 Heath J. Mills1*
Heath J. Mills1* Brandi Kiel Reese1,2 and Cruz St. Peter1
Brandi Kiel Reese1,2 and Cruz St. Peter1
- 1 Department of Oceanography, Texas A&M University, College Station, TX, USA
- 2 Department of Biological Science, University of Southern California, Los Angeles, CA, USA
The objective of this study was to determine shifts in the microbial community structure and potential function based on standard Integrated Ocean Drilling Program (IODP) storage procedures for sediment cores. Standard long-term storage protocols maintain sediment temperature at 4°C for mineralogy, geochemical, and/or geotechnical analysis whereas standard microbiological sampling immediately preserves sediments at −80°C. Storage at 4°C does not take into account populations may remain active over geologic time scales at temperatures similar to storage conditions. Identification of active populations within the stored core would suggest geochemical and geophysical conditions within the core change over time. To test this potential, the metabolically active fraction of the total microbial community was characterized from IODP Expedition 325 Great Barrier Reef sediment cores prior to and following a 3-month storage period. Total RNA was extracted from complementary 2, 20, and 40 m below sea floor sediment samples, reverse transcribed to complementary DNA and then sequenced using 454 FLX sequencing technology, yielding over 14,800 sequences from the six samples. Interestingly, 97.3% of the sequences detected were associated with lineages that changed in detection frequency during the storage period including key biogeochemically relevant lineages associated with nitrogen, iron, and sulfur cycling. These lineages have the potential to permanently alter the physical and chemical characteristics of the sediment promoting misleading conclusions about the in situ biogeochemical environment. In addition, the detection of new lineages after storage increases the potential for a wider range of viable lineages within the subsurface that may be underestimated during standard community characterizations.
Introduction
The Integrated Ocean Drilling Program (IODP) and its predecessor programs Deep Sea Drilling Program and Ocean Drilling Program have led scientific drilling of the marine subsurface for over 40 years. Three main repositories [Gulf Coast Repository, Bremen Core Repository (BCR), and Kochi Core Repository] collect and store drill material from around the world, providing a resource to land based scientists in an effort to expand the research potential of the Program. The main focus of subsurface research has been chemical and physical characterizations. However, recent initiatives have emphasized the importance of understanding the subseafloor biosphere (Bickle et al., 2011). With this new direction has come new technology and awareness to determine the biological effects on subsurface. Previous studies have described some aspects of the physical, chemical, and biological effects of long-term storage on core integrity (König et al., 2000; Lin et al., 2010), however a detailed analysis of changes in the active microbial community during core material storage has not been presented.
Advancements in drilling technology and expeditions dedicated to microbiology have provided an increase in the number of samples suitable for enhanced microbial community characterization. Resulting studies have produced a better understanding of in situ community structure and further described natural diagenetic processes deep beneath the seafloor. Multiple biological processes catalyzed by subsurface microbial populations have been shown to alter the physical and chemical structure of subsurface sediment and rock (reviewed in Fry et al., 2008). With minimal geochemical alteration in subsurface environmental conditions over short time scales (Jørgensen, 2011) the community structure would remain stable in composition and expressed metabolic processes. Therefore, disruptions to in situ fluid flow, temperature, or pressure, such as those created during the drilling process and subsequent core storage would be hypothesized to create shifts in both community structure and overall function. This newly structured population would utilize different chemical compounds at different rates compared to the undisturbed population and thus would change the physical and chemical composition of the stored material. Key to this assumption is the availability of a dormant fraction of the population, or a minor fraction that becomes more metabolically active following a disruption event. Dormancy is a valuable and common trait to microbial populations (Jones and Lennon, 2010) but has been more frequently associated with environments that experience periodic perturbations or shifts in environmental conditions (Fuhrman et al., 2006; Jones and McMahon, 2009). Lin et al. (2010) suggested the presence of potential dormant populations in subsurface sediments by describing a shift in microbial community structure within stored IODP sediment samples. The next step is to determine a shift in metabolically active populations during storage to better describe both the microbial community and associated functional processes threatening core integrity.
This study provides a detailed examination and comparison of the metabolically active fraction of the microbial population at the time of sample recovery, representing near in situ conditions, and active microbial populations detected after 3 months of standard core storage. Total community 16S ribosomal RNA (rRNA) transcripts were sequenced from multiple sediment depths collected during IODP Expedition 325 on the Great Barrier Reef. High throughput sequencing using 454 FLX provided the sequencing capacity to statistically determine shifts in the population over time. This work supports and advances a previous study by Lin et al. (2010) by providing unique insight into the metabolically active microbial populations that would be contributing to alterations in physical and chemical properties of sediment cores during storage. These alterations should be considered during post-expedition sediment analysis. This study also demonstrates that minor and/or dormant lineages can be revived when environmental conditions change, providing evidence for a viable fraction of the community. A viable fraction would represent a sink for genetic material and metabolic processes potentially underestimated in current biosphere characterizations.
Materials and Methods
Sampling Acquisition and Storage
Sediment samples were collected during the IODP Expedition 325 aboard the mission specific platform Greatship Maya. Drilling occurred April 5th, 2010, at Noggin Pass (17°6.3461′S, 146°33.7526′E) drill hole M0058A using the advance piston coring (APC) drilling system. Immediately following recovery, whole round core sections were cut from the drill core liner at three depths below surface at 2, 20, and 40 m below sea floor (mbsf). For the purposes of this manuscript, the samples collected onboard the Greatship Maya are identified as “offshore samples” within this manuscript. These samples were transferred to −80°C freezer and shipped on dry ice to the Mills Laboratory after the completion of the expedition. The remaining core material was then maintained at 4°C during transit, shipping, and storage at the BCR in Bremen, Germany. During expedition onshore activities at the BCR, sediment was collected July 13, 2010 from the same core sections sampled offshore, at locations directly adjacent to the whole round core sections. A 2-cm wide sediment section was collected from the center of the core, homogenized, and frozen at −80°C prior to being shipped on dry ice to the Mills Laboratory. The samples stored at 4°C prior to sub-sampling at the BCR are identified as “onshore samples” within this manuscript.
