 Melissa Bartel1,2,3
Melissa Bartel1,2,3 David Schaffer1,2,3*† and Hildegard Büning4,5*†
David Schaffer1,2,3*† and Hildegard Büning4,5*†- 1 Department of Chemical and Biomolecular Engineering, University of California at Berkeley, Berkeley, CA, USA
- 2 Department of Bioengineering, University of California at Berkeley, Berkeley, CA, USA
- 3 The Helen Wills Neuroscience Institute, University of California at Berkeley, Berkeley, CA, USA
- 4 Department I of Internal Medicine, University Hospital Cologne, Cologne, Germany
- 5 Center for Molecular Medicine Cologne (ZMMK), University of Cologne, Cologne, Germany
Vectors based on adeno-associated viruses (AAV) have shown considerable promise in both preclinical models and increasingly in clinical trials. However, one formidable challenge is pre-existing immunity due to widespread exposure to numerous AAV variants and serotypes within the human population, which affect efficacy of clinical trials due to the accompanying high levels of anti-capsid neutralizing antibodies. Transient immunosuppression has promise in mitigating cellular and humoral responses induced by vector application in naïve hosts, but cannot overcome the problem that pre-existing neutralizing antibodies pose toward the goal of safe and efficient gene delivery. Shielding of AAV from antibodies, however, may be possible by covalent attachment of polymers to the viral capsid or by encapsulation of vectors inside biomaterials. In addition, there has been considerable progress in using rational mutagenesis, combinatorial libraries, and directed evolution approaches to engineer capsid variants that are not recognized by anti-AAV antibodies generally present in the human population. While additional progress must be made, such strategies, alone or in combination with immunosuppression to avoid de novo induction of antibodies, have strong potential to significantly enhance the clinical efficacy of AAV vectors.
Introduction
AAV Biology
Adeno-associated virus (AAV) is a non-pathogenic parvovirus composed of a 4.7-kb single-stranded DNA genome packaged into a non-enveloped, icosahedral capsid (Knipe and Howley, 2006). The viral genome contains three open reading frames (ORF) between two inverted terminal repeats (ITR), which function as the origin of replication and as the packaging signal (Knipe and Howley, 2006). The rep ORF encodes four non-structural proteins that function in viral replication, transcriptional regulation, site-specific integration, and virion assembly (Knipe and Howley, 2006). The cap ORF encodes three structural proteins that assemble to form a 60-mer viral capsid (Knipe and Howley, 2006). An alternative ORF located in the same region of the genome produces the assembly activating protein (AAP; Sonntag et al., 2010), a viral protein that localizes AAV capsid proteins to the nucleolus and functions in the capsid assembly reaction (Sonntag et al., 2010).
The capsid proteins determine the virus’ ability to interact with and infect cells, from their initial binding to various cell surface receptors, to their trafficking inside the cell, to gaining access to the nucleus. Specifically, AAV is internalized rapidly via receptor-mediated endocytosis from clathrin-coated pits (Bartlett et al., 2000). Following cellular internalization, the virion escapes from early endosomes and traffics to the perinuclear area. Evidence exists for AAV uncoating prior to viral DNA entry into the nucleus (Lux et al., 2005), as well as viral trafficking into the nucleus prior to uncoating (Bartlett et al., 2000; Sonntag et al., 2006). In either case, once AAV localizes to the nucleus, second-strand synthesis – i.e., conversion of its single-stranded genome into double-stranded, transcriptionally active DNA – must occur for viral gene expression (Ferrari et al., 1996). In the absence of helper virus co-infection, AAV enters a latent life-cycle with viral genomes integrated in human chromosome 19 or non-integrated as extrachromosomal episomes.
Recombinant versions of AAV can be created in which a gene of interest is inserted between the ITRs, and the ORFs for structural and non-structural proteins are supplied separately (Flotte, 2004). This system allows the gene of interest to be packaged inside the viral capsid and delivered to the cell. Dividing as well as non-dividing cells are transduced, and gene expression – which occurs in the absence of helper virus function – is stable for years in post-mitotic tissue. There are several naturally occurring variants and serotypes of AAV, each of which differs in amino acid sequence, in particular in the hypervariable regions of the capsid proteins, and thus in their gene delivery properties (Wu et al., 2006; Schaffer et al., 2008). Importantly, no AAV has been associated with any human disease, making it a desirable gene delivery vector for clinical applications (Knipe and Howley, 2006).
Clinical Trials Involving AAV
Adeno-associated virus has been employed with promising results in a number of clinical trials. During a Phase I dose-escalation trial for gene therapy of Leber’s congenital amaurosis (LCA), for example, all 12 patients who received a subretinal injection of AAV2 encoding a protein required for the isomerohydrolase activity of retinal pigment epithelium showed sustained improvement in both subjective and objective measurements of vision (Bainbridge et al., 2008; Maguire et al., 2008, 2009). Furthermore, there were no significant adverse events during either the pre-trial efficacy studies or the Phase I trials (Bainbridge et al., 2008; Maguire et al., 2008, 2009). As another example, in a Phase I study for gene therapy of Canavan disease, 10 patients received intracranial infusions of AAV2 encoding human aspartoacylase (McPhee et al., 2006). Of importance with respect to vector re-application, 7 out of 10 patients in this trial had low or undetectable levels of neutralizing antibodies to AAV2 following administration of the gene therapy vector (McPhee et al., 2006).
In contrast to these clinical studies, which targeted immune privileged sites, anti-AAV host responses most likely mediated by cytotoxic T-cells limited the therapeutic efficacy of rAAV vectors following intrahepatic and intramuscular administration. Briefly, a Phase I/II dose-escalation study in which patients received a hepatic artery infusion of AAV2 encoding human Factor IX initially achieved therapeutic levels of Factor IX expression. However, the therapeutic levels were only present for 2 months (Manno et al., 2006), and follow-up experiments concluded that T-cell-mediated immunity to the AAV capsid antigens induced the destruction of AAV2-transduced hepatocytes (Manno et al., 2006). Furthermore, all subjects had a several log increase in neutralizing antibody titer following vector administration (Manno et al., 2006). Similarly, in a gene therapy study on lipoprotein lipase (LPL) deficiency, all eight patients achieved decreased median triglyceride levels and increased local LPL protein levels and activity 12 weeks post-administration of the AAV1 vector encoding LPLS447X (Stroes et al., 2008). But, also in this trial, triglyceride levels returned to baseline levels 18–31 months post-administration (Stroes et al., 2008).
Immune Responses toward AAV
Viruses are, in general, recognized by the innate immune system and induce the production of inflammatory cytokines, chemokines, and/or interferons that foster adaptive immune responses. The latter mainly comprises induction of antibodies produced by B lymphocytes following presentation of viral antigens by antigen-presenting cells, as well as direct cell killing mediated by cytotoxic T-cells (Knipe and Howley, 2006).
Knowledge on the mechanisms that lead to recognition and elimination of AAV in particular is still limited. Only recently, Zhu et al. (2009) provided evidence that AAV’s genome is recognized by the innate immune system through the Toll-like receptor 9-MyD88 pathway. In this study, plasmacytoid dendritic cells (pDCs) were identified as sentinel cells, and activation of the Toll-like receptor 9-MyD88 pathway was shown to elicit humoral and cytotoxic T-lymphocyte immune responses in mice.
