



- 1 Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Oeiras, Portugal
- 2 Biochemistry Department of the University of Missouri, Columbia, MO, USA
- 3 Ecosystems and Networks Integrated with Genes and Molecular Assemblies, Berkeley, CA, USA
The adenosine 5′-phosphosulfate reductase (AprAB) is the enzyme responsible for the reduction of adenosine 5′-phosphosulfate (APS) to sulfite in the biological process of dissimilatory sulfate reduction, which is carried out by a ubiquitous group of sulfate reducing prokaryotes. The electron donor for AprAB has not been clearly identified, but was proposed to be the QmoABC membrane complex, since an aprBA–qmoABC gene cluster is found in many sulfate reducing and sulfur-oxidizing bacteria. The QmoABC complex is essential for sulfate reduction, but electron transfer between QmoABC and AprAB has not been reported. In this work we provide the first direct evidence that QmoABC and AprAB interact in Desulfovibrio spp., using co-immunoprecipitation, cross-linking Far-Western blot, tag-affinity purification, and surface plasmon resonance studies. This showed that the QmoABC–AprAB complex has a strong steady-state affinity (KD = 90 ± 3 nM), but has a transient character due to a fast dissociation rate. Far-Western blot identified QmoA as the Qmo subunit most involved in the interaction. Nevertheless, electron transfer from menaquinol analogs to APS through anaerobically purified QmoABC and AprAB could not be detected. We propose that this reaction requires the involvement of a third partner to allow electron flow driven by a reverse electron bifurcation process, i.e., electron confurcation. This process is deemed essential to allow coupling of APS reduction to chemiosmotic energy conservation.
Introduction
Sulfate respiration is an anaerobic process carried out by a phylogenetically diverse group of organisms including both Bacteria and Archaea. This process is a major contributor to the global cycling of sulfur and carbon in anaerobic habitats, and has very important environmental and economical impacts (Muyzer and Stams, 2008; Barton and Fauque, 2009). Sulfate reducing prokaryotes (SRP) are found ubiquitously in anaerobic environments, and are particularly abundant in marine habitats due to the high concentration of sulfate in sea water. As a group SRP are physiologically versatile and capable of metabolizing a wide variety of substrates, and they can also grow syntrophically with other organisms in the absence of sulfate (Stams and Plugge, 2009; Plugge et al., 2011). Despite its fundamental importance, the mechanism of energy conservation in sulfate respiration remains to be fully elucidated. For many years it was thought that quinones did not play a role in the process, despite their known presence in SRP, and intracellular hydrogen cycling was proposed to account for proton motive force generation. Nowadays, hydrogen cycling is considered as only one of the possible pathways for energy conservation, operating in some, but not all SRP (Keller and Wall, 2011; Pereira et al., 2011). Sulfate reduction is an intracellular process requiring active transport of sulfate, and its activation by reaction with ATP to form adenosine 5′-phosphosulfate (APS). The two terminal reductases, APS reductase (AprAB) and dissimilatory sulfite reductase (DsrAB), are soluble and thus not directly involved in membrane-linked electron transport. One of the key questions remaining about sulfate reduction is the identification of the electron donors to AprAB and DsrAB. The involvement of membrane proteins in the process was first described by Mander et al. (2002) and Pires et al. (2003) through the identification of the DsrMKJOP (initially named Hme) and QmoABC complexes. These two complexes are found both in SRP (Pereira, 2008) and in many anoxygenic phototrophic and chemotrophic sulfur-oxidizing bacteria (SOB; Frigaard and Dahl, 2009), indicating a dedicated role in sulfur metabolism. Furthermore, the two complexes are conserved in the genomes of SRP described to date, with very few exceptions: the archeon Caldivirga maquilingensis lacks the qmoABC genes and in some Gram-positive bacteria the qmoC gene is absent; in both cases also a simpler version of the DsrMKJOP complex occurs, since only the dsrMK genes are present (Junier et al., 2010; Pereira et al., 2011). The QmoABC and DsrMKJOP complexes share an interesting characteristic in that they both contain subunits that are related to heterodisulfide reductases (Hdr) of methanogens (Thauer et al., 2008), and subunits known to interact with quinones. In several organisms, the qmoABC genes cluster with the aprAB genes, and the dsrMKJOP genes cluster with dsrAB, strongly suggesting an involvement of QmoABC in the electron transfer pathway to AprAB and DsrMKJOP in the electron transfer pathway to DsrAB.
The QmoABC complex has one membrane (QmoC) and two cytoplasmic subunits (QmoAB), and the two QmoC hemes b are reduced by quinols, indicating that the Qmo complex participates in electron flow between the quinone pool and the cytoplasm, in a process that may result in energy conservation (Pires et al., 2003). In Desulfovibrio vulgaris Hildenborough a deletion mutant of the qmoABC genes could not grow with sulfate as electron acceptor, but grew normally with sulfite or thiosulfate, providing conclusive evidence that QmoABC is required for reduction of sulfate (Zane et al., 2010). Also, in the green sulfur-oxidizer Chlorobium tepidum the Qmo complex was shown to be involved in oxidation of sulfite as an intermediary in the sulfur oxidation pathway (Chan et al., 2008; Rodriguez et al., 2011). These results show that the Qmo complex is involved in electron flow between the menaquinone pool and APS reduction or oxidation by AprAB. However, direct electron transfer between the isolated Desulfovibrio desulfuricans ATCC 27774 Qmo complex and AprAB could not be detected, which could indicate that additional proteins are involved in the pathway (Pires et al., 2003). In this work we report protein–protein interaction studies that show that there is a direct interaction between QmoABC and AprAB, and that the interaction involves the QmoA subunit. The mechanism of AprAB reduction is further discussed.
Materials and Methods
Protein Purification
Cells of D. desulfuricans ATCC 27774 were grown according to Liu and Peck (1981). The cells were broken and centrifuged and the membrane fraction was used to purify the QmoABC complex in n-Dodecyl-β-D-maltoside (DDM), as previously described by Pires et al. (2003), following its characteristic UV–Visible absorption spectrum. The purification of Qmo was carried out both in aerobic and anaerobic conditions. AprAB was purified from the soluble fraction in anaerobic conditions following the catalytic activity of sulfite oxidation (Fritz et al., 2002a,b). Anaerobic purifications were carried out inside a Coy anaerobic chamber (95% N2, 5% H2) using an AKTA™ Prime plus™ system. The soluble fraction from D. desulfuricans was ultracentrifuged at 140,000 × g for 2 h, and then applied to a Q-Sepharose FF column equilibrated with 50 mM Tris–HCl (pH 7.6) buffer with 10% glycerol (v/v; buffer A). A stepwise gradient of increasing NaCl concentration was performed and fractions were separated according to UV–Visible spectra. The fractions containing highest AprAB activity, which eluted between 180 and 200 mM NaCl, were pooled. After concentration and lowering of ionic strength, this sample was loaded on a Q-Sepharose HP column equilibrated with buffer A. Again, a stepwise gradient of increasing NaCl concentration was performed. The fractions were separated according to the UV–Visible spectra and activity. The pool of fractions with higher activity was diluted in buffer A and applied in a second Q-Sepharose HP column equilibrated with 10 mM potassium phosphate buffer (pH 7) containing 10% glycerol (v/v; Buffer B). A stepwise gradient of increasing NaCl concentration was performed, and fractions containing purified AprAB eluted at 150 mM NaCl. The purified enzyme had a sulfite oxidation activity of 3.3 μmol min−1 mg−1 and displayed the characteristic two subunits on an SDS-PAGE gel.
