisms, the host organism has itself evolved cellular antiviral defense strategies called intrinsic immunity, which block virus replication at the post-entry, post-integration, and virion release steps (Wolf and Goff, 2008), and in addition to well-studied antiviral cellular proteins such as APOBEC and TRIM family proteins, intense efforts have been dedicating to identify novel inhibitory factors against retroviruses (Liu et al., 2011; Tyagi and Kashanchi, 2012). Intriguingly, new cellular inhibitory factors for retroviral integration are also beginning to attract attention. Although viruses may be still capable of surmounting the host defense system, further understanding of the host’s inhibitory machineries could potentially translate into new treatments for retroviral infectious diseases. Here, we focus on the recent findings that illustrate the involvement and molecular mechanisms of potential restriction factors for retroviral integration.
Retroviral IN is expressed and incorporated into virions as the C-terminal part of the Gag-Pol polyprotein precursor during the late phase of retroviral infection. IN comprises three structurally and functionally distinct domains, termed the N-terminal domain (NTD), the catalytic core domain (CCD), and the C-terminal domain (CTD; Figure 1A; Lewinski and Bushman, 2005). The NTD of IN is relatively well conserved amongst retroviruses especially with the respect to a motif consisting of two histidines and two cysteines (HHCC motif). This HHCC motif makes up the zinc-binding site with an overall helix-turn-helix fold commonly found in IN of all retroviruses and retrotransposons (Craigie, 2001). Further studies utilizing mutants containing amino acid mutations in the HHCC motif also elucidated the importance of the NTD in the key steps of integration as well as its involvement in the multimerization of IN (Zheng et al., 1996; Lee et al., 1997; Craigie, 2001). The CCD is composed of a mixture of α-helices and β-sheets folds that bring a triad of critical acidic residues called the DDE motif into close proximity (Lewinski and Bushman, 2005). Crystal structure studies of HIV-1 and avian sarcoma virus (ASV) IN have shown this motif to be important for the binding of divalent metal cations such as Mg2+(Craigie, 2001). Additionally, the CCD was also shown to possess viral DNA recognition and binding ability (Lewinski and Bushman, 2005). Compared to the NTD and CCD of IN, the CTD displays the greatest degree of variation across the retrovirus family. Despite this, studies on the CTD region of HIV-1 have revealed that it has strong DNA-binding activity and also potentially has multimerization capabilities (Woerner et al., 1992; Engelman et al., 1994; Lutzke and Plasterk, 1998; Gao et al., 2001).
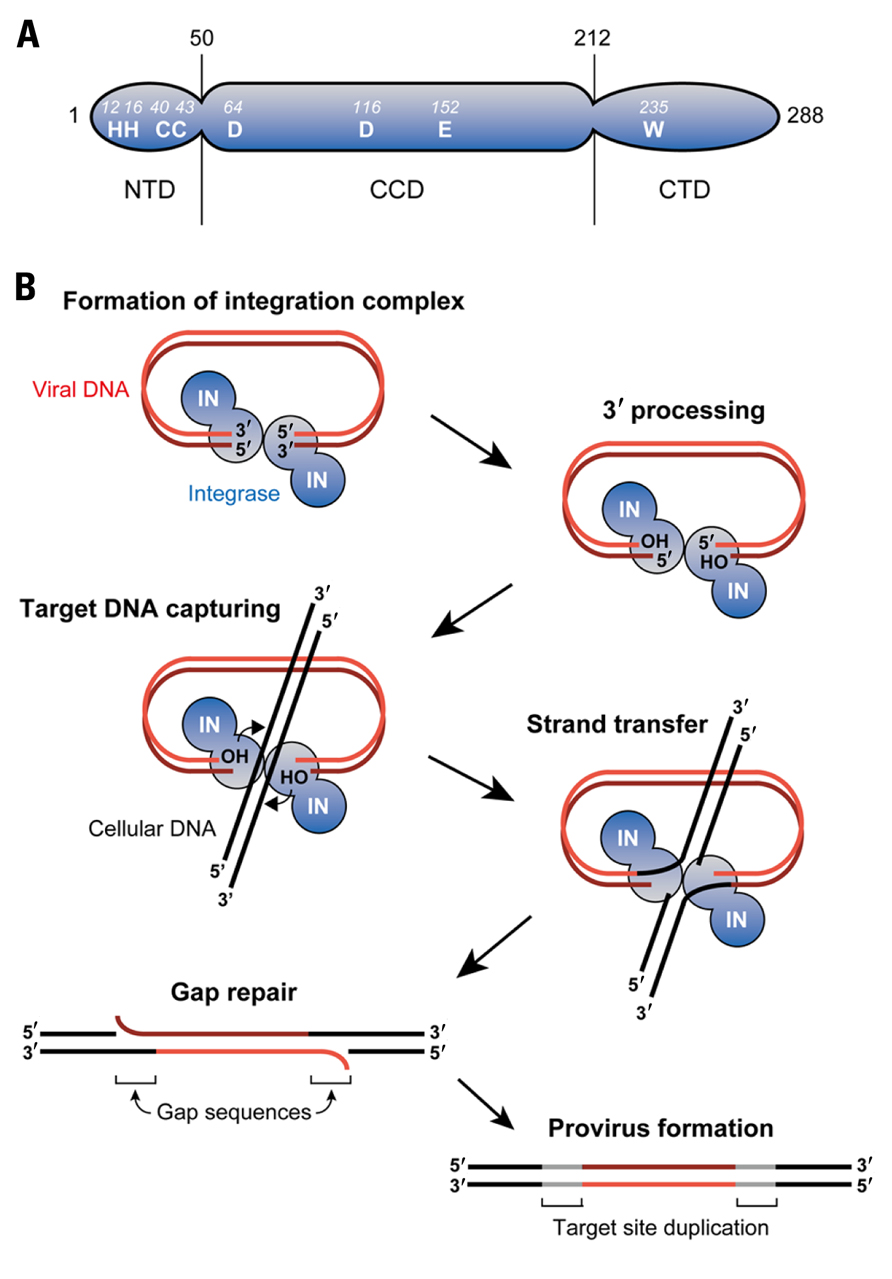
FIGURE 1.Retroviral DNA integration. (A) As an example of retroviral INs, domain organization of HIV-1 IN is illustrated. IN is composed of three domains: N-terminal domain (NTD), catalytic core domain (CCD), and C-terminal domain (CTD). A triad of aspartic (D) and glutamic acid (E) residues termed the DDE motif is found in the CCD, which is highly conserved amongst retroviral INs. Binding of divalent ions such as Mg2+ to the DDE motif domain plays a key role in integration reaction. The NTD contains a well-conserved motif comprising of two histidine (H) and two cysteine (C) residues (HHCC motif) that coordinates Zn2+ binding. In contrast to the NTD and CCD, the CTD is the least conserved domain, although tryptophan (W) is conserved among the retroviral INs. This domain exhibits non-specific DNA-binding activity in vitro. (B) Retroviral integration is initiated by IN’s recognition of both ends of viral DNA and subsequent removal of two (or three) nucleotides from each of the 3′ ends (3′ processing). The target DNA (i.e., chromosomal DNA) captured by IN is cleaved in a staggered fashion via the exposed hydroxy (OH) groups on the viral DNA ends, and the 3′ end of the viral DNA and the 5′ end of the target DNA is simultaneously linked (strand transfer). The 3′ processing and strand transfer steps are reproducible in vitro using recombinant IN, indicating that IN alone suffices to catalyze these steps. However, in infected cells, excision of the mispaired 5′ viral DNA ends and filling in the single-strand gaps are carried out by yet-to-be identified cellular enzymes.
The retrovirus-mediated integration reaction consists of three biochemically distinct steps (Figure 1B; Craigie, 2001). In the first step, IN specifically recognizes the viral attachment (att) sites on the 5′ and 3′ long terminal repeats (LTRs) of newly synthesized viral DNA ends. This interaction between IN and DNA allows for the 3′ processing of viral DNA, where water serves as the nucleophile for the cleavage of viral DNA. In this 3′ processing step, most retroviral IN such as HIV-1 IN catalyze the removal of two nucleotides adjacent to the highly conserved CA dinucleotide from the 3′ end of LTR regions. This chemically activates the viral DNA through the formation of 3′ hydroxyl radicals at the terminal ends of viral DNA, allowing it to participate in the next reaction.
The second step of integration involves the insertion of 3′ processed, activated viral DNA strand into the target DNA through a single transesterification reaction, which is called the strand transfer step (Engelman et al., 1991). IN brings the viral DNA ends into close proximity with the target DNA allowing a nucleophilic attack by the 3′ hydroxyl radical on the target DNA. Subsequently, IN ligates both the 3′ hydroxyl radical terminal of the viral DNA to the 5′ phosphoryl ends of the target DNA and establishes new phosphodiester bond between them. This forms intermediate DNA products with unrepaired gaps between the non-ligated 5′ ends of viral DNA as well as the 3′ ends of target DNA. Additionally, the unrepaired gaps result in the short duplication of target DNA sequences flanking both strand ends, followed by the formation of imperfect inverted repeats upon the sealing of the nick. The length of this inverted repeat sequences can vary from 4 to 6 base pairs (bp) and is highly characteristic of the retrovirus species from which the IN protein is derived. It should be noted that the 3′ processing and strand transfer steps have been shown to be catalyzed by IN proteins through various in vitro studies conducted with E. coli-expressed IN and DNA substrates harboring viral LTR sequence (Turlure et al., 2004).
Lastly, after the ligation reaction, the gaps in the intermediate DNA products are repaired to yield fully functional integrated proviruses (gag repair step). The previous step generates an intermediate product in which viral DNA is joined to the target DNA solely by the 3′ ends of both strands. As such, DNA repair is needed to seal the nick between the 5′ end of viral DNA and the 3′ end of the target DNA to complete the provirus formation. Unlike the earlier steps which are catalyzed by IN, this step is probably mediated by some cellular enzymes involved in a variety of DNA repair pathways including the non-homologous end joining (NHEJ) repair pathway. However, whether or not the NHEJ machinery is involved is controversial, and identification of the specific enzymes conducting this gap repair step is anticipated in future studies (Turlure et al., 2004; Smith and Daniel, 2006; Yoder et al., 2006).