Humans become naturally infected by numerous AAV serotypes, and in particular AAV2, during childhood, as indicated by sero-conversion. As a result of this exposure, memory B and eventually memory T-cells are induced. The latter are assumed to be the cause for the lack of long-term gene expression and loss of therapeutic efficacy in the clinical trials on hemophilia and on LPL mentioned above. Currently, it is not clear how this response is launched, and consequently strategies for its avoidance are lacking. One hypothesis is that memory T-cells were re-activated upon vector administration through presentation of capsid fragments following intracellular degradation of incoming capsids. Alternatively, contamination of the gene therapy vector preparations with cap DNA impurities could result in persistent expression of capsid antigens and thus the destruction of transduced cells, though a detailed analysis was unable to find cap DNA sequences in the vector preparation used for the hemophilia trial (Hauck et al., 2008). Should this prove to be a problem, microRNA approaches or an oversized cap ORF could be employed to avoid de novo capsid production and consequently MHC loading following AAV vector application (Halbert et al., 2011; Lu et al., 2011). Employing these strategies may help to reduce the cytotoxic immune response to successfully transduced cells. Furthermore, immunosuppression is exploited as a strategy to avoid induction of T as well as of B cell responses (Jiang et al., 2006). An as of yet even greater challenge is the high prevalence of pre-existing neutralizing antibodies that significantly hamper the efficacy of de novo cell transduction.
Natural exposure to AAV serotypes 1, 2, 5, 6, 8, and 9 results mainly in production of IgG1, but low amounts of IgG2, IgG3, and IgG4 antibodies are also produced (Boutin et al., 2010). With respect to the total anti-AAV IgG prevalence for each serotype, a recent, comprehensive analysis revealed that AAV2 (72%) and AAV1 (67%) antibodies were the most common, but AAV9 (47%), AAV6 (46%), AAV5 (40%), and AAV8 (38%) antibodies were also present in a large portion of the population studied (Boutin et al., 2010). Furthermore, these anti-AAV antibodies may cross-react with other related AAV serotypes. Differences in antibody prevalence in the human population are likely due to human genetic variation and/or frequency of exposure to the various serotypes.
Animal studies on the mechanisms of anti-AAV antibody induction using class I-deficient (unable to mount a cellular immune response) and class II-deficient (unable to mount a humoral response) mice found that the latter could readily express high levels of transgenes upon a second administration of the vector (Manning et al., 1998). In addition, using several types of immunodeficient mice (RAG1 knock-out, CD40 ligand knock-out, and nude), Chirmule et al. (2000) showed that the AAV neutralizing antibody response is T-cell dependent. Viral capsid antigens are presented to B cells in the lymph nodes, resulting in CD4+ T-cell activation (Bessis et al., 2004). The B cells then differentiate into plasma cells, which produce antibodies against the viral capsid proteins (Bessis et al., 2004). The route of administration has been shown to also have an impact on the induction of immune responses. Rhesus macaques receiving wild-type AAV via intramuscular or intravenous injection developed a humoral immune response to the AAV capsid, even without helper virus co-infection, while animals that received vectors via the intranasal route did not develop an immune response unless co-infected with a helper virus (Hernandez et al., 1999). Analogous results on the role of route of administration have been found in mice. Ge et al. (2001) found that intramuscular vector administration was more effective at inducing a humoral immune response than intraportal vein delivery. Nevertheless, independent of the initial administration route, re-administration of AAV vectors via the tail vein did not lead to transgene expression (Ge et al., 2001).
Recently, an AAV variant derived from rhesus macaques (AAVrh32.33) was found to elicit an immune response in mice similar to that against other AAV vectors seen in primate species (Mays and Wilson, 2009). Specifically, this AAV generates a strong CD8+ T-cell response to the AAV capsid and to the delivered transgene in mice, and its use may thus provide a more accurate murine model of AAV immune activation, and help to better explain the loss of transgene expression in human gene therapy clinical trials. In parallel to developing a deeper mechanistic understanding of anti-vector immune responses, for AAV to effectively function in clinical gene therapy, strategies must be developed to create vectors that evade the body’s immune response.
Strategies to Avoid Neutralization by Humoral Anti-AAV Immune Responses
Transient Immunosuppression
In the naïve host, humoral immune responses are elicited upon AAV vector application. In order to circumvent induction of this response, transient immunosuppression has been exploited. This strategy has been tested in preclinical animal models, using antibodies or small molecule inhibitors against T-cell functionality. Manning et al. (1998) reported that successful vector re-administration was achieved in 60% of mice treated with anti-CD40 antibodies during the first vector administration. However, the mice developed neutralizing antibodies to AAV following the second administration of the vectors, which was conducted without immunosuppression (Manning et al., 1998). Another study by Halbert et al. (1998) attempted transient immunosuppression using MR1 (a monoclonal anti-CD40 ligand antibody) and CTLA4Ig (a CTLA4-immunoglobulin fusion protein) alone or in combination. Mice that received only MR1 or CTLA4Ig were capable of expressing the delivered transgene, but at a lower level than the combination treatment in which the animals also developed low to undetectable levels of neutralizing antibodies to AAV. Also in these animals, neutralizing antibodies were elicited upon re-administration (Halbert et al., 1998).
In response to the above mentioned clinical trial that reported destruction of AAV2-FIX transduced hepatocytes (Manno et al., 2006), Jiang et al. (2006) tested transient immunosuppression as a means to prevent AAV capsid-directed T-cell response against liver cells expressing the FIX transgene. Immunosuppression of rhesus macaques was achieved using a combination of mycophenolate mofetil and tacrolimus (FK506). Neither transaminitis nor expansion of memory T-cells was observed in any of the animals, and consequently only the impact of immunosuppression on humoral, but not on anti-capsid T-cell responses, could be investigated. Of the three animals receiving immunosuppression during vector administration, one developed a strong anti-AAV antibody response. Furthermore, withdrawal of the immunosuppression therapy after 6 weeks lead to a 2-log increase in neutralizing antibody titer, revealing that at least in rhesus macaques long time immunosuppression is required (Jiang et al., 2006).
In summary, immune suppression is an effective strategy to mitigate the body’s immune response long enough to allow the capsid proteins to clear from the cell surface and prevent the formation of neutralizing antibodies in order to facilitate re-administration of the vector. However, transient immunosuppression is not a solution to pre-existing neutralizing antibodies to the AAV capsid. Modifications, either chemical modifications that protect surface exposed parts of the protein capsid or genetic modifications that result in changes to the protein capsid, must be made to the vector to evade these neutralizing antibodies.
Chemical Modification of AAV Capsid to Avoid Antibody Neutralization
One approach to reduce antibody neutralization of vector particles is to graft chemical moieties onto the virion surface to shield neutralizing epitopes, as recently reviewed (Jang et al., 2011). For example, crosslinking synthetic polymers onto the vector can reduce neutralization by anti-virus antibodies as well as enable evasion of innate immune responses, thereby enhancing gene transfer in the presence of existing antibodies and facilitating repeated administration by reducing subsequent adaptive immune responses to the vector (Kreppel and Kochanek, 2008). Polymeric materials that have been explored in conjunction with adenoviral and adeno-associated viral vectors have included polyethylene glycol (PEG; O’Riordan et al., 1999; Croyle et al., 2000, 2005; Cheng et al., 2003; De Geest et al., 2005; Eto et al., 2005; Lee et al., 2005; Mok et al., 2005; Oh et al., 2006; Hofherr et al., 2007), poly-N-(2-hydroxypropyl) methacrylamide (poly-HPMA; Fisher et al., 2001; Green et al., 2008), polysaccharides (Espenlaub et al., 2008), and others (Kasman et al., 2009; Kim et al., 2010). They are typically covalently conjugated through the reaction of active groups on the polymer termini to nucleophilic amino acid side chains on the viral surface, such as lysines and cysteines.
