
 Jamie L. Everman1,2
Jamie L. Everman1,2
- 1 Department of Biomedical Sciences, College of Veterinary Medicine, Oregon State University, Corvallis, OR, USA
- 2 Department of Microbiology, College of Science, Oregon State University, Corvallis, OR, USA
- 3 Program of Molecular and Cell Biology, Oregon State University, Corvallis, OR, USA
Tuberculosis is a disease associated with the infection of a great part of the world’s population and is responsible for the death of two to three million people annually. Mycobacterium tuberculosis infects macrophages and subverts its mechanisms of killing. The pathogen suppresses macrophage apoptosis by many different mechanisms. We describe that, upon uptake by macrophages, M. tuberculosis overexpresses an operon Rv3361c-Rv3365c and secretes Rv3364c. The Rv3365c knockout strain is deficient in apoptosis inhibition. The Rv3364c protein binds to the serine protease cathepsin G on the membrane, inhibiting its enzymatic activity and the downstream activation of caspase-1-dependent apoptosis. In summary, M. tuberculosis prevents macrophage pyroptosis by a novel mechanism involving cytoplasmic surveillance proteins.
Introduction
Tuberculosis is a leading cause of death in the world, with the global significance exacerbated by its common association with HIV-1 infection. Host–pathogen interaction during Mycobacterium tuberculosis infection is a complex process in which the bacterium evades defensive mechanisms (Deretic et al., 2006; Davis et al., 2007). M. tuberculosis’ ability to survive innate mechanisms of host defense has a significant role in the outcome of the infection. M. tuberculosis interferes with host-initiated inflammatory responses and cell death, as part of a complex adaptation to subvert host innate and adaptive responses. Initial detection of M. tuberculosis involves a number of phagocyte receptors, including several Toll-like receptors (TLRs; Bhatt and Salgame, 2007), nucleotide oligomerization domain (NOD)2 cytoplasmic receptor (Ferwerda et al., 2005), complement, and mannose receptors (Hirsch et al., 1994). Upon recognition of pathogen-associated molecular patterns (PAMPs), the host induces an array of responses aimed at limiting growth of the pathogen. Bacteria, however, have been shown to use effector proteins in a pro-active manner to counteract the defense strategies. Previous studies indicate that M. tuberculosis infection of macrophages stimulates TLR-2, subsequently leading to the translocation of NF-κB (Aliprantis et al., 1999) and production of cytokines, chemokines, and the synthesis of nitric oxide (NO; Heldwein and Fenton, 2002). The pathogen also blocks the delivery of nitric oxide synthase (iNOS) to the vacuole membrane and thus avoids the killing effects of NO (Davis et al., 2007).
One of the recently described host mechanisms to eliminate intracellular bacteria is autophagy. Autophagy and apoptosis are interventions used by the human host to control microbial infection. Salmonella, Yersinia, Legionella, Shigella, to cite a few, have been shown to manipulate both autophagy and apoptosis to create a favorable environment inside the host (Gao and Kwaik, 2000; Campoy and Colombo, 2009). M. tuberculosis inhibits both processes, as well. Physiological or pharmacological induction of autophagy limits intracellular survival of M. tuberculosis through maturation of phagolysosomes in infected macrophages (Gutierrez et al., 2004). M. tuberculosis infection also has been shown to protect cells against apoptotic death by modifying the expression of death receptors, such as Fas (CD95), or by triggering the synthesis and release of soluble TNF receptor 2 (TNFR2; Loeuillet et al., 2006). In contrast, other laboratories have also shown that macrophages infected with virulent strains of M. tuberculosis undergo TNF and caspase-independent apoptosis (O’Sullivan et al., 2007). Although discrepancies exist regarding the observations, they are possibly related to the model and timing used in the investigational systems. A few studies, however, have confirmed that, in animal models, the ability to inhibit macrophage apoptosis is directly related to M. tuberculosis strain virulence (Park et al., 2006).
Some mycobacterial effector proteins interfering with host defensive mechanisms have recently been discovered. Using a gain-of-function genetic screen, nuoG was identified as an anti-apoptotic gene in M. tuberculosis (Velmurugan et al., 2007). The deficiency in nuoG gene significantly reduced bacterial virulence in vivo, leading to inhibition of macrophage apoptosis. Inactivation of the M. tuberculosis zmp1 gene encoding putative Zn2+ metalloprotease has been shown to activate inflammasome through increased secretion of IL-1β (Master et al., 2008). The zmp1 gene plays a critical role in M. tuberculosis survival in macrophages and in virulence in vivo. Recently, our group has demonstrated that M. tuberculosis Rv3654c and Rv3655c proteins, which are secreted and transported to the outside of the bacterial phagosome by a novel “type IV pili” apparatus, inhibited TNF-induced apoptosis. The interaction of these proteins with host PSF and ALO17 interfered the caspase post-transcriptional events and blocked the extrinsic pathway of apoptosis (Danelishvili et al., 2010).
By screening a transposon mutant library, we previously identified M. tuberculosis knockout mutants that failed to inhibit macrophage apoptosis in contrast with the wild-type bacterium (Danelishvili et al., 2010). We selected 20A11 out of 10 deficient in inhibition of apoptosis (DIA) mutants, based on the observation that pro- and anti-inflammatory cytokines produced by macrophages upon infection, differed between 20A11-infected macrophages and the wild-type infection or other tested mutants. We investigated the mechanism by which M. tuberculosis interfered with the apoptosis. Here we show that the M. tuberculosis Rv3364c protein (inactivated in the 20A11 clone), which is a component of a signal transduction operon, was capable of inhibiting caspase-1 activation, and consequently the host cell apoptosis (pyroptosis), through interaction with macrophage membrane-associated serine protease, cathepsin G, and suppression of its enzymatic activity.
Materials and Methods
Bacterial Clones and Cell Culture Infection Assay
Mycobacterium tuberculosis strain H37Rv (ATCC 25618) and 20A11 transposon mutant generated with the temperature-sensitive plasmid pTNGJC, as previously described (Danelishvili et al., 2010), were cultured in liquid or solid Middlebrook medium (Hardy Diagnostics, Santa Maria, CA, USA) supplemented with 10% oleic acid, albumin, dextrose, and catalase (OADC) enrichment (Hardy Diagnostics, Santa Maria, CA, USA) and 200 μg/ml kanamycin sulfate (Sigma Chemicals, St. Louis, MO, USA), where appropriate. M. tuberculosis PM638 clone, a derivative of H37Rv strain containing deletion of the β-lactamase gene (Flores et al., 2005), was kindly provided by Dr. Martin S. Pavelka, Jr. at University of Rochester School of Medicine and Dentistry and Department of Microbiology and Immunology, Rochester, NY, USA. Human monocytic cell line U937 (ATCC CRL-1593.2) was purchased from ATCC and cultured in RPMI-1640 (Invitrogen, Carlsbad, CA, USA) supplemented with heat-inactivated 10% fetal bovine serum (FBS; Sigma, St. Louis, MO, USA). Cells were seeded at 80–100% confluence into 75 cm2 tissue culture flasks, in 96-well plates or chamber glass slides as needed. Phorbol-12-myristate-13-acetate (PMA)-activated monolayers, as described previously (Danelishvili et al., 2010), were infected with M. tuberculosis wild-type and 20A11 mutant or complemented strains at MOI of 10:1 for 2 h at 37°C and 5% CO2. Bacteria were subsequently removed by washing the monolayer three times with Hank’s balanced salt solution (HBSS), and the tissue culture wells replenished with RPMI-1640 (Invitrogen) medium + 10% FBS. Macrophage monolayer, macrophage lysates, or supernatants from experimental and control wells were collected at different time points for apoptosis, caspase, or cytokine assays. All infections were carried out in duplicate, and experiments were performed in triplicate.
Infection of macrophages and phagocytosis were assessed with rhodamine (200 μg/ml) labeled M. tuberculosis PM638 clone or PM638 containing β-lactamase constructs, described below, at MOI of 10:1. Macrophages were visualized after 1 and 2 h of post-infection by fluorescein-phalloidin staining of actin, as previously described (Danelishvili et al., 2007). Duplicate experiments were performed, and at least 200 cells per slide were counted to calculate the percentage of infected macrophages. Preparations were visualized under a Leica fluorescence microscope.
Measurement of Cytokines
Supernatants from cell cultures infected with M. tuberculosis wild-type or knockout mutant and from uninfected control wells were harvested at 4, 24, and 48 h, centrifuged at 13,000 × g for 5 min, and assayed for presence of cytokines. TNF, IL-12 (eBioscience Inc., San Diego, CA, USA), and TGF-β, IFN-β (Antigenix America, Inc., New York, NY, USA) were detected by enzyme-linked immunosorbent assay (ELISA) kits, following the instructions provided by the manufacturers. Supernatants from macrophages without infection were used as additional controls. The cytokine concentrations were quantified from standard curves obtained from sequential dilutions of the corresponding recombinant cytokines.
Apoptosis Assay
Quantitative analysis of macrophage apoptosis was performed using TUNEL technology for labeling of DNA strand breaks by Terminal deoxynucleotidyl transferase, according to the manufacture’s protocol (Roche Diagnostics, Indianapolis, IN, USA). Macrophages were seeded in 24-well plates and infected with M. tuberculosis H37Rv, 20A11 mutant, or 20A11 complemented strains. In the duplicate wells of 20A11-infected macrophages, 50 μM of caspase-1 inhibitor or caspase-1 control (Calbiochem, La Jolla, CA, USA) were added to the wells during infection and processed for TUNEL assay after 3 days of infection. In another set of experiments, macrophages seeded in 24-well plates were transfected with PAR4 and NOD1 siRNA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 8 h or treated with 50 nm cathepsin G Inhibitor I (EMD Biosciences, Inc., Madison, WI, USA), then infected with 20A11 mutant and processed for apoptosis assay. In all infection experiments, the MOI was maintained as 10 bacteria to one cell.