The PIC, the key nucleoprotein complex responsible for the integration, is formed in the cytoplasm following the reverse transcription of viral DNA from the RNA genome (Figure 2). Although the full composition of this nucleoprotein complex has yet to be determined, most of the viral protein components of the PIC are thought to be derived from the core of the infecting virion, which includes not only IN but also reverse transcriptase (RT), matrix (MA), capsid (CA), and some HIV-1 accessory proteins (Suzuki and Craigie, 2007). With respect to the cellular components of the PIC, significant efforts have been made to identify them through surveys of IN-interacting proteins using yeast two-hybrid screenings and/or co-immunoprecipitation analysis (Figure 2; Turlure et al., 2004).
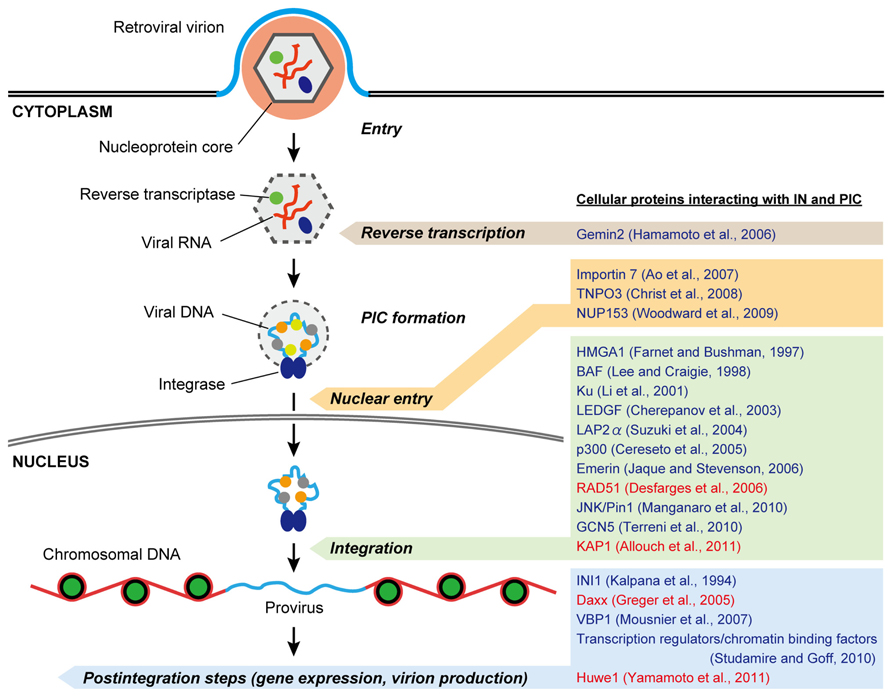
FIGURE 2. Possible roles of IN- and PIC-associated cellular proteins in retrovirus replication. After penetrating the cell membrane, the viral nucleoprotein core containing genomic RNA is released into the cytoplasm, after which reverse transcription of the viral RNA takes place. The synthesis of full-length viral DNA produces the PIC, in which cellular as well as viral proteins including IN are associated. 3′ processing of the viral DNA by IN occurs in the cytoplasmic PIC. Once the PIC reaches the nuclear periphery it must gain entry into the nucleus for integration. Some retroviruses seem to take advantage of nuclear-envelope disassembly during cell division to access chromosomal DNA, whereas the PICs of lentiviruses, including HIV-1, are believed to be capable of crossing the intact nuclear-envelope during interphase. In the nucleus, the PIC gains access to chromosomal DNA and mediates viral DNA integration to produce a provirus, which is acted upon by viral and/or cellular transcription factors to express viral RNAs. To date, a number of host proteins have been reported to interact with the retroviral IN and PIC (representative proteins are listed on the right). Many of them are likely to assist in the integration step, and some other cofactors appear to have functional roles in other steps including reverse transcription, nuclear entry, gene expression, and virion production during virus infection. In addition, recent studies demonstrate that several cellular interactors of IN may negatively regulate retrovirus replication (highlighted in red).
Integrase interactor 1 (INI1), also known as hSNF5, was the first binding partner of HIV-1 IN identified by yeast two-hybrid assay (Kalpana et al., 1994). This nuclear protein is the human homolog of the yeast SNF5 protein, a transcription activator and part of the SWI/SNF complex in mammalian systems (Wang et al., 1996). Interaction with the HIV-1 IN requires a direct imperfect repeat region of INI1 termed Rpt1 (Yung et al., 2001). The function of INI1 in HIV-1 integration was demonstrated in an in vitro integration assay where it stimulated the strand transfer activity of recombinant IN (Kalpana et al., 1994). However, the role of INI1 during virus replication is still a matter of debate. Co-transfection of the minimal IN-binding domain of INI1, including the Rpt1 region, with HIV-1 proviral DNA in HEK293 cells resulted in a nearly 10,000-fold reduction in virus production from the transfected cells (Yung et al., 2001). Furthermore, subsequent studies showed that INI1 is specifically incorporated into HIV-1 virions during virus production (Yung et al., 2004), suggesting a possible role of INI1 in the late stage of HIV-1 replication rather than in the integration step. Interestingly, it was revealed that IN and INI1 selectively recruit the components of the Sin3a–HDAC1 complex into HIV-1 virions, whereafter it is required for the reverse transcription step following the infection of subsequent target cells (Sorin et al., 2009). Thus, INI1 does not seem to be involved in the integration step in spite of its interaction with IN.
Lens epithelium-derived growth factor (LEDGF) is the first cellular protein to have been demonstrated as a bona fide cofactor for HIV-1 integration (Engelman and Cherepanov, 2008). LEDGF is a transcriptional regulatory protein with a molecular mass of 76 kDa from the hepatoma-derived growth factor (HDGF)-related protein (HRP) family, and was found to be strongly associated with chromatin throughout the cell cycle. This protein was identified as an interaction partner of HIV-1 IN through a co-immunoprecipitation study in human cells (Cherepanov et al., 2003) and has been shown to stimulate in vitro integration activity of IN (Cherepanov et al., 2004). LEDGF comprises several functional domains that have been implicated in the integration process. The N-terminal of LEDGF contains the PWWP (proline–tryptophan–tryptophan–proline) domain that is highly conserved amongst the members of the HRP family and was revealed to be crucial for protein–protein interaction and/or DNA-binding. Furthermore, the N-terminal also contains a putative nuclear localization signal (NLS) and dual copies of the AT-hook DNA-binding motif (Engelman and Cherepanov, 2008). In addition, a limited proteolysis analysis of recombinant LEDGF has shown that an evolutionally conserved domain, termed the integrase-binding domain (IBD), is included in its C-terminal and as its name suggests, this domain was found to be crucial for the specificity of the interaction with HIV-1 IN (Engelman and Cherepanov, 2008).
LEDGF is likely to play a critical but not strictly essential role in HIV-1 integration and replication. A study performed using intensified RNA interference demonstrated a significant reduction of HIV-1 replication in human CD4+ T cells in which there was a complete knockdown of endogenous LEDGF (Llano et al., 2006). Moreover, a knockout study done in mouse embryonic fibroblasts (MEFs) cell lines reported a reduction of 90% in HIV-1 infectivity upon the depletion of LEDGF/p75 and that this reduction was ablated upon re-expression of LEDGF (Shun et al., 2007). In these studies, it was found that the block in HIV-1 infection occurs specifically at the integration step, and that both the PWWP and IBD domains are of critical importance for HIV-1 integration and replication (Llano et al., 2006; Shun et al., 2007). Based on these observations, a simple mechanism model is proposed for LEDGF’s function in HIV-1 integration: LEDGF serves as a molecular adaptor that tethers HIV-1 IN to the target DNA (i.e., chromatin). Since LEDGF is a transcriptional coactivator, this tethering activity is thought to be responsible for targeting the integration site of HIV-1 into transcriptionally active regions (Engelman and Cherepanov, 2008).
After the PIC is formed in cytoplasm, it needs to be shuttled into the nucleus to allow the integration process to take place. One of the striking features of HIV-1 is its ability to get across the intact nuclear envelope easily even in non-dividing cells. This phenomenon is believed to be strongly associated with the karyophilic properties of the HIV-1 PIC (Suzuki and Craigie, 2007). Although the main component causing the PICs to be actively imported into the nucleus remains undetermined, some host-encoded proteins that interact with IN have been reported to facilitate the nuclear import of HIV-1 PICs. To date, importin 7 and TNPO3, have been reported as IN-interacting importins that direct the HIV-1 PICs to the nucleus (Fassati et al., 2003; Ao et al., 2007; Christ et al., 2008). Both proteins are members of the importin β family, which act as import receptors for cargo molecules through association with nucleoporin (NUP) of the nuclear pore complex (Suzuki and Craigie, 2007). While a confocal microscopy analysis of digitonin-permeabilized human cells showed the involvement of importin 7 in the nuclear accumulation of HIV-1 integration complexes, the functional role of this protein in virus replication is still unresolved (Fassati et al., 2003; Zaitseva et al., 2009). TNPO3, an HIV-1 IN-binding protein identified by yeast two-hybrid screening, was found to promote viral nuclear import as well (Christ et al., 2008). Although subsequent studies have confirmed the requirement of TNPO3 for efficient replication of HIV-1, the TNPO3–CA interaction, rather than the TNPO3–IN interaction, is likely to be required for the nuclear import of PIC in infected cells (Krishnan et al., 2010; De Iaco and Luban, 2011). By the same token, another cellular protein regulating nucleocytoplasmic trafficking, NUP153, was also shown to interact with HIV-1 IN by a pull-down assay (Woodward et al., 2009), but again, viral CA appears to be the viral determinant for the NUP153-mediated nuclear entry of the PIC (Matreyek and Engelman, 2011).