Geochemical Analyses
Sediment interstitial water was collected shipboard using Rhizon syringes for geochemical analysis. Samples designated for onshore analysis at BCR were preserved immediately upon collection by refrigerated acidified (cations) or by frozen unacidified (anions) and transported on ice. Ammonium (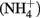 ) was analyzed shipboard using a gas stripping technique described in Hall and Aller (1992). Porewater anions (e.g.,
) was analyzed shipboard using a gas stripping technique described in Hall and Aller (1992). Porewater anions (e.g., 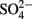 and Cl−,) were measured onshore using ion chromatography at BCR and cations (e.g., Fe, Mn, Ca, S) were measured onshore using inductively coupled plasma–optical emission spectrometry (ICP–OES) at BCR. Total carbon (TC) and total organic carbon (TOC) was analyzed on 50 mg dried sediment taken every 50 cm using a LECO CC-125 carbon–sulfur analyzer onshore at BCR.
and Cl−,) were measured onshore using ion chromatography at BCR and cations (e.g., Fe, Mn, Ca, S) were measured onshore using inductively coupled plasma–optical emission spectrometry (ICP–OES) at BCR. Total carbon (TC) and total organic carbon (TOC) was analyzed on 50 mg dried sediment taken every 50 cm using a LECO CC-125 carbon–sulfur analyzer onshore at BCR.
Nucleic Acid Extraction and RNA Purification
Nucleic acids were extracted from both offshore and onshore sampled sediments using a method modified from Mills et al. (2008). The description below provides details not previously published and represents the most updated version of this versatile extraction method. Key to the method was reducing time spent during the extraction process with extraction yields being inversely proportional to duration of extraction procedure. All chemicals were molecular biology grade (MBG) where available. All water for solutions was autoclaved and treated with diethylpyrocarbonate (DEPC) to remove nucleases. A 0.5-g sample was quickly chipped from frozen sediment under sterile conditions and placed into a 2-ml screw top tube with rubber gasket in lid. All samples remained frozen prior to extraction. Between 100 and 250 μl of extraction buffer [250 μl of 1 M Tris–HCl (MP Biomedicals; Solon, OH, USA; pH 8.0), 200 μl of 500 mM ethylenediamine tetraacetic acid (EDTA; EMD Chemicals; Gibbstown, NJ, USA; pH 8.0), and 230 μl of 40% glucose (Calbiochem; La Jolla, CA, USA) raised to a total 10 ml volume with sterile deionized water] was added to each sample tube. The final concentration of the extraction buffer is as follows: 25 mM Tris–HCl, 10 mM EDTA, and 0.92% glucose. Volume added of the extraction buffer was determined by the amount of porewater in sample and should be adjusted accordingly. Sufficient volume of buffer added is achieved when the sample can be adequately vortexed; forming a soft, well-mixed mud. Samples were cycled five times through a 45-s vortex, frozen in liquid nitrogen, and thawed for 1 min at 55°C. Samples were rapidly and completely thawed during 55°C incubation, but temperatures remained low to avoid increasing activity of DNases and RNases. After five cycles were completed, the following solutions were added: 0–150 μl extraction buffer (total volume of extraction buffer added equaled 250 μl between this and the previous addition steps), 100 μl lysozyme solution [extraction buffer with 4 mg ml−1 lysozyme (Rockland, Inc.; Gilbertsville, PA, USA) added], and 50 μl of 500 mM EDTA (pH 8.0). Samples were incubated for 10 min at 30°C while shaking at 150 rpm. A solution of phenol (EMD Chemicals; Gibbstown, NJ, USA):chloroform (BDH; West Chester, PA, USA):isoamyl alcohol (MP Biomedicals; Solon, OH, USA, 25:24:1) was added at 800 μl along with 50 μl of 10% sodium dodecyl sulfate (SDS; EMD Chemicals; Gibbstown, NJ, USA). Phenol was not buffered with Tris hydrochloric acid so that the pH remained acidic. Acidic phenol is optimal for RNA extraction. Samples were vortexed for 1 min to create an emulsion prior to centrifugation at 15,000 rpm (21,100 × g) for 3 min at room temperature. The top aqueous layer was transferred to a new 2 ml screw top tube containing 800 μl phenol:chloroform:isoamyl alcohol. Samples were again vortexed for 1 min and centrifuged for 3 min at 15,000 rpm (21,100 × g) at room temperature. The aqueous layer was transferred to a new 1.5 ml microcentrifuge tube avoiding any traces of the phenol solution. A total of 50 μl of 3 M sodium acetate (J. T. Baker; Phillipsburg, NJ, USA; pH 6.4) and 1 ml 200 proof MPG ethanol was added to each sample prior to a 15-min centrifuge step at 15,000 rpm (21,100 × g) at 4°C. The supernatant was slowly removed and the pellet dried for 10 min. Pellets were resuspended in 50 μl of sodium citrate (pH 6.4) to optimize for RNA preservation. If DNA is desired, resuspend in autoclaved, deionized water (pH 8.0), and buffer the phenol to raise the pH to 8. For RNA sample preparation, extractions were treated with Turbo DNA Free (Ambion, Inc.; Austin, TX, USA) according to manufacturer’s instructions to remove residual DNA co-extracted with the RNA.
RNA Reverse Transcription
Small subunit (SSU) rRNA was reverse transcribed according to Reese et al. (2012). In brief, complementary DNA (cDNA) was produced using moloney murine leukemia virus (MMLV) reverse transcriptase (Promega, Madison, WI, USA) and 518R (5′-CGT ATT ACC GCG GCT GCT GG-3′; Nogales et al., 1999). Aliquots of RNA extract were screened for the presence of residual DNA using the Bacteria domain-specific SSU rRNA gene primers 27F (5′-AGR GTT TGA TCM TGG CTC AG-3′; Giovannoni et al., 1991) and 518R. Thermocycling conditions are described in Reese et al. (2012). All RNA extracts were determined free of residual DNA (data not shown).
Pyrosequencing
Primers 28F (5′-GAG TTT GAT CNT GGC TCA G-3′) and 519R (5′-GTN TTA CNG CGG CKG CTG-3′) produced amplicons from cDNA that spanned three hyper-variable regions (V1 through V3; Handl et al., 2011). cDNA amplicons from each of the sediment samples were sequenced at the Research and Testing Laboratory (Lubbock, TX, USA). Amplicons from each sample were first labeled with a 10 base unique multiplex identifier (MID) sequence to allow all samples to be sequenced on a single run of a Roche 454 FLX (454 Life Sciences; Branford, CT, USA). Downstream sequence analysis parsed the individual sequences into sample specific libraries. Resulting libraries were screened for reads less than 200 bases, reads lacking a Roche-designed four base key sequence, and non-bacterial reads lacking specific 28F primer recognition site. Sequences were checked for chimeras using the black box chimera check software (B2C2; Gontcharova et al., 2010). All sequences passing quality control were denoized prior to phylogenetic and statistical analysis. Sequences were then deposited to the National Center for Biotechnology Information database using Sequence Read Archive (SRA) under accession number SRA049351.