Polyethylene glycol is a non-toxic material known for its capacity to resist protein binding – likely through steric hindrance and blocking of protein surface charges – and it has been extensively conjugated to proteins to extend their circulatory half-life and reduce immune responses (Delgado et al., 1992; Edwards et al., 2003; Haag and Kratz, 2006; Romberg et al., 2008). Likewise, PEGylation has been utilized to protect viral vectors from neutralizing antibodies (O’Riordan et al., 1999; Croyle et al., 2001; Cheng et al., 2003; De Geest et al., 2005; Eto et al., 2005; Lee et al., 2005; Mok et al., 2005; Hofherr et al., 2007), enable vector retargeting (Oh et al., 2006), and enhance vector stability (Croyle et al., 2000). Several studies have PEGylated AAV, using several different crosslinking chemistries. In one report, high molecular weight PEG conjugated to AAV using a terminal N-hydroxysuccinimidyl ester to attach the polymer to viral surface lysines modestly protected AAV from neutralizing serum in culture (2.3-fold) at intermediate levels of PEG/virus. However, above a key stoichiometric ratio of PEG/AAV, viral infectivity was lost for both high and low molecular weight PEG, presumably due to a loss of key lysine residues and/or steric hindrance of viral surface regions critical for viral infectivity. This loss of infectivity was accompanied by alterations of AAV particles that could be visualized by electron microscopy (i.e., virion shape and size; Lee et al., 2005). Another study, which used different PEGylation chemistries, found more positive results. Specifically, AAV reacted with PEG using succinimidyl succinate chemistry (SSPEG) was partially protected from antibodies. In contrast, AAV coated with PEG via tresyl chloride reactive groups (TMPEG) was more effectively protected from neutralizing antibodies both in vitro and in vivo (Le et al., 2005). Over time, hydrolysis of the SSPEG linkages may progressively unmask antigen binding sites, rendering this conjugation less effective.
Other human viruses also face challenges with pre-existing immunity, and chemical conjugation has, for example, been more extensively explored to shield adenoviral vectors from serum, including both PEG (Croyle et al., 2005; De Geest et al., 2005; Mok et al., 2005; Kasman et al., 2009) and other polymers such as poly-HPMA (Fisher et al., 2001; Green et al., 2008). In one study, adenoviral vectors were coated with a random copolymer containing N-(2-hydroxypropyl)methacrylamide and methacryloyl-Gly-Gly-4-nitrophenoxy (pHPMA–ONp), via reaction with adenoviral surface amino groups. A binding assay involving ELISA measurement of free antibodies found that even at high excess, coated virus did not deplete anti-viral antibodies from solution, indicating the polymer shielded the adenovirus from the antibodies. Furthermore, since polymer coatings can block regions responsible for natural viral tropism, this modification afforded the further opportunity to retarget the virus via addition of fibroblast growth factor (FGF)-2 or vascular endothelial growth factor (VEGF) to the polymer. Adenoviral vectors coated with the polymer plus FGF-2 were 10-fold more resistant to antibody neutralization in vitro (Fisher et al., 2001). In addition, the vectors exhibited selective delivery to cell lines expressing receptors for FGF-2 or VEGF, depending on the ligand attached to the particle surface.
In addition to direct polymer grafting, an alternate strategy for protecting vectors from serum is to encapsulate them inside polymeric gels or particles designed to progressively degrade and release the virus (Jang et al., 2011). In early work, Beer et al. (1998) encapsulated adenovirus in poly-lactic glycolic acid using a double emulsion technique. They found that the encapsulated virus retained 27–50 and 62–65% infectivity in culture at 1:100 and 1:500 dilutions of rat serum, respectively, while non-modified vectors retained less than 1% of infectivity at both dilutions. In addition, they assessed the development of neutralizing antibodies in naïve mice upon administration of protected vector and found that animals receiving encapsulated adenovirus did not develop anti-adenovirus antibodies until after the third dose, and the resulting titers were 45-fold lower than mice receiving unmodified adenovirus (Beer et al., 1998). In another report, adenovirus was encapsulated in microspheres generated from alginate, a linear copolymer of anionic saccharides isolated from brown algae. Upon intranasal or intraperitoneal administration of virus-loaded microspheres, high level LacZ expression was observed in numerous organs in mice (including spleen, liver, lung, kidney, and lymph node) and was not significantly inhibited by the presence of neutralizing anti-adenovirus antibodies. By contrast, administration of un-encapsulated control vector was significantly reduced in animals harboring neutralizing antibodies (Sailaja et al., 2002).
In summary, direct chemical conjugation of protective polymers to AAV shows promise, though with somewhat mixed results, and future work may investigate the application of alternate polymers and controlled release strategies that have shown promise with adenoviral and other vectors.
Genetic Modification of AAV Capsid Proteins to Avoid Antibody Neutralization
As suggested by the term “serotype,” AAV variants with unique serological characteristics have evolved in nature (Gao et al., 2004). Variations in the amino acid composition of the capsid proteins, in particular at protruding sites, can lead to low serum cross-reactivity between serotypes (Lochrie et al., 2006). For example, mice immunized with AAV2 show only a slight reduction in transduction efficacy if subsequently injected with AAV1 (which differs from AAV2 at 16.3% of the capsid amino acids; Gao et al., 2004), while anti-AAV2 antibodies impair re-application of AAV2 (Xiao et al., 1999). In principle, such findings raise the idea that one could harness natural AAV diversity to overcome problems of pre-existing immunity. That is, identification of one or more serotypes to which specific patients have not previously been exposed could allow recombinant vector administration or even re-administration. This idea suffers from several problems, however. First, such personalized therapy poses practical and regulatory concerns. In addition, capsid variability among serotypes affects not only antigenicity but also tropism, so that different serotypes could not be readily alternated. Furthermore, the patterns of pre-existing AAV immunity within the human population are complex (Boutin et al., 2010). As already mentioned, sera are frequently found to neutralize more than one serotype, and for example in >93% of sera positive for AAV2, neutralizing antibodies toward AAV1 are additionally present. Interestingly, antibodies against these two serotypes were also detectable in sera positive for AAV5, 6, 8, or 9 (Boutin et al., 2010). However, the observation that different serotypes exhibit different extents of cross-reactivity raises the idea that a given capsid can be engineered or mutated to retain the natural tropism of, yet evade pre-existing antibodies directed against, its parental serotype. Both rational design and directed evolution approaches have been pursued toward this goal.
Peptide scanning for immunogenic epitopes
The rational design of AAV variants that evade pre-existing humoral immunity necessitates basic knowledge of immunogenic epitopes. In an early study, Moskalenko et al. (2000) utilized peptide scanning to map neutralizing epitopes for antibodies present in human serum samples and found a total of six linear epitopes that are targets of neutralizing antibodies (Table 1). Wobus et al. (2000) mapped additional linear and conformational immunogenic epitopes neutralized by mouse monoclonal antibodies. Linear epitopes in the VP1 unique region, in VP1/VP2, and at the C′-terminus of the VP3 region are recognized by antibodies A1, A69, and B1, respectively (Table 1), while conformational epitopes were bound by C24-B, C37-B, D3, and A20 (Table 2). In order to map the latter, Wobus and colleagues used peptide insertion mutants of AAV2 (Girod et al., 1999) that displayed an integrin binding ligand, L14, at defined and surface exposed positions of the capsid (Wobus et al., 2000).