APS Reductase Activity
The AprAB activity was determined as formation of APS in 50 mM Tris–HCl (pH 7.6), 2 mM Na2SO3, 2 mM AMP, 1 mM K3Fe(CN)6, at room temperature (Fritz et al., 2000, 2002a), or by APS reduction in 80 mM potassium phosphate (pH 7), 30 μM APS, and 0.75 mM methyl viologen as reductant (Fritz et al., 2002a). Methyl viologen was reduced with 0.2 g of metallic zinc granules in the same buffer.
Co-Immunoprecipitation
Antibodies for QmoABC complex and AprAB from D. desulfuricans were produced from the purified proteins by Davids Biotechnology (Regensburg, Germany) and used for Co-immunoprecipitation (Co-IP) experiments with the Thermo Scientific Pierce® Co-IP kit, following the kit instructions. The anti-Qmo antibody did not cross-react with AprAB, and the anti-Apr antibody did not cross-react with QmoABC. Two approaches were used to investigate protein–protein interaction, one based on Anti-QmoABC antibodies and the other based on Anti-AprAB antibodies. In the first case, 500 μg of Anti-QmoABC antibody were immobilized in the AminoLink® Plus Coupling Resin in a small column, and 1 μM of Qmo in the kit Lysis/Wash buffer was added and incubated for 1 h at 4°C. After one washing step with Lysis/Wash buffer 1 μM of AprAB in the same buffer was loaded in the column and incubated for 2 h at 4°C. After five washing steps, the co-IP products were eluted with the kit Elution buffer. The protocol was repeated with 500 μg of Anti-AprAB antibody, 1 μM of AprAB, and 1 μM of QmoABC in the same buffer. Control experiments were run in parallel with no antibody bound to the control resin. The eluates (∼100 μg) were separated in SDS-PAGE gels [12% acrylamide, (v/v)], and transferred to polyvinylidene difluoride (PVDF) membranes (Transfer buffer: 48 mM Tris–HCl pH 9.2 and 39 mM Glycine) using a Mini Trans-Blot® electrophoretic transfer cell (Bio-Rad) during 40 min at 4°C, 100 V, and 350 mA. The membranes were blocked with blocking buffer [20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.05% Tween 20 (v/v), and 5% non-fat milk (w/v)], overnight at room temperature. After two washing steps with TBST [20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.05% Tween 20 (v/v)] anti-QmoABC antibody at 1:200 dilution in TBST or anti-AprAB antibody at 1:1,000 dilution in TBST were incubated with the membranes for 1 h, followed by two washing steps with TBST, and incubation with anti-rabbit IgG antibody (Sigma-Aldrich®) at 1:15,000 dilution in TBST for 45 min. After three washing steps with TBS, protein detection was performed with Alkaline Phosphatase Buffer (100 mM Tris–HCl pH 9.5, 100 mM NaCl, and 5 mM MgCl2), and NBT (nitro-blue tetrazolium chloride)/BCIP (5-bromo-4-chloro-3-indolyl phosphate).
Surface Plasmon Resonance
The surface plasmon resonance (SPR) experiments were performed at 25°C on a BIAcore 2,000 instrument (Biacore Inc., GE HealthCare). The proteins samples were exchanged to the buffer used as running buffer for the SPR experiments [10 mM HEPES pH 7.4 + 150 mM NaCl + 3 mM EDTA + 0.01% DDM (w/v)], using a HiTrap™ Desalting column (Amersham Biosciences). AprAB was immobilized in a CM5 sensor chip (GE® Healthcare) by standard NHS/EDC amine coupling resulting in an immobilization level of 1,000 RU. Flow cell 1 was similarly treated with buffer in the absence of AprAB (control cell). Interaction experiments with QmoABC were performed with duplicate injections of 3.9, 7.8, 15.6, 31.25, 62.5 nM of QmoABC at a flow rate of 15 μl/min. After the end of each injection dissociation was performed with running buffer for 10 min, after which all of the protein completely dissociated from the surface (as indicated by a return to baseline level of the sensorgram) and thus no further regeneration was required. The sensorgrams were processed using the double referencing method to eliminate the non-specific binding from background contribution and the buffer artifacts were removed by subtracting signals from the reference flow cell and from buffer blank injections. The BIA evaluation 3.2 RC1 analysis software was used to determine ka and kd from the processed data sets by globally fitting to a 1:1 biomolecular binding model with drifting baseline. The KD was calculated from the quotient kd/ka.
For the competition experiments, 62.5 nM of QmoABC was incubated with 62.5 or 125 nM AprAB and injected in the chip surface at the same flow rate as before.
Cross-Linking Far-Western Blot
Ten microgram of pure QmoABC were separated in a 12% SDS-PAGE gel and blotted to a PVDF membrane. After overnight blocking, the membrane was incubated with AprAB (1 μM) for 1 h in 20 mM Tris–HCl pH 7.6, 10% Glycerol (v/v), at room temperature. The membrane was washed once with bidistilled water (bDW) and incubated with 32 mM N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC; Sigma-Aldrich®) in bDW for 1 h at room temperature (Sato et al., 2011). After three washing steps with bDW, Western Blot against Anti-AprAB was performed. As positive control we used AprAB and as negative control QmoABC that was not incubated with AprAB.
D. vulgaris Hildenborough Strains and Growth Conditions
A mutant strain lacking the qmoA gene was produced by double homologous recombination in D. vulgaris Hildenborough – IPAR02 – according to Keller et al. (2011), with the exception that following electroporation the cells were recovered and plated in MOYLS3 (lactate 30 mM/sulfite 15 mM) and the electroporation parameters were 1,500 V, 250 Ω, and 25 μF. The pMOIP02 plasmid for the qmoA deletion was obtained by sequence ligation independent cloning (SLIC; Li and Elledge, 2007). Three segments were amplified by PCR with Herculase polymerase II (Stratagene®): 942 bp upstream of qmoA (QmoA Up Fw P1-GCCTTTTGCTGGCCTTTTGCTCACATAAGAGCGCGGTTCTGAAA
TCATGC and QmoA Up Rev P2-CCTGCGTGCAATCCATCTTGTTCAATCATCCTTGGTATCCTCCC
TACGTGT), 932 bp downstream of qmoA (QmoA Dwn Fw P3-CCTTCTATCGCCTTCTTGACGAGTTCTTCTAGACCATAATGGCCA
GCAGAATTGG and QmoA Dwn Rev P4-CGAGGCATTTCTGTCCTGGCTGGAGTGACGTGTTCAGGATGAA
GGCA), and the kanamycin resistance gene from pSC27 (Keller et al., 2011; Kan aa2 Fw-ATTGAACAAGATGGATTGCACGCAGG and Kan aa264 Rev-GAAGAACTCGTCAAGAAGGCGATAGAAGG), and then added into pMO719 background via SLIC. Products from the amplifications were transformed into E. coli α-select Silver Efficiency (Bioline®) and successful transformants were isolated on LC medium (Zane et al., 2010). Correct isolates were identified by the expected PCR amplicons from the plasmids constructs and also by sequencing performed at the DNA Core Facility at the University of Missouri, USA. The pMOIP02 produced was electroporated into D. vulgaris according to Keller et al. (2011), Zane et al. (2010), from which strain IPAR02 was obtained, by selecting with MOYLS3 medium containing 400 μg/ml of geneticin. The deletion of qmoA was confirmed by Southern blot. The IPAR02 mutant strain grows in lactate/sulfite as described previously for the mutant lacking qmoABC (Zane et al., 2010) and is kanamycin resistant.