Phylogenetic and Statistical Analysis
All sequences passing quality control were taxonomically classified (percent of total sequence length that aligned with a given database sequence) using the NCBI Basic Local Alignment Search Tool (BLASTn) .NET algorithm (accessed January 2011; Dowd et al., 2005). Sequences with identity scores greater than 97% identity (<3% divergence) to known or well characterized 16S rRNA sequences were resolved at the species level, between 95 and 97% at the genus level, between 90 and 95% at the family, and between 85 and 90% at the order level, 80 and 85% at the class and 77–80% at phyla (Stackebrandt and Goebel, 1994). Only sequences meeting the genus taxonomic classification were incorporated into statistical analyses. Predictions of functional diversity were derived from genus level taxonomic characterization using previous studies for reference. Lineages with multiple metabolic functions identified were assigned to multiple groups to reduce effects of limited geochemical data. The percentage of sequences identified to individual lineages was determined for each sample providing relative abundance information within and among the samples based upon reads per sample. Sequences were further clustered into operational taxonomic units (OTU) using the Ribosome Database Project (RDP; University of Michigan; Lansing, MI, USA) using a 95% sequence similarity cutoff (corresponding to a genus level classification). The sequences were not trimmed prior to clustering to avoid potential biases for or against indel regions common to 16S rRNA. The authors recognize potential problems with this decision; however, using a lower sequence similarity cutoff (95% compared to a typical 97%) produced a reproducible calculation of OTUs with multiple clustering runs. Chao1, Shannon Wiener, and Evenness were calculated from the OTU clustering using RDP.
Results
Geochemical Characterization
Cations, anions, and TOC were measured onshore at BCR whereas ammonia and alkalinity were measured shipboard. Alkalinity, pH, and chloride concentrations were unchanged with depth into the sediment. Concentrations of ammonia increased from 0.1 mM near the sediment surface to 2.2 mM at 40 m depth (Figure 1). Conversely, sulfate concentrations decreased from 29 mM at the surface to 18 mM at depth. Iron concentrations were low in the top 5 m (1 μM) and increased sharply at 9 m (14 μM) followed by a decrease in concentration until 25 m (0.7 μM). Another sharp increase in iron concentration was observed at 29 m (11.7 μM) and at 36 m (10.9 μM). Manganese followed the same general trend as iron with spikes in concentration at the same depths. Manganese was highest in the surface (0.6 μM) and lowest at 34 m (0.1 μM).
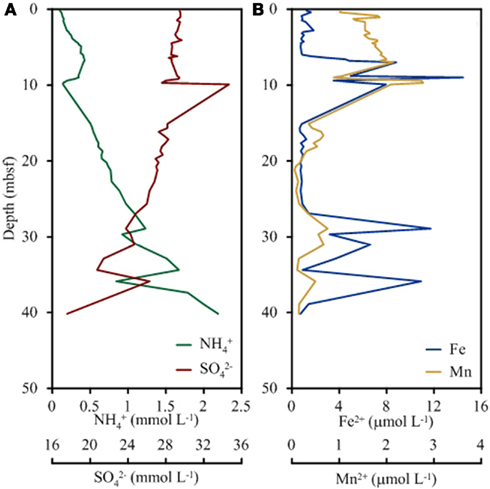
Figure 1. Depth profile of key geochemical species significant to microbial respiration including ammonium and sulfate (A) and total iron and total manganese (B).
Total carbon was 8.7 wt% on average in hole M0058A. Within the TC, TOC comprised 3% and total inorganic carbon (TIC; carbonates) was 97%. The TOC in M0058A averaged 0.3 wt% throughout the sediment column.
Phylogenetic Distribution of Bacterial Lineages
All samples collect onboard the Greatship Maya and immediately stored at −80°C are referred to as “offshore” samples. This sampling strategy is commonly considered indicative of an in situ population characterization. Samples collected at the BCR following 3 months of storage at 4°C prior to being cryogenically frozen are referred to as “onshore” samples.
A total of 14,897 16S rRNA cDNA sequences passed quality control from the two sets of 2, 20, and 40 mbsf sediment samples. Twelve phyla representing 71 families were identified. Sequences derived from offshore samples contained a total of 38 families, while the onshore samples had 54 families. This increased diversity following 3 months storage was also observed in Chao1 and Shannon Wiener calculations (Table 1). With the exception of the 20-mbsf sample collected during post-cruise operations, the evenness increased during storage. Changes in trends in sequence abundance per lineage were observed between the offshore and onshore samples (Figure 2). A majority of the samples had more than 99% of the total sequences represented by lineages that shifted in frequency of detection between sampling time points. Twenty families represented less than 1.0% of the total sequences and were omitted from this analysis. These 20 families accounted for 4.0% of the total sequences from the 2-m sample collected after storage, whereas they incorporated less than 0.8% of the total sequences for four of the other five samples.
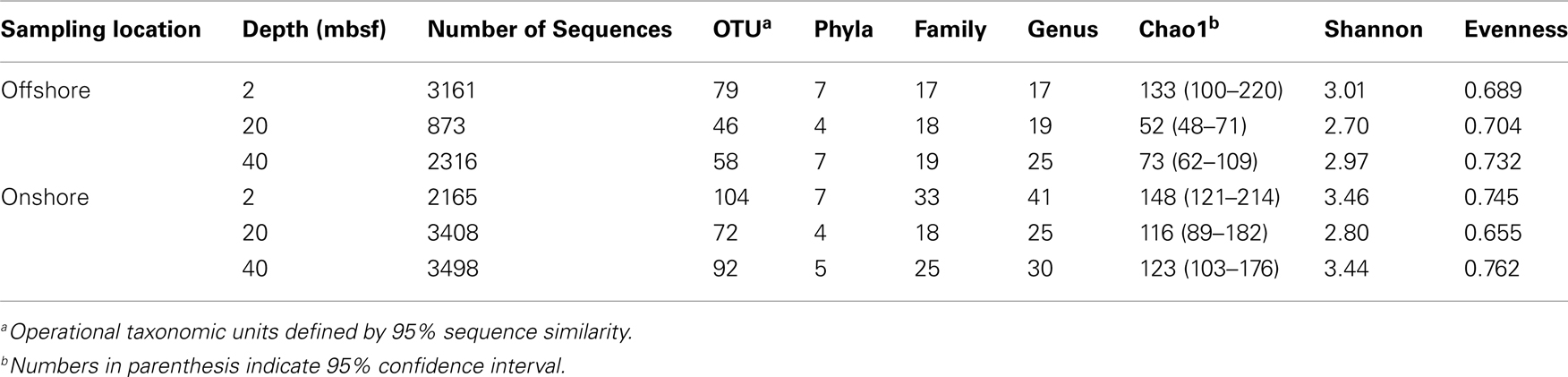
Table 1. Sequence and classification abundances and statistical indices of each sample.