Intriguingly, a certain host protein called Gemin2 may be of importance to the reverse transcription process of HIV-1 via its physical association with IN. Gemin2 is a component of the survival of motor neurons (SMN) complex that is essential for the biogenesis of spliceosomal small nuclear ribonucleoproteins (snRNPs) and was identified as a novel interactor of HIV-1 IN by a yeast two-hybrid screening (Hamamoto et al., 2006). Although this cellular protein is found in the HIV-1 PIC, depletion of endogenous Gemin2 by siRNA in HIV-1-infected primary monocyte-derived macrophages resulted in impairment of the early reverse transcription step rather than the integration step (Hamamoto et al., 2006). Furthermore, a recent study showed that viral DNA synthesis by HIV-1 RT in vitro is enhanced by the concerted action of Gemin2 and IN (Nishitsuji et al., 2009). This augmentation of RT activity by an IN interaction appears to be reasonable because functional interaction between HIV-1 RT and IN has been revealed (Wilkinson et al., 2009). Hence, it would be assumed that Gemin2 serves as cofactor that stimulates and/or stabilizes the formation of the reverse transcription complex to initiate DNA synthesis through its interaction with IN (Masuda, 2011).
Several cellular proteins have been also identified as components of HIV-1 and Moloney murine leukemia virus (MoMLV) PIC by the in vitro reconstitution analysis and immunoprecipitation assays of PICs isolated from virus-infected cells. These include HMG proteins, barrier-to-autointegration factor (BAF, discussed below), Ku, and LEM proteins (Figure 2). Although there are some controversies as to whether these PIC-associated factors are indispensable to retrovirus infection, they may be indirectly involved in the integration process by stabilizing the nucleoprotein complex, promoting the nuclear retention of the PIC, or protecting host cells from viral DNA termini-induced apoptosis (Turlure et al., 2004; Van Maele et al., 2006; Suzuki and Craigie, 2007). Besides the host proteins listed above, more extensive discussions about the other cellular factors interacting with the IN/PIC and their roles in retrovirus replication are presented in previous comprehensive reviews (Turlure et al., 2004; Al-Mawsawi and Neamati, 2007; Goff, 2007; Studamire and Goff, 2010).
In addition to the cellular enhancers for retroviral integration, the existence of potential antiviral proteins controlling the integration activity of IN and PIC has been revealed by several recent studies. Understanding the molecular details by which these factors hamper the integration reaction may potentially lead to future therapeutic inventions against retroviral infectious diseases. Recent findings on the potential restriction factors for integration process are summarized in this section, and their molecular inhibitory mechanisms are discussed.
Although retroviral integration is completed by the gap repair step, which is believed to be mediated by the cellular DNA repair enzymes (Daniel et al., 1999; Lewinski and Bushman, 2005), one of the homologous recombination (HR) proteins, RAD51, has been shown to suppress HIV-1 integration (Desfarges et al., 2006; Cosnefroy et al., 2012). The downregulation of integration by RAD51 was first observed in a unique integration assay using a budding yeast strain in which DNA substrates containing zeocin-resistance gene flanked by HIV-1 U3 and U5 LTR ends can be integrated into genomic DNA by HIV-1 IN expressed by the yeast (Desfarges et al., 2006). When a haploid yeast strain deficient for the RAD51 encoding gene was examined using the integration assay, higher numbers of zeocin-resistant clones were obtained, indicating an increase in integration events in the yeast cells lacking RAD51. This result suggests that RAD51 negatively regulates the HIV-1 IN-catalyzed integration reaction in yeast. In accordance with the yeast integration assay, additional in vitro analyses using purified recombinant proteins showed that human RAD51 (hRAD51) inhibited integration by IN. Interestingly, HIV-1 IN binds hRAD51 in vitro, suggesting that the inhibition of the integration reaction may be mediated through the physical interaction between IN and RAD51 (Desfarges et al., 2006).
Human RAD51 belongs to the RAD52 epistasis group, and members of the RAD52 epistasis group are highly conserved in yeast and humans, which mediate mitotic HR events as well as chromosome segregation during meiosis (San Filippo et al., 2008). RAD51 is also an ortholog of the E. coli RecA (Figure 3A). The RecA family proteins possess a conserved, ~230 amino acid core domain that is involved in the binding and hydrolysis of ATP through Walker A and B motifs, with the human RAD51 also bearing a long N-terminal and a short CTDs (Figure 3A; Lin et al., 2006; Wang et al., 2008). The RAD51 protein exists as a homo-oligomer in solution, and binds to single-stranded (or double-stranded) DNA to form a right-handed helical presynaptic nucleoprotein. In the presence of ATP, RAD51 can polymerize on the DNA to form a nucleofilament that expands for thousands of bases (or base pairs, Figure 3B). This nucleoprotein filament serves as the catalytic center for DNA strand-exchange reactions during HR (San Filippo et al., 2008).
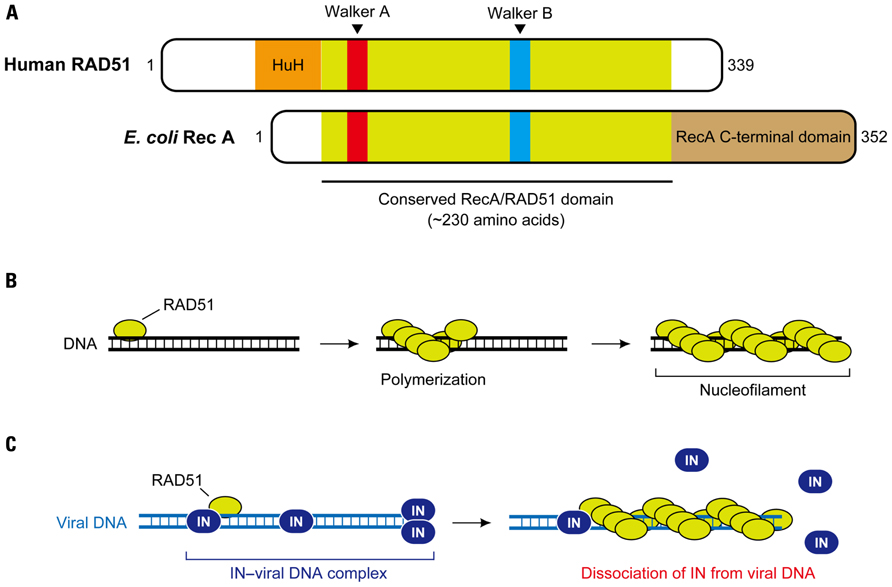
FIGURE 3. RAD51-mediated inhibition of HIV-1 integration. (A) Domain structure of human RAD51. RAD51 and its bacterial counterpart, RecA, share a highly conserved ~ 230 amino acid domain that contains two putative ATP-binding domains, Walker A and Walker B motifs. DNA-binding activity of hRAD51 is attributed to its N-terminal domain, which includes a modified HhH motif, while RecA binds to DNA through its long C-terminal domain. (B) Nucleofilament formation of RAD51. Upon assembly on DNA, RAD51 forms a right-handed helical polymer on the DNA in presence of ATP, and the RAD51-DNA nucleoprotein filament expands to thousands of bases. (C) Disruption of HIV-1 integration complex by the hRAD51 nucleofilament. It is suggested that formation of an active nuclear filament following polymerization of hRAD51 on viral DNA eventually displaces IN, thereby inducing dysfunction of the HIV-1 integration complex prior to the integration reaction. Recruitment of hRAD51 onto viral DNA may be facilitated by its interaction with IN.
The formation of the nucleofilament is also likely to be important for the downregulation of HIV-1 IN activity by RAD51. In a recent report, Cosnefroy et al. (2012) set up an in vitro integration assay for IN in which active nucleofilament formation of RAD51 on substrate DNA was allowed to take place. They revealed that strong inhibition of HIV-1 IN activity was only observed with wild-type hRAD51 in the presence of ATP, but not in the reactions without ATP or with an hRAD51 mutant containing a single amino acid substitution in the Walker A motif (K133A) that ablated hRAD51–ATP-binding and therefore prevented hRAD51 polymerization (Cosnefroy et al., 2012). An additional in vitro experiment using a substrate DNA containing LTR sequences complexed with HIV-1 IN showed that even after the IN–viral DNA complexes were formed, subsequent incubation with hRAD51-induced dissociation of IN from its substrate viral DNA. The dissociation effect was not observed with hRAD51 in the absence of ATP or with the K133A mutant (Cosnefroy et al., 2012). Given that RAD51 can polymerize on DNA in an ATP-dependent manner, these observations suggest that the formation of the nucleofilament on viral DNA is responsible for the RAD51-induced restriction of HIV-1 integration via displacement of IN (Figure 3C). Furthermore, when an allosteric effector of RAD51, RS-1 (RAD51-stimulatory compound 1), which promotes formation of the active RAD51 nucleofilament, was used, HIV-1 integration and replication was significantly impaired in CD4+ cells. This was accompanied in an increase in 2-LTR circles, which is a byproduct from the failure to integrate (Cosnefroy et al., 2012). This in vivo experiment supports the proposed model in which hRAD51 recruited by HIV-1 IN is able to release the IN from viral DNA via formation of the nucleofilament, resulting in dysfunction of the PIC (Figure 3C). Although it is still unclear whether the hRAD51 in a natural setting could restrict HIV-1 integration, this study provides the possibility of using a specific stimulator of RAD51 such as RS-1 for a new antiviral approach against HIV infection.