A complementation plasmid pMOIP05 was produced also by SLIC encoding qmoA with a Strep-TEV-FLAG (STF) tag. To create this vector two segments were amplified by PCR: the qmoA gene (QmoA Exp Vctr P1 Fw-AGGTTGGGAAGCCCTGCAATGCAGTCCCAGGAGGTACCATA
TGTCGAACTCCATACTCGTCGTCG and QmoA Exp Vctr P2 Rev-AATTTTTTCGAACTGCGGGTGGCTCCACCTCCCTCTCACCGTTT
GAATCGC) and the STF-tag gene from pSLIC-DVU0171-STF-Kan-Tag (Chhabra et al., 2011a; STF-Tag Fw-TGGAGCCACCCGCAGTTCGAAAAAATT and STF-Tag Rev-GATCGTGATC CCCTGCGCCATCAGATCCTTGCTACTTGTCATCGTCATCCTTGTA
GTCGATGTCA); and then added into pMO9075 background via SLIC. The amplifications products were transformed into E. coli α-select Silver Efficiency (Bioline®), and cells were plated on spectinomycin (100 μg/ml)-containing agar plates. The correct plasmid construct was screened by colony PCR and later confirmed by sequencing at the DNA Core Facility at the University of Missouri, USA.
The pMOIP05 was successfully introduced in IPAR02 by electroporation (Keller et al., 2011) selecting with MOYLS3 medium containing 400 μg/ml of geneticin and 100 μg/ml of spectinomycin, to generate the complemented strain IPAR03. The plasmid was confirmed by PCR amplification of the insert and also by sequencing performed in GATC Biotech, Germany. The complemented mutant strain IPAR03 was grown either in MOYLS3 lactate/sulfite medium or MOYLS4 lactate/sulfate medium (Zane et al., 2010; Keller et al., 2011) with spectinomycin (100 μg/ml).
Pull-Down Assay
For the pull-down assay, IPAR03 was grown in 100 ml of MOYLS4 with spectinomycin at 37°C for about 24 h. Cells were harvested by centrifuging at 2,500 × g for 15 min at 4°C, washed with 20 mM Tris–HCl buffer (pH 7.6) + 10% glycerol (v/v), and again centrifuged as before. Cells were then disrupted using BugBuster® Protein Extraction Reagent (Novagen®) for 20 min at room temperature and centrifuged at 16,000 × g for 20 min at 4°C. The soluble fraction of IPAR03 was loaded in micro-columns containing Strep®-Tactin resin (IBA GmBH) equilibrated with 50 mM Tris–HCl pH 7.6, 150 mM NaCl, and 10% glycerol (v/v; Buffer W). After five washing steps with Buffer W, the recombinant protein QmoA was eluted with Buffer W containing 2.5 mM desthiobiotin. The elution product was precipitated in acetone and analyzed by SDS-PAGE and Western Blot with Strep-Tactin horse radish peroxidase (HRP) conjugate. The co-elution of AprAB with QmoA was detected by Western Blot with Anti-AprAB from D. desulfuricans. In a control experiment the same conditions were used with wild-type cells of D. vulgaris.
Electron Transfer Experiments
The electron transfer between QmoABC and AprAB was tested in spectrophotometer assays inside the anaerobic chamber, using quartz cuvettes equipped with a magnetic stirrer. The first assay was based on AprAB activity, as previously described (Pires et al., 2003), following reduction of the menaquinone analog 2,3-dimethyl-1,4-naphthoquinone (DMN) with sulfite (reverse reaction) at 350 nm (at 270 nm there is interference from AMP). DMN (500 μM) reduction was followed in 50 mM Tris–HCl (pH 7.6) with 0.0125% DDM (w/v), 2 mM Na2SO3, 2 mM AMP, and 0.5 μM of QmoABC after addition of 1.2 μM AprAB. The second assay was based on oxidation of quinol reduced QmoABC by APS (direct reaction). Qmo (0.3 μM) was reduced with different amounts of menadiol (25, 50, 100, 300, 930 μM) in 10 mM Phosphate buffer (pH 7), 0.0125% DDM (w/v). Qmo heme b oxidation was followed at 424 nm in the presence of different amounts of APS (30, 60, and 120 μM; Sigma®), after addition of 0.1 μM AprAB.
Results
A link between the QmoABC complex and APS reductase was first inferred from the co-localization of their genes in the genomes of several sulfate reducing and SOB. Subsequent deletion of the qmo genes in these organisms proved that the Qmo complex is required for the reduction of sulfate in SRP (Zane et al., 2010), and the oxidation of sulfite in green sulfur bacteria (Rodriguez et al., 2011). However, the fact that no electron transfer could be observed between the two proteins (Pires et al., 2003) raised doubts as to whether there is a direct interaction between them, or if other proteins are involved. Recently, a proteomic study of protein–protein interactions in D. vulgaris Hildenborough was reported, in which several key proteins were used as baits for affinity purification followed by mass spectrometry (Chhabra et al., 2011b). The bait proteins included Strep-tagged AprA and AprB, and again no evidence for a direct interaction with QmoABC proteins was obtained. However, interactions between redox proteins are notably difficult to observe due to their transient nature, which is required for the fast turnover of electron exchange reactions in energy metabolism (Bashir et al., 2011; Martinez-Fabregas et al., 2011). In addition, the fact that Qmo is a membrane-associated complex is likely to further hinder proteomic-based studies. These kind of high-throughput approaches, although invaluable from the amount of information that can be obtained, suffer from the use of the same conditions to evaluate many different types of interactions between many different proteins, so a high number of false negative results is likely to occur. In this work we took advantage of the fact that we can purify both QmoABC and AprAB from D. desulfuricans ATCC 27774 to perform detailed interaction studies between the two proteins.
Co-Immunoprecipitation Experiments
The first approach to evaluate a possible interaction between QmoABC and AprAB complexes was to use co-immunoprecipitation (co-IP). For this we used a Thermo Scientific Pierce® Co-IP kit in which the antibodies are covalently coupled to an amine-reactive resin. Anti-QmoABC or Anti-AprAB specific antibodies were generated using the purified proteins, and immobilized in columns containing the coupling resin. The two antibody-loaded resins were then incubated with the corresponding prey protein (QmoABC or AprAB), washed, and then incubated with the interacting bait partner (AprAB or QmoABC). After several washing steps the retained proteins were eluted and the Co-IP products were separated by SDS-PAGE and blotted to a PVDF membrane. The membranes were treated by Western blot using the antibodies against the bait protein. Two control experiments were run in parallel, where no antibodies were bound to the resin. The Western blot results (Figure 1) show that it was possible to co-immunoprecipitate QmoABC and AprAB, using either of the corresponding antibodies, indicating that there is a direct physical interaction between the two proteins. The control experiments reveal some unspecific retention of both proteins, but the strong difference between the experiments and the controls are indicative of co-immunoprecipitation.
