
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 01 August 2011
Sec. Cellular and Infection Microbiology - closed section
volume 2 - 2011 | https://doi.org/10.3389/fmicb.2011.00162
This article is part of the Research Topic Pseudomonas aeruginosa, Biology, Genetics, and Host-Pathogen Interactions View all 12 articles
 Jayasimha Rao1
Jayasimha Rao1 F. Heath Damron1 Marek Basler2
F. Heath Damron1 Marek Basler2 Antonio DiGiandomenico1
Antonio DiGiandomenico1 Nicholas E. Sherman1,3 Jay W. Fox1,3 John J. Mekalanos2
Nicholas E. Sherman1,3 Jay W. Fox1,3 John J. Mekalanos2 Joanna B. Goldberg1*
Joanna B. Goldberg1*Pseudomonas aeruginosa chronically infects the lungs of cystic fibrosis (CF) patients. The conditions in the CF lung appear to select for P. aeruginosa with advantageous phenotypes for chronic infection. However, the mechanisms that allow the establishment of this chronic infection have not been fully characterized. We have previously reported the transcriptional analysis of two CF isolates strains 383 and 2192. Strain 2192 is a mucoid, alginate overproducing strain whereas strain 383 is non-mucoid. Mucoid strains are associated with chronic infection of the CF lung and non-mucoid strains are the typical initially infecting isolates. To elucidate novel differences between these two strains, we employed two methods of shotgun proteomics: isobaric tags for relative and absolute quantitation (iTRAQ) and two-dimensional gel electrophoresis (2-DE). iTRAQ compares the amount of protein between samples and relies on protein abundance, while 2-DE gel electrophoresis depends on selection of separated protein spots. For both these methods, mass spectrometry was then used to identify proteins differentially expressed between the two strains. The compilation of these two proteomic methods along with Western blot analysis revealed proteins of the HSI-I operon of the type 6 secretion system, showed increased expression in 383 compared to 2192, confirming the our previous transcriptional analysis. Proteomic analysis of other proteins did not fully correlate with the transcriptome but other differentially expressed proteins are discussed. Also, differences were noted between the results obtained for the two proteomic techniques. These shotgun proteomic analyses identified proteins that had been predicted only through gene identification; we now refer to these as “proteins of unknown functions” since their existence has now been established however their functional characterization remains to be elucidated.
Pseudomonas aeruginosa is a Gram-negative bacterium that is both a ubiquitous environmental organism and an important opportunistic pathogen of humans. P. aeruginosa can cause infections in compromised individuals such as those suffering from burns, AIDS, patients undergoing treatment for cancer, or those with cystic fibrosis (CF). P. aeruginosa infection of the CF airway leads to high mortality and morbidity.
The mucoid phenotype of P. aeruginosa is one of the most extensively researched characteristics of this species (Govan and Deretic, 1996; Ramsey and Wozniak, 2005). Mucoid strains overproduce the exopolysaccharide called alginate (Figure 1). Alginate is an unbranched linear polymer of partially acetylated β-D-mannuronic acid and its C5 epimer α-L-guluronic acid. Alginate synthesis begins in the cytoplasm and ends with secretion to the extracellular milieu. The algD alginate biosynthetic operon (containing algD844KEGXLIJFalgA; Chitnis and Ohman, 1993) and gene algC (Zielinski et al., 1991) encode enzymes required for synthesis of alginate. The biosynthetic genes are controlled by the extracytoplasmic function alternative sigma factor σ22 (also known as AlgT or AlgU), which is repressed by the transmembrane anti-sigma factor MucA (Martin et al., 1993). Mutation of mucA causes unregulated σ22 to activate alginate production (Martin et al., 1993). Recently, proteolytic degradation of MucA has also been recognized as a mechanism for alginate production (Qiu et al., 2008; Damron et al., 2009b; Wood and Ohman, 2009; Damron and Yu, 2011). When σ22 is free and active, due to mucA mutation or MucA proteolytic degradation, alginate overproduction will occur. In addition to increased expression of genes, σ22 is also known to repress gene expression as well (Wu et al., 2004; Tart et al., 2005; Jones et al., 2010).
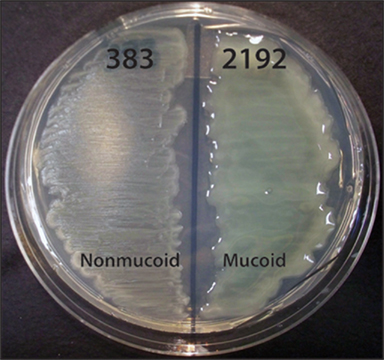
Figure 1. Phenotypes of P. aeruginosa CF isolates 383 and 2192 on L agar media. Streaks of strains 383 and 2192 were cultured on L agar for 24 h at 37°C. These two strains were isolated within 2 days apart from a CF patient. Previous data indicated these strains are isogenic (Hanna et al., 2000). The genome of strain 2192 has been sequenced1 (Mathee et al., 2008).
Ineffective clearance of P. aeruginosa in the environment in the CF lung has been suggested to result in the conversion to the mucoid, alginate over-expressing phenotype, which provides these bacteria additional protection from antibiotics and phagocytic killing (Govan and Deretic, 1996; Lyczak et al., 2002). Mucoid variants of P. aeruginosa effectively establish themselves as dominant pathogens in chronic lung diseases of CF patients (Lyczak et al., 2002). However, many other factors in addition to alginate overproduction have been described which contribute to the lung infection of CF patients (Hogardt and Heesemann, 2010).
Transcriptome analysis of P. aeruginosa via microarray has provided valuable insights into gene regulation of P. aeruginosa on a global level (Goodman and Lory, 2004). In addition, mass spectrometry provides proteomic analysis. In earlier studies with P. aeruginosa, protein identification has been performed using a qualitative approach for the identification of proteins by two-dimensional gel electrophoresis (2-DE) followed by tandem mass spectrometry (MS/MS) method (Quadroni et al., 1999; Malhotra et al., 2000; Sherman et al., 2001; Nouwens et al., 2003; Arevalo-Ferro et al., 2005; Sriramulu et al., 2005; Sharma et al., 2006). A sub-proteomic approach has led to identification and characterization of the periplasmic, membrane, or secreted P. aeruginosa proteins as potential virulence factors (Nouwens et al., 2003; Wehmhoner et al., 2003; Blonder et al., 2004; Imperi et al., 2009). Recently, P. aeruginosa gene products were identified by using collective data from proteomic, transcriptional profiling, and signature-tagged mutagenesis (Platt et al., 2008). Quantitative proteomics have been developed using isotope labeling of peptides in a method known as isobaric tags for relative and absolute quantitation (iTRAQ; Lacerda et al., 2008). Most transcriptome and proteomic analysis of P. aeruginosa have been performed on laboratory strains (Firoved et al., 2002; Wood et al., 2006; Damron et al., 2009a) along with a few studies with CF isolates (Hanna et al., 2000; Rau et al., 2010).
Our laboratory has previously performed proteomic studies of clinical isolates of P. aeruginosa strains 383 (non-mucoid) and 2192 (mucoid; Figure 1), which were isolated from sputum samples obtained from an individual with CF (Hanna et al., 2000). Using PAO1 GeneChip arrays, we determined at least 188 genes differ in expression between non-mucoid strain 383 and mucoid strain 2192 (Rao et al., 2008). Since that time, the genome of 2192 has been sequenced2 (Mathee et al., 2008); pangenome analysis has been performed and indicates niche adaptation is the force that drives the composition of P. aeruginosa genomes (Mathee et al., 2008).
Our hypothesis is that many other factors or systems, which may contribute to the disease in CF are altered in expression between mucoid and non-mucoid strains. In the present study, we have taken a global approach to detect the novel differences between the strains 383 and 2192 by using the shotgun proteomic methods of both iTRAQ and 2-DE. Total whole cell proteins were subjected to iTRAQ for quantification. In addition, total whole cell lysates and Triton X-114 soluble proteins were separated by 2-DE gel electrophoresis. Our data are consistent with the observations of an inverse relationship between the mucoid phenotype and the type 6 secretion system (T6SS). This suggests that the T6SS may be associated with non-mucoid infection strains but repressed in mucoid infection strains. Furthermore, iTRAQ and 2-DE proteomic analysis detected proteins that had only been predicted by gene identification of open reading frames (ORF) with no known homologs. We now refer to these “hypothetical” and “conserved hypothetical” proteins as “proteins of unknown function” (PUF) and determined that some are differentially expressed in the acute and chronic phenotypic strains of P. aeruginosa.
The bacterial strains used in this study, P. aeruginosa strains 383 and 2192, are non-mucoid and mucoid, respectively. These strains were isolated 2 days apart from the sputum of the same CF patient (Hanna et al., 2000). Bacterial cultures were grown overnight in Luria Bertani (LB) medium at 37°C in shaking (200 rpm), diluted to 1:100 in 30 ml of fresh media, and grown to mid-log phase, to an optical density (OD600) of 0.5.