Posttranslational modifications (PTMs) including phosphorylation, ubiquitination, and acetylation is the chemical alternation of a protein’s primary structure after its translation, a process which is well known to have a great impact on various protein functions such as enzymatic activity, protein–protein interaction, and subcellular localization (Walsh et al., 2005). Although the association between PTMs and the retroviral integration process has not been fully addressed, it was demonstrated that the enzymatic activity of HIV-1 IN is positively regulated by histone acetyl transferases (HATs; Cereseto et al., 2005; Terreni et al., 2010). HATs are the cellular enzymes that transfer an acetyl group from acetyl-coenzyme A (acetyl-CoA) to the ε-amino group of certain basic lysine residues of histone’s N-terminal, which modifies the accessibility of DNA to other proteins (Roth et al., 2001). p300, the first HAT protein that was reported to catalyze the acetylation of HIV-1 IN, is a ~300 kDa nuclear phosphoprotein originally isolated as an interaction partner of adenovirus E1A (Sterner and Berger, 2000). This HAT is also a paralog of the CREB-binding protein (CBP), and the p300/CBP family proteins contain several identical domains necessary for their function as HATs, which include three cysteine–histidine rich domains (CH1, CH2, CH3), a KIX domain, a bromodomain, and a HAT domain. (Figure 4A). p300 directly binds to and acetylates three specific lysine residues, Lys-264, Lys-266, and Lys-273, of the C-terminal region of HIV-1 IN (Figure 4C), which is likely to enhance the binding affinity to LTR DNA as well as strand transfer activity of IN in vitro (Cereseto et al., 2005). Importantly, an HIV-1 strain that contained arginine substitutions at the critical C-terminal lysine residues on IN exhibited a significant replication defect in CD4+ cells, and the replication defect occurred at the integration step, indicating the importance of C-terminal acetylation of IN in HIV-1 integration (Cereseto et al., 2005). An another HAT, GCN5 (Figure 4A), was also found to mediate the acetylation of HIV-1 IN at the same C-terminal lysines, and HIV-1 integration was shown to be impaired in GCN5 knockdown cells (Terreni et al., 2010).
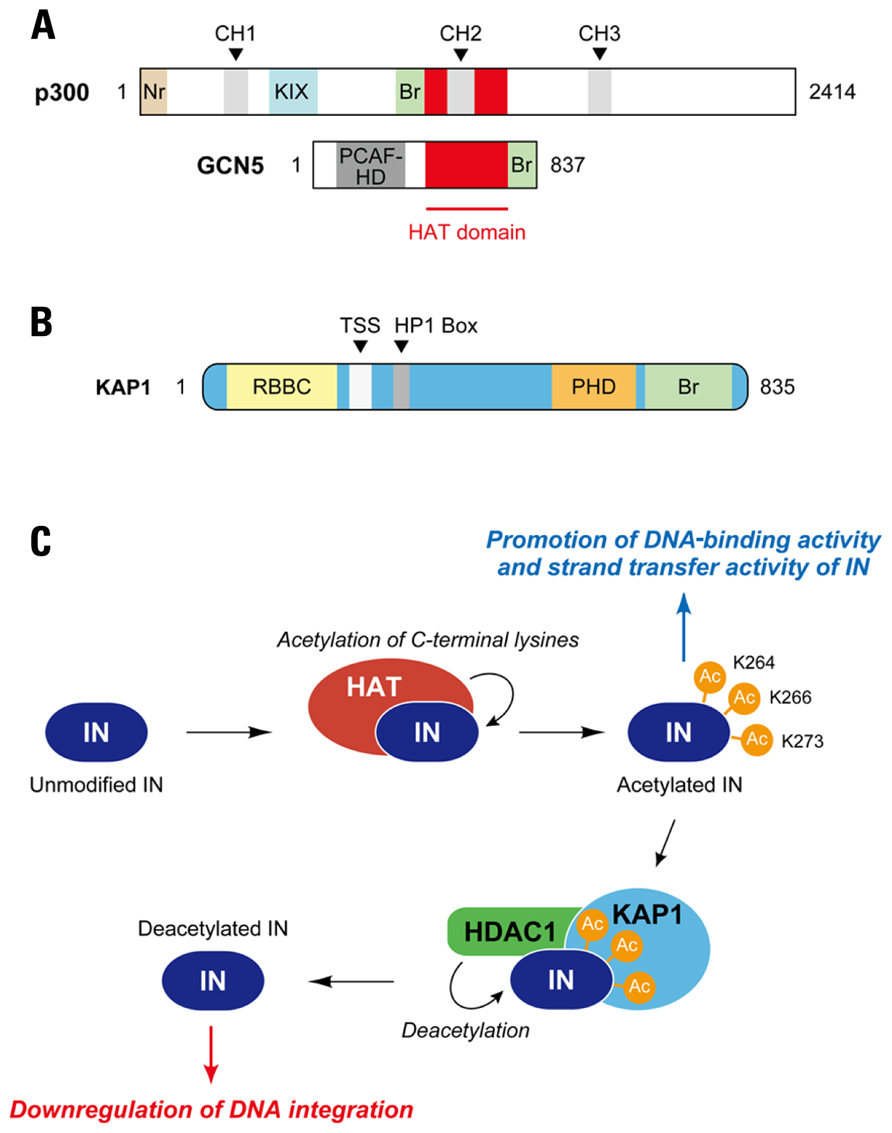
FIGURE 4. Regulation of HIV-1 integration by acetylation and deacetylation of IN. (A) Acetyltransferases for HIV-1 IN. p300 and GCN5 have been identified as cellular enzymes catalyzing acetylation of HIV-1 IN. Both proteins are members of histone acetyltransferase (HAT) family, which transfer an acetyl group from acetyl coenzyme A to specific lysine residues on substrate proteins; HATs function as transcriptional co-activators in the nucleus. Although a conserved HAT domain is indispensable to acetylation activity of HATs, other domains including bromodomain (Br) apparently have effects on their function. p300 is able to bind a variety of transcription factors through cysteine-histidine-rich module (CH1, CH2, and CH3) and KIX domain. Nr, nuclear receptor-interacting box; PCAF-HD, PCAF homology domain. (B) Schematic of KAP1. KAP1, also known as TRIM28, is highly related to three other TRIM proteins, TIF1α, TIF1γ, and TIF1δ. The TIF proteins, including KAP1, exhibit similar domain organization, which includes RBCC (Ring/B-box/coiled coil) domain at the N-terminal, TIF1 signature sequence (TSS) and heterochromatin protein 1 (HP1)-binding domains at central portion, and plant homeodomain (PHD) and bromodomain (Br) at C-terminal. (C) Restriction of HIV-1 integration by KAP1. Cellular acetyltransferase (HAT) such as p300 interacts with HIV-1 IN and catalyzes acetylation of the IN. Three lysine residues in CTD of IN (Lys-264, Lys-266, and Lys-273) have been reported as target sites for p300 and GCN5. The acetylation appears to enhance the DNA-binding activity and strand transfer activity of IN. In a proposed model, KAP1 specifically recognizes the acetylated form of IN and induces a protein complex formation with histone deacetylase (HDAC), resulting in deacetylation of IN and reduced integration activity.
In the case of histones, acetylation of the internal lysines affects several aspects of nucleosome function and generates new protein–protein interactions. However, it is a reversible process, and transcriptional activation of the nucleosome is repressed by deacetylation mechanisms, which are mediated by histone deacetylases (HDAC; Shahbazian and Grunstein, 2007). It is likely that deacetylation of HIV-1 IN also takes place. To find a cellular partner that selectively recognizes the acetylated IN, Cereset and colleagues developed a unique yeast two-hybrid screening assay, in which acetylated HIV-1 IN was constitutively expressed as the bait by fusion of the p300 HAT domain to IN (Allouch et al., 2011). From the screening of a human lymphocyte cDNA library, they identified that a cellular protein, KAP1 was able to interact with acetylated IN (Figure 4B; Allouch et al., 2011). KAP1 was first identified as a transcriptional corepressor that is recruited to its target genes via interaction with the Kruppel-associated box-domain-containing (KRAB) zinc-finger DNA proteins (Friedman et al., 1996; Peng et al., 2000). Interestingly, KAP1 is a member of the human TRIM family, and hence contains a RBCC (Ring [really interesting new gene] finger, two B-box zinc fingers and a coiled coil) domain at the N-terminal, which is a characteristic feature of the TRIM family protein domains (Figure 4B; Iyengar and Farnham, 2011). Additionally, KAP1, also called TRIM28 and transcription intermediary factor (TIF) 1β, is composed of a plant homeodomain (PHD) and bromodomain at the C-terminus, and contains a central TIF1 signature sequence (TSS) domain and a heterochromatin protein 1 (HP1)-binding domain, which are found in other TIF1 subfamily members (Iyengar and Farnham, 2011). In accordance with the yeast two-hybrid screen, preferential binding of KAP1 to acetylated HIV-1 IN was confirmed by in vitro and in vivo analysis. Importantly, knockdown of KAP1 increased the level of integration in HIV-1-infected cells, which was accompanied by a decrease in 2-LTR circles (Allouch et al., 2011). When the infection experiments were performed with HIV-1 carrying mutations in the C-terminal lysines (Lys-264, Lys-266, Lys-273) of IN, knockdown of KAP1 did not significantly increase the levels of integrated DNA, indicating that KAP1-mediated inhibition was related to the C-terminal acetylation of IN. Consistent with this, the level of acetylated IN was shown to be decreased by expression of KAP1 in cells (Allouch et al., 2011). KAP1 has been reported to form complexes with HDAC to regulate transcription at specific target sites on the genome via modification of histone structure (Iyengar and Farnham, 2011). Together with the observation that KAP1 has a higher affinity to acetylated IN, the evidence raises a possibility that KAP1 may serve as a scaffolding mediator that recruits HDAC to acetylated HIV-1 IN (Figure 4C). Indeed, HIV-1 IN formed a complex with KAP1 and HDAC1, and HIV-1 integration was enhanced by treatment with the HDAC inhibitors (Allouch et al., 2011). These evidences therefore support the proposed model that KAP1 recruits the HDAC to acetylated IN allowing for the HDAC1-mediated deacetylation of IN to take place, which results in the subsequent reduction of integration efficiency in HIV-1 infection (Figure 4C).