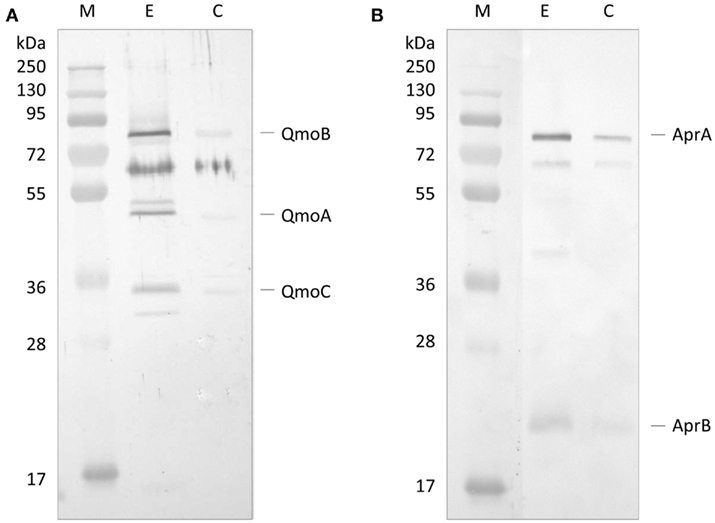
Figure 1. Western Blot analysis of co-immunoprecipitation experiment. (A) Western Blot with Anti-QmoABC of the elution products of: E – the Co-IP using immobilized Anti-AprAB antibody; C – control resin with no antibody. (B) Western Blot with Anti-AprAB of the elution products of: E – the Co-IP using immobilized Anti-QmoABC antibody; C – control resin with no antibody. M – Pre-stained molecular mass markers.
Surface Plasmon Resonance Experiments
Since an interaction between QmoABC and AprAB was detected we next sought to quantify the kinetics and affinity parameters of this interaction. For this we used SPR, which is a gold standard for studying protein–protein interactions, since it can provide direct quantitative measurements of binding kinetics and affinities, without the need for any labeling methods. Using a CM5 sensor chip we tested covalent immobilization of either QmoABC or AprAB. Considerable loss of immobilized material was observed in the case of QmoABC, likely due to gradual dissociation of subunits, whereas this was not observed with immobilized AprAB. Further studies proceeded using AprAB as the ligand and QmoABC as the analyte. An interaction was again observed between the two proteins, which could be detected even at low concentrations of QmoABC. The dissociation of QmoABC was complete after injection stopped, and did not require special regeneration conditions, which confirms the transient nature of the interaction between the two proteins. The sensorgrams obtained (Figure 2A) were used to calculate the binding rate constants, by fitting the results to a 1:1 interaction model with drifting baseline, yielding an association rate constant ka = (3.0 ± 0.1) × 105 M−1 s−1, a dissociation rate constant kd = (2.7 ± 0.4) × 10−2 s−1, and an equilibrium affinity constant KD = 90 ± 3 nM. These values reveal a high affinity for the AprAB–QmoABC complex in steady-state conditions, and that the complex dissociation is very fast, as it is to be expected for an electron transfer interaction. To further validate these results we carried out a competition assay in which QmoABC was pre-incubated with two different concentrations of free AprAB in solution before the SPR measurement (Figure 2B). This experiment confirmed a reduced interaction between QmoABC and the immobilized AprAB protein, due to the competition of AprAB in solution.
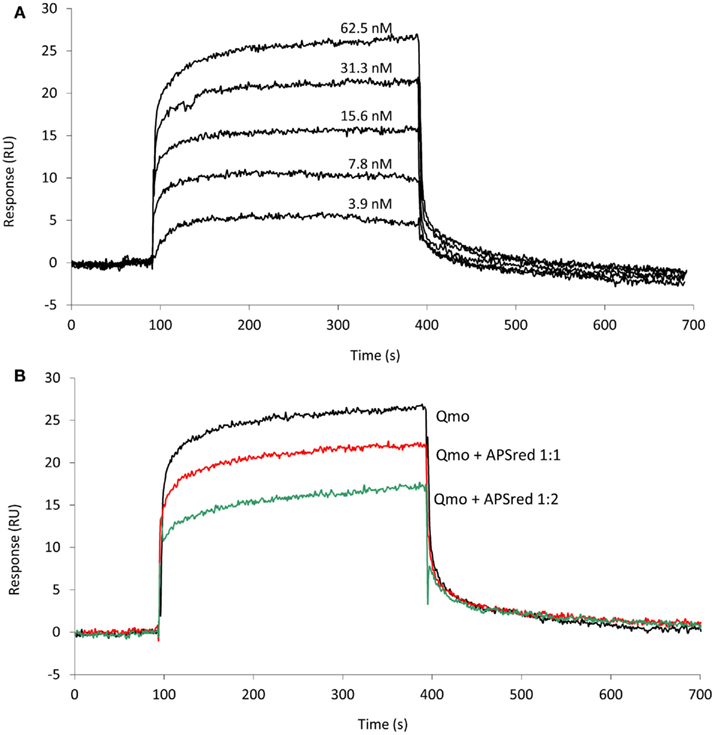
Figure 2. Surface plasmon resonance analysis of the interaction between QmoABC (analyte) and immobilized AprAB (ligand). (A) Sensorgrams obtained from injection of serial dilutions of 62.5, 31.25, 15.6, 7.8, 3.9 nM QmoABC at 15 μl/min flow rate and 25°C. (B) Competition experiment where QmoABC (62.5 nM) was mixed with 62.5 (1:1) or 125 nM (1:2) of AprAB before injection.
Cross-Linking Far-Western Blot
Recently, a modification of the Far-Western protocol to include a cross-linking step was described, which allows for the detection of weak or transient interactions (Sato et al., 2011). Since a strong steady-state interaction was detected between QmoABC and AprAB, we used cross-linking Far-Western blot to try to elucidate which subunits are involved in this interaction. In this experiment, the QmoABC subunits were separated in a SDS-PAGE gel and blotted to a PVDF membrane. The membrane was incubated with AprAB, washed, and EDC was then added to promote cross-linking to the retained protein, following which detection was performed by Western blot with Anti-AprAB antibodies (Figure 3). This showed a positive signal for the QmoA band and a weaker signal for the QmoC band. A shift in the molecular mass of the Qmo subunits is not expected to occur since they are already fixed in the membrane upon incubation with AprAB. No signals were detected when the experiment was run in the absence of cross-linker. In the reverse experiment where AprAB was run in the gel and the membrane was incubated with QmoABC, followed by cross-linking and detection with Anti-QmoABC antibodies, no signals could be detected. This indicates that in this case the denaturation of the AprAB subunits in SDS-PAGE prevents the interaction with QmoABC.
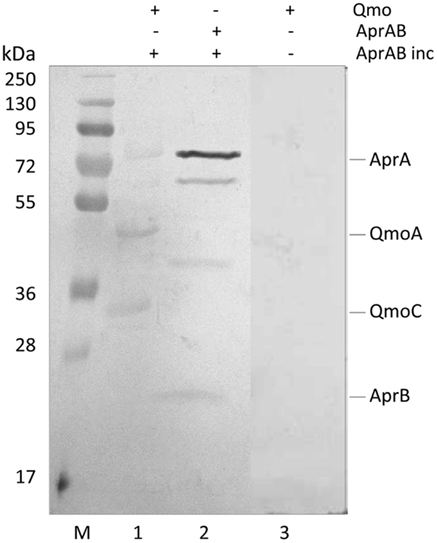
Figure 3. Detection of interacting subunits by cross-linking Far-Western Blot with Anti-AprAB antibodies. From left to right: M – Pre-stained molecular mass markers; (1) QmoABC in PVDF membrane was incubated with AprAB (AprAB inc), cross-linked with EDC and detected; (2) positive control with AprAB in PVDF membrane; (3) negative control with QmoABC in PVDF membrane not incubated with AprAB.