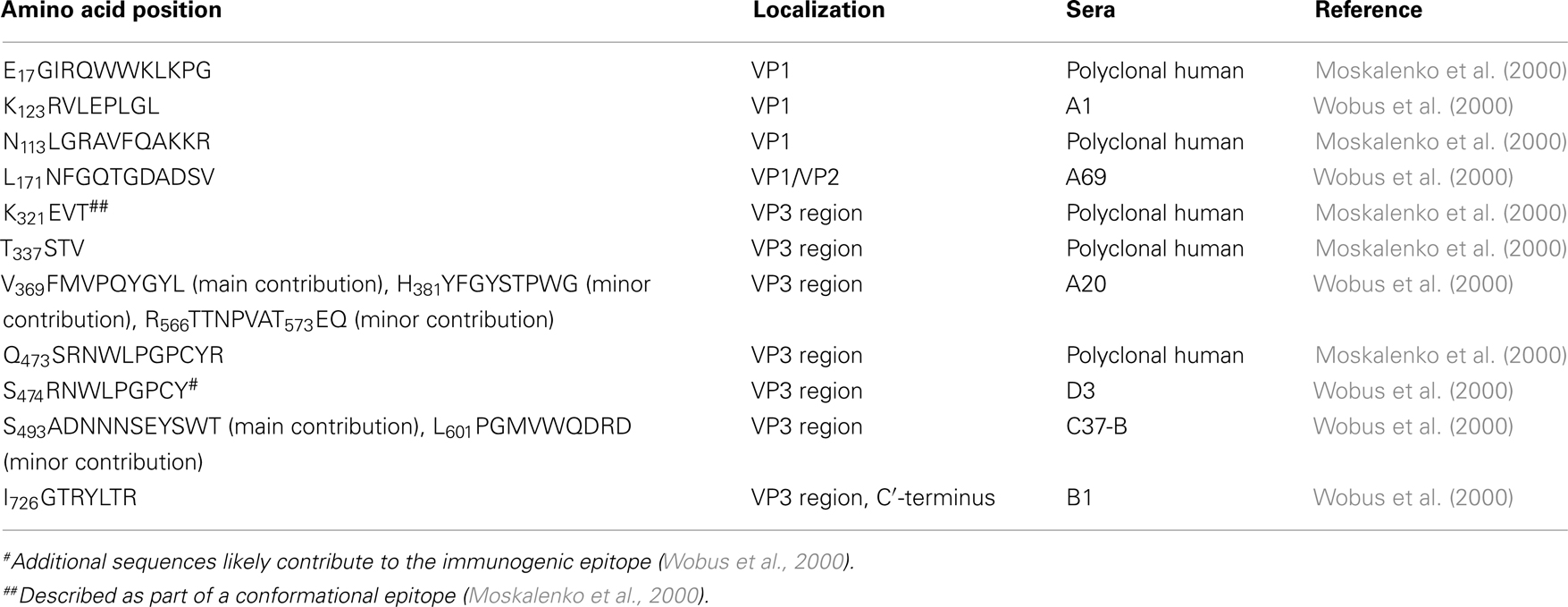
Table 1. Immunogenic sites of the AAV2 capsid mapped by peptide scan.
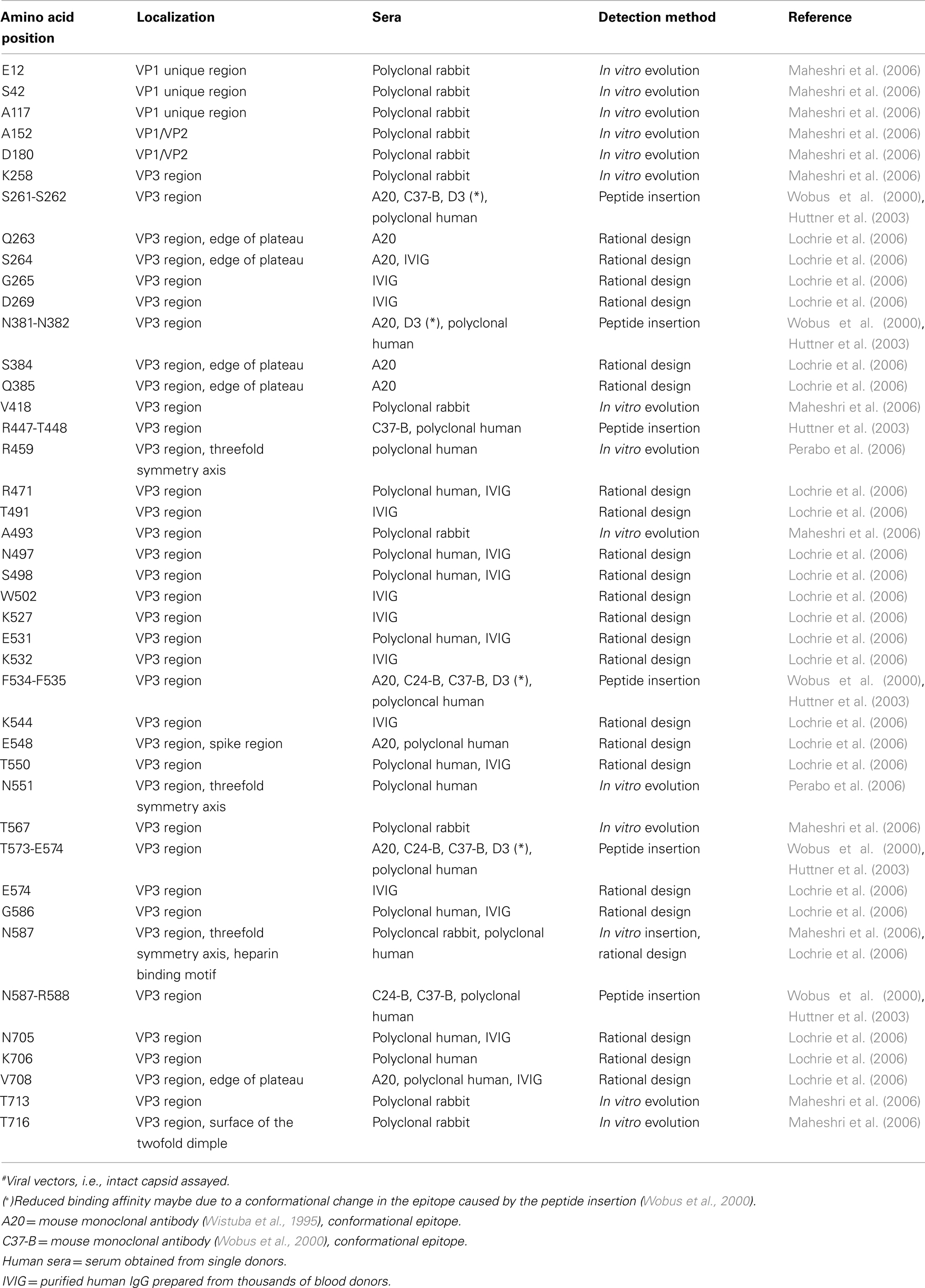
Table 2. Immunogenic sites at the AAV2 capsid#.