Total proteins were extracted from strains 383 and 2192 using Tris–EDTA buffer (10 mM Tris; pH 8.3, 5 mM EDTA). Briefly, 10 ml of each bacterial culture of 0.5 (OD600) were harvested by centrifugation and cell pellets were resuspended in 1.0 ml of Tris–EDTA buffer containing 50 μl of cocktail protease inhibitors (Sigma-Aldrich, St. Louis, MO, USA), and 1 μl (5000 units) of benzonase (Sigma). The cells were disrupted by sonication on ice four to five times for 30 s with a micro-tip probe, then incubated at room temperature for 1 h, followed by centrifugation at 6000 × g for 15 min at 4°C. Supernatant solution was transferred to a microfuge tube and the protein concentration was estimated by Bradford method using Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA, USA). Approximately 1 mg of protein was precipitated with 10% trichloroacetic acid (TCA) at 4°C for 1 h and centrifuged at 20,800 × g for 10 min, followed by washing three times with cold acetone. Analytical replicate samples from each strain were used for iTRAQ labeling [Applied Biosystems Inc. (ABI), Foster City, CA, USA].
One analytical separation was performed with replicate samples of each strain (383 and 2192). Samples were reduced, digested, labeled, and combined per ABI’s standard protocol. Pooled samples were subjected to MS/MS for peptide/protein identification after strong cation exchange (SCX) chromatographic fractionation. iTRAQ reagents are isobaric in nature and thus yield similar m/z for the same peptide for the first stage of MS, when labeled with either 114, 115, 116, or 117 tags. Briefly, 100 μg of proteins from each strain were resuspended in dissolution buffer (0.5 M triethyl ammonium bicarbonate, TEAB), and then reduced, alkylated, digested, and labeled according to ABI’s standard protocol. Briefly, 20 μl of dissolution buffer, 1 μl of denaturing solution (2% SDS), and 2 μl of the reducing agent (Tris 2-carboxyethyl phosphine) were added to each protein-containing tube and incubated for 1 h at 60°C. At the end of this reduction process, proteins were brought to room temperature and the samples were alkylated by addition of 1 μl of cysteine blocking reagent [200 mM methyl methanethiosulfonate (MTT) in isopropanol] and incubation for 10 min at room temperature. The reduced and alkylated protein samples were digested with sequencing-grade modified porcine trypsin that was reconstituted with Milli Q water; 5 mg trypsin was added to each tube and incubated overnight at 37°C. The peptide samples were labeled as follows: 383-A peptides were labeled with 114, 2192-A with 115, 383-B with 116, and 2192-B with 117. Each labeling was done in 70 μl of ethanol at room temperature for 1 h (according to manufacturer’s instructions). Equivalent aliquots (77 μg) of each labeled peptide solution were pooled in a fresh tube and then dried completely in a vacuum centrifuge. These pooled, tryptic-labeled peptide samples were desalted as per manufacturer’s protocol prior to SCX chromatographic fractionation. The entire sample was reconstituted in buffer A [10 mM KH2PO4, pH 2.93, 25% acetonitrile (ACN)] and loaded onto a Pharmacia Mono S, 5 mm × 50 mm (Pharmacia, Uppsala, Sweden) column, and peptides were eluted with a linear gradient of 0–100% buffer B (10 mM KH2PO4, pH 2.73, 25% ACN, and 1 M KCl) over 20 min. For the MS/MS analysis, four fractions (8–11) were selected based on the UV peptide elution profile recorded at absorbance of OD280, and dried samples were stored at −80°C (Figure S1 in Supplementary Material).
Each SCX fraction was reconstituted in loading buffer (98:2, water/ACN, 0.1% acetic acid) and peptides were separated by reverse phased chromatography. Two microliter of each fraction were loaded onto an in-house LC column packed with 7.5 cm of Gemini 5 μm packing material; the column was washed for 10 min with 1% acetic acid, and the sample was introduced into a QStar mass spectrometer (ABI) via a Tempo nano-LC pumping system. The LC gradient moved from 98% solvent A (0.1 M acetic acid) to 80% solvent B (a mixture of 90% ACN, 10% solvent A) over 120 min, with a flow rate of 250 nL/min. The QStar Elite was run with 3300 V electrospray voltage and a curtain gas setting of 17. The scans were set to collect one MS scan followed by the product ion scans of the three most intense ions from the MS scan.
During MS/MS analysis, peptides are fragmented with appropriate collision energy, and generate a product that corresponded to reporter ions from 114, 115, 116, and 117 m/z and peptide fragment ions, from which amino acid sequences were deduced. This MS/MS spectrum provides the data for protein identification and quantification. The resulting data were analyzed using ProteinPilot software 4.0 (ABI) with the Paragon search engine to search the Pseudomonas protein database of strain PAO13. The default search settings used for quantitative processing and protein identification allowed for trypsin cleavage sites lysine and arginine, and the cysteine residues were blocked and alkylated with methyl methanethiosulfonate as described in the iTRAQ protocol. Protein detection threshold was set at 95% peptide confidence level. All four of the data files were concatenated into a single file for analysis and quantitation. Peptide quantification was carried out using ProteinPilot, which computes the area under the peaks at 114, 115, 116, and 117 Da, corresponding to the four iTRAQ reagents. The ratio between proteins was calculated by using each dataset as a denominator and shown in File S1 in Supplementary Material.
For 2-DE proteome analysis, total cellular proteins were extracted from strains 383 and 2192 by using Chaotropic Membrane Extraction Reagent 2 (Sigma; 5.0 M Urea, 2.0 M thiourea 40 mM Trizma™ base, 2.0% CHAPS, and 2.0% SB3-10, pH 10.1), followed by reduction and alkylation procedures, as previously described (Herbert et al., 1998; Molloy et al., 1998). Briefly, 10 ml of 0.5 (OD600) cultures were harvested by centrifugation and cell pellets were resuspended in 2.0 ml of Chaotropic Membrane Extraction Reagent 2 containing 100 μl of protease inhibitors (Sigma), 1 μl of benzonase (Sigma), and 80 μl of tributylphosphine (TBP; Sigma) to get a final concentration of 5 mM in the total volume. Cell suspensions were disrupted by sonication with a micro-tip probe for 4–5 min on ice, followed by centrifugation at 6000 × g for 15 min at 4°C for supernatant collection, and removal of unbroken cells. Further, the reduced proteins were alkylated by adding 23 μl of iodoacetamide (Sigma) to a final concentration of 15 mM. This mixture was incubated for 90 min at room temperature in the dark. Protein concentration was determined by the Bradford method, and sample aliquots were stored at −20°C until use. Triton X-114 extractions were performed as described previously (Bordier, 1981), with slight modifications. Two ml bacterial pellets from 0.5 OD600 cultures were collected from strains 383 and 2192. Proteins were extracted by solubilizing in 2 ml of 1.7% Triton X-114 (Sigma) in 1× phosphate buffered saline (pH 7.4; PBS) with 100 μl of protease inhibitors (Sigma) by gentle rocking at 4°C for 2 h. Insoluble cell debris was removed by centrifugation at 6000 × g for 15 min at 4°C and the Triton X-114 solubilized proteins were collected by transferring supernatants to fresh microcentrifuge tubes, adding PBS to reduce the concentration of Triton X-114 to 1%, and incubating at 30°C for 3 min. The resulting cloudy solutions were partitioned into aqueous and detergent phases by centrifugation at 300 × g for 3 min at room temperature. The upper aqueous phase was transferred to a fresh tube and re-extracted three times following the above procedure. The lower partitioned detergent phase was pooled from each extract and washed with cold 1× PBS. Finally, total extract, aqueous partitioned, and detergent partitioned proteins were precipitated with cold acetone (1:10, v/v) overnight at −70°C. Acetone precipitated proteins were collected after centrifugation at 20,800 × g for 15 min at 4°C and protein samples were subjected to 2-DE gel electrophoresis. The protein samples were prepared for electrophoresis in Chaotropic Membrane Extraction Reagent 2 (Sigma) by reduction and alkylation procedures performed as described earlier. Protein concentrations of each sample were determined by the Bradford method and samples were stored at −20°C until use.
All protein preparations were subjected to 2-DE analysis by using PROTEAN® IEF Cell (Bio-Rad). One hundred fifty microgram of proteins were solubilized in rehydration buffer (Bio-Rad) containing 9 M urea, 2% Triton X-100, 2% Pharmalyte pH 3–10 (Pharmacia), 2% β-mercaptoethanol, bromophenol blue, and TBP (20 mM) was added. Total proteins were applied on each immobilized pH gradient (IPG) strip (isoelectric point [pI] of 3–10 NL, 11 cm) from Bio-Rad, and active rehydration was carried out for 12–15 h under low-viscosity paraffin oil. Proteins were first separated by IEF with the following voltage/time profile: a linear step for 0–500 V for 15 min, 500–8000 V up to a total of 30,000 V/h. After completing the isoelectrofocusing (IEF), strips were incubated with equilibration buffer (3.7 g of Tris base, 150 ml of 10% SDS, 57.5 ml of glycerol, 7.71 g of DTT and 0.2 g of bromophenol blue, and made to 500 ml distilled H2O) for 15–30 min. For the second dimension, proteins were resolved on 8–16% SDS-PAGE (Criterion gels) with Dodeca cell system from Bio-Rad at 100 V. Gels were stained with silver or with Bio-Safe Coomassie (Bio-Rad). Stained gels were scanned using HP scanner and images were stored as tagged image files (TIFF) files. All the cored spots from silver and Coomassie were characterized by either peptide mass mapping (PMM) or tandem mass spectrometry (MS/MS) for protein identification.