RNA Extraction, Probe Synthesis, and Quantification of Bacterial- and Host-Gene Expression
U937 macrophages were exposed to M. tuberculosis wild-type for 30 min or infected for 4 h with MOI 100 bacteria:1 cell and used as a source of total experimental RNA. M. tuberculosis, growing in Middlebrook 7H9 broth to the exponential phase of growth, was exposed to RPMI-1640 medium + 10% FBS for 30 min or 4 h and used as a source of control RNA. Bacterial RNAs from control and experimental (exposed and intracellular) samples were extracted and processed for Real-Time PCR, as previously described (Danelishvili et al., 2004). Briefly, macrophage monolayers were lysed with 0.1% sodium dodecyl sulfate (SDS, Sigma Chemicals, St. Louis, MO, USA) and differentially centrifuged to recover the intracellular bacteria. Bacterial pellets were resuspended in a guanidine thiocyanate-based buffer (Trisol, Invitrogen, San Diego, CA, USA) and lysed with rapid mechanical agitation in a bead-beater. RNA was cleaned with RNA Cleanup kit (QIAGEN, Valencia, CA, USA). The quality and concentrations of DNase-treated RNA samples were analyzed spectrophotometrically, as well as verified on a 1% denaturing agarose gel.
Host cell RNAs obtained from uninfected, as well as M. tuberculosis-infected macrophages, at 4 and 24 h time points were extracted using the Atlas Pure Total RNA Labeling System, according the manufacturer instruction (Clontech, Palo Alto, CA, USA). Briefly, cell cultures were lysed in denaturing solution, vortexed, and centrifuged at 12,000 × g for 10 min at 4°C to remove cellular debris, extracted three times with saturated phenol–chloroform, precipitated in isopropanol, and washed with 80% ethanol. DNase treatment of total RNA samples was carried out according to the manufacturer’s recommendations. The quality of RNA was verified on a 1% denaturing agarose gel and the concentration was measured at 260 nm absorbance.
Mycobacterial and host cell total RNAs (1 μg) were reverse transcribed with 100 U of Superscript III Plus RNase H− Reverse Transcriptase, using RT primers according to the manufacturer’s instruction (Invitrogen, Carlsbad, CA, USA). Quantitation of the expression of M. tuberculosis and macrophage genes was carried out on the iCycler (Bio-Rad, Hercules, CA, USA) with SYBR Green I assay by Real-Time PCR detection system using gene-specific primers. Approximately 100–150 bp target cDNAs from experimental and control samples were amplified in separate PCR reactions. The calculated threshold (Ct) cycle for each bacterial gene amplification was normalized to the Ct of the 16S rRNA gene amplified from the corresponding sample. The host-gene expression was normalized with an internal control of β-actin. The fold change in gene expression was quantified as described in User Bulletin #2 for the ABI PRISM 7700 sequence detection system (Applied-Biosystems, 1997).
Generation of Complemented Strains and Screening
To complement the defect of M. tuberculosis 20A11 mutant, M. tuberculosis Rv3365c, Rv3364c, Rv3363c-61c, and Rv3364c-61c genes were amplified from the wild-type H37Rv strain using Fidelitaq PCR system (US Biochemicals, Inc.). PCR-Generated fragments were cloned into EcoRI and HindIII sites of E. coli–Mycobacterium shuttle vector pMV261 + AprII, containing hsp60 promoter and encoding apramycin resistance. The resulting plasmids pLDRv3365c, pLDRv3364c, pLDRv3363-61c, and pLDRv3364-61c were propagated in DH10B E. coli, following the transformation into M. tuberculosis 20A11 mutant by electroporation. Transformants were selected on Middlebrook 7H10 agar plates containing apramycin 200 μg/ml and screened by PCR for apramycin gene (50).
β-Lactamase (BlaC) Assay for Protein Secretion
We have constructed the β-lactamase reporter vector for Mycobacterium and visualized bacterial protein translocation (secretion) in the cytoplasm of macrophages. The assay uses technology based on β-lactamase-catalyzed hydrolysis of the substrate, which changes the fluorescence of substrate molecules from green to blue by disrupting resonance energy transfer. Rv3364c gene was fused with BlaC gene lacking the first 69 nucleotides important for β-lactamase enzyme secretion, over-expressed in pLDG13 plasmid and transformed into M. tuberculosis PM638 clone. Human macrophages (106) were seeded in 96-well plates or in two-chamber slides and infected with M. tuberculosis PM638 or PM638 clones expressing BlaC(+) with secretion segment and Rv3364c:BlaC(−) fusion lacking BlaC secretion segment at a multiplicity of infection of 10:1. Cells were incubated at 37°C in 5% CO2 for 2 h and washed three times with Hank’s balanced salt solution (Invitrogen) to remove extracellular bacteria. After 48 h infection, macrophages were loaded with the fluorescent substrate CCF2-AM at room temperature using manufacturer’s protocol (Invitrogen). Readings were recorded with two filter sets: excitation 405 ± 20 nm/emission 460 ± 40 nm and excitation 405 ± 20 nm/emission 530 ± 30 nm using Tecan Infinity 200 cytofluorometer. Fluorescence micrographs were captured with CCF2 filter sets (Leica).
Western Blot Analysis of M. tuberculosis Rv3364c Protein
The pLDG13:His:Rv3364c construct was introduced into M. tuberculosis H37Rv and grown for 21 days, after which cells were harvested, disturbed in a bead-beater, and processed for western blot to insure that His:Rv3364c protein was expressed in M. tuberculosis. Over-expressed clone was washed two times with phosphate-buffered saline (PBS) and used for infection of U937 macrophages. After 48 h, cell cultures were lysed with 0.1% Triton X-100 and centrifuged at 3,000 × g for 15 min to remove bacterial pellet from the suspension. Pre-cleared cell lysate was incubated with His-tag primary agarose conjugate antibody, overnight at 4°C. Samples were resolved by electrophoresis on 12% Tris–HCl gels, transferred to nitrocellulose membranes and blocked with 5% non-fat milk solution. Membrane was exposed to His primary antibody at a dilution of 1:1,000 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 1 h, followed by incubation with anti-rabbit IgG linked to AlexaFluor680 secondary antibody (1:5,000; Li-Cor, Lincoln, NE, USA) for 30 min. Reactivity was assessed with Odyssey Imager (Li-Cor).
Caspase-1 Activity and Inhibition Assays
Uninfected and infected macrophages with the 20A11 mutant, M. tuberculosis H37Rv or 20A11 complemented (Rv3361c-65c) strains were assayed for in situ labeling and detection of active caspase-1, using the APO LOGIX™-Carboxyfluorescein (FAM) kit (Bachem, Torrance, CA, USA). Staurosporine-treated U937 cells were used as a positive control. Briefly, 10 μl of 30× Caspase-1 detection reagent (FAM–YVAD–FMK) were added to the cells in 300 μl medium of 96-well plate and incubated at 37°C for 2 h. Wells were gently washed two times. After the final wash, 100 μl of wash buffer were added to experimental wells, and immediately processed for fluorescence microscopy or fluorometric analysis. Readings were taken on a Cytofluorometer II (Biosearch, Bedford, MA, USA) with excitation at 488 nm and emission at 515–530 nm. A cell-permeable inhibitor I of caspase-1 at concentration of 10 μM and a caspase-inhibitor negative control at a 10-μM concentration (irreversible cathepsin B inhibitor; CalBiochem, Los Angeles, CA, USA) were used in caspase-1 inhibition assay. Macrophage monolayers in two-chamber slides were treated with caspase-1 inhibitor or negative control and then were infected with the 20A11 mutant strain. Apoptosis was quantified with TUNEL assay.
Immunoprecipitation and Immunoblot Analysis
U937 cells, infected with M. tuberculosis wild-type and 20A11 mutant, were scraped from tissue culture plates in 25 cm2 flasks after 24 h, 2 or 4 days of infection, and lysed with 0.1% SDS. Pre-cleared cell lysates were subjected to immunoprecipitation for 5 h at 4°C using A/G beads, and NOD1, NOD2, and PAR4 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) primary antibodies, according to the method recommended by the manufacturer. The captured proteins were washed three times with PBS and resolved on 12% Tris–HCl gels. The Western blot analysis for the cathepsin G protein was processed without immunoprecipitation; cell lysates were separated by electrophoresis on 12% Tris–HCl gels and transferred onto nitrocellulose membranes, using a semidry transfer apparatus (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 3% non-fat milk solution, following washes with PBS-0.1% Tween-20. Proteins were probed with anti-cathepsin G, PAR4, NOD1, and NOD2 (1:1,000) primary antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 1 h, followed by incubation with anti-mouse, rabbit, or goat IgG linked to AlexaFluor680 secondary antibodies (Li-Cor, Lincoln, NE, USA) at a dilution of 1:5,000 for 30 min. Membranes were scanned using Odyssey Imager (Li-Cor).
Purification of Rv3364c and Target Host Protein Pull-Up Assay
The M. tuberculosis Rv3364c gene was cloned into pMV261 vector containing His-tag red fluorescent protein (RFP) fusion, transformed into M. smegmatis and grown for 8–10 days on 7H10 agar plates containing 50 μg/ml km. M. smegmatis red colonies expressing His-tag-RFP–Rv3364c fusion were harvested in PBS, disrupted with bead-beater, and isolated directly from a crude cell lysate using 30 μl of paramagnetic pre-charged His–nickel particles, according to the manufacturer’s protocol (Promega, Fitchburg, WI, USA). The activated U937 cell extracts were prepared, as previously described (Danelishvili et al., 2007). The macrophage lysates were mixed with captured His-tag-RFP–Rv3364c or His-tag-RFP (control) proteins overnight at 4°C and next day pulled-out with magnetic holder. The captured complex was washed three times with PBS, resuspended, and boiled in Laemmli sample buffer containing mercaptoethanol for 5 min. Samples were electrophoresed on 12% Tris–HCl gel and stained with Coomassie blue staining. The target protein was cut from the gel and processed for In-Gel Tryptic digestion (Thermo Scientific, Rockford, IL, USA). Digested protein was analyzed at Environmental Health Science (EHS) Center of Mass Spectrometry Facility, Oregon State University, Corvallis, by chromatography and electrospray ionization mass spectrometry (ESI-MS/MS). A database search was performed using Mascot (Matrix Science, London, UK) and IPI human (Bioinformatics Solutions Inc., PEAKS Studio, Canada) software.