The same mutants were subsequently screened for their immune escape phenotype by Huttner et al. (2003) using a panel of human polyclonal sera. Forty-two percentage of the sera showed a significant reduction (∼31%) in antibody binding affinity when the capsid position 534 or 573 were subjected to peptide insertion, while 21% of the sera additionally showed a reduced binding affinity for the other six mutants (Table 2). These findings for polyclonal human sera agreed with the results of Wobus et al. (2000) obtained with mouse monoclonal antibodies and identified positions 534 and 573 as major antigenic determinants in humans. Peptide insertion not only impacts antibody binding affinity, but also the transduction ability. Further investigations by Huttner et al. (2003) revealed that the peptide insertion mutant I-587, which displayed the L14 peptide at position 587 as ligand for targeting B16F10 cells, transduced this cell line despite the presence of neutralizing antibodies. Exchanging the peptide for a 7-mer peptide targeting Mec1 cells (Perabo et al., 2003) yielded similar results, demonstrating that insertion of peptides at 587 can modulate both cell tropism and antibody neutralization (Huttner et al., 2003).
Rational design of AAV variants via peptide insertion or site-directed mutagenesis
Lochrie et al. (2006) utilized extensive site-directed mutagenesis of the AAV2 capsid to develop variants with immune escaping properties. In silico structural analysis of potential docking for a murine IgG2a antibody with the AAV2 surface yielded a number of sterically accessible, candidate positions that were then subjected to point mutagenesis. The resulting collection of mutants were assessed for antibody binding using A20 (mouse anti-AAV2 capsid antibody), human sera (three different donors), and IVIG (pooled human IgG isolated from thousands of blood donors). As may be anticipated for polyclonal antibody mixtures, in contrast to work with A20 that identified a set of variants with mutations at a specific epitope, analysis with human sera (single donors) and IVIG did not map a single epitope. At any rate, point mutations at distinct sites spread across the capsid reduced immune neutralization.
Directed evolution of AAV variants via random mutagenesis
In contrast to rational design based approaches, directed evolution strategies can be exploited in the absence of knowledge on the capsid biology or on immunogenic sites. This strategy is based on the generation and high-throughput selection of diverse genetic libraries to create variants with enhanced biological functions (Neylon, 2004). Maheshri et al. (2006) and Perabo et al. (2006) applied this technology to AAV to overcome the obstacle of pre-existing immunity. Both approaches used error-prone mutagenesis to randomize the AAV2 capsid. Random point mutations to the AAV cap ORF are introduced using a “sloppy” polymerase chain reaction (PCR) to amplify and mutate the cap DNA sequence at a defined rate. The PCR conditions can be adjusted to tune the average number of mutations in each cap gene sequence (Figure 1A; Pritchard et al., 2005). Viral capsid mutants are then packaged by standard production protocols and screened for infectivity despite the presence of neutralizing antibodies. Progeny production is induced by helper virus co-infection and followed by a new round of selection.
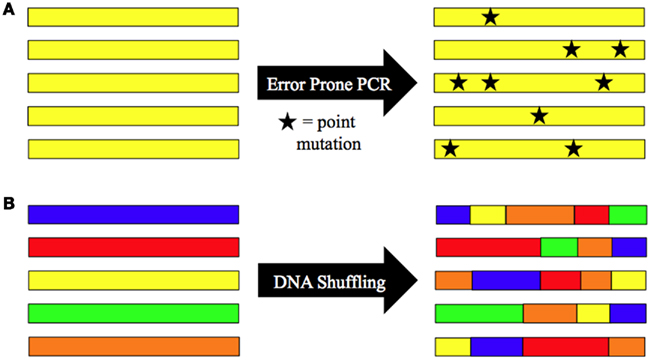
Figure 1. Schematic of library AAV capsid protein engineering strategies for evasion of antibody neutralization. Methods for generation of highly diverse viral libraries include (A) random point mutagenesis (error-prone PCR) and (B) in vitro recombination (DNA shuffling). Directed evolution strategies use these approaches as part of an iterative strategy to increase AAV’s ability to avoid antibody neutralization.
Perabo et al. (2006) selected in presence of human sera on a highly permissive cell line, HeLa. In contrast to selections in the presence of non-neutralizing sera, selections in the presence of neutralizing antibodies yielded viral mutants in which 73% of point mutations clustered in the same, surface exposed region. The most frequent selected clones carried mutations at capsid positions 459 and 551. Introduction of selected amino acid substitution at position 459 and/or 551 of capsids of recombinant AAV vectors conferred the vectors with an improved ability to evade neutralization. The highest immune evasion was observed for the sera used to select the mutants, but with each of the seven different sera assayed, a notable higher serum concentration was required to halve the transduction efficacy of the mutants compared to AAV2 (up to 5.3-fold).
The viral library used by Perabo et al. (2006) possess on average 0.9 mutations per clone making it unlikely that multiple combinations of mutations are found on a single clone. The latter, however, increases the likelihood of selecting potent immune escaping variants. An efficient strategy to achieve this goal was exploited by Maheshri et al. (2006), who combined error-prone PCR mutagenesis and DNA shuffling, in their approach to establish directed evolution strategies for AAV. Random DNA recombination (Figure 1B) can be achieved using several different methods, including the staggered extension process (StEP) recombination (Zhao et al., 1998; used by Maheshri et al., 2006) and DNase I digestion followed by fragment reassembly with DNA polymerase (Stemmer, 1994; used by Grimm et al., 2008; Koerber et al., 2008). The StEP consists of repeated cycles of denaturation, annealing, and short polymerase-catalyzed extension steps. The abbreviated extension step results in short fragments of DNA that can then anneal to new templates and further extend, creating genes that contain sequence information from multiple templates. DNA shuffling uses DNase I digestion to create small fragments of DNA similar to the fragments created in StEP. These fragments are then reassembled into new genes through repeated annealing cycles in the presence of DNA polymerase. Both methods can be used to create point mutations (when all cap gene templates are from the same serotype) or to generate chimeric capsids (when cap gene templates are from different serotypes).
The viral library produced by Maheshri et al. (2006) was selected for mutants that productively infect HEK293 in the presence of a strongly neutralizing rabbit anti-AAV2 serum. Interestingly, each mutant selected in this screen contained a threonine to alanine substitution at position 716, near the C-terminus of the common VP3 region. In order to further optimize the selection procedure, the viral library was subjected to a further round of mutagenesis. While transduction of AAV2 in the presence of rabbit sera was neutralized at a 1:1,500 dilution, the most promising candidate of the second screen was only mildly neutralized at a 1:2.35 serum dilution. More importantly, this mutant mediated transgene expression in vivo following pre-incubation with anti-AAV serum at levels 2–3 orders of magnitude higher than serum concentrations that neutralized AAV2 (Maheshri et al., 2006).
To date, AAV libraries based on error-prone PCR mutagenesis possess a diversity of 107–108 clones (Maersch et al., 2009). However, in order to screen every possible amino acid substitution for their immune escape properties, a diversity of 3.2 × 1017 would be required (Maersch et al., 2009). A possibility to overcome this technical limitation and identify the most effective amino acid substitution for a given position, and hence improve the antibody evasion capacity of immune escape mutants was proposed by Maersch et al. (2009). In this procedure, only regions implicated as immunogenic sites are randomized, and these mutants are subjected to high-throughput selections for viral infectivity in the presence of neutralizing sera. In their proof-of-principle study, Märsch and colleagues focused on five immunogenic sites: positions 449, 558, 459, 551, and 493 (Maheshri et al., 2006; Perabo et al., 2006; Maersch et al., 2009), which were subjected to saturation mutagenesis. Due to this restriction, the library purportedly contained all possible amino acid substitutions for these positions (required diversity 205, obtained diversity 6 × 106). From the mutants selected in the presence of human sera on HEK293 cells, six mutants were analyzed in comparison to AAV1, AAV2, and AAV2 R459K/N551D, a double mutant selected in the previous study of Perabo et al. (2006). Two of these mutants were significantly less neutralized by human sera from single donors and by IVIG compared to AAV2 and AAV2 R459K/N551D. In agreement with the initial hypothesis that the level of immune evasion can be improved if an optimal amino acid substitution is introduced to disrupt an immunogenic epitope, both mutants contained novel substitutions at positions 459, 493, and 551. In addition, the relatively minor sequence changes – compared for example to the 120 amino acid differences between AAV2 and AAV1 – likely did not change the mutant’s tropism relative to AAV2.