The selected individual protein spots were cut out of the stained gels and were submitted to the W. M. Keck Biomedical Mass Spectrometry Laboratory at the University of Virginia Biomedical Research Facility for analysis, as described elsewhere (Hanna et al., 2000). Data was analyzed by using the SEQUEST (ThermoFinnigan) search algorithm against Pseudomonas genome deposited with National Center for Biotechnology Information (NCBI).
Rabbit polyclonal antibodies were raised and purified against synthetic peptides CLASAPKPKDDEPQA for TssB1 (PA0083), CVDGDPAHSTETTKA for TssC1 (PA0084), KYIDKSTPNLMMAC for Hcp1 (PA0085; GenScript, Piscataway, NJ, USA).
Pseudomonas aeruginosa strains (383 and 2192) were grown on L agar overnight at 37°C. Cells were scraped of the plates and suspended in L broth. Starting cultures were matched to an OD600 of 0.1 and then diluted 1:1 with L broth. Five milliliter of culture were rotated at 100 rpm 37°C for 3 h until the strains were at OD600 of 0.5. Cultures were streaked onto L agar to ensure non-mucoid and mucoid phenotypes were maintained during log growth. Cell suspensions were matched and diluted in 2× Laemmli buffer. Samples were frozen at −80°C until separated on 12% polyacrylamide SDS-PAGE and then transferred to 0.2 μm nitrocellulose. Blocking and probing of membranes were performed with 5% skim milk in phosphate buffered saline. Polyclonal antibodies for T6SS proteins were diluted 1:500 and monoclonal antibodies to RpoA (Neoclone). Secondary antibodies with horseradish peroxidase were diluted 1:3000. Amersham Advanced ECL chemiluminescent substrate utilized for T6SS blots and Amersham ECL was used for RpoA blots. Amersham ECL hyper film was exposed for 1 min to probed membranes and developed.
Early P. aeruginosa isolates taken from the CF airway are generally non-mucoid and resemble environmental strains. Strains 383 and 2192 have been classified as isogenic (Hanna et al., 2000). Strain 383 is non-mucoid and thus represents an initially infecting isolate, while 2192 is mucoid and represents a chronically infecting isolate. To discover the proteomic differences in these two CF isolates, iTRAQ analysis and quantitation was performed. This approach is a “gel-free” mass spectrometry technique that uses isobaric amine-specific tags to compare the peptide intensities between samples and infer quantitative values for corresponding proteins. Two analytical replicates of protein preparations from each strain were subjected to digestion and iTRAQ labeling, followed by MS/MS analyses. Expression data were compiled by scoring of peptides with 99% confidence using ProteinPilot software version 4. Proteins were identified on the basis of having at least one distinct peptide with threshold of 1% false positive rate using the ProteinPilot search algorithm against all bacterial and non-redundant database entries deposited with NCBI. In this study we report the identification of 399 proteins (above 95% confidence) from 5690 peptides. The relative quantification data were obtained for 297 proteins between strains 383 and 2192. Of the quantified 297, 81 proteins showed significant p values in either one or both iTRAQ replicates (File S1 in Supplementary Material). In comparison with earlier studies of other P. aeruginosa strains (Guina et al., 2003; Wu et al., 2005), here we have observed similar numbers differentially expressed proteins.
Isobaric tags for relative and absolute quantitation analysis of the proteomes of 383 and 2192 identified 297 proteins differentially regulated (Table S1A and File S1 in Supplementary Material). The 297 quantified proteins can be grouped into three categories: (1) 67 proteins (23%) showed higher expression in the non-mucoid strain, 383, (2) 77 proteins (26%) in showed higher expression in the mucoid strain, 2192, and (3) 153 proteins (51%) showed no difference in relative expression between the non-mucoid and mucoid strains (Table S1A in Supplementary Material).
Bioinformatics analysis of the 297 proteins recognized from iTRAQ analysis allowed functional classifications as defined by the Pseudomonas Genome Database4 (Winsor et al., 2009; Table 1 and File S2 in Supplementary Material). The distribution of proteins observed showed representatives of almost all of the functional classes identified suggest iTRAQ analysis. However, this may represent a shallow sampling of the proteome and may also define the limits of detection. No proteins corresponding to the functional classes of antibiotic resistance and susceptibility, chemotaxis, or phage proteins were detected in the analysis, but proteins assigned to the other 23 functional classes were identified. Proteins representing 16% the amino acid biosynthesis and metabolism class were observed. Interestingly most of the proteins of this class were expressed at equal levels between the two strains. We would expect that a mucoid P. aeruginosa strain, such as 2192 would have increased expression of proteins corresponding to the cell wall/LPS/capsule class. iTRAQ analysis indicated four proteins of this class were upregulated in strain 2192 (Table 1). Furthermore the mucoid phenotype would place a demand on metabolism pathways to overproduce alginate. Eight proteins of the central intermediary metabolism class were upregulated in mucoid strain 2192 (Table 1). However, the protein class of energy metabolism has an equal number of increased and decreased proteins and 13 proteins equally expressed. If iTRAQ analysis properly represents the proteome, it would be expected that some classes of proteins would not be discordant. Seven proteins of the associated with DNA maintenance were equal expressed between the two strains (Table 1).
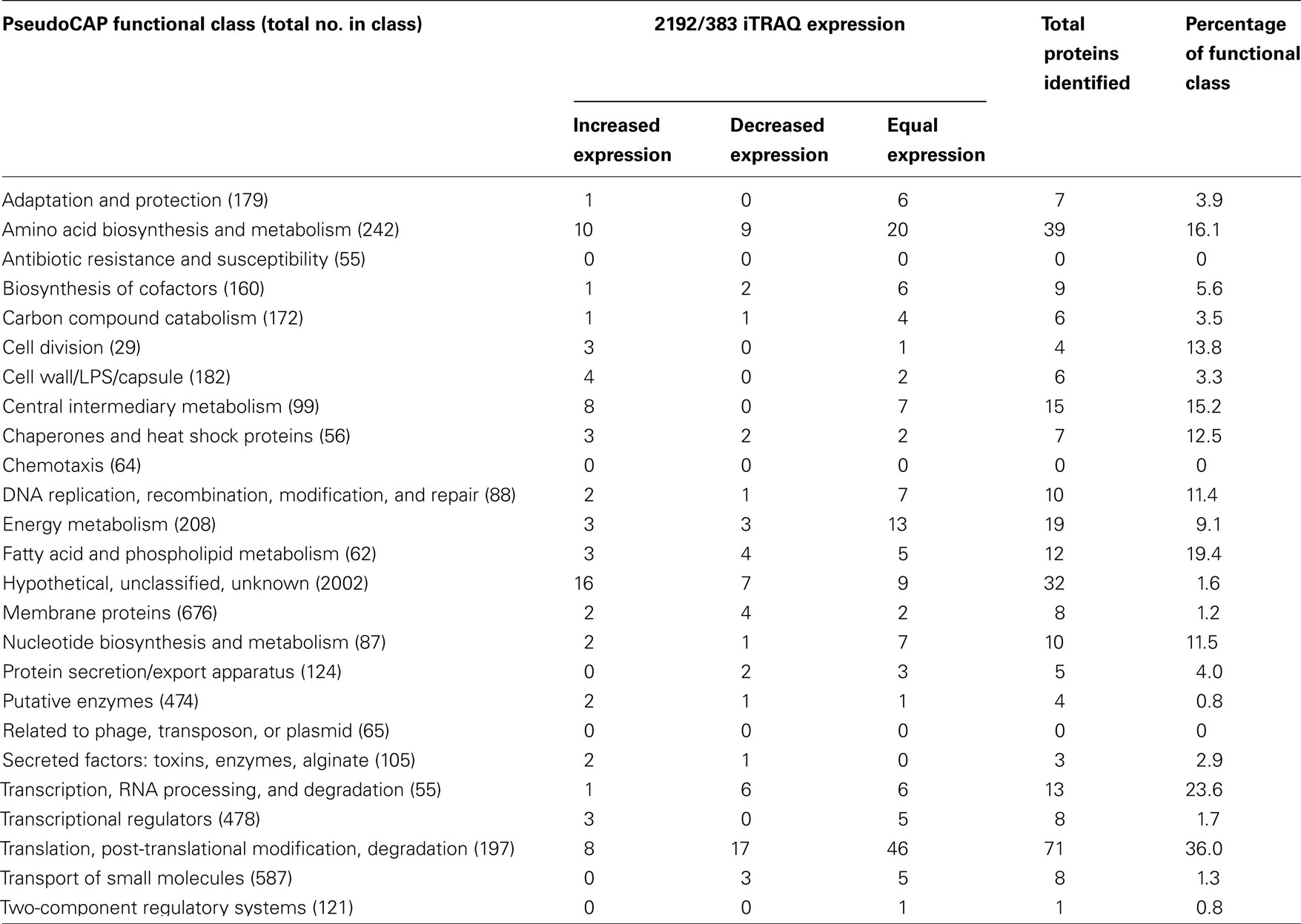
Table 1. Distribution and PseudoCAP functional classification of proteins identified in CF strains 383 and 2192 by iTRAQ analysis.