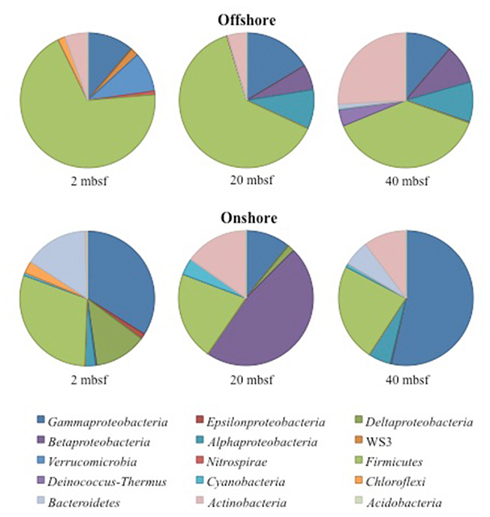
Figure 2. Phyla and class level distribution of bacterial lineages. Sequences were annotated using RDP Classifier. Identification was determined with a confidence interval of 70%. Sequence data supports a high taxonomic level alteration of the community composition during 4°C storage.
Microbial Community Structure – Offshore Samples
A total of 40 families were detected from samples immediately frozen shipboard at −80°C. A subset of 19 families (83.4% of the offshore sequences) was more frequently detected at all depths compared to the samples collected during the onshore portion of Expedition 325 (Table 2). These families represented 94.5% at 2 mbsf, 87.5% at 20 mbsf, and 68.2% at 40 mbsf of the in situ sequences detected. In contrast, these families represented only 4.3, 14.8, and 23.8%, respectively, of the total sequences obtained from the corresponding depths in the onshore sediment samples.
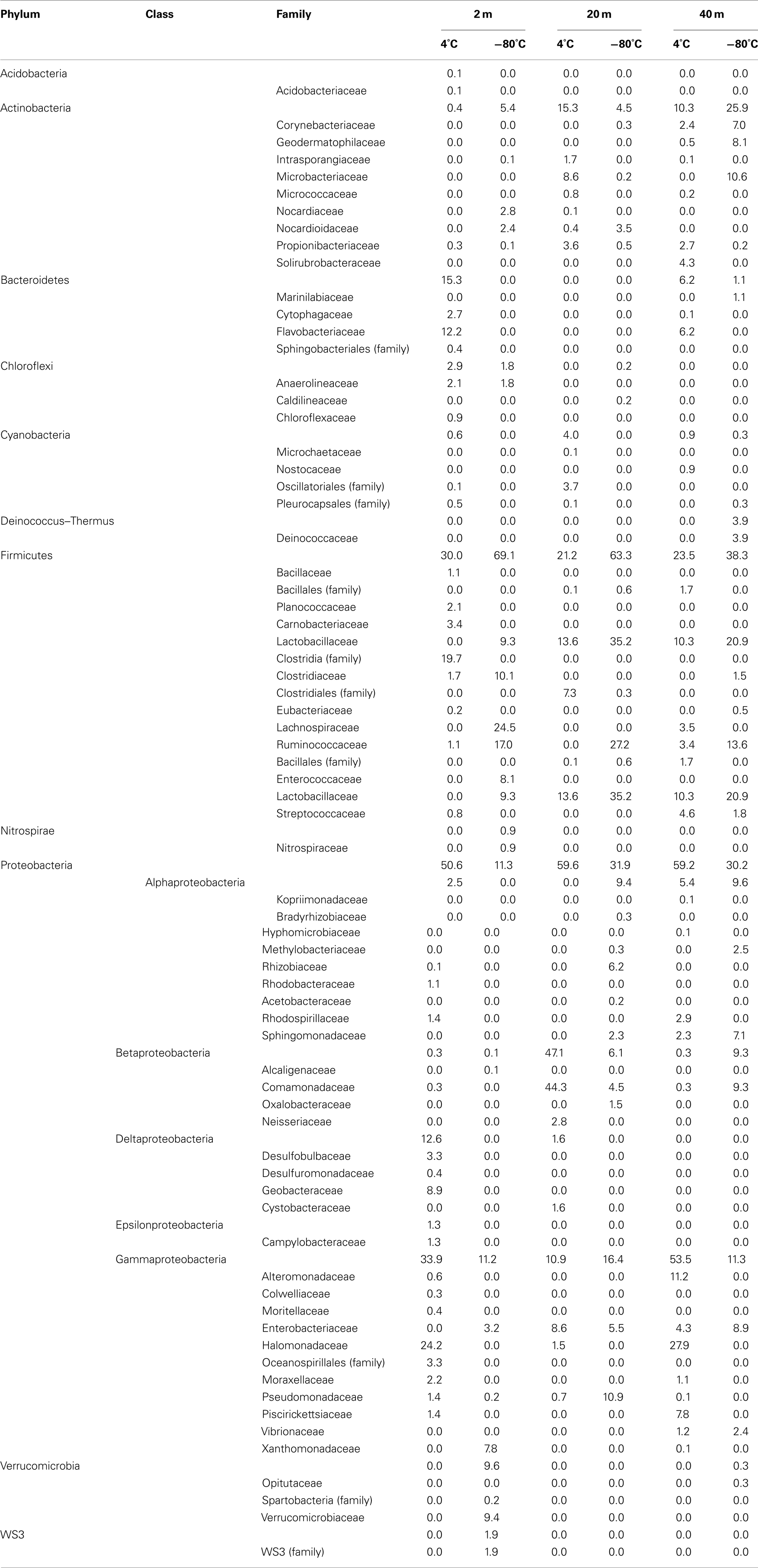
Table 2. Phylogenetic classification of bacterial sequences obtained from Expedition 325.