A different inhibitory mechanism of KAP1 has been also demonstrated in other retroviruses and endogenous retroviruses, which involves the transcriptional repression of the provirus in embryonic carcinoma and embryonic stem cells (Wolf and Goff, 2007, 2009; Wolf et al., 2008; Matsui et al., 2010; Rowe et al., 2010). The KAP1-mediated inhibition of viral transcription appeared to be largely attributable to the recruitment of KAP1 to certain types of primer binding sites (PBS) near the 5′ end of the provirus containing PBS sequences complementary to tRNAPro (MoMLV and human T-lymphotropic virus type I [HTLV-1]) and tRNALys-1,2 (spumavirus and Mason–Pfeizer monkey virus; Wolf et al., 2008; Wolf and Goff, 2009). In contrast, HIV-1 is not susceptible to the transcriptional silencing induced by KAP1 as its PBS is complementary to neither tRNAPro nor tRNALys-1,2 (Wolf et al., 2008). Since the study by Cereset and colleagues showed that MoMLV integration was not affected by depletion of KAP1 (Allouch et al., 2011), the KAP1-mediated retroviral integration block should only occur in HIV-1 replication due to the specific interaction between KAP1 and HIV-1 IN. Therefore, it can be concluded that KAP1 is an intrinsic restriction factor against a broad range of retroviruses, which inhibits either the integration or post-integration step. However, despite the ubiquitous expression of KAP1 in many cell types, substantial levels of HIV-1 integration can still be detected, indicating KAP1 may not be a very powerful integration inhibitor. Yet, it would shed light on a potentially novel KAP1-based strategy for the treatment of retrovirus infections.
Phosphorylation is another type of PTM that is also likely to influence in HIV-1 integration. Recently, phosphorylation of HIV-1 IN by c-Jun NH2-terminal kinase (JNK) was reported (Manganaro et al., 2010). JNK is one of the major groups of mitogen-activated protein kinases (MAPKs), a family of well-studied serine/threonine kinases involved in signal transduction from extracellular stimuli such as growth factors, cytokines, infection, and stress (Raman et al., 2007; Cargnello and Roux, 2011). JNK consists of three variants, JNK1, JNK2, and JNK3, which are generated through alternative splicing but exhibit distinct expressions and functions (Sabapathy et al., 2004). The MAPK signaling pathway can also be activated by the interaction of T cell receptors with antigen-major histocompatibility complex (MHC) class II molecules, resulting in T cell activation (Weiss and Littman, 1994). In HIV infection, the requirement for the activation states of CD4+ T cells for efficient initiation and maintenance of replication has been well established (Stevenson et al., 1990; Pope and Haase, 2003; Stevenson, 2003; Chiu et al., 2005). In particular, it has been demonstrated that HIV-1 infection is restricted at the post-entry stages in resting T cells, following reverse transcription and prior to integration (Bukrinsky et al., 1992; Pierson et al., 2002; Coiras et al., 2009).
Manganaro et al. (2010) reported that although expression levels of JNK was low in the resting CD4+ T cells, it became activated upon stimulation, and that even in activated CD4+ T cells, HIV-1 infection was impaired by treatment with a specific JNK inhibitor, SP600125. Furthermore, decreased amounts of integrated DNA was detected in the SP600125-treated activated CD4+ T cells, indicating JNK contributes to the efficient infection and integration of HIV-1. Importantly, IN was found to be phosphorylated, and a conserved serine residue in the CCD, Ser-57, was identified as the target site of phosphorylation. Since the phosphorylated IN could not be detected in cells treated with SP600125, JNK appeared to be the kinase responsible for the phosphorylation of IN (Manganaro et al., 2010).
Next question to be asked was how the IN phosphorylation augmented the efficiency of HIV-1 integration. In vitro integration activity assays using recombinant IN showed that an alanine substitution at the Ser-57 residue did not affect the strand transfer activity of HIV-1 IN, suggesting that this serine residue itself is not involved in the enzymatic activity of IN (Manganaro et al., 2010). Meanwhile, Manganaro et al. (2010) found that the Ser-57 and a flanking Pro-58 are conserved amongst most of the HIV-1 clades and subtypes, and that the phosphorylated of the serine/proline motif can potentially be recognized by peptidyl-prolyl cis-trans isomerase (PPIase), Pin1 (Lu and Zhou, 2007). Pin1 is a regulator of phosphorylation signaling which specifically recognizes phosphorylated serine or threonine residues preceding a proline. Pin1 then catalyzes the structural rearrangement of a target molecule through cis-trans isomerization of its specific proline (Lu and Zhou, 2007). Pin1 binds wild-type HIV-1 IN, but not the IN mutant with a single amino acid mutant on the phosphorylation site (S57A). As expected, treatment of IN-expressing cells with a Pin1 inhibitor, Pib, decreased the stability of HIV-1 IN. Moreover, in vitro incubation of recombinant IN with Pin1 increased IN resistance against a protease, subtilisin, suggesting that Pin1 induced a conformational change in phosphorylated IN, thereby rendering it less sensitive to protein degradation (Manganaro et al., 2010). Supporting this notion, treatment of cells with a Pin1 inhibitor or the depletion of endogenous Pin1 by siRNA reduced the level of integrated DNA in HIV-1-infected cells without any impairment of the reverse transcription step (Manganaro et al., 2010). More important, whereas HIV-1 carrying the S57A IN mutant exhibited decreased integration activity, treatment with the proteasome inhibitor MG132 restored integration activity (Manganaro et al., 2010), demonstrating that JNK-mediated phosphorylation of HIV-1 IN at Ser-57 and subsequent prolyl cis-trans isomerization by Pin1 increased the stability of IN and thereby enhanced integration efficiency in infected CD4+ T cells. Taken together, resting CD4+ T cells could have lower expression/activation levels of JNK, leading to decreased IN phosphorylation and consequently decreased IN stability, resulting in inefficient HIV-1 replication. Also, one could speculate that extracellular stimulations, including cytokines and chemokines, may enhance integration in resting T cells via activation of the JNK pathway (Cameron et al., 2010; Guo et al., 2011).
Of particular interest is that the study by Manganaro et al. (2010) also implies yet-to-be identified host factor(s) participating the degradation of IN, particularly unphosphorylated form of IN, in the resting T cells. HIV-1 IN is well known to undergo proteasome-mediated degradation via ubiquitination (Devroe et al., 2003). A previous study has reported that the von Hippel–Lindau binding protein 1 (VBP1) and cullin2-based von Hippel–Lindau (Cul2/VHL) ubiquitin ligase cooperate to mediate the polyubiquitination and subsequent proteasomal degradation of IN, but this event was likely to occur at a post-integration step and also to promote gene expression from the integrated provirus in the nucleus (Mousnier et al., 2007). Therefore, further work will be required to identify novel ubiquitin–proteasome proteolytic pathways that drive the cytoplasmic degradation of HIV-1 IN in a manner that restricts integration.
Integration reactions mediated by PICs can be categorized into two pathways: (i) intermolecular integration into a target DNA and (ii) intramolecular integration into the viral DNA, which is also called autointegration (Lee and Craigie, 1994). In order to establish productive infection, the retrovirus (or retroviral PIC) needs to avoid the suicidal autointegration pathway. Hence, a strong preference for intermolecular integration is one of the key features of the retroviral PIC (Lee and Craigie, 1994). BAF was identified as a host factor responsible for blocking the autointegration of MoMLV PICs (Lee and Craigie, 1998). BAF is a small dimeric protein (Margalit et al., 2007) and shown as a cellular component of HIV-1 and MoMLV PICs (Suzuki and Craigie, 2002; Lin and Engelman, 2003). Crystal structure analysis has revealed that BAF binds double-stranded DNA through its helix-hairpin-helix (HhH) motif (Bradley et al., 2005; Figure 5A). Importantly, the dimerization of BAF appears to cross-bridge DNA, thereby preventing the autointegration of retroviral PICs by compacting viral DNA into rigid structure (Lee and Craigie, 1998; Suzuki and Craigie, 2002; Bradley et al., 2005).