The bacterial membrane is the largest single structure of a bacterium. Moreover, homeostasis is maintained by upholding the integrity of this structure. In our iTRAQ analysis of the two CF isolates, we noted a total of 12 proteins (19.4% of the entire class), which correspond to the fatty acid and phospholipid metabolism class (Table 1). Since membrane integrity is paramount for bacteria, we would predict this class would be highly represented in a distribution of the total proteome. Another validation of the iTRAQ analysis was the identification of differential expression of transcriptional regulators and transcriptional machinery (Table 1). Transcriptional regulators are rarely expressed at high levels. Therefore, identification of these regulators via iTRAQ suggests the analysis was able to detect at least some proteins that may be of relatively low abundance. Conversely translational machinery would likely represent a high amount of the peptide concentration within a total protein lysate. iTRAQ analysis identified 36% of the PseudoCAP functional class of translation, post-translational modification and degradation. Seventy-one of the 197 proteins belonging to this class were identified. However, most (46) were equally expressed by the two strains (Table 1).
We have also compared the proteins detected by iTRAQ with the location of the genes on the chromosome. As anticipated by our microarray analysis (Rao et al., 2008), in some cases proteins expression seemed to correspond to genes organized in operons. A particularly prominent region of the chromosome from PA4239 to PA4274, which encodes most of the 50S and 30S ribosomal subunits, were identified in our iTRAQ analysis (Table S1B in Supplementary Material). In vitro growth analysis, as determined by ribosomal content, has previously suggested that P. aeruginosa isolated from chronically infected patients grow as much as three-fold slower than laboratory strains such as PAO1 (Yang et al., 2008). Here in our iTRAQ analysis, we observed similar down-regulation of ribosomal proteins in strain 2192 (Table S1A in Supplementary Material).
The high detection of ribosomal proteins in both samples likely decreases the depth of detection of the total proteome and is a limitation of the current study. In light of this, one way to increase the depth of iTRAQ analysis may be to deplete the amount of translation associated proteins in a sample. Fractionations that remove the translational machinery may result in more unique analysis of a proteome. This idea is reminiscent of the recent push to deplete rRNA from total RNA samples for RNA-seq analysis. With the majority of a total RNA sample being comprised of rRNA, removal of rRNA can provide more unique sequence analysis. Application of ribosomal depletion to total protein samples may increase the depth of analysis of a proteome and is currently being investigated in our laboratory.
Table 2 indicates selected proteins identified in iTRAQ analysis. Additional data representing the entire iTRAQ analysis is supplied in Files S1 and S2 in Supplementary Material. Since strain 2192 is mucoid, we would expect to see proteins such as those that play a role in alginate overproduction. Our proteomic analysis indicated that alginate motility regulator Z (AmrZ), AlgF, AlgC, and AlgD were upregulated in mucoid strain 2192 (Table 2). AmrZ, is a ribbon–helix–helix DNA-binding protein (Baynham et al., 2006) that can act as a transcriptional repressor (Tart et al., 2006) or enhancer of algD expression (Baynham et al., 1999). It was expected that AmrZ would be present as we had also observed its increased expression in our previous transcriptome analysis (Rao et al., 2008). AlgC is a phosphomannomutase enzyme which catalyzes the second step in the alginate pathway (Zielinski et al., 1991). AlgC converts mannose 6-phosphate into mannose 1-phosphate. Furthermore, AlgC is a phosphoglucomutase involved in LPS synthesis (Coyne et al., 1994). Here we observed AlgC significantly upregulated in strain 2192 (Table 2). AlgD, GDP-mannose 6-dehydrogenase, converts GDP-mannose into GDP-mannuronate which is the precursor of polymannuronic acid (Tatnell et al., 1994). Another alginate enzyme, AlgF was also upregulated by not identified as significant due to low number of peptide identifications (Table 2). AlgF is one of three proteins that function to acetylate O-2 and or O-3 of the mannuronic acid residues (Franklin and Ohman, 2002). Acetylation changes the physical and immunological properties of the alginate (Pier et al., 2001).
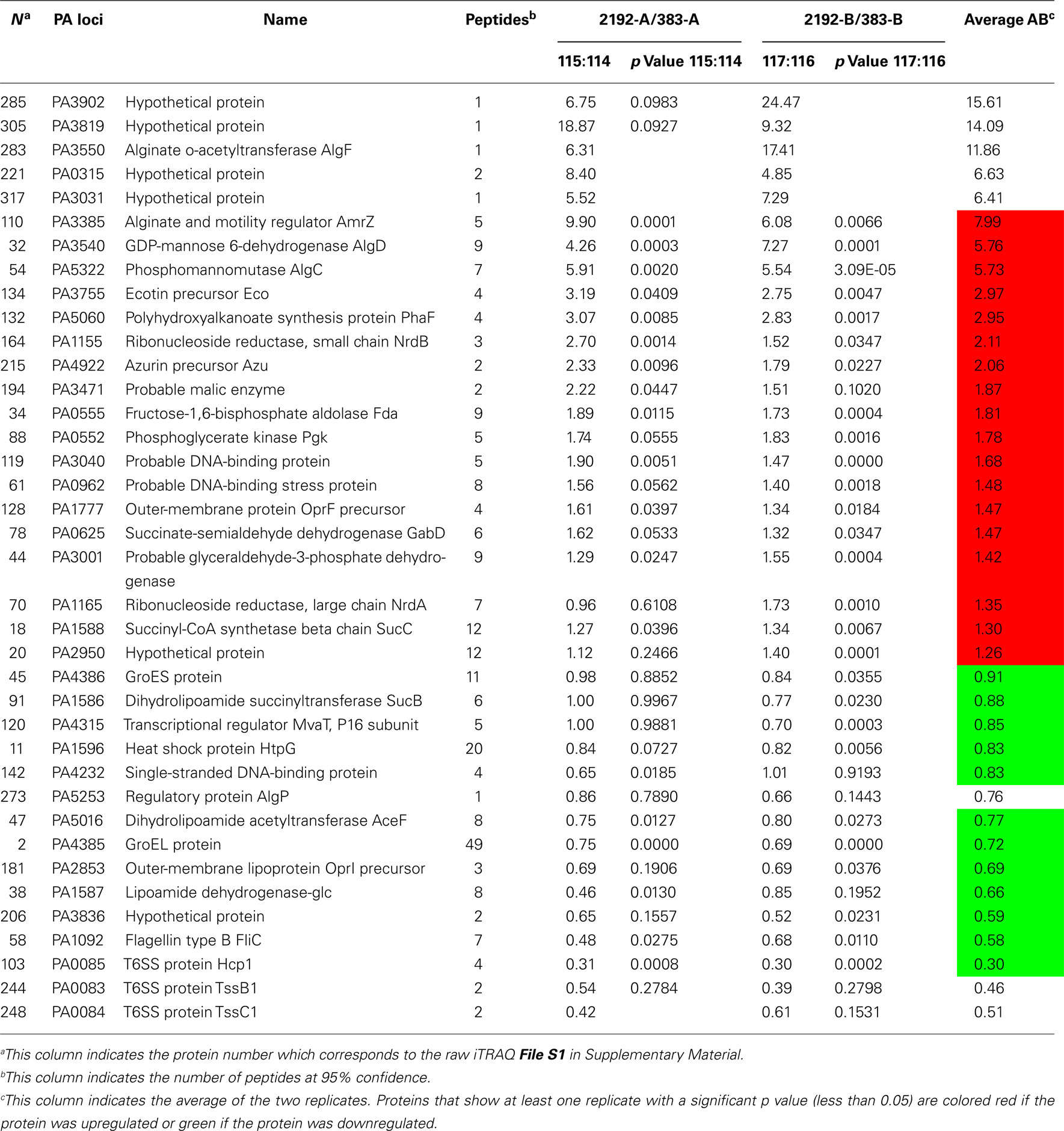
Table 2. Selected proteins with differential expression between strains 383 and 2192 as observed by iTRAQ analysis.
In addition to identifying σ22-dependent alginate production proteins, outer-membrane protein F (PA1777) was also upregulated in strain 2192 (Table 2). Previous studies have shown that OprF is upregulated in mucoid strains (Hanna et al., 2000; Malhotra et al., 2000) and is directly controlled by σ22 (Firoved et al., 2002). Thus, our shotgun proteomic analysis was validated by the identification of some known alginate biosynthesis proteins and AlgU-dependent proteins such as OprF that were upregulated in the mucoid 2192 strain.