Lentiviral Transduction for Colocalization of M. tuberculosis and Host Proteins
The interactions of bacterial Rv3364c protein with Homo sapiens cathepsin G in macrophages were investigated by using the Lenti-X™ Lentiviral Expression and Delivery Systems (Clontech). M. tuberculosis Rv3364c was cloned in fusion with dtTomato gene in pLVX–dtTomato–C1 vector and cathepsin G in fusion with ZsGreen1 gene in pLVX–ZsGreen1–C1 vector. The pLVX–dtTomato–C1 vector was used as a control for colocalization assays.
The packaging mixes of above constructs were prepared in accordance with the supplied protocol (Clontech), added to 293FT cells and incubated for overnight at 37°C in 5% CO2 incubator. Next day, medium were replenished with the complete medium and incubated for additional 48 h. Virus containing supernatants were collected and titers were determined according to the manufacturer’s protocol (Clontech). Cultures of U937 cells were coinfected with lentiviruses containing both bacterial and human genes, and immunofluorescence microscopy was performed 24 h following transduction.
Cathepsin G Enzymatic Activity Assay
U937 macrophages were seeded onto six-well plates and infected or not infected with M. tuberculosis wild-type or 20A11 bacteria for 2, 12, and 24 h. Lysed cells were pre-cleared and processed for detection of active cathepsin G, as previously described (Peterszegi et al., 1997). Fifty micro liters of samples were mixed with 200 μl Tris–Hcl buffer (100 nM) containing 0.05% of CaCl2, 0.02% of NaNO3, and 0.01% of Brij 35, pH 8.0. A 100-nM water-soluble peptide MeOSuc-Ala-Ala-Pro-Met-pNA (CalBioChem, Los Angeles, CA, USA) was added to samples as a substrate for cathepsin G. The rates of nitroanilide (pNA) hydrolysis were measured at 25°C on a microplate reader (Bio-Rad, Hercules, CA, USA), and cleavage of pNA was monitored colorimetrically at 405 nm. Enzyme activity was expressed as % of cathepsin G inhibition in 106 cells. In addition, the effects of purified His-tag-RFP–Rv3364c protein at the concentration range of 0.1–100 μg/ml was measured on cathepsin G activity with the same procedure. The following controls were used, together with the experimental samples: water alone, water with substrate, His-tag-RFP alone, His-tag-RFP with substrate, and His-tag-RFP–Rv3364c protein alone.
Inactivation of PAR4 and NOD1 with SiRNA and Cathepsin G Inhibition Assay
The PAR4 and NOD1 siRNAs, as well as control siRNA (scrabbled sequences), were purchased from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). To examine the contribution of both PAR4 and NOD1 expressions to caspase-1 activation and apoptosis, macrophages were transfected with PAR4 or NOD1, and then infected with the M. tuberculosis clone 20A11. The transfection was performed according to the manufacturer’s recommendations. In brief, 1 × 105/ml cells were seeded onto six-well tissue culture plates, washed once with siRNA transfection medium, and replaced with transfection medium containing PAR4 or NOD1 siRNA transfection reagent mixture containing 6 μl of each siRNA duplex or negative control siRNA, 8 μl siRNA transfection reagent, and 200 μl medium. After 8 h of transfection, cells were infected with the 20A11 mutant, and the next day, cells were processed for detection of apoptosis by TUNEL assay or active caspase-1 using APO LOGIX™-Carboxyfluorescein (FAM–YVAD–FMK detection assay) kit, as described above. Cathepsin G Inhibitor I (CalBiochem) at the 20-μM concentration was added to 1 × 105/ml cells into six-well plates and then infected with 20A11 mutant for 24 h. Apoptosis was quantified with TUNEL assay and active caspase-1 with FAM–YVAD–FMK detection kit. Apoptosis and caspase-1 activation in 20A11 mutant infected or uninfected cells were assayed in parallel experiments.
Statistical Analysis
Experiments were repeated at least three times and the results analyzed using the Student’s t-test or ANOVA. Results were considered statistically significant if the p value was <0.05.
Results
Gene Organization and Complementation of M. tuberculosis 20A11 Transposon Mutant
Mycobacterium tuberculosis DIA mutant 20A11 was identified in macrophage assays as previously described (Danelishvili et al., 2010). The sequencing of 20A11 with non-specific nested suppression PCR method (Tamme et al., 2000) identified the interrupted Rv3365c (2,631 bp) gene at 2,624 bp location (Figure 1A). The gene Rv3365c is part of an operon that consists of five genes that are highly conserved in mycobacteria. Although M. tuberculosis Rv3361c, Rv3363, Rv3364, and Rv3365c genes are annotated as conserved hypothetical proteins, significant homologies to other bacterial proteins were found, based on the domain search. The Rv3365c belongs to the histidine kinase sensory family of proteins, possibly initiating the signal transduction. The Rv3364c gene contains the Roadblock/LC7 domain associated with the outer/inner flagellar dynein and cytoplasmic dynein of eukaryotic cells, and possibly plays a structural or regulatory role. The Rv3363c gene encodes a protein that has the DUF472 domain. This class of proteins belongs to the helix-turn-helix (HTH) family proteins that contain a DNA-binding helix-turn-helix domain. Rv3363c most likely is the partner protein for the sensory histidine kinase Rv3365c. The downstream genes in the operon are an ATP/GTP binding protein Rv3362c and putative Rv3361c protein containing five pentapeptide repeat domains. The Rv3361c protein has been shown to have an inhibitory effect on fluoroquinolone resistance through binding to DNA gyrase and suppressing its activity. The crystal structure of this protein has been well investigated (Hegde et al., 2005). Genes Rv3365c-Rv3361c are highly expressed during M. tuberculosis exposure to macrophages, as well as within the cells, established by Real-Time PCR (Figure 1B). The complementation of 20A11 using the pMV261–AprII plasmid with Rv3365c, Rv3364c, or Rv3364c-Rv3362c genes did not restore the anti-apoptotic phenotype. As shown in the Table 1, complete recovery of the phenotype was observed when the deficient mutant 20A11 was complemented with Rv3364c-Rv3361c genes, suggesting the transposon inactivating effect on downstream region of Rv3365c gene.
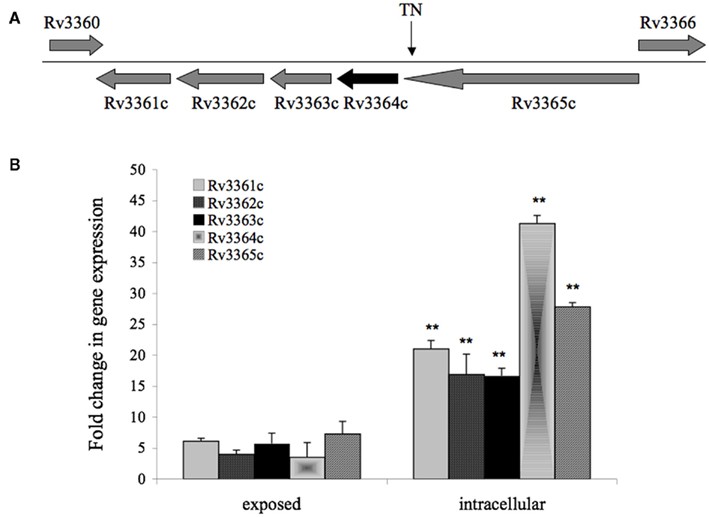
Figure 1. (A) Gene organization of 20A11 mutant associated with induction of macrophage apoptosis. The operon consists of five genes, including histidine kinase sensor Rv3365c, possible structural or regulatory Rv3364c, DNA-binding Rv3363c, ATP/GTP binding Rv3362c, and putative Rv3361c that most likely forms pore. The site disrupted by the insertion of Tn5367 transposon (TN) is indicated with an arrow. (B) Real-Time PCR quantification of M. tuberculosis histidine kinase operon at a 30-min exposure or 4-h infection of macrophages. RNAs of intracellular or macrophage-exposed bacteria, and broth-grown bacteria were assessed to determine cDNA copy numbers for target and reference genes. Ct values were normalized with an internal housekeeping gene (16S) and fold change calculated between differentially expressed experimental and control transcripts, as previously reported (1). The data represent the average of three independent experiments ± SD. A p value of <0.01 indicated by ** was calculated between gene expressions in intracellular and exposed bacteria.
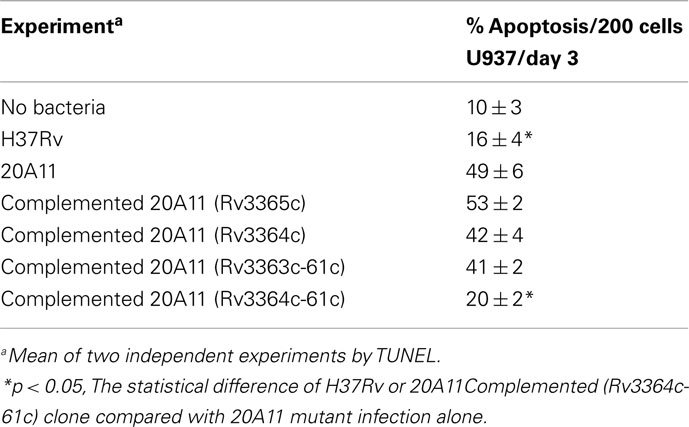
Table 1. Comparisons of the levels of apoptosis in macrophages infected with different clones of M. tuberculosis.