Of the 19 families detected in the 2-mbsf offshore sediment samples, Lachnospiraceae (24.5%), Ruminococcaceae (17.0%), Clostridiaceae (10.1%), Lactobacillaceae (9.3%), Enterococcaceae (8.1%), and Xanthomonadaceae (7.8%) were detected the most frequently (Table 2). These lineages collectively only represented 2.8% of the sequences obtained from the 2-mbsf sediments collected during on shore sampling. Functional classification suggest over 66% of these sequences were associated with lineages capable of fermentative processes (Figure 3). Aerobic lineages comprised 5.2% of the sequences while groups capable of carbonate dissolution represented 17.0%.
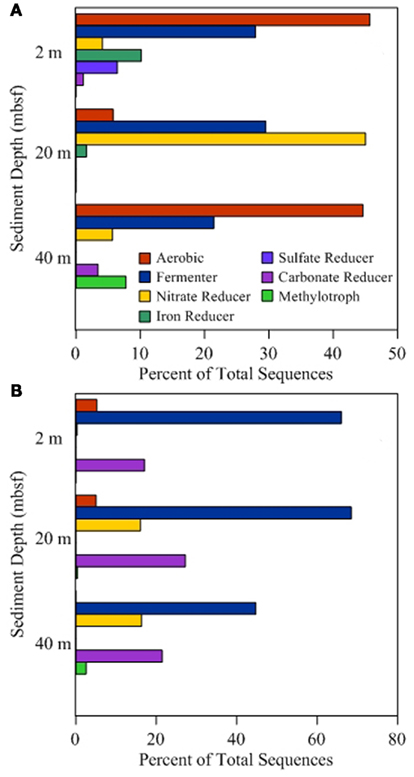
Figure 3. Functional group distribution. Sequence annotation at the genus level was used to identify functional groups associated with different metabolic processes. Lineages associated with multiple processes were placed in multiple categories to avoid assumptions of activity. Percentages presented for onshore samples (A) and offshore samples (B) are based on the total number of sequences obtained and are independent of percentages reported for other categories.
A total of 19 families were detected at 20 mbsf depth. Although the number of families detected was similar to the 19 families at 2 mbsf sample, a population shift was observed at 20 mbsf with no Lachnospiraceae, Clostridiaceae, Enterococcaceae, and Xanthomonadaceae detected (Table 2). Lactobacillaceae and Ruminococcaceae increased in abundance to represent 35.2 and 27.2% of the sequences obtained from this depth, respectively. Ruminococcaceae was not detected in the sediments after the storage period at 4°C. Similarly, Pseudomonadaceae represented 10.9% of the total sequences at this depth, but only 0.7% after the 3-month storage. Lactobacillaceae was detected at 13.6% of the total sequences obtained from the 20-mbsf sediment stored at 4°C. Rhizobiaceae (6.1%) and Enterobacteriaceae (5.5%) were also detected at greater than 5% abundance of total sequences (Table 2). Similar to the 2-mbsf sample, fermentative lineages were the most frequently detected (68.5% of total sequences; Figure 3). Aerobic lineages were again 5.0% of the sequences however groups capable of carbonate dissolution represented 27.2% of the total sequences. Nitrate reducing lineages increased in frequency detected (16.1% of the total sequences).
At 40 mbsf, 19 families were identified. Both Lactobacillaceae and Ruminococcaceae decreased in abundance (20.1 and 13.6%, respectively) compared to the 20-mbsf sample (Table 2). Other lineages detected at 40 mbsf included Microbacteriaceae (10.6%), Comamonadaceae (9.3%), Enterobacteriaceae (8.9%), Sphingomonadaceae (7.1%), and Corynebacteriaceae (7.0%). A general trend of increasing abundance with depth was noted with all of these lineages. In addition, these lineages were detected over three times more frequently in the offshore samples compared to the onshore samples. A similar distribution of functional process was observed compared to the 20-m sample with the exception of no aerobic lineages detected (Figure 3).
Microbial Community Structure – Onshore Samples
A total of 57 families were detected in the three samples collected during the onshore portion of Expedition 325 following 3 months of storage at 4°C (Table 2). There were 24 families with a higher sequence abundance (62.1% of the total sequences) at all three depths following the 3-month storage at 4°C (Table A1 in Appendix) compared to the samples collected and preserved at −80°C on the ship. These families accounted for 91.4% at 2 mbsf, 23.3% at 20 mbsf, and 71.6% mbsf of the total classified sequences from sediments collected during the onshore portion of Expedition 325. In contrast, the same 24 families represented only 0.2, 1.4, and 2.3% of the sequences obtained from 2, 20, and 40 mbsf, respectively, from sediments stored immediately at −80°C.
At 2 mbsf, a total of 33 families were detected with 20 families representing 90.3% of the sequences (Table 2). The most frequently detected lineages included Halomonadaceae (24.2%), Clostridia (19.7%), Flavobacteriaceae (12.2%), Geobacteraceae (8.9%), Carnobactericeae (3.4%), Oceanospirillales (3.3%), and Desulfobulbaceae (3.3%). No other family represented more than 3.0% of the total sequences. Aerobic lineages were the most frequently detected with 45.7% of the sequences attributed to this group (Figure 3). While fermentative lineages were also detected after the 3-months, the frequency decreased over 50% compared to the offshore samples. Nitrate, iron, and sulfate-reducing lineages represented 4.1, 10.2, and 6.4%, respectively. In contrast to the offshore 2 mbsf sample, carbonate reducing lineages accounted for only 1.1% of the sequences.
A total of 20 families were detected in the 20 mbsf sediment samples collected during the onshore portion of the Expedition (Table 2). Within this set, 23.3% of the total sequences were represented by 10 families that were more abundant in the stored sediments at all depths compared to the offshore samples. An additional 62.5% of the sequences from this sample were related to Comamonadaceae (44.3%), Microbacteriaceae (8.6%), and Enterobacteriaceae (8.6%; Table 2). Sequences related to Comamonadaceae increased in abundance by an order of magnitude between the offshore sampling (4.5%) and the post-cruise sampling period at 20 mbsf (44.3%). However, this lineage was not included in the initial percent calculations due to the abundance decreasing at 40 mbsf from 9.3 to 0.3% after 3 months at 4°C. A similar trend was determined for Microbacteriaceae with an increase at 20 mbsf from 0.2 to 8.6% during the 3-month period at 4°C, whereas in samples at 40 mbsf the abundance decreased from 10.6 to 0.0%. The 20 mbsf sample was the only depth where Enterobacteriaceae was more frequently detected in onshore sampled sediment (8.6%) compared to the offshore sampled sediment (4.3%). Collectively, the 10 families plus Comamonadaceae, Microbacteriaceae, and Enterobacteriaceae equal 84.9% of the sequences from the 20-m sample collected onshore, compared to 11.6% of the sequences from the corresponding offshore sample. Additional families detected frequently at 20 mbsf included Clostridia (7.3%), Oscillatoriales (3.7%), and Propionibacteriaceae (3.6%). In contrast to the 2-mbsf sediments, Halomonadaceae, Clostridia, Flavobacteriaceae, Geobacteraceae, Carnobactericeae, Oceanospirillales, and Desulfobulbaceae were not detected as this depth. Nitrate reducing lineages incorporated 45.0% of the total sequences while fermenters remained constant with depth (Figure 3).