When P. aeruginosa overproduces alginate, there would be high demands on the central metabolism to produce metabolic intermediates and such to fuel the high energy demand for alginate overproduction. In Table 2, a number of proteins are shown that have direct roles in metabolism. Additionally, we noticed a number of hypothetical proteins upregulated in 2192 some of which are shown in Table 2.
Several heat shock proteins were downregulated in strain 2192 (Table 2). GroES, HtpG, and GroEL were significantly downregulated. Previous work has shown that CF patients produce antibodies to GroEL (Ulanova et al., 1997). Other recent work has shown that heat shock proteins such as GroES and HtpG are actually downregulated in biofilms (Patrauchan et al., 2007). It is interesting that heat shock proteins may be differentially expressed depending on the growth conditions such as planktonic versus biofilm modes of growth.
Many total transcriptome analyses have examined the effect of σ22 expression (Firoved et al., 2002, 2004a,1991; Firoved and Deretic, 2003; Wood et al., 2006; Wood and Ohman, 2009). However, here unlike other studies, we were also interested in proteins that have decreased expression in mucoid strains. In our iTRAQ analysis, three proteins which are components of one (HIS-I) of the three T6SS (Mougous et al., 2006) were downregulated in the mucoid strain. These proteins, TssB1 (PA0083), TssC1 (PA0084), Hcp1 (PA0085), have greater expression levels in non-mucoid strain 383 than in mucoid strain 2192 (Table 2). Hcp1 is secreted by the T6SS and has been found in CF sputum (Mougous et al., 2006). CF patients also have antibodies in their serum to Hcp1 (Mougous et al., 2006). Although the exact role of Hcp1 is not clear, it has been suggested that Hcp1 may form a channel through which macromolecules might pass and may be used for communication with the host (Mougous et al., 2006). Recently, the HSI-I system was shown to secrete Tse2 (PA2702) which is a toxin that arrests both prokaryotes and eukaryotes; however, Tse2 is targeted toward prokaryotes (Hood et al., 2010). We do not observe expression of Tse2 in either of our proteomic analysis or differential expression in transcriptomic analysis (Rao et al., 2008); however, tse2 is present in the 2192 genome5.
RetS is a hybrid sensor kinase that controls expression of the T6SS in P. aeruginosa. Without the retS, all of the HSI-I genes are highly expressed (Goodman et al., 2004; Mougous et al., 2006; Ventre et al., 2006; Figure 2). Comparison of our previous transcriptome analysis to the transcriptome analysis of ΔretS is shown in Figure 2. It seems than non-mucoid strain 383 has increased expression of most of the HIS-I operon and the HSI-II genes hsiA2, hsiC2, and hsiF1 (Figure 2). Furthermore, as mentioned, T6SS proteins TssB1, TssC1, Hcp1 were detected in iTRAQ analysis (Table 2). Also of interest, HSI-II genes were not affected by deletion of retS, but strain 383 has increased HSI-II expression of hsiA2, hsiC2, and hsiF2 (Figure 2). These data suggest that mucoidy may suppress expression of Type 6 secretion machinery in P. aeruginosa. To further corroborate this, examination of the literature revealed that inactivation of σ22 in a mucA mutant strain caused increased expression of HSI-I and HSI-II genes (Tart et al., 2005). In a recent study, extensive transcriptome analysis of early and late CF isolates revealed that, tssB1, tssC1, and hcp1 were upregulated in non-mucoid derivatives of mucoid CF strains (Rau et al., 2010). Collectively, our data along with others suggested an inverse relationship between the T6SS and the mucoid phenotype of P. aeruginosa. It has been previously theorized that the T6SS may be used to out compete other bacteria and increase the fitness of P. aeruginosa for infection of the CF lung (Hood et al., 2010). The addition of mucoid phenotype to this model paints a picture whereby early non-mucoid isolates compete to initiate infection and then mucoid phenotype emerges, after which chronic infection can be established, negating the necessity of T6SS.
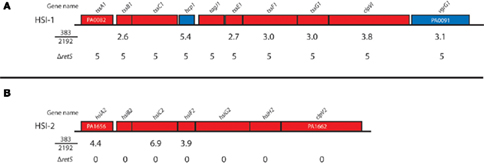
Figure 2. Type 6 secretion operons and their expression in strain 383 and 2192. The relative expression of the type 6 secretion system (T6SS) genes are indicated for the comparisons of strain 383 to 2192 from Rao et al. (2008) and also the relative expression from a retS deletion mutant where T6SS genes are uniformly upregulated (Goodman et al., 2004; Mougous et al., 2006; Ventre et al., 2006). Genes are named and colored according to Pseudomonas Genome Database. (A,B) Indicates the P. aeruginosa HSI-I and HSI-II operons of T6SS genes, respectively.
One inherent drawback to the use of chromatography-based proteomics is potential bias due to association and elution of peptides during liquid separation. Theoretically, some peptides could lost due to interactions during separation. The advantage of 2-DE analysis is that it gives the size and pI of the isolated proteins, whereas only protein fragments are detected by iTRAQ. To visualize protein differences between the non-mucoid strain 383 and the mucoid strain 2192, independent biological samples for each strain, 383 and 2192, were separated and representative examples are shown in Figure S2 in Supplementary Material. Spots with differential expression were visualized (Figure S3 in Supplementary Material) and quantified for each strain. The predicted size and pI were compared to that of the full-length protein sequences (Table S2A in Supplementary Material). A protein spot on 2-DE can contain multiple co-migrating proteins (Table S2A in Supplementary Material). To determine the proteins that make up a 2-DE spot, either PMM or mass spectral analysis or both were performed (Table S2A in Supplementary Material). PMM indicates the proteins that are the most abundant in a cored spot (Table S2B in Supplementary Material). MS/MS does not suggest a protein is the most abundant but rather identifies peptides that may correspond to the any protein in the cored spot. As expected half of the protein spots analyzed contained multiple proteins and some proteins were identified in more than one location (Table S2A in Supplementary Material).
Total protein lysates, as well as membrane, and extracellular protein enriched preparations, were utilized for 2-DE analysis. For membrane/extracellular preparations, Triton X-114 treatments as indicated in Section “Materials and Methods” were also employed to enrich for detergent-soluble hydrophobic membrane proteins (Bordier, 1981). This method has been used in other bacterial proteomic studies (Sinha et al., 2005). 2-DE analysis revealed two outer-membrane proteins, OprH and OprF were differentially expressed between strains 383 and 2192 (Figure 3A). OprH and OprF spots were visible in the Triton X-114 enrichment as expected (Figure 3A). Previously, it has been shown that OprH protein is expressed in both mucoid and non-mucoid strains (Kelly et al., 1990; Hanna et al., 2000). Here, an OprH protein spot was observed in both strains but was visually more abundant in the mucoid 2192 strain than non-mucoid strain 383 (Figure 3A) however our densitometry did not corroborate this observation (Table 3).
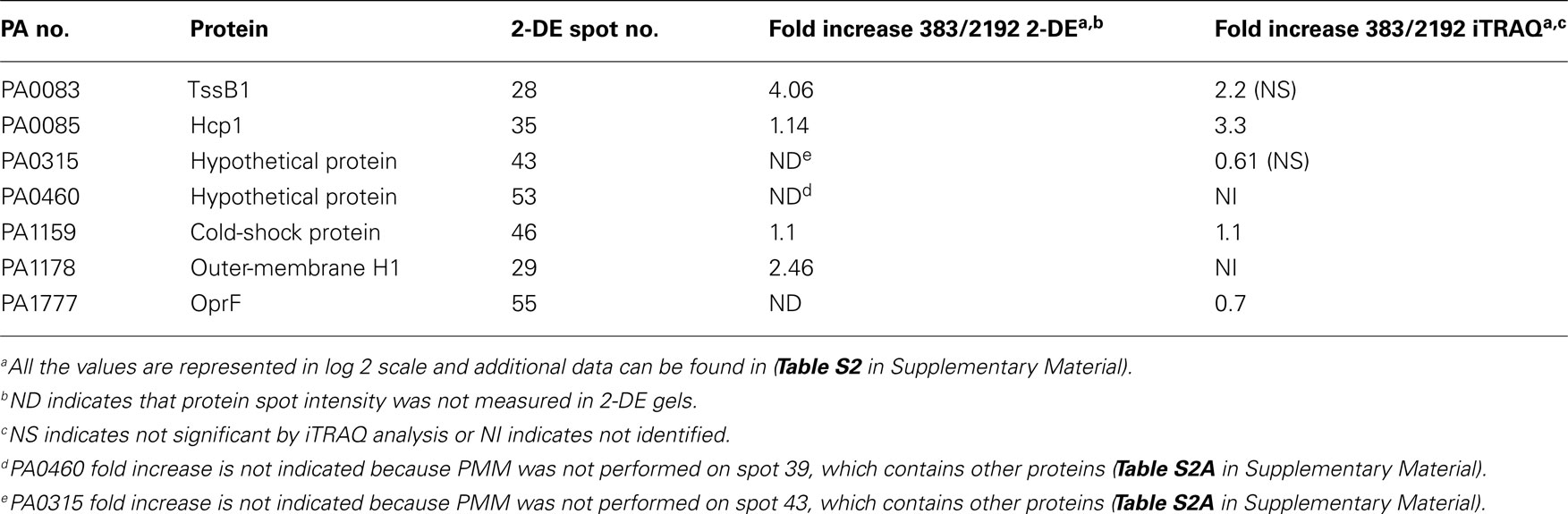
Table 3. Selected proteins quantified from cored protein spots of 2-DE.