M. tuberculosis Rv3364c Protein is Translocated into the Cytoplasm of Macrophages
In an effort to identify if M. tuberculosis Rv3364c protein was exported in the cytoplasm of host cells, we created a β-lactamase gene reporter vector. Using a cell-permeable fluorescent substrate CCF2-AM for β-lactamase enzyme, assay was performed on U937 cells infected with M. tuberculosis PM638 (BlaC knockout) strain (Figure 2A), PM638 clones either expressing BlaC(+) protein containing a secretion region (Figure 2B) or BlaC(−) protein lacking a secretion region (Figure 2C), and PM638 clone containing Rv3364c fusion with BlaC(−) (Figure 2D). Upon endogenous β-lactamase hydrolysis, CCF2-AM substrate is rapidly converted to a product that lacks the FRET and fluoresces blue. In the absence of the cytoplasmic β-lactamase enzyme charged form of the CCF2-AM fluoresces green. The strong changes were observed in the emission between control [PM638, PM638 BlaC(−)] and experimental [PM638 BlaC(+), PM638 Rv3364c:BlaC(−)] groups recorded with the cytofuorometer (Figure 2E). Analysis of the intracellular distribution of bacteria indicated 59–64% infection rate of U937 macrophages in all groups (Figure 2F). In addition, western blot analysis was performed on macrophages infected with M. tuberculosis over-expressed His:Rv3364c clone, confirming Rv3364c protein secretion in U937 cells (Figure 2G).
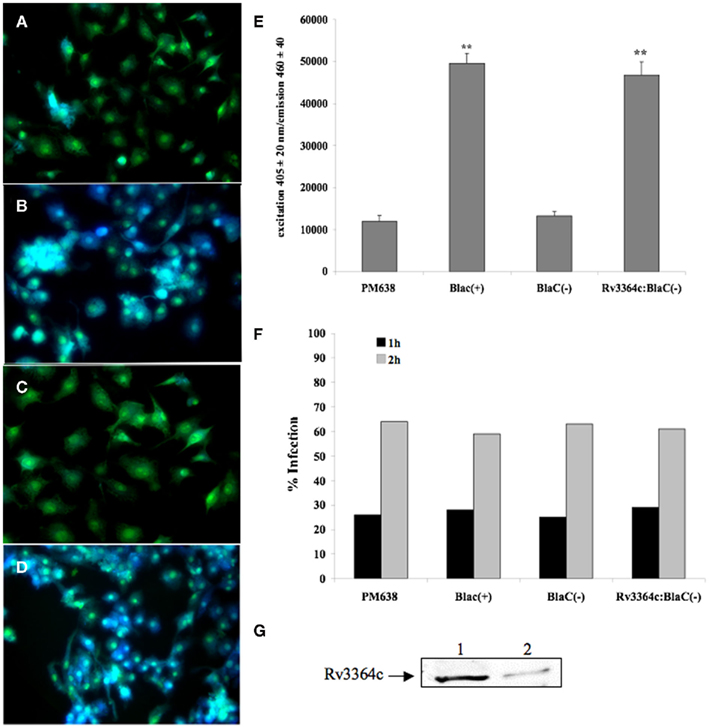
Figure 2. Mycobacterium tuberculosis Rv3364c protein translocation into cytoplasm of U937 cells visualized by β-lactamase-catalyzed hydrolysis of cell-permeable CCF2 (FRET-based) substrate. β-Lactamase hydrolysis in (A) macrophages infected with M. tuberculosis BlaC deletion clone PM638; (B) cells infected with M. tuberculosis clone expressing full length BlaC protein; (C) macrophages infected with PM638 clone containing BlaC(−) lacking a signal sequence; and (D) U937 cells infected with M. tuberculosis PM638 clone expressing Rv3364c:BlaC(−). While uncleaved CCF2 substrate emits green fluorescence, cleaved substrate by translocated β-lactamase from bacteria emits blue fluorescence. (E) The readings from CCF2-AM hydrolysis were also recorded using Infinity 200 cytofluorometer (Tecan). The values are means ± SD of three separate experiments performed in duplicate. **p < 0.01, The significance of differences between PM638 BlaC(+) and PM638; and between Rv3364c:BlaC(−) clone and PM638 BlaC(−). (F) Percentage of infected macrophages after 1 or 2 h of incubation as a function of the number of ingested PM638, PM638 BlaC(+), PM638 BlaC(−), or Rv3364c:BlaC(−) clone. Data from one experiment performed in duplicate and representative of three independent experiments. (G) Western blot analysis of Rv3364c protein in the cytoplasm of infected macrophages. U937 cells were infected with M tuberculosis containing pLDG13:His:Rv3364c over-expressed vector at an MOI 1:10. Over-expressed bacterial lysate and cell supernatants were subjected to immunoprecipitation and western blotting using His-tag antibody. Line 1, His:Rv3364c protein extracted from the over-expressed M. tuberculosis; Line 2, His:Rv3364c protein identified in the cytoplasmic fraction of macrophages.
Effect of M. tuberculosis Wild-Type and 20A11 Infection on Cytokine Production
Macrophage-derived cytokines are important players in the control of M. tuberculosis infection. To examine if there were any differences in cytokine induction activating or inhibiting anti-mycobacterial activity in macrophages, U937 mononuclear cells were infected with M. tuberculosis H37Rv or DIA mutant 20A11, and the production of IL-12, TNF, IFN-β, and TGF-β cytokines was then analyzed in the supernatants after 4, 24, and 48 h (Figure 3A). The results indicate that infection of the 20A11 mutant was associated with significantly greater IFN-β and lower TGF-β production, but no significant changes in IL-12 and TNF production were observed, when compared to the wild-type infection.
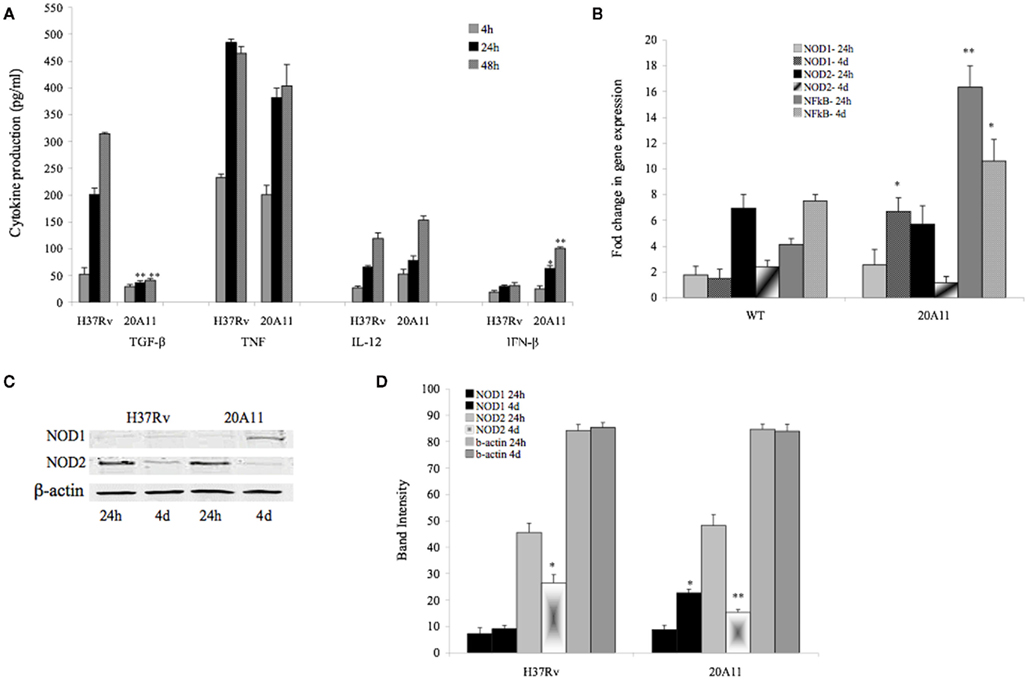
Figure 3. (A)Production of IL-12, TNF, IFN-β, and TGF-β cytokines over time by macrophages infected with M. tuberculosis H37Rv and 20A11 mutant. Cells were infected with MOI of 10. The numbers represent the mean ± SD of three independent experiments. The significance of differences between the 20A11 mutant and the wild-type strain was evaluated *p < 0.05 and **p < 0.01 at the same time point infections. (B) Expression profile of intracellular pattern recognition receptor NOD1 and NOD2 and NF-κB in H37Rv- or 20A11-infected macrophages analyzed by Real-Time quantitative PCR. Bars represent the fold change between differentially expressed transcripts from wild-type and mutant infected cells. Positive values indicate relative increased expression levels over uninfected control. The numbers represent the mean ± SD of three independent experiments. The differences between the 20A11 mutant and H37Rv strain was evaluated *p < 0.05 and **p < 0.01 at the same times of post-infection. (C) Western blot analysis of NOD1 and NOD2 proteins. U937 cells were infected with M. tuberculosis H37Rv or 20A11 mutant (MOI 1:10) and cultured for 24 h and 4 days. Immunoprecipitated cell lysates were subjected to Western blotting, as described in Section “Materials and Methods.” (D) NOD1, NOD2 and β-actin protein levels were quantified via semi-quantitative Western blot analysis on the Li-Cor Odyssey Platform. The data represent the average of three independent experiments ± SD. A *p < 0.05 for NOD1 and NOD2 protein levels at day 4 compared with 24 h post-infection with 20A11 and H37Rv, respectively. **p < 0.01 Difference in NOD2 protein levels at day 4 compared with 24 h time post-infection with 20A11 mutant.
Expression Profile of NF-κB and Intracellular Pattern Recognition Receptors of Macrophages
Previously shown is the importance of IFN-β initiating signaling in activation of the nuclear factor-κB (NF-κB; Yang et al., 2000), a transcription factor that controls many among host immune response mechanisms, such as cytokine synthesis, apoptosis, and cell cycle. Real-Time PCR was performed to assess the NF-κB gene expression levels in M. tuberculosis H37Rv and 20A11-infected macrophages. Higher levels of NF-κB expression were observed in both wild-type and mutant infected cells, but the expression level was significantly higher in 20A11-infected macrophages, compared to the wild-type infection at 24 h (p < 0.01; Figure 3B).