Directed evolution of AAV variants via serotype shuffling
Grimm et al. (2008) implemented an in vitro evolution strategy that utilized a library of chimeric capsids. DNA shuffling through fragment reassembly was exploited to randomly combine cap sequences of eight AAV serotypes [AAV2, 4, 5, 8, and 9, and caprine (CAAV), avain (AAAV), and bovine AAV (BAAV)]. The library was firstly selected for the ability to transduce human hepatoma cell lines, followed by further selection in the presence of IVIG. The result was a single mutant, AAV-DJ, a chimera of AAV2, 8, and 9 in which the majority of non-AAV2 amino acid sequences were found in immunogenic epitopes of the capsid. For in depth analysis of the immune profile of AAV-DJ, Grimm et al. (2008) passively immunized mice with IVIG followed by vector injection. At lower IVIG levels, AAV-DJ possessed an in vivo immune evasion capacity comparable to AAV8 and AAV9 as indicated by the comparable level of transgene expression, though both serotypes clearly outperformed AAV-DJ at a higher IVIG concentration. Interestingly, although re-administration of AAV-DJ was impaired, and although mice pre-immunized with AAV8 or 9 could not be transduced by AAV-DJ, AAV2, 8, and 9 were able to mediate transgene expression in AAV-DJ pre-immunized mice.
In another study involving DNA shuffling of AAV, Koerber et al. (2008) performed a detailed analysis of seven chimeric AAV vectors, which had been selected in the absence of antibodies in a single selection round for viability. The library used for their selection contained DNA shuffling based virus chimeras of AAV1, 2, 4, 5, 6, 8, and 9, i.e., they included additional non-human primate serotypes but not more distantly related serotypes. Sequencing of candidate chimeras revealed a greater than 90% similarity to AAV2 for three of the mutants, and to AAV1 and 6 for the other four mutants. In addition, all clones contained a high proportion of sequences originating from non-AAV2 and non-AAV1/6 serotypes, respectively, in the surface exposed regions of the capsid. Of note, four of these seven mutants showed, in the absence of a selection pressure, a naturally greater resistance to neutralization by IVIG than the parental serotypes. The highest immune evasion capability was detected for a mutant with a greater than 90% similarity to AAV1/6. This mutant, cB4, was 400-fold more resistant to neutralization by IVIG than AAV2 and is as of yet one of the most potent immune escaping AAV vectors. This work by Koerber et al. (2008) and Grimm et al. (2008) clearly reveal the great potential of chimeric virions. The immune evasion phenotype is, however, due to a new combination of cap sequences of different serotypes and thus requires the selection of chimeric mutants which in addition to immune evasion possess a desired tropism in order to fully exploit this strategy to improve AAV’s in vivo application.
Conclusion
Adeno-associated virus possesses several characteristics that have contributed to its growing popularity as a gene delivery vector for clinical gene therapy applications. Unfortunately, pre-existing neutralizing antibodies against the AAV capsid and humoral immune responses following vector administration have prevented AAV from reaching its full potential as a gene therapy vector. Chemical modifications that protect exposed immunogenic epitopes of the capsid and genetic modifications that mutate these epitopes have demonstrated that AAV vectors can be altered to decrease antibody neutralization, while still effectively delivering genetic material, both in vitro and in vivo. The modification methods described above can be used to isolate new AAV variants with more improved immune evasion characteristics and thus possibly stronger clinical potential.
Conflict of Interest Statement
The authors declare that this literature review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bainbridge, J. W., Smith, A. J., Barker, S. S., Robbie, S., Henderson, R., Balaggan, K., Viswanathan, A., Holder, G. E., Stockman, A., Tyler, N., Petersen-Jones, S., Bhattacharya, S. S., Thrasher, A. J., Fitzke, F. W., Carter, B. J., Rubin, G. S., Moore, A. T., and Ali, R. R. (2008). Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 358, 2231–2239.
Bartlett, J. S., Wilcher, R., and Samulski, R. J. (2000). Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol. 74, 2777–2785.
Beer, S. J., Matthews, C. B., Stein, C. S., Ross, B. D., Hilfinger, J. M., and Davidson, B. L. (1998). Poly (lactic-glycolic) acid copolymer encapsulation of recombinant adenovirus reduces immunogenicity in vivo. Gene Ther. 5, 740–746.
Bessis, N., GarciaCozar, F. J., and Boissier, M. C. (2004). Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 11, S10–S17.
Boutin, S., Monteilhet, V., Veron, P., Leborgne, C., Benveniste, O., Montus, M. F., and Masurier, C. (2010). Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 21, 704–712.
Cheng, X., Ming, X., and Croyle, M. A. (2003). PEGylated adenoviruses for gene delivery to the intestinal epithelium by the oral route. Pharm. Res. 20, 1444–1451.
Chirmule, N., Xiao, W., Truneh, A., Schnell, M. A., Hughes, J. V., Zoltick, P., and Wilson, J. M. (2000). Humoral immunity to adeno-associated virus type 2 vectors following administration to murine and nonhuman primate muscle. J. Virol. 74, 2420–2425.
Croyle, M. A., Chirmule, N., Zhang, Y., and Wilson, J. M. (2001). “Stealth” adenoviruses blunt cell-mediated and humoral immune responses against the virus and allow for significant gene expression upon readministration in the lung. J. Virol. 75, 4792–4801.
Croyle, M. A., Le, H. T., Linse, K. D., Cerullo, V., Toietta, G., Beaudet, A., and Pastore, L. (2005). PEGylated helper-dependent adenoviral vectors: highly efficient vectors with an enhanced safety profile. Gene Ther. 12, 579–587.
Croyle, M. A., Yu, Q. C., and Wilson, J. M. (2000). Development of a rapid method for the PEGylation of adenoviruses with enhanced transduction and improved stability under harsh storage conditions. Hum. Gene Ther. 11, 1713–1722.
De Geest, B., Snoeys, J., Van Linthout, S., Lievens, J., and Collen, D. (2005). Elimination of innate immune responses and liver inflammation by PEGylation of adenoviral vectors and methylprednisolone. Hum. Gene Ther. 16, 1439–1451.
Delgado, C., Francis, G. E., and Fisher, D. (1992). The uses and properties of PEG-linked proteins. Crit. Rev. Ther. Drug Carrier Syst. 9, 249–304.
Edwards, C. K. III, Martin, S. W., Seely, J., Kinstler, O., Buckel, S., Bendele, A. M., Ellen Cosenza, M., Feige, U., and Kohno, T. (2003). Design of PEGylated soluble tumor necrosis factor receptor type I (PEG sTNF-RI) for chronic inflammatory diseases. Adv. Drug Deliv. Rev. 55, 1315–1336.
Espenlaub, S., Wortmann, A., Engler, T., Corjon, S., Kochanek, S., and Kreppel, F. (2008). Reductive amination as a strategy to reduce adenovirus vector promiscuity by chemical capsid modification with large polysaccharides. J. Gene Med. 10, 1303–1314.