At 40 mbsf, sequence annotation identified 29 total families within the sediments. A subset of 14 families represented 71.6% of the total sequences at this depth (Table 2). These families included Halomonadaceae (27.9%), Altermonadaceae (11.2%), Piscirickettsiaceae (7.7%), Flavobacteriaceae (6.2%), Streptococcaceae (4.6%), and Solirubrobacteraceae (4.3%). Only Streptococcaceae was also detected (1.8%) in the offshore samples. Although Lactobacillaceae (10.3%) and Enterobacteriaceae (4.6%) were detected frequently in the onshore samples, they were two times less that the number of sequences detected in sediments preserved on the ship. Aerobic lineages represented 44.6% of the sequences detected while fermenters accounted for 21.4% (Figure 3). Lineages associated with methylotrophs incorporated 7.8% of the total sequences while carbonate dissolution groups represented 3.4% of the sequences, the most at any depth in the onshore samples.
Discussion
This study describes a shift in population structure within sediment core material obtained from borehole M00058A during IODP Expedition 325 to the Great Barrier Reef following 3 months storage at 4°C. These data indicate subsurface microbial populations remain metabolically active under these conditions and are thus capable of altering sediment geochemistry during storage. Unique to this analysis is the use of RNA-based targets to determine the metabolically active fraction of the total population. The concentration of ribosomes, and thus copies of SSU rRNA within a cell, is linearly correlated to cellular metabolic activity (Delong et al., 1989; Kerkhof and Ward, 1993; Lee and Kemp, 1994), with dormant and dead cells having few to no ribosomes present (Fegatella et al., 1998). The active population both responds to and changes the geochemistry in the environment. Therefore, characterizing this population provided a proxy for identifying potential shifts in the geochemistry and assessment of the integrity of the core. In addition, a change in this population determined a fraction of the population that was below detection limits or dormant during in situ geochemical conditions, but active during storage conditions. This viable fraction of the community represented a source for different metabolic processes available to the community when the environment changed and highlighted a potentially underestimated portion of the community. Further enrichment-based culturing has the potential to define a larger amount of diversity and function within this and other communities.
A shift at the family taxonomic level following 3 months of storage between the offshore and onshore sampling time points incorporated as much as 90% of the sequence data set from the active fraction of the population. Active microbial processes alter both the chemical and physical composition of subsurface sediments. These diagenetic processes are common and have been well studied in situ, however the potential for alteration has been less understood when the sediments were removed from the environment and placed in cold storage conditions. The integrity of core material is critical for post-expedition research and sediment curation efforts for programs like IODP. This manuscript serves to better understand the microbial populations that are metabolically active during sediment core storage and provide descriptions of potential processes acting on those sediments. In addition, increased diversity in the stored sediments compared to in situ analysis suggests that species diversity in the subsurface may be underestimated. Although sediments selected here are from an IODP expedition, results describing microbial activity at 4°C should be considered when storing any non-sterilized samples for subsequent analysis.
Shifts in Community Structure
Metabolically active microbial populations alter their surrounding environments through the consumption and production of chemical compounds. As populations shift, the consumption and production rates change and are no longer representative of in situ conditions. During initial characterization of the in situ metabolically active community structure, lineages associated with fermentation, including Lactobacillaceae, Clostridiaceae, and Ruminococcaceae, were detected at highest frequency compared to other lineages (Figure 3). In total, lineages associated with fermentation processes accounted for nearly 70% of the 2 and 20 mbsf samples and over 40% for the 40-mbsf sample. The prevalence of fermentative lineages in the shallow subsurface has been previously noted (Toffin et al., 2005), providing support for the annotation presented. These lineages may play a significant role in the conversion of organic matter present in the shallow sediments (Toffin et al., 2005).
Following storage for 3 months, the number of groups associated with fermentative sequences decreased over two times. This discrepancy may be attributed to a reduction of substrate due to disrupted in situ fluid flow. As the geochemical conditions changed, the niche advantage decreased allowing other lineages to become more abundant. In the 2- and 40-mbsf samples, fermentation capable lineages were in part replaced by an increase in aerobic lineages (Figure 3). While this group was detected in the offshore samples at 2 mbsf, no aerobic lineages were detected at the 40-mbsf sample. After the 3-months at 4°C, 44.6% of the sequences obtained related to aerobic organisms compared to 0.0% in the offshore sample at the same depth. This suggests oxygen intrusion into the core. Oxygen can affect both the biology as well as the chemistry of the sediments and should be avoided where possible. Lin et al. (2010) observed sulfide oxidation to sulfate when sediment cores experienced oxygen penetration. Additionally, the increase in sulfate was predicted to promote a community shift favoring sulfate-dependent methane oxidizers. While the Lin et al. (2010) study was DNA based, this current RNA-based study supports their conclusion as methylotrophs were detected in the metabolically active fraction of the total populations. The depletion of sulfate by these lineages may inhibit geochemical detection of the sulfide oxidation process, stressing the importance of molecular analyses coupled to geochemical measurements to better environmental characterizations. In contrast to the 40-mbsf sample, it is hypothesized that oxygen remained limiting in the sample collected at 20 mbsf as a rise in nitrate reducers were observed. An increase in nitrogen cycling populations, including Comamonadaceae, Rhodospirillaceae, and Rhodobacteraceae, would affect ammonia, nitrate, and nitrite analysis to levels not reflective of the in situ conditions. Differences between the 20- and 40-mbsf biogeochemical profiles suggest variable storage affects between core sections, adding to the unpredictability of geochemical and microbiological results from stored sediments.