The cytosolic proteins nucleotide-binding oligomerization domain 1 and 2 (NOD1 and NOD2) are involved in intracellular recognition of microbes and their products (Carneiro et al., 2004), and have been described as activators of NF-κB pathway (Athman and Philpott, 2004). Alternatively, since NOD proteins interact or enhance caspase-1 and caspase-9 activation, a role for these proteins in apoptosis has been proposed (Hong and Jung, 2002; Franchi et al., 2009). Because the 20A11 mutant is associated with significant macrophage apoptosis, higher IFN-β production and NF-κB synthesis in macrophages, we investigated the expression of NOD1 and NOD2, as well as caspase-1 activation, and compared them with macrophages infected with the wild-type bacterium. Figure 3B shows that, while high levels of NOD2 expression were observed in both, M. tuberculosis H37Rv and 20A11-infected macrophages after 24 h post-infection, NOD1 expression was significantly reduced in wild-type-infected macrophages at day 4 after infection. In fact, NOD1 expression in 20A11-infected phagocytic cells was sixfold higher than the expression in wild-type-infected cells at day 4 (p < 0.05). Western blot analysis of U937 mononuclear phagocytes upon the wild-type or mutant infection confirmed the Real-Time observations. Results revealed the high levels of NOD2 protein in H37Rv- and 20A11-infected cells. The NOD1 protein levels were significantly lower during the wild-type infection at 24 h and 4 days post-infection, compared with the NOD1 protein level after 20A11 infection at day 4 (Figures 3C,D).
Caspase-1 Activation during M. tuberculosis Wild-Type and 20A11 Mutant Infection
Caspase-1 has been shown to be a potent activator of NF-κB (Bauernfeind et al., 2009). In situ labeling and APO LOGIX™-FAM detection system for active caspase-1 were performed to assess any changes in caspase-1 activation during M. tuberculosis wild-type and mutant infection. Our results indicate that, while infection of macrophages with 20A11 mutant or treatment with staurosporine resulted in sixfold to eightfold greater caspase-1 activity than uninfected or untreated controls, significant inhibition of caspase-1 activation was seen in macrophages infected with M. tuberculosis H37Rv or complemented 20A11 clone at the 24-h time point infection (Figures 4A,B). We then examined whether 20A11-associated caspase-1 activation was related to the induction of macrophage apoptosis. Host cells were treated with caspase-1 inhibitor, infected with M. tuberculosis 20A11 mutant, and the levels of apoptosis were determined after 3 days. Inhibition of caspase-1 had a direct correlation to the decreased levels of apoptosis during 20A11 infection of U937 cells (Figure 4C).
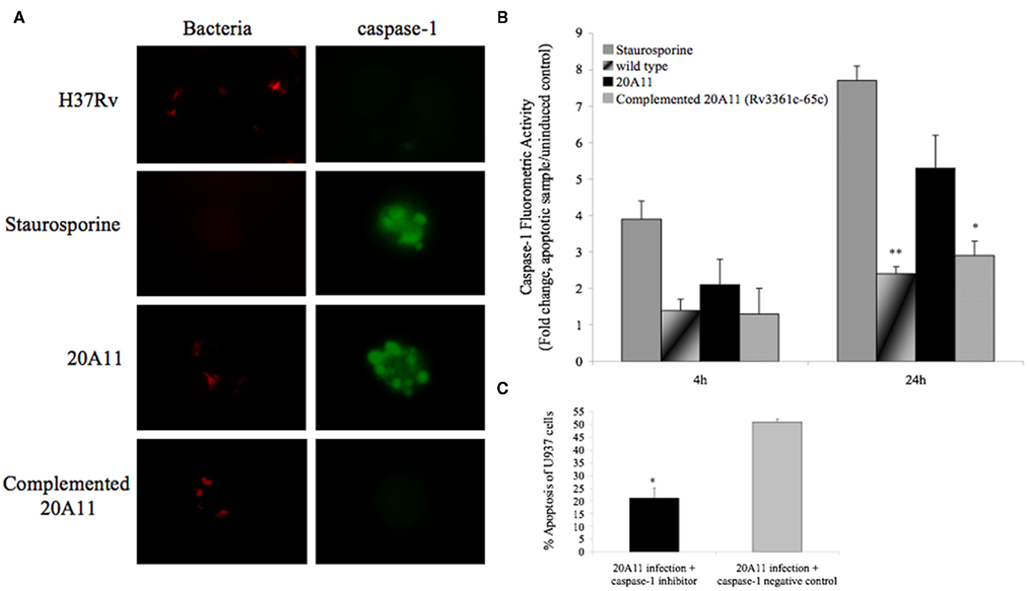
Figure 4. Role of caspase-1-dependent apoptosis. (A) In situ analysis of U937 macrophages for caspase-1 activation during M. tuberculosis H37Rv and 20A11 infection (MOI 10:1). While staurosporine treatment and 20A11 infection triggered caspase-1 activation of macrophages, M. tuberculosis H37Rv and complemented 20A11 clone were able to suppress caspase-1 activation. Bar, 10 μm. (B) M. tuberculosis infection prevents caspase-1 activation in macrophages. Cells were infected with wild-type or mutant strains, and fold change of caspase-1 activation was calculated according the manufacturer’s protocol. The data represent the average of three independent experiments ± SD. *p < 0.05 And **p < 0.01 statistically significant differences between complemented 20A11 and wild-type infection versus 20A11 mutant after 24 h post-infection, respectively. Active caspase-1 was quantified in situ using FAM–YVAD–FMK caspase-1 detection assay (Bachem). (C) Apoptosis level in caspase-1 inhibited macrophages after 24 h of 21A11 mutant post-infection examined by TUNEL. The values are means ± SD of two separate experiments performed in duplicate. The significance of differences between caspase-1 inhibitor and the caspase-1 negative control groups during 20A11 mutant infection was evaluated *p < 0.05.
M. tuberculosis Rv3364c Interaction with Cathepsin G Protein
In order to identify Rv3364c interacting host protein, we performed His pull-up assay. The bacterial His-tagged RFP–Rv3364c protein was incubated with activated macrophage lysates for overnight and after 24 h captured with the paramagnetic pre-charged His–nickel particles, following the isolation of interacting host protein present in the U937 cell lysate (Figure 5A). The mass spectrometric analysis of captured target protein identified 29 kDa plasma-membrane-associated protease cathepsin G (Trypsin-like serine protease; Table 2).
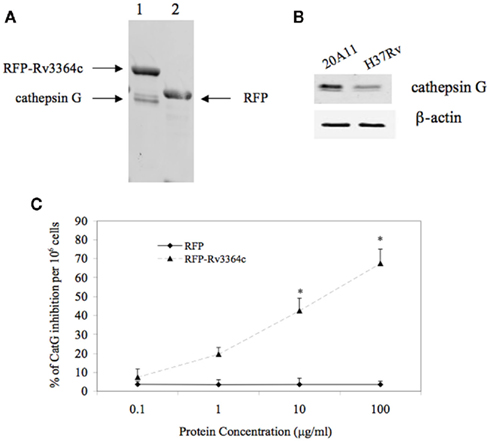
Figure 5. Host target protein for Rv3364c. (A) The His-paramagnetic pre-charged nickel-pulling assay of macrophage proteins interacting with M. tuberculosis Rv3364c identified 29 kDa plasma-membrane-associated serine protease cathepsin G. (1) His-tag-RFP–Rv3364c pull-up sample with cathepsin G; (2) His-tag-RFP control pull-up sample. (B) Cathepsin G protein expression levels by Western blot analysis: (1) 20A11 mutant infection; (2) M. tuberculosis H37Rv infection. (C) The effects of the Rv3364c protein on cathepsin G activity. Purified His-tag-RFP–Rv3364c protein was exposed for 4 h to pre-cleared macrophage lysates at 0.1–100 μg/ml concentration range and changes in cathepsin G activity were measured as% of cathepsin G inhibition per 106 cells. The data represent the average of three independent experiments ± SD. *p < 0.05 for the comparison between His-tag-RFP–Rv3364c experimental and His-tag-RFP control treatments at the same concentration of proteins.
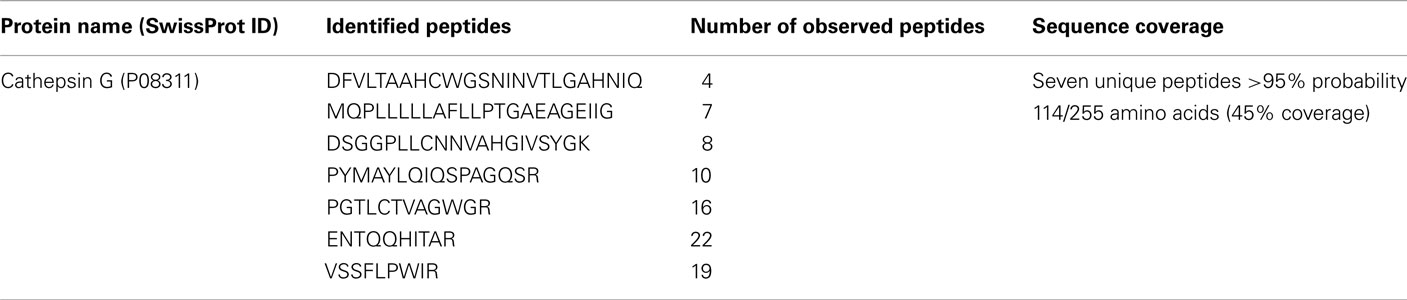
Table 2. Mass spectrometry analysis of host protein interacting with M. tuberculosis Rv3364c.
Western blot analysis of macrophages infected with M. tuberculosis H37Rv and 20A11 mutant showed differences in cathepsin G protein expression levels. While the cathepsin G protein was presented at a high level in 20A11-infected cells, M. tuberculosis H37Rv infection significantly reduced the levels of cathepsin G protein at the 4-h time point (Figure 5B).