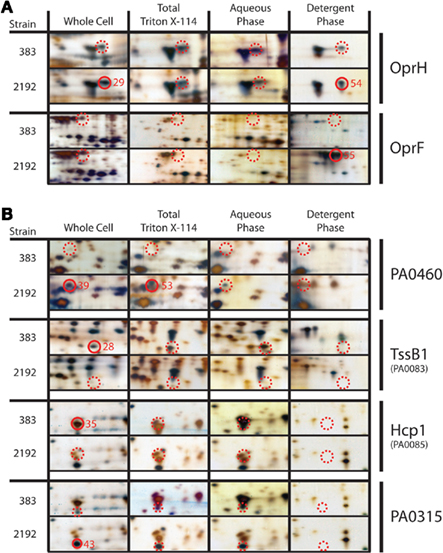
Figure 3. Two-dimensional gel protein analysis of strains 383 and 2192. (A) Indicates outer-membrane proteins that were analyzed and (B) indicates other proteins of interest. Samples are designated as follows: whole cell total soluble extract, total Triton X-114 soluble extract, aqueous phase extract and detergent phase extract. Approximately 15 μg of total protein was focused in a 3–10 (NL) IPG strips using a PROTEAN IEF cell (Bio-Rad). Second dimension, focused IPG strips were placed onto 8–16% SDS-PAGE Criterion gel and proteins were separated by molecular size. Following electrophoresis gels were silver stained and stored in distilled water with 0.5% acetic acid. Closed red circles represent cored spots with the core numbers indicated whereas dashed red circles indicate the location of spots for comparison that were not cored or MS identified. Closed and dashed red circles are indicated smaller on PA0315 panels to indicate the protein spot precisely due to a closely co-migrating spot. All cored protein spots indicated were the most abundant protein in the sample as determined by peptide mass mapping (Table S2 in Supplementary Material) except spot 43, which corresponds to PA0315.
One differential stained protein spot between mucoid strain 2192 and non-mucoid strain 383 (whole cell extract spot #39 and #53 Triton X-114 preparation) was a conserved hypothetical protein (PA0460; Figure 3B). Little to no information is available for PA0460 except that it has been shown to be five-fold upregulated when the sensor kinase phoQ is deleted from strain PAO1 (Gooderham et al., 2009). Based on our data presented here (Figure 3B), PA0460 may be a secreted protein, as it was predicted with signal peptide at N-terminal 21 amino acids followed by a cleavage site (data not shown). Spot #43 from mucoid strain (2192) whole cell extract identified PA0315, another conserved hypothetical protein (Figure 3B). PA0315 was upregulated in 2192 and our densitometry corroborated this observation. Recent data suggests PA0315 may be a homolog to a quorum sensing regulated protein of Burkholderia cepacia (Riedel et al., 2003).
Our previous transcriptome analysis had indicated a differential expression of T6SS proteins as described above. Furthermore, iTRAQ analysis revealed a significant decrease in Hcp1 in strain 2192. As expected, 2-DE gel electrophoresis confirmed our iTRAQ analysis (Table 2) that Hcp1 and TssB1 (PA0083) were increased in expression in 383 (Figure 3B and Table 3). However, more direct confirmation of this observation was necessary. For this, antibodies specific to TssB1 (PA0083), TssC1 (PA0084), and Hcp1 (PA0085) were used to quantify expression of the T6SS in CF strains 383 and 2192. In Figure 4, Western blot analysis is shown for TssB1, TssC1, and Hcp1. Antibodies specific for the alpha subunit of RNA polymerase (RpoA) were utilized as a loading control for the analysis. Western blot analysis confirmed iTRAQ and 2-DE and showed that TssB1 and Hcp1 were more highly expressed in the non-mucoid strain 383 than mucoid strain 2192 (Figure 4). Of interest antibodies for TssC1 (PA0084) suggested that TssC1 correlates with TssB1 and Hcp1 expression. These data suggest that strain 383 has increased T6SS protein expression than strain 2192 (Figure 4). Ultimately our data corroborate both the microarray and the two shotgun proteomic methods and show that the T6SS is differentially regulated in CF strains 383 and 2192.
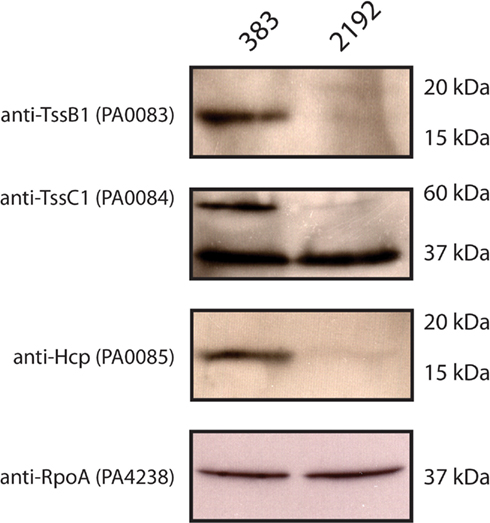
Figure 4. Western blot analysis of Type 6 secretion system (T6SS) proteins in strains 383 and 2192. L broth cultures were inoculated with the strains 383 and 2192 and grow at 37°C into log phase (OD600 O.5). Log phase whole cells suspended in L broth were added to equal volumes of 2× Laemmli buffer and subjected to separation by 12% SDS-PAGE and electro-transferred to 0.2 μm nitrocellulose membranes. Membranes were probed with the antibodies indicated. Molecular weight markers are indicated showing the relative migration of proteins detected. Shown is a representative of three independent experiments.
The goal of this study was to obtain the proteomic profiles of P. aeruginosa non-mucoid strain 383 and mucoid strain 2192 to discover novel differences. To achieve this goal, we utilized a two different complementary proteomic techniques (iTRAQ and 2-DE) followed by MS analysis and correlated these results with our previous transcriptome analysis (Rao et al., 2008). Microarray analysis elucidates the relative gene expression profiles between RNA samples; proteomic approaches such as iTRAQ and 2-DE/MS can validate microarray analysis and provide insightful information. As we expected these techniques identified proteins differentially regulated between strains 383 and 2192. However, we have also noted some limitations of these proteomic approaches. For example, proteomic techniques such as iTRAQ and 2-DE currently do not give a complete protein profile. Our iTRAQ proteomic analysis gave expression data for only 4.8% of the ORF of strain 2192 consistent with other studies. In spite of this limitation, we were able to identify novel biological differences between isolates of P. aeruginosa from a single CF patient. iTRAQ and 2-DE analysis revealed a differential expression of the T6SS in strains 383 and 2192. In non-mucoid strain 383, proteins TssB1 and Hcp1 were observed with increased spot intensities. iTRAQ quantification data also indicated an increase in TssB1 and Hcp1. iTRAQ and 2-DE results were corroborated by Western blot analysis using T6SS antibodies for TssB1, TssC1, Hcp1 (Figure 4). Most of T6SS analysis has been performed with laboratory strains. Our data suggest 383 and 2192 may be useful prototype strains to further elucidate T6SS. Collectively, our data indicates an inverse relationship between the T6SS and the chronic mucoid phenotype which is controlled by σ22.
Previously, among the 99 proteins that we identified in our proteomic analysis of PAO1, 9% were “hypothetical” and 6% were “conserved hypothetical” (Sherman et al., 2001). Furthermore, the P. aeruginosa pangenome (Mathee et al., 2008) and recent genome sequencing efforts indicate there are many ORF left to be characterized. Here among the proteins identified at least 25% were “hypothetical” or “conserved hypothetical”. We now refer to these as PUFs; since they have been confirmed by mass spectral analysis, the existence of these proteins are no longer hypothetical. This intermediate classification between hypothetical and characterized protein is important because it confirms the annotation of an organism. The P. aeruginosa reference strain, PAO1, currently contains 2002 hypothetical ORF. It is likely many of these proteins have been identified by proteomic approaches by various studies (66 were identified here) but searching the literature for each hypothetical can be difficult given the body of data in the public domain. We believe that one potential way to facilitate the elucidation of PUFs into characterized proteins is to begin database compilation of the PUFs and the studies in which they were identified.
In conclusion, iTRAQ, 2-DE, and Western blot analyses indicated differential expression of many proteins between these two CF isolates. Multiple approaches directed focus on a differential regulation of T6SS proteins between the non-mucoid strain 383 and the mucoid strain 2192. We recognized that one limitation of this approach was the use of standard laboratory growth conditions. Future studies could utilize synthetic growth media that better mimic CF conditions.