Eto, Y., Gao, J. Q., Sekiguchi, F., Kurachi, S., Katayama, K., Maeda, M., Kawasaki, K., Mizuguchi, H., Hayakawa, T., Tsutsumi, Y., Mayumi, T., and Nakagawa, S. (2005). PEGylated adenovirus vectors containing RGD peptides on the tip of PEG show high transduction efficiency and antibody evasion ability. J. Gene Med. 7, 604–612.
Ferrari, F. K., Samulski, T., Shenk, T., and Samulski, R. J. (1996). Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 70, 3227–3234.
Fisher, K. D., Stallwood, Y., Green, N. K., Ulbrich, K., Mautner, V., and Seymour, L. W. (2001). Seymour, Polymer-coated adenovirus permits efficient retargeting and evades neutralising antibodies. Gene Ther. 8, 341–348.
Flotte, T. R. (2004). Gene therapy progress and prospects: recombinant adeno-associated virus (rAAV) Vectors. Gene Ther. 11, 805–810.
Gao, G., Vandenberghe, L. H., Alvira, M. R., Lu, Y., Calcedo, R., Zhou, X., and Wilson, J. M. (2004). Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 78, 6381–6388.
Ge, Y., Powell, S., Van Roey, M., and McArthur, J. G. (2001). Factors influencing the development of an anti-factor IX (FIX) immune response following administration of adeno-associated virus-FIX. Blood 97, 3733–3737.
Girod, A., Ried, M., Wobus, C., Lahm, H., Leike, K., Kleinschmidt, J., Deléage, G., and Hallek, M. (1999). Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 5, 1052–1056.
Green, N. K., Morrison, J., Hale, S., Briggs, S. S., Stevenson, M., Subr, V., Ulbrich, K., Chandler, L., Mautner, V., Seymour, L. W., and Fisher, K. D. (2008). Retargeting polymer-coated adenovirus to the FGF receptor allows productive infection and mediates efficacy in a peritoneal model of human ovarian cancer. J. Gene Med. 10, 280–289.
Grimm, D., Lee, J. S., Wang, L., Desai, T., Akache, B., Storm, T. A., and Kay, M. A. (2008). In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 82, 5887–5911.
Haag, R., and Kratz, F. (2006). Polymer therapeutics: concepts and applications. Angew. Chem. Int. Ed. Engl. 45, 1198–1215.
Halbert, C. L., Metzger, M. J., Lam, S. L., and Miller, A. D. (2011). Capsid-expressing DNA in AAV vectors and its elimination by use of an oversize capsid gene for vector production. Gene Ther. 18, 411–417.
Halbert, C. L., Standaert, T. A., Wilson, C. B., and Miller, A. D. (1998). Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J. Virol. 72, 9795–9805.
Hauck, B., Murphy, S. L., Smith, P. H., Qu, G., Liu, X., Zelenaia, O., Mingozzi, F., Sommer, J. M., High, K. A., and Wright, J. F. (2008). Undetectable transcription of cap in a clinical AAV vector: implications for preformed capsid in immune responses. Mol. Ther. 17, 144–152.
Hernandez, Y. J., Wang, J., Kearns, W. G., Loiler, S., Poirier, A., and Flotte, T. R. (1999). Latent adeno-associated virus infection elicits humoral but not cell-mediated immune responses in a nonhuman primate model. J. Virol. 73, 8549–8558.
Hofherr, S. E., Mok, H., Gushiken, F. C., Lopez, J. A., and Barry, M. A. (2007). Polyethylene glycol modification of adenovirus reduces platelet activation, endothelial cell activation, and thrombocytopenia. Hum. Gene Ther. 18, 837–848.
Huttner, N. A., Girod, A., Perabo, L., Edbauer, D., Kleinschmidt, J. A., Büning, H., and Hallek, M. (2003). Genetic modifications of the adeno-associated virus type 2 capsid reduce the affinity and the neutralizing effects of human serum antibodies. Gene Ther. 10, 2139–2147.
Jang, J. H., Schaffer, D. V., and Shea, L. D. (2011). Engineering biomaterial systems to enhance viral vector gene delivery. Mol. Ther. 19, 1407–1415.
Jiang, H., Couto, L. B., Patarroyo-White, S., Liu, T., Nagy, D., Vargas, J. A., Zhou, S., Scallan, C. D., Sommer, J., Vijay, S., Mingozzi, F., High, K. A., and Pierce, G. F. (2006). Effects of transient immunosuppression on adeno-associated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood 108, 3321–3328.
Kasman, L. M., Barua, S., Lu, P., Rege, K., and Voelkel-Johnson, C. (2009). Polymer-enhanced adenoviral transduction of CAR-negative bladder cancer cells. Mol. Pharm. 6, 1612–1619.
Kim, P. H., Kim, T. I., Yockman, J. W., Kim, S. W., and Yun, C. O. (2010). The effect of surface modification of adenovirus with an arginine-grafted bioreducible polymer on transduction efficiency and immunogenicity in cancer gene therapy. Biomaterials 31, 1865–1874.
Knipe, D. M., and Howley, P. M. (ed.). (2006). Field’s Virology, 5th Edn. (Philadelphia: Lippincott Williams & Wilkins).
Koerber, J. T., Jang, J. H., and Schaffer, D. V. (2008). DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol. Ther. 16, 1703–1709.
Kreppel, F., and Kochanek, S. (2008). Modification of adenovirus gene transfer vectors with synthetic polymers: a scientific review and technical guide. Mol. Ther. 16, 16–29.
Le, H. T., Yu, Q. C., Wilson, J. M., and Croyle, M. A. (2005). Utility of PEGylated recombinant adeno-associated viruses for gene transfer. J. Control Release 108, 161–177.
Lee, G. K., Maheshri, N., Kaspar, B., and Schaffer, D. V. (2005). PEG conjugation moderately protects adeno-associated viral vectors against antibody neutralization. Biotechnol. Bioeng. 92, 24–34.
Lochrie, M. A., Tatsuno, G. P., Christie, B., McDonnell, J. W., Zhou, S., Surosky, R., Pierce, G. F., and Colosi, P. (2006). Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J. Virol. 80, 821–834.
Lu, H., Qu, G., Yang, X., Xu, R., and Xiao, W. (2011). Systemic elimination of de novo capsid protein synthesis from replication-competent AAV contamination in the liver. Hum. Gene Ther. 22, 625–632.
Lux, K., Goerlitz, N., Schlemminger, S., Perabo, L., Goldnau, D., Endell, J., Leike, K., Kofler, D. M., Finke, S., Hallek, M., and Büning, H. (2005). Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 79, 11776–11787.
Maersch, S., Huber, A., Büning, H., Hallek, M., and Perabo, L. (2009). Optimization of stealth adeno-associated virus vectors by randomization of immunogenic epitopes. Virology 397, 167–175.
Maguire, A. M., High, K. A., Auricchio, A., Wright, J. F., Pierce, E. A., Testa, F., Mingozzi, F., Bennicelli, J. L., Ying, G. S., Rossi, S., Fulton, A., Marshall, K. A., Banfi, S., Chung, D. C., Morgan, J. I., Hauck, B., Zelenaia, O., Zhu, X., Raffini, L., Coppieters, F., De Baere, E., Shindler, K. S., Volpe, N. J., Surace, E. M., Acerra, C., Lyubarsky, A., Redmond, T. M., Stone, E., Sun, J., McDonnell, J. W., Leroy, B. P., Simonelli, F., and Bennett, J. (2009). Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 374, 1597–1605.