Pull-Down Assay
Since the QmoA protein is the subunit showing stronger interaction with AprAB, we set up an endogenous pull-down assay using single-epitope tag-affinity purification based on tagged QmoA. No genetic tools are available for the organism D. desulfuricans ATCC 27774, but D. vulgaris Hildenborough can be genetically manipulated and extensive tools have been developed allowing chromosomal deletion and tagging of specific genes (Chhabra et al., 2011a,b; Keller et al., 2011). A D. vulgaris Hildenborough mutant strain lacking the qmoA gene (IPAR02) was produced by double homologous recombination, as previously described (Zane et al., 2010), and was complemented with plasmid pMOIP05 encoding qmoA with a Strep-TEV-FLAG (STF) tag to give strain IPAR03. This strain could grow on lactate/sulfate, in contrast to IPAR02 that only grew on lactate/sulfite, confirming that the complementation was successful. The QmoA protein was detected both in the membrane and in the soluble fraction of strain IPAR03 grown in lactate/sulfate. We took advantage of this fact to perform affinity tag purification of the soluble fraction using Strep-Tactin resin. The desthiobiotin elution fraction was analyzed by SDS-PAGE gel followed by Western blot with antibodies against D. desulfuricans AprAB (it was previously confirmed that these antibodies recognized the AprAB protein from D. vulgaris). A band for AprA was detected in the Western blot (Figure 4), confirming the ability of QmoA to interact and pull-down AprAB from the soluble fraction. In a parallel control experiment with wild-type cells of D. vulgaris Hildenborough no band was detected for AprAB.
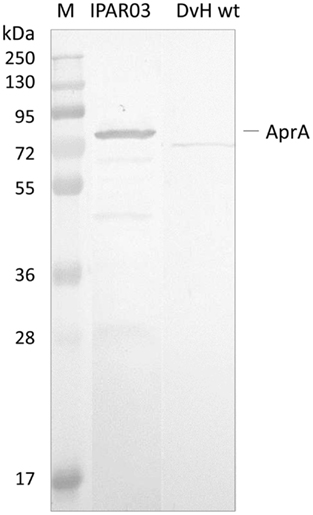
Figure 4. Analysis of Pull-down assay. Strep-Tactin desthiobiotin elution products of soluble fraction from cells expressing STF-tagged QmoA (IPAR03) or wild-type D. vulgaris (DvH wt, negative control) analyzed by Western Blot with Anti-AprAB. M – Pre-stained molecular mass markers.
Electron Transfer Experiments
Since it was established that QmoABC can interact directly with AprAB we attempted again to observe electron transfer using anaerobically purified proteins. Previous experiments had been carried out with proteins purified aerobically (Pires et al., 2003), which could have suffered some damage to their iron–sulfur centers thus preventing electron transfer. We tested reduction of a menaquinone analog (DMN) with sulfite (reverse reaction), or oxidation of quinol reduced QmoABC by APS (direct reaction; Scheme 1). Despite a screening of different conditions, no evidence for electron transfer could be obtained.
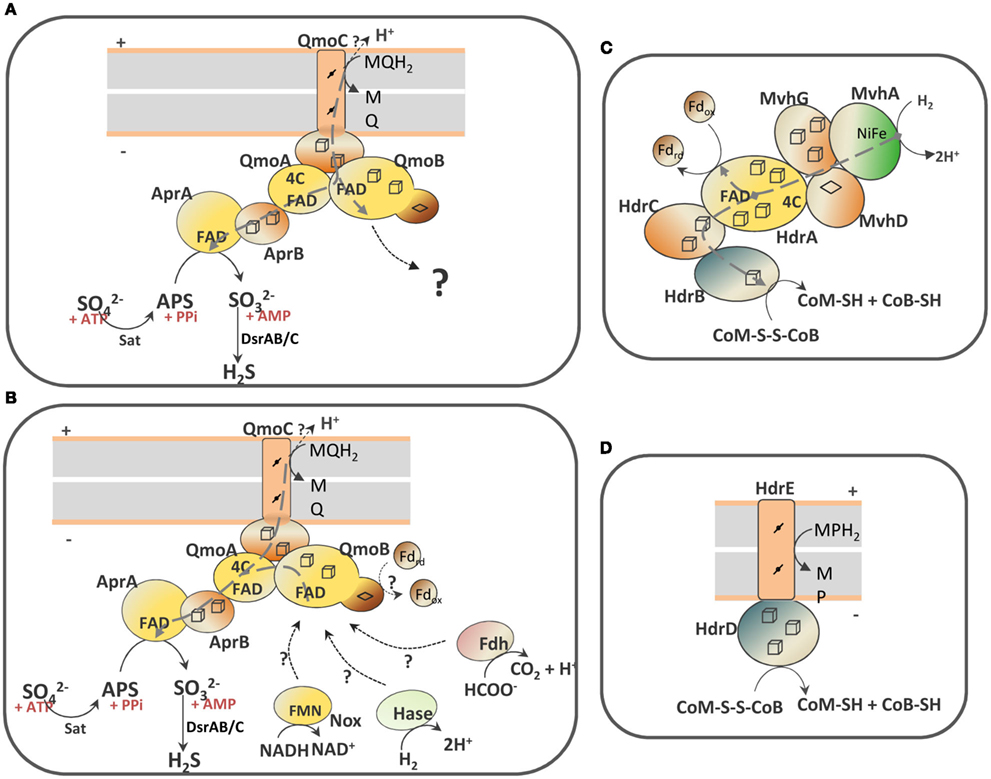
Scheme 1. Schematic representation of the QmoABC–AprAB interaction and the proposed involvement of third partners. (A) In the hypothesis of an electron bifurcation process the putative electron acceptor of QmoB with a high redox potential is represented by a question mark. (B) In the hypothesis of an electron confurcating mechanism several possible co-electron donors for the Qmo complex are considered: ferredoxin (Fd), hydrogenase (Hase), formate dehydrogenase (Fdh) or NADH dehydrogenase (Nox). The soluble HdrABC–MvhGAD complex (C) and the membrane-bound HdrED (D) of methanogens are shown for comparison. The gray dashed arrows represent electron bifurcation in (A,C), or electron confurcation in (B). The gray boxes represent the cytoplasmic membrane with + indicating the periplasm and − the cytoplasm.