A large percentage of offshore sequence data sets were associated with lineages capable of carbonate dissolution (Figure 3). This was expected given the location of the sediments being in close proximity to the Great Barrier Reef. However, following incubation, the frequency of detection for these groups dropped an order of magnitude on average with the 20-mbsf sample populations shifting from 27.2% of the total sequences to undetectable. This shift suggests a major alteration of the geochemical environment, requiring specific analysis to determine the cause and implications. Due to shipboard limitations, this analysis could not be preformed and thus was beyond the capacity of this study. These data represent the first step to being able to identify metabolic processes within stored sediments and provide a more clear prediction of sediment alteration capacity.
A significant rise in the number of sequences related to iron and sulfate-reducing lineages was observed in the sediments obtained onshore compared to the offshore samples. Both iron and sulfate-reducing functional groups were only detected after sediment storage and accounted for over 10 and 6% of the total sequences. These populations becoming metabolically active would affect any post-expedition analysis of sulfur and iron chemistry. They have the potential to reduce sulfate and Fe(III) concentrations in marine systems to below detection limits while forming multiple iron sulfur mineral compounds. Secondary effects would change the pH of the sediment and alter organic matter content and concentration. In addition, the paleomagnetic record within the sediments, or other material would be altered by both iron reducing and oxidizing populations (Kostka and Nealson, 1995; Zachara et al., 1998 reviewed in Kopp and Kirschvink, 2008). An active microbial population would alter multiple characterization efforts within the sediment profile. Understanding these processes and how to better reducing them is required.
Viable Fraction of the Population – Metabolic Sink
A community shift within a closed system implies that a portion of the population was either metabolically active but below detection limits, or dormant in situ. The implications of these shifts are critical for the core preservation strategies and down stream analysis reliability, as well as, for understanding the overall viable fraction of the subsurface biosphere. To verify a closed system, sediments collected were examined for contamination resulting from the drilling process. Results suggest that the contamination potential was very low, but the authors recognize that this possibility cannot be fully ruled out. A general contamination during drilling or storage would have resulted in a homogenization of the detected community structure. Variations in the detection frequency of multiple lineages suggest a general contamination did not occur. Aerobic populations were detected in the 40-mbsf onshore sample. These two lineages, Alteromonadaceae and Flavobacteriaceae were detected in the 2-mbsf sample as well, but not in the 20-mbsf sample. The increase in abundance of related sequences suggests oxygen intrusion into the sediment may have occurred, however these populations can be facultatively anaerobic as well. These two populations were not detected at 20 mbsf, reducing the potential for this to be a drilling contamination or air borne contamination during storage. Additional testing beyond the scope of this paper would be required to determine the true origin of this population.
These results support a fraction of the population remaining viable but dormant within the subsurface. The lower amount of diversity reported in the metabolically active in situ population (offshore sample) compared to the higher level of diversity observed on shore supports niche diversification during storage and the revival of dormant populations. The coring process disrupted multiple environmental factors, including resource availability, pressure, and temperature. These stressors have contributed to shifts in community composition in other environments (Hunter et al., 2006; Lewis, 2007). Within these samples, the environmental changes revived microbial populations in sediments dated to over 50 kyr cal before present (Yusuke et al., 2011). The subsurface is considered geochemically stable with slow change over time; a characteristic that has the potential to limit diversity (Orcutt et al., 2011) and the effectiveness of dormancy as a strategy to maintain a viable population (Jones and Lennon, 2010). However, the population characterized in these samples potentially provided a source of functional diversity for community activity over geologic timescales. This expands the potential range of such survival strategies beyond environments with periodic shifts in geochemical and geotechnical conditions (Cohen, 1970; Fuhrman et al., 2006; Jones and Lennon, 2010), and should be considered in estimates of community functional capacity. Future studies will quantify the depth of the functional sink available to respond to changing environments. In addition, the length of time these dormant populations remain viable will be determined to better understand the effect of temporal isolation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the science party of IODP Expedition 325 and the crew aboard the Greatship Maya. Additionally, we thank Scot Dowd, Alicia Shepard, and Laura Zinke with data processing. The Consortium of Ocean Leadership provided funding for this project. This paper is contribution number 126 to the Center for Dark Energy Biosphere Investigations (CDEBI).
References
Bickle, M., Arculus, R., Barrett, P., Decanto, R., Camoin, G., Edwards, K., Fisher, A., Inagaki, F., Ravelo, C., Saffer, D., and Teagle, D. (2011). Illuminating Earth’s Past, Present, and Future: International Ocean Discovery Program Science Plan 2013–2023. Washington DC: Integrated Ocean Drilling Program Management International.
Delong, E. F., Wickham, G. S., and Pace, N. R. (1989). Phylogenetic stains – ribosomal RNA-based probes for the identification of single cells. Science 243, 1360–1363.
Dowd, S. E., Zaragoza, J., Rodriguez, J. R., Oliver, M. J., and Payton, P. R. (2005). Windows.NET network distributed Basic Local Alignment Search Toolkit (W.ND-BLAST). BMC Bioinformatics 6, 93. doi: 10.1186/1471-2105-6-93
Fegatella, F., Lim, J., Kjelleberg, S., and Cavicchioli, R. (1998). Implications of rRNA operon copy number and ribosome content in the marine oligotrophic ultramicrobacterium Sphingomonas sp. strain RB2256. Appl. Environ. Microbiol. 64, 4433–4438.
Fry, J. C., Parkes, R. J., Cragg, B. A., Weightman, A. J., and Webster, G. (2008). Prokaryotic biodiversity and activity in the deep subseafloor biosphere. FEMS Microbiol. Ecol. 66, 181–196.
Fuhrman, J. A., Hewson, I., Schwalbach, M. S., Steele, J. A., Brown, M. V., and Naeem, S. (2006). Annually reoccurring bacterial communities are predictable from ocean conditions. Proc. Natl. Acad. Sci. U.S.A. 103, 13104–13109.
Giovannoni, J. J., Wing, R. A., Ganal, M. W., and Tanksley, S. D. (1991). Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res. 19, 6553–6558.
Gontcharova, V., Youn, E., Wolcott, R. D., Hollister, E. B., Gentry, T. J., and Dowd, S. E. (2010). Black box chimera check (B2C2): a windows-based software for batch depletion of chimeras from bacterial 16S rRNA gene datasets. Open Microbiol. J. 4, 47–52.
Hall, P. O., and Aller, R. C. (1992). Rapid, small-volume, flow-injection analysis for sigma CO2 and NH4+ in marine and fresh waters. Limnol. Oceanogr. 37, 1113–1119.
Handl, S., Dowd, S. E., Garcia-Mazcorro, J. F., Steiner, J. M., and Suchodolski, J. S. (2011). Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiol. Ecol. 76, 1–10.