Our study suggests that iTRAQ analysis provides insightful proteomic data, but we suggest it may be possible in the future to deplete translational machinery from the protein sample to increase the depth of analysis. Since proteomic analysis by iTRAQ or 2-DE requires known protein sequences, the recent explosion of deep sequencing may quickly increase power of shotgun proteomic analysis, which may assist in the determination of the functions of many PUFs in organisms such as P. aeruginosa.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by grants from the University of Virginia Fund for Excellence in Science and Technology (FEST) Program and the National Institutes of Health (NIH; R21-AI053842) to Joanna B. Goldberg. F. Heath Damron was supported by a Cystic Fibrosis Post Doctoral Fellowship (DAMRON10F0). Marek Basler was supported by EMBO fellowship ALTF 350-2008. John J. Mekalanos was supported by R01-AI026289 from NIH.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/cellular_and_infection_microbiology/10.3389/fmicb.2011.00162/abstract
Arevalo-Ferro, C., Reil, G., Gorg, A., Eberl, L., and Riedel, K. (2005). Biofilm formation of Pseudomonas putida IsoF: the role of quorum sensing as assessed by proteomics. Syst. Appl. Microbiol. 28, 87–114.
Baynham, P. J., Brown, A. L., Hall, L. L., and Wozniak, D. J. (1999). Pseudomonas aeruginosa AlgZ, a ribbon-helix-helix DNA-binding protein, is essential for alginate synthesis and algD transcriptional activation. Mol. Microbiol. 33, 1069–1080.
Baynham, P. J., Ramsey, D. M., Gvozdyev, B. V., Cordonnier, E. M., and Wozniak, D. J. (2006). The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J. Bacteriol. 188, 132–140.
Blonder, J., Goshe, M. B., Xiao, W., Camp, D. G. II, Wingerd, M., Davis, R. W., and Smith, R. D. (2004). Global analysis of the membrane subproteome of Pseudomonas aeruginosa using liquid chromatography-tandem mass spectrometry. J. Proteome Res. 3, 434–444.
Bordier, C. (1981). Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256, 1604–1607.
Chitnis, C. E., and Ohman, D. E. (1993). Genetic analysis of the alginate biosynthetic gene cluster of Pseudomonas aeruginosa shows evidence of an operonic structure. Mol. Microbiol. 8, 583–593.
Coyne, M. J. Jr., Russell, K. S., Coyle, C. L., and Goldberg, J. B. (1994). The Pseudomonas aeruginosa algC gene encodes phosphoglucomutase, required for the synthesis of a complete lipopolysaccharide core. J. Bacteriol. 176, 3500–3507.
Damron, F. H., Napper, J., Teter, M. A., and Yu, H. D. (2009a). Lipotoxin F of Pseudomonas aeruginosa is an AlgU-dependent and alginate-independent outer membrane protein involved in resistance to oxidative stress and adhesion to A549 human lung epithelia. Microbiology 155, 1028–1038.
Damron, F. H., Qiu, D., and Yu, H. D. (2009b). The Pseudomonas aeruginosa sensor kinase KinB negatively controls alginate production through AlgW-dependent MucA proteolysis. J. Bacteriol. 191, 2285–2295.
Damron, F. H., and Yu, H. D. (2011). Pseudomonas aeruginosa MucD regulates alginate pathway through activation of MucA degradation via MucP proteolytic activity. J. Bacteriol. 193, 286–291.
Firoved, A. M., Boucher, J. C., and Deretic, V. (2002). Global genomic analysis of AlgU (sigma(E))-dependent promoters (sigmulon) in Pseudomonas aeruginosa and implications for inflammatory processes in cystic fibrosis. J. Bacteriol. 184, 1057–1064.
Firoved, A. M., and Deretic, V. (2003). Microarray analysis of global gene expression in mucoid Pseudomonas aeruginosa. J. Bacteriol. 185, 1071–1081.
Firoved, A. M., Ornatowski, W., and Deretic, V. (2004a). Microarray analysis reveals induction of lipoprotein genes in mucoid Pseudomonas aeruginosa: implications for inflammation in cystic fibrosis. Infect. Immun. 72, 5012–5018.
Firoved, A. M., Wood, S. R., Ornatowski, W., Deretic, V., and Timmins, G. S. (2004b). Microarray analysis and functional characterization of the nitrosative stress response in nonmucoid and mucoid Pseudomonas aeruginosa. J. Bacteriol. 186, 4046–4050.
Franklin, M. J., and Ohman, D. E. (2002). Mutant analysis and cellular localization of the AlgI, AlgJ, and AlgF proteins required for O acetylation of alginate in Pseudomonas aeruginosa. J. Bacteriol. 184, 3000–3007.
Gooderham, W. J., Gellatly, S. L., Sanschagrin, F., McPhee, J. B., Bains, M., Cosseau, C., Levesque, R. C., and Hancock, R. E. (2009). The sensor kinase PhoQ mediates virulence in Pseudomonas aeruginosa. Microbiology 155, 699–711.
Goodman, A. L., Kulasekara, B., Rietsch, A., Boyd, D., Smith, R. S., and Lory, S. (2004). A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 7, 745–754.
Goodman, A. L., and Lory, S. (2004). Analysis of regulatory networks in Pseudomonas aeruginosa by genomewide transcriptional profiling. Curr. Opin. Microbiol. 7, 39–44.
Govan, J. R., and Deretic, V. (1996). Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60, 539–574.
Guina, T., Wu, M., Miller, S. I., Purvine, S. O., Yi, E. C., Eng, J., Goodlett, D. R., Aebersold, R., Ernst, R. K., and Lee, K. A. (2003). Proteomic analysis of Pseudomonas aeruginosa grown under magnesium limitation. J. Am. Soc. Mass Spectrom. 14, 742–751.
Hanna, S. L., Sherman, N. E., Kinter, M. T., and Goldberg, J. B. (2000). Comparison of proteins expressed by Pseudomonas aeruginosa strains representing initial and chronic isolates from a cystic fibrosis patient: an analysis by 2-D gel electrophoresis and capillary column liquid chromatography-tandem mass spectrometry. Microbiology 146, 2495–2508.
Herbert, B. R., Molloy, M. P., Gooley, A. A., Walsh, B. J., Bryson, W. G., and Williams, K. L. (1998). Improved protein solubility in two-dimensional electrophoresis using tributyl phosphine as reducing agent. Electrophoresis 19, 845–851.
Hogardt, M., and Heesemann, J. (2010). Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int. J. Med. Microbiol. 300, 557–562.
Hood, R. D., Singh, P., Hsu, F., Guvener, T., Carl, M. A., Trinidad, R. R., Silverman, J. M., Ohlson, B. B., Hicks, K. G., Plemel, R. L., Li, M., Schwarz, S., Wang, W. Y., Merz, A. J., Goodlett, D. R., and Mougous, J. D. (2010). A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37.
Imperi, F., Ciccosanti, F., Perdomo, A. B., Tiburzi, F., Mancone, C., Alonzi, T., Ascenzi, P., Piacentini, M., Visca, P., and Fimia, G. M. (2009). Analysis of the periplasmic proteome of Pseudomonas aeruginosa, a metabolically versatile opportunistic pathogen. Proteomics 9, 1901–1915.
Jones, A. K., Fulcher, N. B., Balzer, G. J., Urbanowski, M. L., Pritchett, C. L., Schurr, M. J., Yahr, T. L., and Wolfgang, M. C. (2010). Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. J. Bacteriol. 192, 5709–5717.
Kelly, N. M., Macdonald, M. H., Martin, N., Nicas, T., and Hancock, R. E. (1990). Comparison of the outer membrane protein and lipopolysaccharide profiles of mucoid and nonmucoid Pseudomonas aeruginosa. J. Clin. Microbiol. 28, 2017–2021.
Lacerda, C. M., Xin, L., Rogers, I., and Reardon, K. F. (2008). Analysis of iTRAQ data using Mascot and Peaks quantification algorithms. Brief. Funct. Genomic. Proteomic. 7, 119–126.
Lyczak, J. B., Cannon, C. L., and Pier, G. B. (2002). Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15, 194–222.
Malhotra, S., Silo-Suh, L. A., Mathee, K., and Ohman, D. E. (2000). Proteome analysis of the effect of mucoid conversion on global protein expression in Pseudomonas aeruginosa strain PAO1 shows induction of the disulfide bond isomerase, DsbA. J. Bacteriol. 182, 6999–7006.
Martin, D. W., Schurr, M. J., Mudd, M. H., Govan, J. R., Holloway, B. W., and Deretic, V. (1993). Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 90, 8377–8381.
Mathee, K., Narasimhan, G., Valdes, C., Qiu, X., Matewish, J. M., Koehrsen, M., Rokas, A., Yandava, C. N., Engels, R., Zeng, E., Olavarietta, R., Doud, M., Smith, R. S., Montgomery, P., White, J. R., Godfrey, P. A., Kodira, C., Birren, B., Galagan, J. E., and Lory, S. (2008). Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 3100–3105.