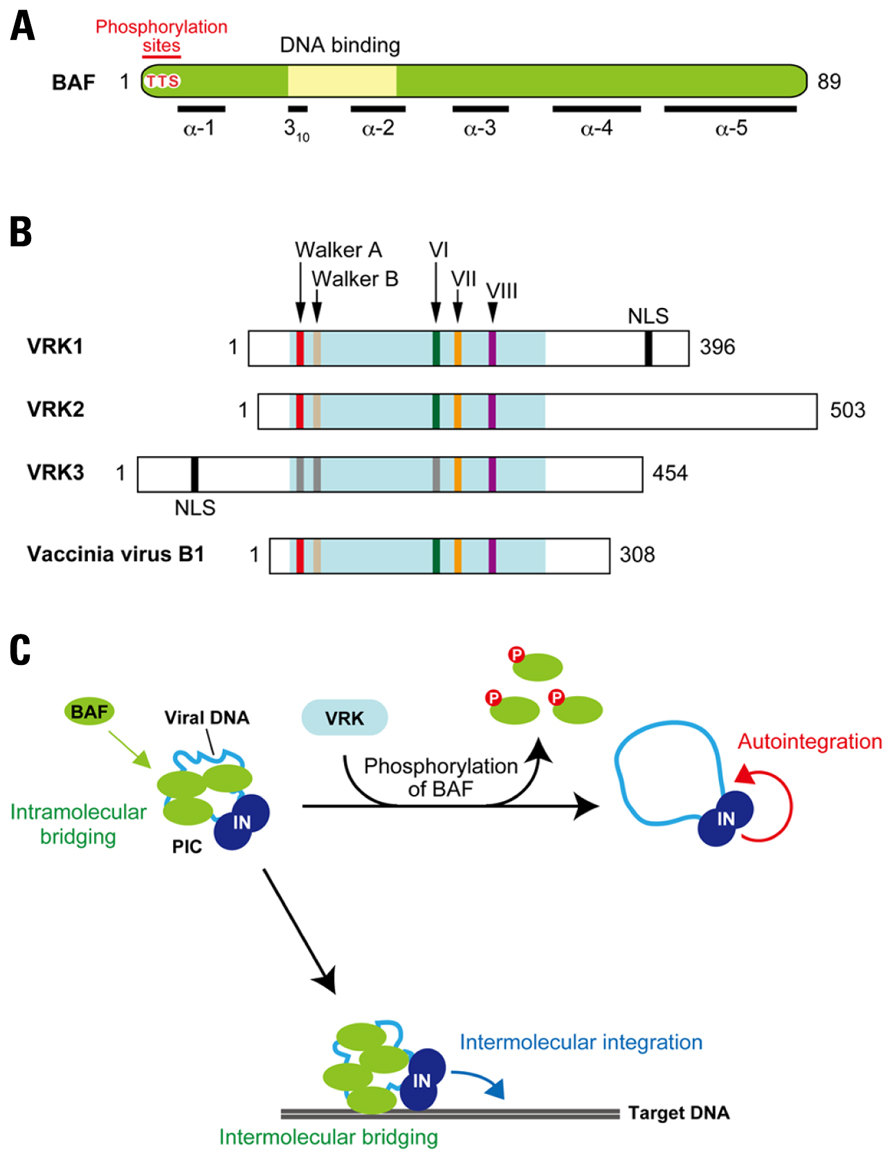
FIGURE 5. Disruption of the retroviral PIC by phosphorylation of BAF. (A) Features of BAF. BAF is highly conserved among multicellular eukaryotes, and 86 out of 89 amino acid residues are identical between humans and mice. This protein is composed of five helices (α-1 to -5) and a 310-helical turn, and DNA-binding activity of BAF is thought to be attributed to the HhH motif. N-terminal threonine (T) and serine (S) residues (Thr-2, Thu-3, and Ser-4) indicated in red are reported as phosphorylation sites of BAF. (B) VRK family proteins. Domain organization of mouse VRKs and their vaccinia viral homolog, B1 kinase, are shown. All the VRK/B1 family proteins contain highly conserved enzymatic domain involved in the phosphotransfer reaction, which is consists of the ATP binding motifs (Walker A and Walker B), catalytic domain (subdomain VI), active site (subdomain VII), and substrate recognition domain (subdomain VIII). However, some substitutions in several residues are found in Walker A, Walker B, and subdomain VI of VRK3, which may affect kinase activity of this ortholog. Additionally, VRK1 and VRK3 possess putative NLS at the C-terminal and N-terminal portions, respectively. (C) Reciprocal regulation of PIC integration activity by BAF and VRK. As the viral double-stranded DNA is synthesized by reverse transcription, BAF is recruited into the PIC in cytoplasm through direct binding with viral DNA. Once BAF is incorporated into the PIC, viral DNA is intramolecularly bridged by BAF’s dimerization and DNA-binding activities. This intramolecular bridging by BAF compacts the viral DNA, making it less accessible as a target for autointegration. In addition, PIC associated BAF also anchors to the target DNA by its DNA bridging property (intermolecular bridging), thereby stimulating the intermolecular integration activity of the PIC. On the other hand, VRK is able to induce release of BAF from the PIC via phosphorylation of BAF. By the dissociation of BAF, viral DNA may adopt a more open structure, resulting in its increased availability for autointegration.
Barrier-to-autointegration factor is involved in the organization of nuclear architecture through interaction with lamin A and lamina-associated polypeptide 2-emerin-MAN1 (LEM) family proteins, and therefore predominantly localizes in the nucleus during interphase (Margalit et al., 2007). On the other hand, in the mitotic phase, BAF is diffusely localized and subsequently assembles on the chromatin core region at anaphase to form a scaffold for the gathering of lamin A and LEM proteins to reconstitute the nuclear envelope (Segura-Totten and Wilson, 2004; Margalit et al., 2007). Intriguingly, these cell cycle-dependent actions of BAF appear to be regulated by the phosphorylation (Margalit et al., 2007). So far, vaccinia-related kinases (VRKs) have been identified as the cellular kinases that participate in the BAF phosphorylation (Figure 5B; Nichols et al., 2006). VRKs are a family of serine/threonine kinases, and three alternative splicing isoforms, VRK1, VRK2, and VRK3, are found in human and murine cells (Nichols et al., 2006; Klerkx et al., 2009). They share several domains comprised of ATP-binding motifs (Walker A and Walker B), catalytic domains (subdomain VI), active sites (subdomain VII), and substrate recognition domains (subdomain VIII; Figure 5B; Boyle and Traktman, 2004). VRK was first identified as cellular homolog of the poxvirus (vaccinia virus) B1 kinase that plays an essential role in viral DNA replication in the early stage of vaccinia virus infections (Figure 5B; Nichols et al., 2006). Importantly, VRK-mediated phosphorylation of BAF causes the loss of its DNA-binding ability (Nichols et al., 2006). Depletion of VRK1 in Caenorhabditis elegans results in several mitotic defects and BAF delocalization (Gorjanacz et al., 2007), suggesting that the release of BAF from chromatin during mitosis is regulated by the VRK family proteins in a phosphorylation-dependent manner.
Since the DNA-binding property of BAF is crucial to the authentic integration activity of the retroviral PIC, down-modulation of the BAF activity by phosphorylation may disrupt the integration function of the PIC through dissociation of BAF from viral DNA. Indeed, our recent study revealed that BAF could be removed from the PIC following treatment with recombinant VRK1 in vitro (Suzuki et al., 2010). More important, the intermolecular integration activity of the PIC assay was strongly inhibited by the VRK1 treatment, and this was accompanied by an increase in autointegration activity (Lee and Craigie, 1994; Suzuki et al., 2010). Little inhibition of the PIC activity was observed in following treatment with mutant VRK1 harboring alanine substitutions in the catalytic subdomain VI, and also in PICs treated with wild-type VRK1 in the presence of a well-known kinase inhibitor, staurosporine, indicating that disruption of PIC function is attributed to the kinase activity of VRK1 (Suzuki et al., 2010). Based on these findings, a potential inhibitory mechanism by which VRK1 restricts PIC integration activity can be proposed: phosphorylation by VRK1 dissociates BAF from the PIC, resulting in destructive autointegration of viral DNA (Figure 5C).
When other members of the VRK family of proteins were assessed for their ability to inhibit the intermolecular integration activity of PICs, VRK2, but not VRK3, was found to abrogate the function of PICs in a similar manner with VRK1 (Suzuki et al., 2010). This raised the question of why VRK3 was unable to inhibit PIC activity. Phosphorylation of BAF exhibits biphasic phases (hypophosphorylation and hyperphosphorylation), with the phosphorylation taking place on the Thr-2, Thr-3, and Ser-4 located on the N-terminus of BAF (Figure 5A). VRK1 and VRK2 are able to catalyze the hyperphosphorylation (Nichols et al., 2006). However, the BAF product phosphorylated by VRK3 is the hypophosphorylated form, and an in vitro kinase assay using BAF mutants, in which Thr-2, Thr-3, and/or Ser-4 had been substituted with alanine, revealed that Ser-4 is the sole site for the VRK3-mediated hypophosphorylation (Suzuki et al., 2010). Ser-4 has been also indicated as initial phosphorylation site by VRK1 and VRK2 (Nichols et al., 2006) and our preliminary results of an in vitro kinase assay using BAF mutants showed that subsequent phosphorylation likely takes place on Thr-3 (Suzuki and Suzuki, unpublished data). These results suggest that Ser-4 is the primary target of all VRK family proteins, but that Thr-3 is an additional preferred target of VRK1 and VRK2. The hyperphosphorylation of BAF at both the N-terminal Thr-3 and Ser-4 should therefore be a prerequisite for the functional disruption of PIC function. Although the findings of our in vitro studies propose that VRK is a cellular kinase harboring potentially restrictive activity against retroviral integration, delineating the precise role of the VRK family of proteins in retrovirus infection and integration in vivo requires further experimentation.
As discussed in this review, the processes leading up to integration are assisted by a number of host proteins. Furthermore, it has become evident that there are some cellular proteins that act as potential restriction factors. Research into intracellular effectors and inhibitors of retroviral integration therefore provides an important insight for the development of novel classes of anti-HIV drugs that can act synergistically with existing drugs targeting the active sites of viral enzymes or the binding pockets of cellular receptors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Wei Xin Chin for proofreading and commenting on the manuscript.
Allouch, A., Di Primio, C., Alpi, E., Lusic, M., Arosio, D., Giacca, M., and Cereseto, A. (2011). The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 9, 484–495.
Al-Mawsawi, L. Q., and Neamati, N. (2007). Blocking interactions between HIV-1 integrase and cellular cofactors: an emerging anti-retroviral strategy. Trends Pharmacol. Sci. 28, 526–535.
Ao, Z., Huang, G., Yao, H., Xu, Z., Labine, M., Cochrane, A. W., and Yao, X. (2007). Interaction of human immunodeficiency virus type 1 integrase with cellular nuclear import receptor importin 7 and its impact on viral replication. J. Biol. Chem. 282, 13456–13467.
Bowerman, B., Brown, P. O., Bishop, J. M., and Varmus, H. E. (1989). A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev. 3, 469–478.
Boyle, K. A., and Traktman, P. (2004). Members of a novel family of mammalian protein kinases complement the DNA-negative phenotype of a vaccinia virus ts mutant defective in the B1 kinase. J. Virol. 78, 1992–2005.
Bradley, C. M., Ronning, D. R., Ghirlando, R., Craigie, R., and Dyda, F. (2005). Structural basis for DNA bridging by barrier-to-autointegration factor. Nat. Struct. Mol. Biol. 12, 935–936.
Brown, P. O., Bowerman, B., Varmus, H. E., and Bishop, J. M. (1989). Retroviral integration: structure of the initial covalent product and its precursor, and a role for the viral IN protein. Proc. Natl. Acad. Sci. U.S.A. 86, 2525–2529.