Maguire, A. M., Simonelli, F., Pierce, E. A., Pugh EN, J. r., Mingozzi, F., Bennicelli, J., Banfi, S., Marshall, K. A., Testa, F., Surace, E. M., Rossi, S., Lyubarsky, A., Arruda, V. R., Konkle, B., Stone, E., Sun, J., Jacobs, J., Dell’Osso, L., Hertle, R., Ma, J. X., Redmond, T. M., Zhu, X., Hauck, B., Zelenaia, O., Shindler, K. S., Maguire, M. G., Wright, J. F., Volpe, N. J., McDonnell, J. W., Auricchio, A., High, K. A., and Bennett, J. (2008). Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 358, 2240–2248.
Maheshri, N., Koerber, J. T., Kaspar, B. K., and Schaffer, D. V. (2006). Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat. Biotechnol. 24, 198–204.
Manning, W. C., Zhou, S., Bland, M. P., Escobedo, J. A., and Dwarki, V. (1998). Transient immunosuppression allows transgene expression following readministration of adeno-associated viral vectors. Hum. Gene Ther. 9, 477–485.
Manno, C. S., Pierce, G. F., Arruda, V. R., Glader, B., Ragni, M., Rasko, J. J., Ozelo, M. C., Hoots, K., Blatt, P., Konkle, B., Dake, M., Kaye, R., Razavi, M., Zajko, A., Zehnder, J., Rustagi, P. K., Nakai, H., Chew, A., Leonard, D., Wright, J. F., Lessard, R. R., Sommer, J. M., Tigges, M., Sabatino, D., Luk, A., Jiang, H., Mingozzi, F., Couto, L., Ertl, H. C., High, K. A., and Kay, M. A. (2006). Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347.
Mays, L. E., and Wilson, J. M. (2009). Identification of the murine AAVrh32.33 capsid-specific CD8+ T cell epitopes. J. Gene Med. 11, 1095–1102.
McPhee, S. W., Janson, C. G., Li, C., Samulski, R. J., Camp, A. S., Francis, J., Shera, D., Lioutermann, L., Feely, M., Freese, A., and Leone, P. (2006). Immune responses to AAV in a phase i study for canavan disease. J. Gene Med. 8, 577–588.
Mok, H., Palmer, D. J., Ng, P., and Barry, M. A. (2005). Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol. Ther. 11, 66–79.
Moskalenko, M., Chen, L., van Roey, M., Donahue, B. A., Snyder, R. O., McArthur, J. G., and Patel, S. D. (2000). Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure. J. Virol. 74, 1761–1766.
Neylon, C. (2004). Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: library construction methods for directed evolution. Nucleic Acids Res. 32, 1448–1459.
Oh, I. K., Mok, H., and Park, T. G. (2006). Folate immobilized and PEGylated adenovirus for retargeting to tumor cells. Bioconjug. Chem. 17, 721–727.
O’Riordan, C. R., Lachapelle, A., Delgado, C., Parkes, V., Wadsworth, S. C., Smith, A. E., and Francis, G. E. (1999). PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum. Gene Ther. 10, 1349–1358.
Perabo, L., Büning, H., Kofler, D. M., Ried, M. U., Girod, A., Wendtner, C. M., Enssle, J., and Hallek, M. (2003). In vitro selection of viral vectors with modified tropism: the adeno-associated virus display. Mol. Ther. 8, 151–157.
Perabo, L., Endell, J., King, S., Lux, K., Goldnau, D., Hallek, M., and Büning, H. (2006). Combinatorial engineering of a gene therapy vector: directed evolution of adeno-associated virus. J. Gene Med. 8, 155–162.
Pritchard, L., Corne, D., Kell, D., Rowland, J., and Winson, M. (2005). A general model of error-prone PCR. J. Theor. Biol. 234, 497–509.
Romberg, B., Hennink, W. E., and Storm, G. (2008). Sheddable coatings for long-circulating nanoparticles. Pharm. Res. 25, 55–71.
Sailaja, G., HogenEsch, H., North, A., Hays, J., and Mittal, S. K. (2002). Encapsulation of recombinant adenovirus into alginate microspheres circumvents vector-specific immune response. Gene Ther. 9, 1722–1729.
Schaffer, D. V., Koerber, J. T., and Lim, K. I. (2008). Molecular engineering of viral gene delivery vehicles. Annu. Rev. Biomed. Eng. 10, 169–194.
Sonntag, F., Bleker, S., Leuchs, B., Fischer, R., and Kleinschmidt, J. A. (2006). Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 80, 11040–11054.
Sonntag, F., Schmidt, K., and Kleinschmidt, J. A. (2010). A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. U.S.A. 107, 10220–10225.
Stemmer, W. P. (1994). DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. U.S.A. 91, 10747–10751.
Stroes, E. S., Nierman, M. C., Meulenberg, J. J., Franssen, R., Twisk, J., Henny, C. P., Maas, M. M., Zwinderman, A. H., Ross, C., Aronica, E., High, K. A., Levi, M. M., Hayden, M. R., Kastelein, J. J., and Kuivenhoven, J. A. (2008). Intramuscular administration of AAV1-lipoprotein lipaseS447X lowers triglycerides in lipoprotein lipase-deficient patients. Arterioscler. Thromb. Vasc. Biol. 28, 2303.
Wistuba, A., Weger, S., Kern, A., and Kleinschmidt, J. A. (1995). Intermediates of adeno-associated virus type 2 assembly: identification of soluble complexes containing rep and cap proteins. J. Virol. 69, 5311–5319.
Wobus, C. E., Hügle-Dörr, B., Girod, A., Petersen, G., Hallek, M., and Kleinschmidt, J. A. (2000). Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 74, 9281–9293.
Wu, Z., Asokan, A., and Samulski, R. J. (2006). Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol. Ther. 14, 316–327.
Xiao, W., Chirmule, N., Berta, S. C., McCullough, B., Gao, G., and Wilson, J. M. (1999). Gene therapy vectors based on adeno-associated virus type 1. J. Virol. 73, 3994–4003.
Zhao, H., Giver, L., Shao, Z., Affholter, J. A., and Arnold, F. H. (1998). Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 16, 258–261.
Keywords: adeno-associated virus, viral vector, immune response, neutralizing antibodies, bioconjugation, mutagenesis, directed evolution
Citation: Bartel M, Schaffer D and Büning H (2011) Enhancing the clinical potential of AAV vectors by capsid engineering to evade pre-existing immunity. Front. Microbio. 2:204. doi: 10.3389/fmicb.2011.00204
Received: 23 August 2011; Paper pending published: 31 August 2011;
Accepted: 09 September 2011; Published online: 04 October 2011.
Edited by:
Roland W. Herzog, University of Florida, USACopyright: © 2011 Bartel, Schaffer and Büning. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: David Schaffer, Departments of Chemical and Biomolecular Engineering, Bioengineering, The Helen Wills Neuroscience Institute, University of California at Berkeley, 176 Stanley Hall, Berkeley, CA 94720-3220, USA. e-mail:c2NoYWZmZXJAYmVya2VsZXkuZWR1;
Hildegard Büning, Department I of Internal Medicine, Center for Molecular Medicine Cologne (ZMMK), ZMMK Research Building, University of Cologne, Robert-Koch-Str. 21, 50931 Cologne, Germany. e-mail:aGlsZGVnYXJkLmJ1ZW5pbmdAdWsta29lbG4uZGU=
†Joint senior authorship.