Mycobacterium tuberculosis strains H37Rv and 20A11-infected macrophages were further analyzed for cathepsin G enzymatic activity, which showed a significantly lower cathepsin G activity in wild-type-infected cells, in opposition to 20A11 mutant infection at both 12- and 24-h time point infections (Table 3). In order to identify whether the Rv3364c protein was capable of inhibiting cathepsin G activity, we exposed different concentrations of purified bacterial protein to macrophage cell lysates and the percentage of the decreased cathepsin G activity was measured after 4 h of incubation. As shown in the Figure 5C, the high inhibitory effects of the Rv3364c protein on cathepsin G activity were found in macrophage lysates exposed to 1, 10, and 100 μg/ml concentrations compared to the RFP treatment alone.
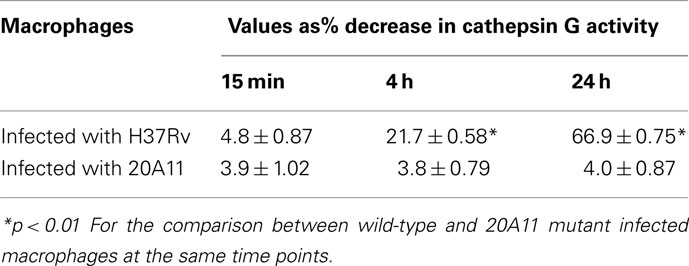
Table 3. Cathepsin G activity from samples of cell lysates of U937 macrophages.
The further interaction studies of Rv3364c with cathepsin G using the lentiviral expression system confirmed the colocalization of these over-expressed proteins in macrophages (Figure 6).
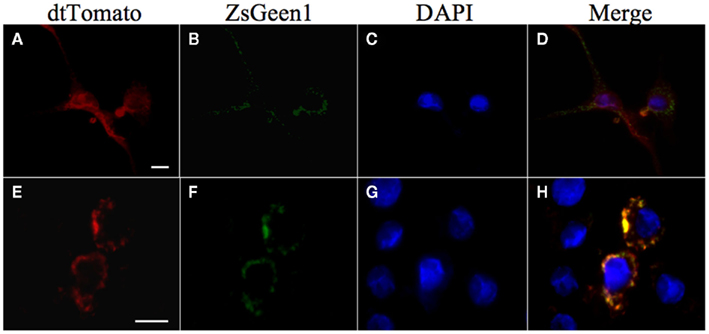
Figure 6. Colocalization of Rv3364c with the cathepsin G protein. U937 cells were transiently coinfected with Lenti-X viruses produced from a pLVX–ZsGreen1–cathepsin G vector and either a pLVX–dtTomato (A–D) or pLVX–dtTomato-Rv3364c fusion vector (E–H). ZsGgreen1-labeled cathepsin G visualization show punctate pattern in transduced cells (B,F). dtTomato labeled Rv3364c protein is also visible as punctate pattern (E), whereas dtTomato plane protein is diffused in the cytoplasm of macrophages (A). Nuclei are stained with DAPI (C,G). The overlap between cathepsin G and Rv3364c immunoreactivity appears yellow in the merged micrographs indicating colocalization of studied proteins (H). Scale bar, 10 μm.
Inhibition of Cathepsin G during M. tuberculosis 20A11 Infection Decreased Caspase-1 Activation
Cathepsin G inhibition assay was conducted in macrophages infected with 20A11 mutant to identify the relationship between cathepsin G expression and caspase-1 activation, which consequently led to the host cell apoptosis during 20A11 mutant infection. As shown in the Table 4, cathepsin G inhibition in M. tuberculosis 20A11-infected cells significantly decreased macrophage apoptosis levels compared with 20A11 mutant infection.
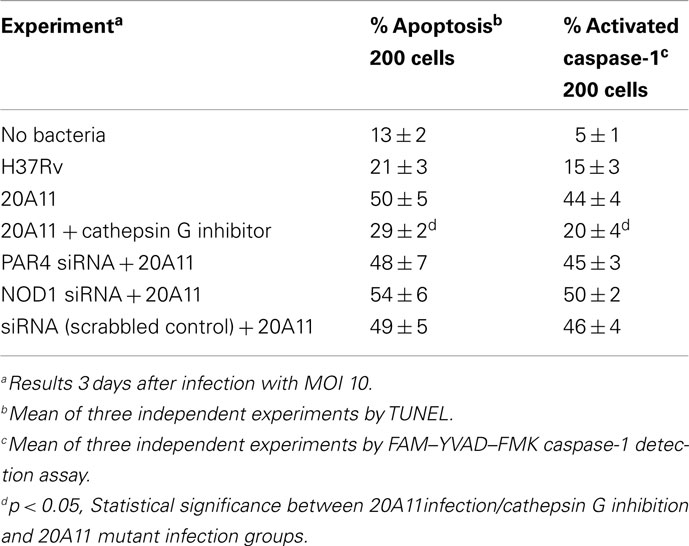
Table 4. Inhibition of cathepsin G in M. tuberculosis 20A11-infected macrophages reduces caspase-1-induced apoptosis.
Differential Expression of Cathepsin G Changed the Expression Level of Protease-Activated Receptor-4 (PAR4) during M. tuberculosis Infection
It has been recently discovered that the protease-activated receptor family member PAR4 is activated by cathepsin G (Sambrano et al., 2000). PAR4 is considered an effective mediator of platelet activation and inflammation (Ramachandran et al., 2007). Alternatively, PAR receptors have been shown to modulate the program cell death (Borensztajn and Spek, 2008). To investigate the possible mechanism by which the inhibition of cathepsin G by M. tuberculosis wild-type inhibited activation of caspase-1, we examined the PAR4 protein expression levels in wild-type and 20A11 mutant infected cells (Figure 7). Western blot analysis of immunoprecipitated PAR4 protein showed a detectible level of this protein in 20A11-infected macrophages. The PAR4 protein was not presented in wild-type-infected or uninfected cells.
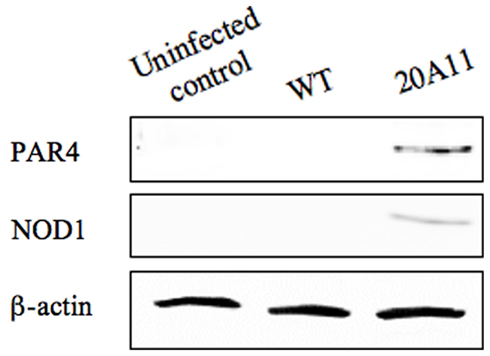
Figure 7. Western blot analysis of PAR4 and NOD1 proteins. M. tuberculosis H37Rv or 20A11-infected U937 cell lysates were processed for immunoprecipitation and then subjected to protein analysis. Detectable levels of both proteins were observed just after 4 days of post-infection.
We observed the changes in PAR4 and NOD1 protein expression levels during 20A11 infection. To determine if either protein expression in macrophages was the reason for increased levels of apoptosis and caspase-1 activation, using interference siRNA system, we inactivated PAR4 and NOD1. Cells were then infected with M. tuberculosis 20A11 and apoptosis levels and caspase-1 activations were determined. There were similar high levels of macrophage apoptosis in PAR4- or NOD1 siRNA-transfected cells compared to 20A11-infected macrophages alone or siRNA scrambled control, as shown on Table 4, suggesting that none of these protein had direct role in caspase-1 activation.
Discussion
Early inflammatory and innate responses during M. tuberculosis infection play essential roles in the host defense mechanisms. Apoptotic response of macrophages to mycobacterial infection appears to be an important aspect of the host innate immunity. Inhibition of apoptosis results in M. tuberculosis survival in macrophages both in tissue culture and in vivo (Danelishvili et al., 2003; Park et al., 2006; Velmurugan et al., 2007), and in infected mice apoptosis of macrophages is indirectly related to virulence (Park et al., 2006). TNF, IL-12, and IFN-γ are essential cytokines that contribute to protective immunity against M. tuberculosis (Bhatt and Salgame, 2007); whereas, cytokines, such as IL-10 and TGF-β, down-regulate the immune response and inhibit excessive inflammatory response (Toossi et al., 1995).
We evaluated a M. tuberculosis clone, with a transposon in the gene Rv3365c, an inactivation that lead to a polar effect on a chromosomal region with five genes. The first gene in the operon, Rv3365c, encodes for a putative regulatory protein that may be able to sense the environment. Rv3364c encodes most similar protein to the Roadblock/LC7 family of proteins, which in Myxococcus xanthus is associated with gliding motility, and in eukaryotes, with dynein (Koonin and Aravind, 2000). Rv3361c, the last gene in the operon, encodes for a protein involved in fluoroquinolone resistance (Hegde et al., 2005). The gene just upstream (Rv3362c) encodes for a probable ATP-binding protein. The Rv3363c most likely is a partner protein to Rv3365c and is activated by this sensory histidine kinase. Therefore, the operon is suggestive to be composed of genes encoding for proteins to sense the surrounding environment. The expression of whole operon is highly upregulated inside phagocytic cells, which again supports the hypothesis that the secreted protein(s) would have a function within the host cell. The finding that Rv3364c inactivation and consequent lack of function in the region was associated with a significant increased production of IFN-β by infected macrophages raised the possibility of an association between M. tuberculosis Rv3361c-Rv3364c and host cytosolic sensors. Type I IFNs are an important class of cytokines, responsible for a number of diverse immune responses to pathogens (Monroe et al., 2009). This group of cytokines is transcribed in response to signals from surveillance pathways, such as caspase recruitment domain (CARD)-containing sensors like RIG-1 and MDA5 and NOD proteins (Franchi et al., 2008). These latter proteins, also known as NOD-like family receptors (NLR) recognize primarily microbial molecules. The cytosolic receptors, NOD1 and NOD2, bind to meso-diaminopimelic acid and muramyl dipeptide, respectively. Once activated, NOD1 and NOD2 undergo conformational changes and activate NF-κB downstream. Many bacteria were demonstrated to activate NOD1, including Shigella flexneri (Girardin et al., 2001), and Listeria monocytogenes (Park et al., 2007). Staphylococcus aureus (Kapetanovic et al., 2007) and L. monocytogenes (Kobayashi et al., 2005) have been associated with NOD2 activation. Ferwerda et al. (2005) showed that NOD2 is a recognition system for M. tuberculosis. This intracellular receptor has been shown to regulate the host response to M. tuberculosis and BCG infection in human macrophages (Brooks et al., 2011). Our results also confirm the previous observation that M. tuberculosis infection inhibits NOD2 expression. The reason NOD2 transcript and protein were down-regulated in macrophages after 4 days of infection is not known, but it indicates that the phagocytic cell does not transmit any message using that pathway, silencing a recognition path. Since NOD2 is constitutively expressed in cells, in contrast to NOD1, one would expect that NOD2 production would be sustained during infection. NOD1 expression, however, was only triggered at late time points by the mutant 20A11 (Rv3364c-inactivated), but not by the wild-type bacterium. Considering that the bacterial structure responsible for the activation of NOD1 is almost certainly present on the 20A11 mutant strain (diaminopimelic acid), since the synthetic pathway is present, one wonders about the reasons why NOD1 is only expressed late in the infection. Although this may be related with the late release of diaminopimelic acid from the vacuole, further studies are necessary to confirm it. Alternatively, M. tuberculosis or bacterial antigens derived from the pathogen may suppress NOD1 expression by unknown mechanisms.