Bukrinsky, M. I., Sharova, N., Dempsey, M. P., Stanwick, T. L., Bukrinskaya, A. G., Haggerty, S., and Stevenson, M. (1992). Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. U.S.A. 89, 6580–6584.
Bushman, F. D., Fujiwara, T., and Craigie, R. (1990). Retroviral DNA integration directed by HIV integration protein in vitro. Science 249, 1555–1558.
Cameron, P. U., Saleh, S., Sallmann, G., Solomon, A., Wightman, F., Evans, V. A., Boucher, G., Haddad, E. K., Sekaly, R. P., Harman, A. N., Anderson, J. L., Jones, K. L., Mak, J., Cunningham, A. L., Jaworowski, A., and Lewin, S. R. (2010). Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 107, 16934–16939.
Cargnello, M., and Roux, P. P. (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83.
Cereseto, A., Manganaro, L., Gutierrez, M. I., Terreni, M., Fittipaldi, A., Lusic, M., Marcello, A., and Giacca, M. (2005). Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J. 24, 3070–3081.
Cherepanov, P., Devroe, E., Silver, P. A., and Engelman, A. (2004). Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 279, 48883–48892.
Cherepanov, P., Maertens, G., Proost, P., Devreese, B., Van Beeumen, J., Engelborghs, Y., De Clercq, E., and Debyser, Z. (2003). HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 278, 372–381.
Chiu, Y. L., Soros, V. B., Kreisberg, J. F., Stopak, K., Yonemoto, W., and Greene, W. C. (2005). Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435, 108–114.
Christ, F., Thys, W., De Rijck, J., Gijsbers, R., Albanese, A., Arosio, D., Emiliani, S., Rain, J. C., Benarous, R., Cereseto, A., and Debyser, Z. (2008). Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 18, 1192–1202.
Coiras, M., Lopez-Huertas, M. R., Perez-Olmeda, M., and Alcami, J. (2009). Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 7, 798–812.
Cosnefroy, O., Tocco, A., Lesbats, P., Thierry, S., Calmels, C., Wiktorowicz, T., Reigadas, S., Kwon, Y., De Cian, A., Desfarges, S., Bonot, P., San Filippo, J., Litvak, S., Le Cam, E., Rethwilm, A., Fleury, H., Connell, P. P., Sung, P., Delelis, O., Andreola, M. L., and Parissi, V. (2012). Stimulation of the human RAD51 nucleofilament restricts HIV-1 integration in vitro and in infected cells. J. Virol. 86, 513–526.
Craigie, R. (2001). HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 276, 23213–23216.
Craigie, R., Fujiwara, T., and Bushman, F. (1990). The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 62, 829–837.
Daniel, R., Katz, R. A., and Skalka, A. M. (1999). A role for DNA-PK in retroviral DNA integration. Science 284, 644–647.
De Iaco, A., and Luban, J. (2011). Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 8, 98.
Desfarges, S., San Filippo, J., Fournier, M., Calmels, C., Caumont-Sarcos, A., Litvak, S., Sung, P., and Parissi, V. (2006). Chromosomal integration of LTR-flanked DNA in yeast expressing HIV-1 integrase: down regulation by RAD51. Nucleic Acids Res. 34, 6215–6224.
Devroe, E., Engelman, A., and Silver, P. A. (2003). Intracellular transport of human immunodeficiency virus type 1 integrase. J. Cell Sci. 116, 4401–4408.
Engelman, A. (2003). The roles of cellular factors in retroviral integration. Curr. Top. Microbiol. Immunol. 281, 209–238.
Engelman, A., and Cherepanov, P. (2008). The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 4, e1000046. doi: 10.1371/journal.ppat.1000046
Engelman, A., Hickman, A. B., and Craigie, R. (1994). The core and carboxyl-terminal domains of the integrase protein of human immunodeficiency virus type 1 each contribute to nonspecific DNA binding. J. Virol. 68, 5911–5917.
Engelman, A., Mizuuchi, K., and Craigie, R. (1991). HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 67, 1211–1221.
Farnet, C. M., and Bushman, F. D. (1997). HIV-1 cDNA integration: requirement of HMG I(Y) protein for function of preintegration complexes in vitro. Cell 88, 483–492.
Farnet, C. M., and Haseltine, W. A. (1990). Integration of human immunodeficiency virus type 1 DNA in vitro. Proc. Natl. Acad. Sci. U.S.A. 87, 4164–4168.
Fassati, A., Gorlich, D., Harrison, I., Zaytseva, L., and Mingot, J. M. (2003). Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. EMBO J. 22, 3675–3685.
Friedman, J. R., Fredericks, W. J., Jensen, D. E., Speicher, D. W., Huang, X. P., Neilson, E. G., and Rauscher, F. J. III. (1996). KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 10, 2067–2078.
Fujiwara, T., and Mizuuchi, K. (1988). Retroviral DNA integration: structure of an integration intermediate. Cell 54, 497–504.
Gao, K., Butler, S. L., and Bushman, F. (2001). Human immunodeficiency virus type 1 integrase: arrangement of protein domains in active cDNA complexes. EMBO J. 20, 3565–3576.
Gorjanacz, M., Klerkx, E. P., Galy, V., Santarella, R., Lopez-Iglesias, C., Askjaer, P., and Mattaj, I. W. (2007). Caenorhabditis elegans BAF-1 and its kinase VRK-1 participate directly in post-mitotic nuclear envelope assembly. EMBO J. 26, 132–143.
Greger, J. G., Katz, R. A., Ishov, A. M., Maul, G. G., and Skalka, A. M. (2005). The cellular protein daxx interacts with avian sarcoma virus integrase and viral DNA to repress viral transcription. J. Virol. 79, 4610–4618.
Guo, J., Wang, W., Yu, D., and Wu, Y. (2011). Spinoculation triggers dynamic actin and cofilin activity that facilitates HIV-1 infection of transformed and resting CD4 T cells. J. Virol. 85, 9824–9833.
Hamamoto, S., Nishitsuji, H., Amagasa, T., Kannagi, M., and Masuda, T. (2006). Identification of a novel human immunodeficiency virus type 1 integrase interactor, Gemin2, that facilitates efficient viral cDNA synthesis in vivo. J. Virol. 80, 5670–5677.
Iyengar, S., and Farnham, P. J. (2011). KAP1 protein: an enigmatic master regulator of the genome. J. Biol. Chem. 286, 26267–26276.
Jacque, J. M., and Stevenson, M. (2006). The inner-nuclear-envelope protein emerin regulates HIV-1 infectivity. Nature 441, 641–645.
Kalpana, G. V., Marmon, S., Wang, W., Crabtree, G. R., and Goff, S. P. (1994). Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science 266, 2002–2006.
Katz, R. A., Merkel, G., Kulkosky, J., Leis, J., and Skalka, A. M. (1990). The avian retroviral IN protein is both necessary and sufficient for integrative recombination in vitro. Cell 63, 87–95.
Klerkx, E. P., Lazo, P. A., and Askjaer, P. (2009). Emerging biological functions of the vaccinia-related kinase (VRK) family. Histol. Histopathol. 24, 749–759.
Krishnan, L., Matreyek, K. A., Oztop, I., Lee, K., Tipper, C. H., Li, X., Dar, M. J., Kewalramani, V. N., and Engelman, A. (2010). The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J. Virol. 84, 397–406.
Lee, M. S., and Craigie, R. (1994). Protection of retroviral DNA from autointegration: involvement of a cellular factor. Proc. Natl. Acad. Sci. U.S.A. 91, 9823–9827.
Lee, M. S., and Craigie, R. (1998). A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl. Acad. Sci. U.S.A. 95, 1528–1533.
Lee, S. P., Xiao, J., Knutson, J. R., Lewis, M. S., and Han, M. K. (1997). Zn2+ promotes the self-association of human immunodeficiency virus type-1 integrase in vitro. Biochemistry 36, 173–180.
Lewinski, M. K., and Bushman, F. D. (2005). Retroviral DNA integration – mechanism and consequences. Adv. Genet. 55, 147–181.
Li, L., Olvera, J. M., Yoder, K. E., Mitchell, R. S., Butler, S. L., Lieber, M., Martin, S. L., and Bushman, F. D. (2001). Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 20, 3272–3281.
Lin, C. W., and Engelman, A. (2003). The barrier-to-autointegration factor is a component of functional human immunodeficiency virus type 1 preintegration complexes. J. Virol. 77, 5030–5036.
Lin, Z., Kong, H., Nei, M., and Ma, H. (2006). Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl. Acad. Sci. U.S.A. 103, 10328–10333.
Liu, L., Oliveira, N. M., Cheney, K. M., Pade, C., Dreja, H., Bergin, A. M., Borgdorff, V., Beach, D. H., Bishop, C. L., Dittmar, M. T., and McKnight, A. (2011). A whole genome screen for HIV restriction factors. Retrovirology 8, 94.
Llano, M., Saenz, D. T., Meehan, A., Wongthida, P., Peretz, M., Walker, W. H., Teo, W., and Poeschla, E. M. (2006). An essential role for LEDGF/p75 in HIV integration. Science 314, 461–464.
Lu, K. P., and Zhou, X. Z. (2007). The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916.
Lutzke, R. A., and Plasterk, R. H. (1998). Structure-based mutational analysis of the C-terminal DNA-binding domain of human immunodeficiency virus type 1 integrase: critical residues for protein oligomerization and DNA binding. J. Virol. 72, 4841–4848.
Manganaro, L., Lusic, M., Gutierrez, M. I., Cereseto, A., Del Sal, G., and Giacca, M. (2010). Concerted action of cellular JNK and Pin1 restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nat. Med. 16, 329–333.
Margalit, A., Brachner, A., Gotzmann, J., Foisner, R., and Gruenbaum, Y. (2007). Barrier-to-autointegration factor – a BAFfling little protein. Trends Cell Biol. 17, 202–208.
Masuda, T. (2011). Non-enzymatic functions of retroviral integrase: the next target for novel anti-HIV drug development. Front. Microbiol. 2:210. doi: 10.3389/fmicb.2011.00210
Matreyek, K. A., and Engelman, A. (2011). The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 85, 7818–7827.