Discussion
The AprAB APS reductase from SRP is a heterodimeric iron–sulfur flavoenzyme, which catalyzes the reversible reduction of APS to sulfite and AMP (Lampreia et al., 1994). It binds FAD, which is the site of APS reduction, and two [4Fe–4S] clusters that serve to transfer electrons from the protein surface to the catalytic site (Fritz et al., 2002a,b). Its physiological electron donor has not been unequivocally identified, but in many SRP and SOB the aprAB genes are part of a sat–aprBA–qmoABC gene cluster (Meyer and Kuever, 2007a,b; Frigaard and Dahl, 2009; Gregersen et al., 2011; Pereira et al., 2011; sat codes for the sulfate adenylyltransferase), which together with other indirect evidence (Pires et al., 2003; Haveman et al., 2004) led to the general conviction that QmoABC is the missing electron donor to AprAB, linking the quinone pool to sulfate reduction. The essential role of QmoABC in sulfate reduction has been recently established (Zane et al., 2010), but a direct connection between the two proteins has not been reported and direct electron transfer could not be observed (Pires et al., 2003). In some SOB lineages the qmoABC genes are absent and instead an aprM gene coding for a membrane protein is present (Hipp et al., 1997; Meyer and Kuever, 2007a; Frigaard and Dahl, 2009), suggesting that AprM can replace QmoABC in electron exchange between AprAB and the quinone pool. Homology modeling of AprAB from the different SOB lineages highlighted differences in the AprB structure that correlate with the presence of either the qmo or aprM genes. This points to adaptation of the electron transfer protein AprB as a result of docking to either Qmo or AprM proteins (Meyer and Kuever, 2008), and further substantiates a direct interaction.
In this work we report the first evidence that in SRP the QmoABC complex interacts directly with the APS reductase. This interaction could be detected by co-IP, and SPR showed that the two proteins are involved in a transient interaction that has a strong affinity (KD = 90 ± 4 nM) in equilibrium conditions, and which has a fast dissociation rate. This property allowed the cross-linking of two proteins and detection by Far-Western blot, which revealed that the QmoA subunit, and to a less extent QmoC, is involved in the interaction. The reverse experiment gave no results, but in the case of AprAB it is known that AprA is the catalytic subunit and AprB the electron transfer subunit (Fritz et al., 2002a,b), so that interaction with the electron donor should involve AprB. Expression of a tagged version of QmoA in D. vulgaris Hildenborough, followed by affinity purification allowed the detection of co-eluting AprAB, further confirming a specific direct interaction between the two proteins in a physiological setting.
However, reduction of APS with a menaquinol analog in the presence of QmoABC and AprAB could not be detected. The QmoABC subunits bind two hemes b, two FAD groups and several iron–sulfur centers, and are homologous to subunits of soluble (HdrABC) and membrane-bound (HdrED) heterodisulfide reductases from methanogens (Scheme 1; Pires et al., 2003; Thauer et al., 2008). QmoA and QmoB are both soluble iron–sulfur flavoproteins homologous to HdrA, the flavin-containing subunit of soluble HDRs. The function of HdrA has not been completely established, but it has been proposed to be involved in flavin-based electron bifurcation carried out by a complex between HdrABC and the F420-non-reducing MvhADG hydrogenase (Scheme 1C), which allows the coupling between the exergonic reduction of the CoM-S-S-CoB heterodisulfide by H2 to the endergonic reduction of ferredoxin by H2 (Thauer et al., 2008; Kaster et al., 2011). This bifurcation process is believed to involve the HdrA FAD cofactor, which transfers one electron to the heterodisulfide through HdrBC and another electron to ferredoxin. Such process may also occur with formate instead of H2, with a formate dehydrogenase replacing the Mvh hydrogenase (Costa et al., 2010). QmoB includes also a domain similar to MvhD, the [2Fe–2S] subunit of the Mvh hydrogenase that is responsible for electron transfer to HdrABC (Scheme 1C; Stojanowic et al., 2003). QmoC is a fusion protein that contains a cytochrome b transmembrane domain related to HdrE (Scheme 1D) and a hydrophilic iron–sulfur domain related to electron transfer subunit HdrC. Thus, QmoC fuses in a single protein the two subunits that in many trimeric respiratory oxidoreductases (composed of membrane subunit, electron transfer subunit, and catalytic subunit) are responsible for electron exchange with the quinone pool and electron transfer to the catalytic subunit (Rothery et al., 2008; Simon et al., 2008). This leaves two subunits, QmoA and QmoB, with an unknown function and which will likely interact with other physiological partners. QmoA is shown here to interact with AprAB, but the function of QmoB remains enigmatic. Its similarity to HdrA and MvhD suggests the involvement of a third physiological partner for the Qmo complex. We must also consider that menaquinol (E0′ −75 mV) cannot serve as sole electron donor to reduce APS due to the small difference in redox potentials, and to the fact that the membrane potential (∼150 mV) has to be overcome when transferring electrons from the quinone binding site in QmoC (likely situated toward the periplasmic side of the membrane) to AprAB in the cytoplasm. Thus, the reduction of APS by menaquinol has to be driven by coupling it to a second more favorable reaction. The idea that an electron bifurcation or confurcation mechanism, originally proposed by Buckel and coworkers (Herrmann et al., 2008), could be operating in the reduction of APS then appears as a very attractive and plausible hypothesis. Two possibilities can be envisioned: in the first one (Scheme 1A) the QmoB subunit reduced by menaquinol could bifurcate electrons to QmoA/AprAB and to a second electron acceptor with a high redox potential. The energetically favorable reduction of such electron acceptor by menaquinol could drive the unfavorable reduction of APS by menaquinol. The only problem with this hypothesis is that we cannot identify a candidate in SRB with a high enough reduction potential to drive this reaction.
The second possibility, that we favor, is to consider a reverse electron bifurcation mechanism, which has been referred to as electron confurcation. In such a process menaquinol and a cytoplasmic reductant of low redox potential could both serve as electron donors to the Qmo complex, which would confurcate electrons to the APS reductase (Scheme 1B). The favorable reduction of APS by this low potential electron donor would drive the unfavorable reduction of APS by menaquinol. The process of bifurcation/confurcation requires the presence of a two-electron center, such as a flavin, as the coupling site. In Qmo there are two FAD cofactors that can perform this process. According to the idea of crossed potentials at the flavin proposed by Nitschke and Russell (2011), the reduction of FAD at QmoA or QmoB by the low potential electron donor could generate a “hot” flavosemiquinone with a high redox potential that would then be a favorable electron acceptor for a second electron coming from menaquinol, and in practice “pulling” this electron from the quinone. Electron confurcation has been reported in the reduction of NADP+ with both reduced ferredoxin and NADH by Clostridium kluyveri NfnAB (Wang et al., 2010), and also in a multimeric soluble [FeFe] hydrogenase from Thermotoga maritima, which uses both NADH and reduced ferredoxin to produce H2 (Schut and Adams, 2009). This process has also been implicated in the energy metabolism of syntrophic organisms (Müller et al., 2010; Sieber et al., 2010).