Molloy, M. P., Herbert, B. R., Walsh, B. J., Tyler, M. I., Traini, M., Sanchez, J. C., Hochstrasser, D. F., Williams, K. L., and Gooley, A. A. (1998). Extraction of membrane proteins by differential solubilization for separation using two-dimensional gel electrophoresis. Electrophoresis 19, 837–844.
Mougous, J. D., Cuff, M. E., Raunser, S., Shen, A., Zhou, M., Gifford, C. A., Goodman, A. L., Joachimiak, G., Ordonez, C. L., Lory, S., Walz, T., Joachimiak, A., and Mekalanos, J. J. (2006). A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312, 1526–1530.
Nouwens, A. S., Beatson, S. A., Whitchurch, C. B., Walsh, B. J., Schweizer, H. P., Mattick, J. S., and Cordwell, S. J. (2003). Proteome analysis of extracellular proteins regulated by the las and rhl quorum sensing systems in Pseudomonas aeruginosa PAO1. Microbiology 149, 1311–1322.
Patrauchan, M. A., Sarkisova, S. A., and Franklin, M. J. (2007). Strain-specific proteome responses of Pseudomonas aeruginosa to biofilm-associated growth and to calcium. Microbiology 153, 3838–3851.
Pier, G. B., Coleman, F., Grout, M., Franklin, M., and Ohman, D. E. (2001). Role of alginate O acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect. Immun. 69, 1895–1901.
Platt, M. D., Schurr, M. J., Sauer, K., Vazquez, G., Kukavica-Ibrulj, I., Potvin, E., Levesque, R. C., Fedynak, A., Brinkman, F. S., Schurr, J., Hwang, S. H., Lau, G. W., Limbach, P. A., Rowe, J. J., Lieberman, M. A., Barraud, N., Webb, J., Kjelleberg, S., Hunt, D. F., and Hassett, D. J. (2008). Proteomic, microarray, and signature-tagged mutagenesis analyses of anaerobic Pseudomonas aeruginosa at pH 6.5, likely representing chronic, late-stage cystic fibrosis airway conditions. J. Bacteriol. 190, 2739–2758.
Qiu, D., Damron, F. H., Mima, T., Schweizer, H. P., and Yu, H. D. (2008). PBAD-based shuttle vectors for functional analysis of toxic and highly-regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl. Environ. Microbiol. 74, 7422–7426.
Quadroni, M., James, P., Dainese-Hatt, P., and Kertesz, M. A. (1999). Proteome mapping, mass spectrometric sequencing and reverse transcription-PCR for characterization of the sulfate starvation-induced response in Pseudomonas aeruginosa PAO1. Eur. J. Biochem. 266, 986–996.
Ramsey, D. M., and Wozniak, D. J. (2005). Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol. Microbiol. 56, 309–322.
Rao, J., Digiandomenico, A., Unger, J., Bao, Y., Polanowska-Grabowska, R. K., and Goldberg, J. B. (2008). A novel oxidized low-density lipoprotein-binding protein from Pseudomonas aeruginosa. Microbiology 154, 654–665.
Rau, M. H., Hansen, S. K., Johansen, H. K., Thomsen, L. E., Workman, C. T., Nielsen, K. F., Jelsbak, L., Hoiby, N., Yang, L., and Molin, S. (2010). Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ. Microbiol. 12, 1643–1658.
Riedel, K., Arevalo-Ferro, C., Reil, G., Gorg, A., Lottspeich, F., and Eberl, L. (2003). Analysis of the quorum-sensing regulon of the opportunistic pathogen Burkholderia cepacia H111 by proteomics. Electrophoresis 24, 740–750.
Sharma, S., Sundaram, C. S., Luthra, P. M., Singh, Y., Sirdeshmukh, R., and Gade, W. N. (2006). Role of proteins in resistance mechanism of Pseudomonas fluorescens against heavy metal induced stress with proteomics approach. J. Biotechnol. 126, 374–382.
Sherman, N. E., Stefansson, B., Fox, J. W., and Goldberg, J. B. (2001). Pseudomonas aeruginosa and a proteomic approach to bacterial pathogenesis. Dis. Markers 17, 285–293.
Sinha, S., Kosalai, K., Arora, S., Namane, A., Sharma, P., Gaikwad, A. N., Brodin, P., and Cole, S. T. (2005). Immunogenic membrane-associated proteins of Mycobacterium tuberculosis revealed by proteomics. Microbiology 151, 2411–2419.
Sriramulu, D. D., Nimtz, M., and Romling, U. (2005). Proteome analysis reveals adaptation of Pseudomonas aeruginosa to the cystic fibrosis lung environment. Proteomics 5, 3712–3721.
Tart, A. H., Blanks, M. J., and Wozniak, D. J. (2006). The AlgT-dependent transcriptional regulator AmrZ (AlgZ) inhibits flagellum biosynthesis in mucoid, nonmotile Pseudomonas aeruginosa cystic fibrosis isolates. J. Bacteriol. 188, 6483–6489.
Tart, A. H., Wolfgang, M. C., and Wozniak, D. J. (2005). The alternative sigma factor AlgT represses Pseudomonas aeruginosa flagellum biosynthesis by inhibiting expression of fleQ J. Bacteriol. 187, 7955–7962.
Tatnell, P. J., Russell, N. J., and Gacesa, P. (1994). GDP-mannose dehydrogenase is the key regulatory enzyme in alginate biosynthesis in Pseudomonas aeruginosa: evidence from metabolite studies. Microbiology 140, 1745–1754.
Ulanova, M., Petersen, T. D., Ciofu, O., Jensen, P., Hahn-Zoric, M., Hanson, L. A., and Hoiby, N. (1997). The clonal antibody response to Pseudomonas aeruginosa heat shock protein is highly diverse in cystic fibrosis patients. APMIS 105, 449–456.
Ventre, I., Goodman, A. L., Vallet-Gely, I., Vasseur, P., Soscia, C., Molin, S., Bleves, S., Lazdunski, A., Lory, S., and Filloux, A. (2006). Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc. Natl. Acad. Sci. U.S.A. 103, 171–176.
Wehmhoner, D., Haussler, S., Tummler, B., Jansch, L., Bredenbruch, F., Wehland, J., and Steinmetz, I. (2003). Inter- and intraclonal diversity of the Pseudomonas aeruginosa proteome manifests within the secretome. J. Bacteriol. 185, 5807–5814.
Winsor, G. L., Van Rossum, T., Lo, R., Khaira, B., Whiteside, M. D., Hancock, R. E., and Brinkman, F. S. (2009). Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Res. 37, D483–D488.
Wood, L. F., Leech, A. J., and Ohman, D. E. (2006). Cell wall-inhibitory antibiotics activate the alginate biosynthesis operon in Pseudomonas aeruginosa: roles of sigma (AlgT) and the AlgW and Prc proteases. Mol. Microbiol. 62, 412–426.
Wood, L. F., and Ohman, D. E. (2009). Use of cell wall stress to characterize sigma 22 (AlgT/U) activation by regulated proteolysis and its regulon in Pseudomonas aeruginosa. Mol. Microbiol. 72, 183–201.
Wu, M., Guina, T., Brittnacher, M., Nguyen, H., Eng, J., and Miller, S. I. (2005). The Pseudomonas aeruginosa proteome during anaerobic growth. J. Bacteriol. 187, 8185–8190.
Wu, W., Badrane, H., Arora, S., Baker, H. V., and Jin, S. (2004). MucA-mediated coordination of type III secretion and alginate synthesis in Pseudomonas aeruginosa. J. Bacteriol. 186, 7575–7585.
Yang, L., Haagensen, J. A., Jelsbak, L., Johansen, H. K., Sternberg, C., Hoiby, N., and Molin, S. (2008). In situ growth rates and biofilm development of Pseudomonas aeruginosa populations in chronic lung infections. J. Bacteriol. 190, 2767–2776.
Keywords: Pseudomonas aeruginosa, cystic fibrosis, alginate, iTRAQ, 2-DE, type 6 secretion
Citation: Rao J, Damron FH, Basler M, DiGiandomenico A, Sherman NE, Fox JW, Mekalanos JJ and Goldberg JB (2011) Comparisons of two proteomic analyses of non-mucoid and Mucoid Pseudomonas aeruginosa clinical isolates from a cystic fibrosis patient. Front. Microbio. 2:162. doi: 10.3389/fmicb.2011.00162
Received: 09 March 2011;
Accepted: 14 July 2011;
Published online: 01 August 2011.
Edited by:
Dara Frank, Medical College of Wisconsin, USAReviewed by:
Sam Moskowitz, Massachusetts General Hospital, USACopyright: © 2011 Rao, Damron, Basler, DiGiandomenico, Sherman, Fox, Mekalanos and Goldberg. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Joanna B. Goldberg, Department of Microbiology, University of Virginia, 7230 Jordan Hall, 1300 Jefferson Park Avenue, Charlottesville, VA 22908-0734, USA. e-mail:amJnMmJAdmlyZ2luaWEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.