Matsui, T., Leung, D., Miyashita, H., Maksakova, I. A., Miyachi, H., Kimura, H., Tachibana, M., Lorincz, M. C., and Shinkai, Y. (2010). Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464, 927–931.
Mousnier, A., Kubat, N., Massias-Simon, A., Segeral, E., Rain, J. C., Benarous, R., Emiliani, S., and Dargemont, C. (2007). von Hippel Lindau binding protein 1-mediated degradation of integrase affects HIV-1 gene expression at a postintegration step. Proc. Natl. Acad. Sci. U.S.A. 104, 13615–13620.
Nichols, R. J., Wiebe, M. S., and Traktman, P. (2006). The vaccinia-related kinases phosphorylate the N′ terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol. Biol. Cell 17, 2451–2464.
Nishitsuji, H., Hayashi, T., Tkahashi, T., Miyano, M., Kannagi, M., and Masuda, T. (2009). Augmentation of reverse transcription by integrase through an interaction with host factor, SIP1/Gemin2 is critical for HIV-1 infection. PLoS ONE 4, e7825. doi: 10.1371/journal.pone.0007825
Peng, H., Begg, G. E., Schultz, D. C., Friedman, J. R., Jensen, D. E., Speicher, D. W., and Rauscher, F. J. III. (2000). Reconstitution of the KRAB-KAP-1 repressor complex: a model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J. Mol. Biol. 295, 1139–1162.
Pierson, T. C., Zhou, Y., Kieffer, T. L., Ruff, C. T., Buck, C., and Siliciano, R. F. (2002). Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J. Virol. 76, 8518–8531.
Pope, M., and Haase, A. T. (2003). Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat. Med. 9, 847–852.
Raman, M., Chen, W., and Cobb, M. H. (2007). Differential regulation and properties of MAPKs. Oncogene 26, 3100–3112.
Roth, S. Y., Denu, J. M., and Allis, C. D. (2001). Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120.
Rowe, H. M., Jakobsson, J., Mesnard, D., Rougemont, J., Reynard, S., Aktas, T., Maillard, P. V., Layard-Liesching, H., Verp, S., Marquis, J., Spitz, F., Constam, D. B., and Trono, D. (2010). KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240.
Sabapathy, K., Hochedlinger, K., Nam, S. Y., Bauer, A., Karin, M., and Wagner, E. F. (2004). Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 15, 713–725.
San Filippo, J., Sung, P., and Klein, H. (2008). Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257.
Segura-Totten, M., and Wilson, K. L. (2004). BAF: roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol. 14, 261–266.
Shahbazian, M. D., and Grunstein, M. (2007). Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100.
Sherman, P. A., and Fyfe, J. A. (1990). Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc. Natl. Acad. Sci. U.S.A. 87, 5119–5123.
Shun, M. C., Raghavendra, N. K., Vandegraaff, N., Daigle, J. E., Hughes, S., Kellam, P., Cherepanov, P., and Engelman, A. (2007). LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 21, 1767–1778.
Smith, J. A., and Daniel, R. (2006). Following the path of the virus: the exploitation of host DNA repair mechanisms by retroviruses. ACS Chem. Biol. 1, 217–226.
Sorin, M., Cano, J., Das, S., Mathew, S., Wu, X., Davies, K. P., Shi, X., Cheng, S. W., Ott, D., and Kalpana, G. V. (2009). Recruitment of a SAP18-HDAC1 complex into HIV-1 virions and its requirement for viral replication. PLoS Pathog. 5, e1000463. doi: 10.1371/journal.ppat.1000463
Sterner, D. E., and Berger, S. L. (2000). Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459.
Stevenson, M., Stanwick, T. L., Dempsey, M. P., and Lamonica, C. A. (1990). HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 9, 1551–1560.
Studamire, B., and Goff, S. P. (2010). Interactions of host proteins with the murine leukemia virus integrase. Viruses 2, 1110–1145.
Suzuki, Y., and Craigie, R. (2002). Regulatory mechanisms by which barrier-to-autointegration factor blocks autointegration and stimulates intermolecular integration of Moloney murine leukemia virus preintegration complexes. J. Virol. 76, 12376–12380.
Suzuki, Y., and Craigie, R. (2007). The road to chromatin – nuclear entry of retroviruses. Nat. Rev. Microbiol. 5, 187–196.
Suzuki, Y., Ogawa, K., Koyanagi, Y., and Suzuki, Y. (2010). Functional disruption of the Moloney murine leukemia virus preintegration complex by vaccinia-related kinases. J. Biol. Chem. 285, 24032–24043.
Suzuki, Y., Yang, H., and Craigie, R. (2004). LAP2α and BAF collaborate to organize the Moloney murine leukemia virus preintegration complex. EMBO J. 23, 4670–4678.
Terreni, M., Valentini, P., Liverani, V., Gutierrez, M. I., Di Primio, C., Di Fenza, A., Tozzini, V., Allouch, A., Albanese, A., Giacca, M., and Cereseto, A. (2010). GCN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology 7, 18.
Turlure, F., Devroe, E., Silver, P. A., and Engelman, A. (2004). Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 9, 3187–3208.
Tyagi, M., and Kashanchi, F. (2012). New and novel intrinsic host repressive factors against HIV-1: PAF1 complex, HERC5 and others. Retrovirology 9, 19.
Van Maele, B., Busschots, K., Vandekerckhove, L., Christ, F., and Debyser, Z. (2006). Cellular co-factors of HIV-1 integration. Trends Biochem. Sci. 31, 98–105.
Walsh, C. T., Garneau-Tsodikova, S., and Gatto, G. J. Jr. (2005). Protein posttranslational modifications: the chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 44, 7342–7372.
Wang, T. F., Chen, L. T., and Wang, A. H. (2008). Right or left turn? RecA family protein filaments promote homologous recombination through clockwise axial rotation. Bioessays 30, 48–56.
Wang, W., Cote, J., Xue, Y., Zhou, S., Khavari, P. A., Biggar, S. R., Muchardt, C., Kalpana, G. V., Goff, S. P., Yaniv, M., Workman, J. L., and Crabtree, G. R. (1996). Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 15, 5370–5382.
Weiss, A., and Littman, D. R. (1994). Signal transduction by lymphocyte antigen receptors. Cell 76, 263–274.
Wilkinson, T. A., Januszyk, K., Phillips, M. L., Tekeste, S. S., Zhang, M., Miller, J. T., Le Grice, S. F., Clubb, R. T., and Chow, S. A. (2009). Identifying and characterizing a functional HIV-1 reverse transcriptase-binding site on integrase. J. Biol. Chem. 284, 7931–7939.
Woerner, A. M., Klutch, M., Levin, J. G., and Marcus-Sekura, C. J. (1992). Localization of DNA binding activity of HIV-1 integrase to the C-terminal half of the protein. AIDS Res. Hum. Retroviruses 8, 297–304.
Wolf, D., and Goff, S. P. (2007). TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 131, 46–57.
Wolf, D., and Goff, S. P. (2008). Host restriction factors blocking retroviral replication. Annu. Rev. Genet. 42, 143–163.
Wolf, D., and Goff, S. P. (2009). Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458, 1201–1204.
Wolf, D., Hug, K., and Goff, S. P. (2008). TRIM28 mediates primer binding site-targeted silencing of Lys1,2 tRNA-utilizing retroviruses in embryonic cells. Proc. Natl. Acad. Sci. U.S.A. 105, 12521–12526.
Woodward, C. L., Prakobwanakit, S., Mosessian, S., and Chow, S. A. (2009). Integrase interacts with nucleoporin NUP153 to mediate the nuclear import of human immunodeficiency virus type 1. J. Virol. 83, 6522–6533.
Yamamoto, S. P., Okawa, K., Nakano, T., Sano, K., Ogawa, K., Masuda, T., Morikawa, Y., Koyanagi, Y., and Suzuki, Y. (2011). Huwe1, a novel cellular interactor of Gag-Pol through integrase binding, negatively influences HIV-1 infectivity. Microbes Infect. 13, 339–349.
Yoder, K., Sarasin, A., Kraemer, K., McIlhatton, M., Bushman, F., and Fishel, R. (2006). The DNA repair genes XPB and XPD defend cells from retroviral infection. Proc. Natl. Acad. Sci. U.S.A. 103, 4622–4627.
Yung, E., Sorin, M., Pal, A., Craig, E., Morozov, A., Delattre, O., Kappes, J., Ott, D., and Kalpana, G. V. (2001). Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat. Med. 7, 920–926.
Yung, E., Sorin, M., Wang, E. J., Perumal, S., Ott, D., and Kalpana, G. V. (2004). Specificity of interaction of INI1/hSNF5 with retroviral integrases and its functional significance. J. Virol. 78, 2222–2231.
Zaitseva, L., Cherepanov, P., Leyens, L., Wilson, S. J., Rasaiyaah, J., and Fassati, A. (2009). HIV-1 exploits importin 7 to maximize nuclear import of its DNA genome. Retrovirology 6, 11.
Keywords: retrovirus, integration, host factors, RAD51, KAP1, VRK, posttranslational modifications
Citation: Suzuki Y, Chew ML and Suzuki Y (2012) Role of host-encoded proteins in restriction of retroviral integration. Front. Microbio. 3:227. doi: 10.3389/fmicb.2012.00227
Received: 13 April 2012; Paper pending published: 08 May 2012;
Accepted: 02 June 2012; Published online: 22 June 2012.
Edited by:
Atsushi Koito, Kumamoto University, JapanReviewed by:
Akihide Ryo, Yokohama City University, JapanCopyright: © 2012 Suzuki, Chew and Suzuki. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Youichi Suzuki, Department of Microbiology, Yong Loo Lin School of Medicine, National University of Singapore, Center for Translational Research, 14 Medical Drive, #15-02, Singapore 117599. e-mail:bWljeXNAbnVzLmVkdS5zZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.