Several coupling partners for Qmo can be considered in the confurcation hypothesis. The first is a hydrogenase or a formate dehydrogenase by analogy to what happens with HdrABC of methanogens (Costa et al., 2010; Kaster et al., 2011). An analysis of SRP genomes showed that a cytoplasmic version of either one of the two enzymes is always present (Pereira et al., 2011), except in C. maquilingensis where the qmoABC genes are also absent. In several organisms an MvhADG homolog is present, which in the archeal and in some bacterial organisms is part of an mvhADG–hdrABC gene cluster, suggesting this was acquired by lateral gene transfer from methanogenic organisms. In other bacteria the mvhADG genes are isolated, which may indicate subsequent loss of the hdrABC genes. In Desulfovibrio organisms no mvhADG genes are present, but genes coding for a membrane-associated hydrogenase (Ech or Coo) or a soluble [FeFe] hydrogenase are detected. The second possible partner for QmoB is a ferredoxin, also by analogy to HdrA. Ferredoxins are present in the genomes of all SRP, often in multiple copies (Pereira et al., 2011). Several proteins in SRP are known to reduce ferredoxin, including hydrogenases and formate dehydrogenases, pyruvate:ferredoxin oxidoreductase and the Rnf complex, which is also present in several Desulfovibrio spp. (Pereira et al., 2011). Finally, a third possible partner of QmoB is the mononuclear NADH oxidoreductase, Nox, which has been reported to reduce AprAB (Chen et al., 1994). Nox homologs (DVU3212 in D. vulgaris Hildenborough) are also present in the 25 genomes of SRP analyzed, except Thermodesulfovibrio yellowstonii. Recently, a study of protein–protein interactions failed to detect a link between Nox and energy metabolism proteins (Chhabra et al., 2011b), but such a negative result is not entirely conclusive due to the possibility of transient interactions not being detected in the conditions used. In these hypotheses H2 (E0′ −414 mV), formate (E0′ −430 mV), NADH (E0′ −320 mV), or ferredoxin (E0′ ∼−400 mV), would all be favorable reductants for APS . It is conceivable that more than one of these compounds may be used depending on the metabolic conditions, as observed for HdrABC (Costa et al., 2010), which could explain why no genes for interacting partners are co-localized with the sat–aprBA–qmoABC gene cluster. Any of these reductants could serve as a sole electron donor for the reduction of APS on its own, but in such situation the cells would get no energy benefit from this step. Coupling of APS reduction with oxidation of the menaquinone pool allows for energy conservation, considering that the oxidation of menaquinol by QmoC occurs at the periplasmic side of the membrane, with release of protons to the periplasm. In conclusion, the confurcation mechanism proposed here effectively allows the coupling of sulfate reduction with chemiosmotic energy conservation, a process long known to occur in SRP, but for which the molecular basis has been hard to identify. Clearly, further experiments will be required to test this hypothesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported (for Ana Raquel Ramos and Inês A. Cardoso Pereira) by research grants PTDC/QUI-BIQ/100591/2008 funded by Fundação para a Ciência e Tecnologia and FEDER program, and by grant PEst-OE/EQB/LA0004/2011; and (for Judy D. Wall and Kimberly L. Keller) by ENIGMA – Ecosystems and Networks Integrated with Genes and Molecular Assemblies supported by the Office of Science, Office of Biological, and Environmental Research, of the U.S. Department of Energy (DOE) under Contract No. DE-AC02-05CH11231 and by DOE grant DE-FG02-08ER64691. A. R. Ramos was supported by FCT Ph.D. fellowship SFRH/BD/60500/2009. We thank Manuela M. Pereira, Wolfgang Nitschke, and Wolfgang Buckel for helpful discussions.
References
Barton, L. L., and Fauque, G. D. (2009). Biochemistry, physiology and biotechnology of sulfate-reducing bacteria. Adv. Appl. Microbiol. 68, 41–98.
Bashir, Q., Scanu, S., and Ubbink, M. (2011). Dynamics in electron transfer protein complexes. FEBS J. 278, 1391–1400.
Chan, L. K., Weber, T. S., Morgan-Kiss, R. M., and Hanson, T. E. (2008). A genomic region required for phototrophic thiosulfate oxidation in the green sulfur bacterium Chlorobium tepidum (syn. Chlorobaculum tepidum). Microbiology 154, 818–829.
Chen, L., Le Gall, J., and Xavier, A. V. (1994). Purification, characterization and properties of an NADH oxidase from Desulfovibrio vulgaris (Hildenborough) and its coupling to adenylyl phosphosulfate reductase. Biochem. Biophys. Res. Commun. 203, 839–844.
Chhabra, S. R., Butland, G., Elias, D. A., Chandonia, J. M., Fok, O. Y., Juba, T. R., Gorur, A., Allen, S., Leung, C. M., Keller, K. L., Reveco, S., Zane, G. M., Semkiw, E., Prathapam, R., Gold, B., Singer, M., Ouellet, M., Szakal, E. D., Jorgens, D., Price, M. N., Witkowska, H. E., Beller, H. R., Arkin, A. P., Hazen, T. C., Biggin, M. D., Auer, M., Wall, J. D., and Keasling, J. D. (2011a). Generalized schemes for high-throughput manipulation of the Desulfovibrio vulgaris genome. Appl. Environ. Microbiol. 77, 7595–7604.
Chhabra, S. R., Joachimiak, M. P., Petzold, C. J., Zane, G. M., Price, M. N., Reveco, S. A., Fok, V., Johanson, A. R., Batth, T. S., Singer, M., Chandonia, J. M., Joyner, D., Hazen, T. C., Arkin, A. P., Wall, J. D., Singh, A. K., and Keasling, J. D. (2011b). Towards a rigorous network of protein-protein interactions of the model sulfate reducer Desulfovibrio vulgaris Hildenborough. PLoS ONE 6, e21470. doi:10.1371/journal.pone.0021470
Costa, K. C., Wong, P. M., Wang, T., Lie, T. J., Dodsworth, J. A., Swanson, I., Burn, J. A., Hackett, M., and Leigh, J. A. (2010). Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc. Natl. Acad. Sci. U.S.A. 107, 11050–11055.
Frigaard, N. U., and Dahl, C. (2009). Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 54, 103–200.
Fritz, G., Büchert, T., Huber, H., Stetter, K. O., and Kroneck, P. M. H. (2000). Adenylylsulfate reductases from archaea and bacteria are 1:1 αβ-heterodimeric iron-sulfur flavoenzymes – high similarity of molecular properties emphasizes their central role in sulfur metabolism. FEBS Lett. 473, 63–66.
Fritz, G., Büchert, T., and Kroneck, P. M. H. (2002a). The function of the [4Fe-4S] clusters and FAD in bacterial and archaeal adenylylsulfate reductases. Evidence for flavin-catalyzed reduction of adenosine 5′-phosphosulfate. J. Biol. Chem. 277, 26066–26073.
Fritz, G., Roth, A., Schiffer, A., Buchert, T., Bourenkov, G., Bartunik, H. D., Huber, H., Stetter, K. O., Kroneck, P. M., and Ermler, U. (2002b). Structure of adenylylsulfate reductase from the hyperthermophilic Archaeoglobus fulgidus at 1.6-A resolution. Proc. Natl. Acad. Sci. U.S.A. 99, 1836–1841.
Gregersen, L. H., Bryant, D. A., and Frigaard, N. U. (2011). Mechanisms and evolution of oxidative sulfur metabolism in green sulfur bacteria. Front. Microbiol. 2:116. doi:10.3389/fmicb.2011.00116
Haveman, S. A., Greene, E. A., Stilwell, C. P., Voordouw, J. K., and Voordouw, G. (2004). Physiological and gene expression analysis of inhibition of Desulfovibrio vulgaris Hildenborough by nitrite. J. Bacteriol. 186, 7944–7950.
Herrmann, G., Jayamani, E., Mai, G., and Buckel, W. (2008). Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190, 784–791.