Hunter, E. M., Mills, H. J., and Kostka, J. E. (2006). Microbial community diversity associated with carbon and nitrogen cycling in permeable marine sediments. Appl. Environ. Microbiol. 72, 5689–5701.
Jones, S. E., and Lennon, J. T. (2010). Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. U.S.A. 107, 5881–5886.
Jones, S. E., and McMahon, K. D. (2009). Species-sorting may explain an apparent minimal effect of immigration on freshwater bacterial community dynamics. Environ. Microbiol. 11, 905–913.
Jørgensen, B. B. (2011). Deep subseafloor microbial cells on physiological standby. Proc. Natl. Acad. Sci. U.S.A. 108, 18193–18194.
Kerkhof, L. J., and Ward, B. B. (1993). Comparison of nucleic acid hybridization and fluorometry for measurement of RNA/DNA relationship with growth rate in a marine bacterium. Appl. Environ. Microbiol. 59, 1303–1307.
König, I., Lougear, A., Bruns, P., Gru¨tzner, J., Trautwein, A. X., and Dullo, W.-C. (2000). “Iron oxidation in sediment cores (site 1062) during six months of storage in the Ocean Drilling Program archive,” in Proceedings of the Ocean Drilling Program, Scientific Results, eds L. D. Keigwin, D. Rio, G. D. Acton, and E. Arnold (College Station: Ocean Drilling Program).
Kopp, R. E., and Kirschvink, J. L. (2008). The identification and biogeochemical interpretation of fossil magnetotactic bacteria. Earth Sci. Rev. 86, 42–61.
Kostka, J. E., and Nealson, K. H. (1995). Dissolution and reduction of magnetite by bacteria. Environ. Sci. Technol. 29, 2535–2540.
Lee, S. H., and Kemp, P. F. (1994). Single cell RNA content of natural marine planktonic bacteria measured by hybridization with multiple 16S ribosomal RNA targeted fluorescent probes. Limnol. Oceanogr. 39, 869–879.
Lin, Y. S., Biddle, J. F., Lipp, J. S., Orcutt, B. N., Holler, T., Teske, A., and Hinrichs, K. U. (2010). Effect of storage conditions on archaeal and bacterial communities in subsurface marine sediments. Geomicrobiol. J. 27, 261–272.
Mills, H. J., Hunter, E., Humphrys, M., Kerkhof, L., Mcguinness, L. M., Huettel, M., and Kostka, J. E. (2008). Characterization of nitrifying, denitrifying, and overall bacterial communities in permeable marine sediments of the northeastern Gulf of Mexico. Appl. Environ. Microbiol. 74, 4440–4453.
Nogales, B., Moore, E. R. B., Abraham, W. R., and Timmis, K. N. (1999). Identification of the metabolically active members of a bacterial community in a polychlorinated biphenyl-polluted moorland soil. Environ. Microbiol. 1, 199–212.
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422.
Reese, B. K., Mills, H. J., Dowd, S., and Morse, J. W. (2012). Benthic biogeochemistry of microbial iron and sulfate reduction in the Gulf of Mexico hypoxic zone. Geomicrobiology (in press).
Stackebrandt, E., and Goebel, B. M. (1994). A place for DNA-DNA reassociation and 16S ribosomal RNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44, 846–849.
Toffin, L., Zink, K., Kato, C., Pignet, P., Bidault, A., Bienvenu, N., Birrien, J. L., and Prieur, D. (2005). Marinilactibacillus piezotolerans sp nov., a novel marine lactic acid bacterium isolated from deep sub-seafloor sediment of the Nankai Trough. Int. J. Syst. Evol. Microbiol. 55, 345–351.
Yusuke, Y., Webster, J. M., Cotterill, C., Braga, J. C., Jovane, J., Mills, H. J., Morgan, S., Suzuki, A., and Scientists, T. I. E. (2011). IODP Expedition 325: Great Barrier Reefs reveals past sea-level, climate and environmental changes since the last ice age. Sci. Drilling 12, 32–45.
Zachara, J. M., Fredrickson, J. K., Li, S. M., Kennedy, D. W., Smith, S. C., and Gassman, P. L. (1998). Bacterial reduction of crystalline Fe3+ oxides in single phase suspensions and subsurface materials. Am. Mineral. 83, 1426–1443.
Appendix
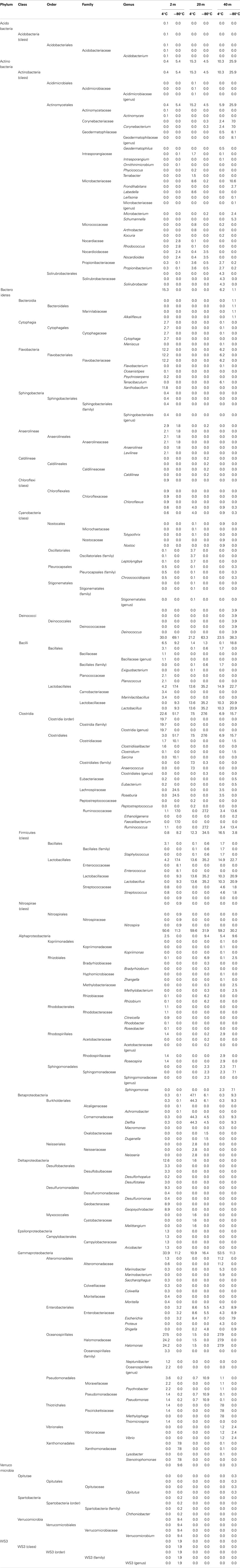
Table A1. Taxonomic identification of the Great Barrier Reef subsurface sequence data set.
Keywords: sediment microbial ecology, pyrosequencing, geobiology, sediment core storage
Citation: Mills HJ, Reese BK and Peter CS (2012) Characterization of microbial population shifts during sample storage. Front. Microbio. 3:49. doi: 10.3389/fmicb.2012.00049
Received: 23 November 2011; Paper pending published: 16 December 2011;
Accepted: 30 January 2012; Published online: 17 February 2012.
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, USAReviewed by:
Rebecca Gast, Woods Hole Oceanographic Institution, USAKesen Ma, University of Waterloo, Canada
Copyright: © 2012 Mills, Reese and Peter. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Heath J. Mills, Department of Oceanography, Texas A&M University, 716A Eller O&M Building, College Station, TX 77843-3146, USA. e-mail: hmills@ocean.tamu.edu