Protein–protein interaction assay demonstrated that the host protein that binds to Rv3364c is cathepsin G, a serine protease found in the lysosome and associated with membranes of phagocytic cells and epithelial cells (Maison et al., 1991; Moriuchi et al., 2000). Both cathepsin G and B have been linked to apoptosis of epithelial cells and phagocytic cells (Sabri et al., 2003; Averette et al., 2009; Chung et al., 2010). In addition, cathepsin D activation during pneumococcal infection was shown to induce apoptosis of macrophages killing this pathogen (Bewley et al., 2011). Due to its direct proteolytic activities, cathepsin G has been shown to have antimicrobial killing effects against S. aureus (Reeves et al., 2002). Cleavage of Pseudomonas aeruginosa flagellin by cathepsin G has been shown to inactivate the biological function of this virulence factor (Lopez-Boado et al., 2004). The interaction of membrane-associated serine protease of U937 monocytes with gp120 protein during HIV infection has been described to help the virus to enter host cells and spread the infection (Avril et al., 1994).
How membrane cathepsin G is activated is still unknown, but it has been shown that cathepsin G cleaves and activates IL-1-β in vivo (Hazuda et al., 1990). This serine protease has also been shown to stimulate protease-activated receptor-4 (PAR4; Sambrano et al., 2000; Ramachandran et al., 2007), belonging to a family of transmembrane domain G-protein-coupled receptor mediating cellular responses to proteases (Rohani et al., 2010). PARs function on epithelial cells and macrophages as a part of the immuno-surveillance system and trigger caspase-1 activation through the activation of NOD proteins (Chung et al., 2010; Rohani et al., 2010). Cathepsin G is also described as stimulating the intrinsic pathway of apoptosis, secondary to oxidative stress (Blomgran et al., 2007), and caspase-8-mediated apoptosis (Baumgartner et al., 2007).
Our results suggest that the wild-type M. tuberculosis inhibits expression and activation of cathepsin G. The inhibition has important effect downstream, with consequent suppression of caspase-1 activation. These findings are quite intriguing and bring to light the complexity of the process. M. tuberculosis has been shown to inhibit apoptosis by interfering with the eicosanoid pathways (Divangahi et al., 2010) and the extrinsic pathway of apoptosis by secreting nuoG (Velmurugan et al., 2007). The bacterium also suppresses the extrinsic pathway of apoptosis by blocking post-transcription mechanisms of caspase activation (Danelishvili et al., 2010). In addition, a recent study showed that M. tuberculosis prevents the inflammasome activation, probably by secreting Zmp-1, a putative metalloprotease, and inhibiting the stimulation of caspase-1-dependent apoptosis (Master et al., 2008). We now describe a different pathway that the bacterium interferes with and, consequently, blocks pyroptosis. It is clear that the success of M. tuberculosis as a pathogen depends on the ability to interfere with a number of redundant mechanisms of apoptosis in macrophages. Cathepsin G most likely plays a relevant role in the early host defense against mycobacterial infection. The activation of cathepsin G in the membrane triggers effects downstream that lead to the stimulation of the inflammatory process. Whether the reason for blocking this pathway is only related to apoptosis suppression or is also aimed to regulate the inflammatory process is currently unknown. The significant down-regulation of the expression of cathepsin G by M. tuberculosis virulent strain infection of macrophages has been reported to increase bacterial survival (Rivera-Marrero et al., 2004). Cathepsin G was also found strongly upregulated during M. bovis BCG infection of alveolar macrophages in vivo (Srivastava et al., 2007). Adding this to our data explains that sensor mechanisms in M. tuberculosis are in place, which stimulate the synthesis of a number of downstream proteins, including release of Rv3364c and inactivation of one of many pathways leading to apoptosis. Apparently, the puzzle is beginning to come together, and we are once more reminded of how fascinating this pathogen is.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the NIH grant # AI064018A and the Microbiology Foundation. We would like to thank Brian Arbogast for assistance with mass spectrometric sequencing and analysis. The mass spectrometric sequencing was supported in part by the National Institute of Environmental Health Science National Institute of Health Grant P30 ES00210. We thank Dr. Martin Pavelka for providing M. tuberculosis strain PM638. We also thank Denny Weber for editing the text and helping with the typing.
References
Aliprantis, A. O., Yang, R. B., Mark, M. R., Suggett, S., Devaux, B., Radolf, J. D., Klimpel, G. R., Godowski, P., and Zychlinsky, A. (1999). Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285, 736–739.
Applied-Biosystems. (1997). Relative quantitation of gene expression. ABI PRISM 7700 sequence detection system user bulletin #2. London: ABI Company Publication.
Athman, R., and Philpott, D. (2004). Innate immunity via toll-like receptors and Nod proteins. Curr. Opin. Microbiol. 7, 25–32.
Averette, K. M., Pratt, M. R., Yang, Y., Bassilian, S., Whitelegge, J. P., Loo, J. A., Muir, T. W., and Bradley, K. A. (2009). Anthrax lethal toxin induced lysosomal membrane permeabilization and cytosolic cathepsin release is Nlrp1b/Nalp1b-dependent. PLoS ONE 4, e7913. doi:10.1371/journal.pone.0007913
Avril, L. E., Di Martino-Ferrer, M., Pignede, G., Seman, M., and Gauthier, F. (1994). Identification of the U-937 membrane-associated proteinase interacting with the V3 loop of HIV-1 gp120 as cathepsin G. FEBS Lett. 345, 81–86.
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., Fernandes-Alnemri, T., Wu, J., Monks, B. G., Fitzgerald, K. A., Hornung, V., and Latz, E. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791.
Baumgartner, H. K., Gerasimenko, J. V., Thorne, C., Ashurst, L. H., Barrow, S. L., Chvanov, M. A., Gillies, S., Criddle, D. N., Tepikin, A. V., Petersen, O. H., Sutton, R., Watson, A. J., and Gerasimenko, O. V. (2007). Caspase-8-mediated apoptosis induced by oxidative stress is independent of the intrinsic pathway and dependent on cathepsins. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G296–G307.
Bewley, M. A., Marriott, H. M., Tulone, C., Francis, S. E., Mitchell, T. J., Read, R. C., Chain, B., Kroemer, G., Whyte, M. K., and Dockrell, D. H. (2011). A cardinal role for cathepsin d in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog. 7, e1001262. doi:10.1371/journal.ppat.1001262
Bhatt, K., and Salgame, P. (2007). Host innate immune response to Mycobacterium tuberculosis. J. Clin. Immunol. 27, 347–362.
Blomgran, R., Zheng, L., and Stendahl, O. (2007). Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J. Leukoc. Biol. 81, 1213–1223.
Borensztajn, K. S., and Spek, C. A. (2008). Protease-activated receptors, apoptosis and tumor growth. Pathophysiol. Haemost. Thromb. 36, 137–147.
Brooks, M. N., Rajaram, M. V., Azad, A. K., Amer, A. O., Valdivia-Arenas, M. A., Park, J. H., Nunez, G., and Schlesinger, L. S. (2011). NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cell. Microbiol. 13, 402–418.
Campoy, E., and Colombo, M. I. (2009). Autophagy subversion by bacteria. Curr. Top. Microbiol. Immunol. 335, 227–250.
Carneiro, L. A., Travassos, L. H., and Philpott, D. J. (2004). Innate immune recognition of microbes through Nod1 and Nod2: implications for disease. Microbes Infect. 6, 609–616.
Chung, W. O., An, J. Y., Yin, L., Hacker, B. M., Rohani, M. G., Dommisch, H., and DiJulio, D. H. (2010). Interplay of protease-activated receptors and NOD pattern recognition receptors in epithelial innate immune responses to bacteria. Immunol. Lett. 131, 113–119.
Danelishvili, L., McGarvey, J., Li, Y. J., and Bermudez, L. E. (2003). Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell. Microbiol. 5, 649–660.
Danelishvili, L., Poort, M. J., and Bermudez, L. E. (2004). Identification of Mycobacterium avium genes up-regulated in cultured macrophages and in mice. FEMS Microbiol. Lett. 239, 41–49.
Danelishvili, L., Wu, M., Stang, B., Harriff, M., Cirillo, S., Cirillo, J., Bildfell, R., Arbogast, B., and Bermudez, L. E. (2007). Identification of Mycobacterium avium pathogenicity island important for macrophage and amoeba infection. Proc. Natl. Acad. Sci. U.S.A. 104, 11038–11043.