Hipp, W. M., Pott, A. S., Thum-Schmitz, N., Faath, I., Dahl, C., and Truper, H. G. (1997). Towards the phylogeny of APS reductases and sirohaem sulfite reductases in sulfate-reducing and sulfur-oxidizing prokaryotes. Microbiology 143(Pt 9), 2891–2902.
Junier, P., Junier, T., Podell, S., Sims, D. R., Detter, J. C., Lykidis, A., Han, C. S., Wigginton, N. S., Gaasterland, T., and Bernier-Latmani, R. (2010). The genome of the Gram-positive metal- and sulfate-reducing bacterium Desulfotomaculum reducens strain MI-1. Environ. Microbiol. 12, 2738–2754.
Kaster, A. K., Moll, J., Parey, K., and Thauer, R. K. (2011). Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc. Natl. Acad. Sci. U.S.A. 108, 2981–2986.
Keller, K. L., and Wall, J. D. (2011). Genetics and molecular biology of the electron flow for sulfate respiration in Desulfovibrio. Front. Microbiol. 2:135. doi:10.3389/fmicb.2011.00135
Keller, K. L., Wall, J. D., and Chhabra, S. (2011). Methods for engineering sulfate reducing bacteria of the genus Desulfovibrio. Meth. Enzymol. 497, 503–517.
Lampreia, J., Pereira, A. S., and Moura, J. J. G. (1994). Adenylylsulfate reductases from sulfate-reducing bacteria. Meth. Enzymol. 243, 241–260.
Li, M., and Elledge, S. J. (2007). Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256.
Liu, M. C., and Peck, H. D. Jr. (1981). The isolation of hexaheme cytochrome from Desulfovibrio desulfuricans and its identification as a new type of nitrite reductase. J. Biol. Chem. 256, 13159–13164.
Mander, G. J., Duin, E. C., Linder, D., Stetter, K. O., and Hedderich, R. (2002). Purification and characterization of a membrane-bound enzyme complex from the sulfate-reducing archaeon Archaeoglobus fulgidus related to heterodisulfide reductase from methanogenic archaea. Eur. J. Biochem. 269, 1895–1904.
Martinez-Fabregas, J., Rubio, S., Diaz-Quintana, A., Diaz-Moreno, I., and De la Rosa, M. A. (2011). Proteomic tools for the analysis of transient interactions between metalloproteins. FEBS J. 278, 1401–1410.
Meyer, B., and Kuever, J. (2007a). Phylogeny of the alpha and beta subunits of the dissimilatory adenosine-5′-phosphosulfate (APS) reductase from sulfate-reducing prokaryotes – origin and evolution of the dissimilatory sulfate-reduction pathway. Microbiology 153, 2026–2044.
Meyer, B., and Kuever, J. (2007b). Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5′-phosphosulfate reductase-encoding genes (aprBA) among sulfur-oxidizing prokaryotes. Microbiology 153, 3478–3498.
Meyer, B., and Kuever, J. (2008). Homology modeling of dissimilatory APS reductases (AprBA) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE 3, e1514. doi:10.1371/journal.pone.0001514
Müller, N., Worm, P., Schink, B., Stams, A. J. M., and Plugge, C. M. (2010) Syntrophic butyrate and propionate oxidation processes: from genomes to reaction mechanisms. Environ. Microbiol. Rep. 2, 489–499.
Muyzer, G., and Stams, A. J. M. (2008). The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 6, 441–454.
Nitschke, W., and Russell, M. J. (2011) Redox bifurcations: mechanisms and importance to life now, and at its origin: a widespread means of energy conversion in biology unfolds. Bioessays 34, 106–109.
Pereira, I. A. C. (2008). “Membrane complexes in Desulfovibrio,” in Microbial Sulfur Metabolism, eds C. Friedrich and C. Dahl (Berlin: Springer-Verlag), 24–35.
Pereira, I. A. C., Ramos, A. R., Grein, F., Marques, M. C., da Silva, S. M., and Vesceslau, S. S. (2011). A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol. 2:69. doi:10.3389/fmicb.2011.00069
Pires, R. H., Lourenco, A. I., Morais, F., Teixeira, M., Xavier, A. V., Saraiva, L. M., and Pereira, I. A. (2003). A novel membrane-bound respiratory complex from Desulfovibrio desulfuricans ATCC 27774. Biochim. Biophys. Acta 1605, 67–82.
Plugge, C. M., Zhang, W., Scholten, J. C., and Stams, A. J. (2011). Metabolic flexibility of sulfate-reducing bacteria. Front. Microbiol. 2:81.
Rodriguez, J., Hiras, J., and Hanson, T. E. (2011). Sulfite oxidation in Chlorobaculum tepidum. Front. Microbiol. 2:112. doi:10.3389/fmicb.2011.00112
Rothery, R. A., Workun, G. J., and Weiner, J. H. (2008). The prokaryotic complex iron-sulfur molybdoenzyme family. Biochim. Biophys. Acta 1778, 1897–1929.
Sato, Y., Kameya, M., Arai, H., Ishii, M., and Igarashi, Y. (2011). Detecting weak protein-protein interactions by modified far-western blotting. J. Biosci. Bioeng. 112, 304–307.
Schut, G. J., and Adams, M. W. (2009) The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J. Bacteriol. 191, 4451–4457.
Sieber, J. R., Sims, D. R., Han, C., Kim, E., Lykidis, A., Lapidus, A. L., McDonnald, E., Rohlin, L., Culley, D. E., Gunsalus, R., and McInerney, M. J. (2010) The genome of Syntrophomonas wolfei: new insights into syntrophic metabolism and biohydrogen production, Environ. Microbiol. 12, 2289–2301.
Simon, J., van Spanning, R. J., and Richardson, D. J. (2008). The organisation of proton motive and non-proton motive redox loops in prokaryotic respiratory systems. Biochim. Biophys. Acta 1777, 1480–1490.
Stams, A. J., and Plugge, C. M. (2009). Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat. Rev. Microbiol. 7, 568–577.
Stojanowic, A., Mander, G. J., Duin, E. C., and Hedderich, R. (2003). Physiological role of the F-420-non-reducing hydrogenase (Mvh) from Methanothermobacter marburgensis. Arch. Microbiol. 180, 194–203.
Thauer, R. K., Kaster, A. K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591.
Wang, S., Huang, H., Moll, J., and Thauer, R. K. (2010) NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J. Bacteriol. 192, 5115–5123.
Keywords: sulfate reducing bacteria, sulfur-oxidizing bacteria, electron bifurcation, membrane complex, protein–protein interactions, surface plasmon resonance
Citation: Ramos AR, Keller KL, Wall JD and Pereira IAC (2012) The membrane QmoABC complex interacts directly with the dissimilatory adenosine 5′-phosphosulfate reductase in sulfate reducing bacteria. Front. Microbio. 3:137. doi: 10.3389/fmicb.2012.00137
Received: 16 February 2012; Paper pending published: 02 March 2012;
Accepted: 22 March 2012; Published online: 23 April 2012.
Edited by:
Niels-Ulrik Frigaard, University of Copenhagen, DenmarkReviewed by:
Ulrike Kappler, University of Queensland, AustraliaBen Berks, University of Oxford, UK
Copyright: © 2012 Ramos, Keller, Wall and Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Inês A. Cardoso Pereira, Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Av. República, EAN, 2780-157 Oeiras, Portugal. e-mail:aXBlcmVpcmFAaXRxYi51bmwucHQ=