Danelishvili, L., Yamazaki, Y., Selker, J., and Bermudez, L. E. (2010). Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c proteins participate in the suppression of macrophage apoptosis. PLoS ONE 5, e10474. doi:10.1371/journal.pone.0010474
Davis, A. S., Vergne, I., Master, S. S., Kyei, G. B., Chua, J., and Deretic, V. (2007). Mechanism of inducible nitric oxide synthase exclusion from mycobacterial phagosomes. PLoS Pathog. 3, e186. doi:10.1371/journal.ppat.0030186
Deretic, V., Singh, S., Master, S., Harris, J., Roberts, E., Kyei, G., Davis, A., de Haro, S., Naylor, J., Lee, H. H., and Vergne, I. (2006). Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell. Microbiol. 8, 719–727.
Divangahi, M., Desjardins, D., Nunes-Alves, C., Remold, H. G., and Behar, S. M. (2010). Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat. Immunol. 11, 751–758.
Ferwerda, G., Girardin, S. E., Kullberg, B. J., Le Bourhis, L., de Jong, D. J., Langenberg, D. M., van Crevel, R., Adema, G. J., Ottenhoff, T. H., Van der Meer, J. W., and Netea, M. G. (2005). NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog. 1, e34. doi:10.1371/journal.ppat.0010034
Flores, A. R., Parsons, L. M., and Pavelka, M. S. Jr. (2005). Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to beta-lactam antibiotics. Microbiology 151, 521–532.
Franchi, L., Park, J. H., Shaw, M. H., Marina-Garcia, N., Chen, G., Kim, Y. G., and Nunez, G. (2008). Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 10, 1–8.
Franchi, L., Warner, N., Viani, K., and Nunez, G. (2009). Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128.
Gao, L. Y., and Kwaik, Y. A. (2000). The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol. 8, 306–313.
Girardin, S. E., Tournebize, R., Mavris, M., Page, A. L., Li, X., Stark, G. R., Bertin, J., DiStefano, P. S., Yaniv, M., Sansonetti, P. J., and Philpott, D. J. (2001). CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2, 736–742.
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766.
Hazuda, D. J., Strickler, J., Kueppers, F., Simon, P. L., and Young, P. R. (1990). Processing of precursor interleukin 1 beta and inflammatory disease. J. Biol. Chem. 265, 6318–6322.
Hegde, S. S., Vetting, M. W., Roderick, S. L., Mitchenall, L. A., Maxwell, A., Takiff, H. E., and Blanchard, J. S. (2005). A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308, 1480–1483.
Heldwein, K. A., and Fenton, M. J. (2002). The role of toll-like receptors in immunity against mycobacterial infection. Microbes Infect. 4, 937–944.
Hirsch, C. S., Ellner, J. J., Russell, D. G., and Rich, E. A. (1994). Complement receptor-mediated uptake and tumor necrosis factor-alpha-mediated growth inhibition of Mycobacterium tuberculosis by human alveolar macrophages. J. Immunol. 152, 743–753.
Hong, G. S., and Jung, Y. K. (2002). Caspase recruitment domain (CARD) as a bi-functional switch of caspase regulation and NF-kappaB signals. J. Biochem. Mol. Biol. 35, 19–23.
Kapetanovic, R., Nahori, M. A., Balloy, V., Fitting, C., Philpott, D. J., Cavaillon, J. M., and Adib-Conquy, M. (2007). Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus-activated macrophages. Infect. Immun. 75, 830–837.
Kobayashi, K. S., Chamaillard, M., Ogura, Y., Henegariu, O., Inohara, N., Nunez, G., and Flavell, R. A. (2005). Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734.
Koonin, E. V., and Aravind, L. (2000). Dynein light chains of the Roadblock/LC7 group belong to an ancient protein superfamily implicated in NTPase regulation. Curr. Biol. 10, R774–R776.
Loeuillet, C., Martinon, F., Perez, C., Munoz, M., Thome, M., and Meylan, P. R. (2006). Mycobacterium tuberculosis subverts innate immunity to evade specific effectors. J. Immunol. 177, 6245–6255.
Lopez-Boado, Y. S., Espinola, M., Bahr, S., and Belaaouaj, A. (2004). Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J. Immunol. 172, 509–515.
Maison, C. M., Villiers, C. L., and Colomb, M. G. (1991). Proteolysis of C3 on U937 cell plasma membranes. Purification of cathepsin G. J. Immunol. 147, 921–926.
Master, S. S., Rampini, S. K., Davis, A. S., Keller, C., Ehlers, S., Springer, B., Timmins, G. S., Sander, P., and Deretic, V. (2008). Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3, 224–232.
Monroe, K. M., McWhirter, S. M., and Vance, R. E. (2009). Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5, e1000665. doi:10.1371/journal.ppat.1000665
Moriuchi, H., Moriuchi, M., and Fauci, A. S. (2000). Cathepsin G, a neutrophil-derived serine protease, increases susceptibility of macrophages to acute human immunodeficiency virus type 1 infection. J. Virol. 74, 6849–6855.
O’Sullivan, M. P., O’Leary, S., Kelly, D. M., and Keane, J. (2007). A caspase-independent pathway mediates macrophage cell death in response to Mycobacterium tuberculosis infection. Infect. Immun. 75, 1984–1993.
Park, J. H., Kim, Y. G., Shaw, M., Kanneganti, T. D., Fujimoto, Y., Fukase, K., Inohara, N., and Nunez, G. (2007). Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J. Immunol. 179, 514–521.
Park, J. S., Tamayo, M. H., Gonzalez-Juarrero, M., Orme, I. M., and Ordway, D. J. (2006). Virulent clinical isolates of Mycobacterium tuberculosis grow rapidly and induce cellular necrosis but minimal apoptosis in murine macrophages. J. Leukoc. Biol. 79, 80–86.
Peterszegi, G., Texier, S., Robert, A. M., Moulias, R., and Robert, L. (1997). Increased elastase and cathepsin G activity in activated lymphocytes from aged patients: role of denutrition and dementia. Arch. Gerontol. Geriatr. 25, 285–298.
Ramachandran, R., Sadofsky, L. R., Xiao, Y., Botham, A., Cowen, M., Morice, A. H., and Compton, S. J. (2007). Inflammatory mediators modulate thrombin and cathepsin-G signaling in human bronchial fibroblasts by inducing expression of proteinase-activated receptor-4. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L788–798.
Reeves, E. P., Lu, H., Jacobs, H. L., Messina, C. G., Bolsover, S., Gabella, G., Potma, E. O., Warley, A., Roes, J., and Segal, A. W. (2002). Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416, 291–297.
Rivera-Marrero, C. A., Stewart, J., Shafer, W. M., and Roman, J. (2004). The down-regulation of cathepsin G in THP-1 monocytes after infection with Mycobacterium tuberculosis is associated with increased intracellular survival of bacilli. Infect. Immun. 72, 5712–5721.
Rohani, M. G., Beyer, R. P., Hacker, B. M., Dommisch, H., Dale, B. A., and Chung, W. O. (2010). Modulation of expression of innate immunity markers CXCL5/ENA-78 and CCL20/MIP3alpha by protease-activated receptors (PARs) in human gingival epithelial cells. Innate Immun. 16, 104–114.
Sabri, A., Alcott, S. G., Elouardighi, H., Pak, E., Derian, C., Andrade-Gordon, P., Kinnally, K., and Steinberg, S. F. (2003). Neutrophil cathepsin G promotes detachment-induced cardiomyocyte apoptosis via a protease-activated receptor-independent mechanism. J. Biol. Chem. 278, 23944–23954.
Sambrano, G. R., Huang, W., Faruqi, T., Mahrus, S., Craik, C., and Coughlin, S. R. (2000). Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem. 275, 6819–6823.
Srivastava, M., Meinders, A., Steinwede, K., Maus, R., Lucke, N., Buhling, F., Ehlers, S., Welte, T., and Maus, U. A. (2007). Mediator responses of alveolar macrophages and kinetics of mononuclear phagocyte subset recruitment during acute primary and secondary mycobacterial infections in the lungs of mice. Cell. Microbiol. 9, 738–752.
Tamme, R., Camp, E., Kortschak, R. D., and Lardelli, M. (2000). Nonspecific, nested suppression PCR method for isolation of unknown flanking DNA. Biotechniques 28, 895–899, 902.
Toossi, Z., Gogate, P., Shiratsuchi, H., Young, T., and Ellner, J. J. (1995). Enhanced production of TGF-beta by blood monocytes from patients with active tuberculosis and presence of TGF-beta in tuberculous granulomatous lung lesions. J. Immunol. 154, 465–473.
Velmurugan, K., Chen, B., Miller, J. L., Azogue, S., Gurses, S., Hsu, T., Glickman, M., Jacobs, W. R., Porcelli, S. A., and Briken, V. (2007). Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 3, e110. doi:10.1371/journal.ppat.0030110
Keywords: Mycobacterium tuberculosis, macrophages, apoptosis, pyroptosis, inhibition, caspase-1-dependent
Citation: Danelishvili L, Everman JL, McNamara MJ and Bermudez LE (2012) Inhibition of the plasma-membrane-associated serine protease cathepsin G by Mycobacterium tuberculosis Rv3364c suppresses caspase-1 and pyroptosis in macrophages. Front. Microbio. 2:281. doi: 10.3389/fmicb.2011.00281
Received: 19 September 2011;
Accepted: 28 December 2011;
Published online: 11 January 2012.
Edited by:
Adel M. Talaat, University of Wisconsin Madison, USAReviewed by:
Jordi Torrelles, Ohio State University, USASteve Blanke, University of Illinois Urbana-Champaign, USA
Copyright: © 2012 Danelishvili, Everman, McNamara and Bermudez. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Luiz E. Bermudez, Biomedical Sciences, College of Veterinary Medicine, 105 Magruder Hall, Oregon State University, Corvallis, OR 97331, USA. e-mail:bHVpei5iZXJtdWRlekBvcmVnb25zdGF0ZS5lZHU=