 Yongjing Wang†
Yongjing Wang† Yuan Jiang
Yuan Jiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 26 March 2025
Sec. Intensive Care Medicine and Anesthesiology
Volume 12 - 2025 | https://doi.org/10.3389/fmed.2025.1559264
Delayed encephalopathy after acute carbon monoxide poisoning (DEACMP) is one of the severe complications that can occur after acute carbon monoxide poisoning (ACOP). The pathogenesis of DEACMP is complex, featuring a delitescence onset and poor prognosis. As a result, many scholars are concentrating on identifying predictors of DEACMP and evaluating their effects, including clinical characteristics, laboratory indicators, neuroelectrophysiology, imaging examination, and genetic susceptibility. However, current identified predictors lack consensus and their clinical application is limited. Therefore, we need to explore new predictors. Exosomes, the smallest extracellular vesicles (EVs) with nano-size, participate in both the physiological and pathological processes of the brain, and the changes in their content can provide valuable information for clinical diagnosis and evaluation of neurodegenerative diseases, suggesting that they may serve as a potential biomarker. However, the practicability of exosomes as biomarkers of DEACMP remains unclear. In the present review, we first introduced the pathogenesis of DEACMP and the currently identified predictors. Then, we also discussed the possibility of exosomes as the biomarkers of DEACMP, aiming to stimulate more attention and discussion on this topic, thereby providing meaningful insights for future research.
Acute carbon monoxide poisoning (ACOP) is a hypoxic disease caused by excessive carbon monoxide (CO) inhalation and is one of the most common causes of poisoning-related deaths worldwide (1). According to statistics, the global cumulative incidence rate of ACOP is 137 cases per million, and mortality is 4.6 deaths per million (2). In many countries, especially in winter, CO exposure increases due to stove heating, coal gas leak from a water heater, or inhalation during a fire, resulting in a higher incidence rate (1–4). CO is a tasteless, colorless, odorless, and nonirritating but highly toxic gas, so the patients may not realize they are poisoned CO unless they experience symptoms such as dizziness, headache, nausea, vomiting, consciousness change, or even coma (5). There have been reports that mortality has significantly declined during the last 25 years due to continued public education, enhanced efficacy of residential CO alarms, and more efficient therapeutic management of patients with CO poisoning (2). However, inhaled CO mainly binds with hemoglobin to form carboxyhemoglobin (COHb), leading to tissue hypoxia, oxidative stress, and inflammation in the heart, brain, nerves, and other tissues after ACOP (3). About 10–30% of surviving patients may develop delayed encephalopathy after acute carbon monoxide poisoning (DEACMP), with symptoms including cognitive dysfunction, personality changes, psychosis, and even Parkinsonism symptoms after a lucid interval (also known as the pseudo-recovery period) ranging from a few days up to months from the ACOP (5, 6).
So far, the pathogenesis of DEACMP is still unclear, involving multiple aspects such as ischemia and hypoxia, oxidative stress injury, reperfusion-oxygenation injury, and so on. As a result, effective treatment options for DEACMP are limited. In addition, the disability rate of DEACMP can be as high as 78%, and the case fatality rate is 31%, causing a heavy burden to families and society (7). DEACMP is characterized by neuronal apoptosis and necrosis, leading to irreversible damage to neural function. Timely identification of DEACMP can help patients receive treatment as early as possible. Scholars have attempted to identify predictors of DEACMP based on clinical characteristics, laboratory indicators, neuroelectrophysiology, imaging examination, and even genetic susceptibility. Although some valuable findings have emerged, current identified predictors lack consensus and their clinical application is limited due to specificity, feasibility, and cost. Therefore, there is a need to explore new predictors for DEACMP.
Mounting evidence indicates that exosomes play a vital role in the pathogenesis of various brain diseases, such as neurodegenerative diseases, ischemic and hemorrhagic stroke, and brain cancer (8–10). Exosomes, a subtype of extracellular vesicles (EVs), are secreted from various cells and transport proteins, lipids, and nucleic acids for intercellular communication. Brain cells can release exosomes to regulate physiological and pathologic processes in the brain (11). The contents of exosomes reflect the physiological and pathological properties of the cell of origin, suggesting that brain cell-derived exosomes may be a source of brain disease biomarkers. Notably, these exosomes can cross the blood-brain barrier (BBB) and circulate in peripheral blood and cerebrospinal fluid, allowing researchers to isolate the exosomes from harvested blood serum and cerebrospinal fluid for brain disease screening, diagnosis, prognosis, and monitoring of therapeutic efficacy. For example, many scholars suggest that peripheral blood neuronal-derived exosomes may be the potential biomarkers for Alzheimer's disease (AD) (12, 13). By comparison, reports on exosomes and DEACMP are scarce, indicating that this area has not yet gained widespread attention. The practicality of exosomes as the biomarkers of DEACMP remains unclear. Therefore, in the present article, we will first introduce the pathogenesis of DEACMP and discuss the advantages and disadvantages of the currently identified predictors. Then, we also discussed the possibility of exosomes as the biomarkers of DEACMP, aiming to stimulate more attention and discussion on this topic, thereby providing meaningful insights for future research.
DEACMP is a type of encephalopathy that occurs in patients after ACOP. After exposure to excessive CO, brain cells experience a series of changes, including ischemia and hypoxia, reperfusion-oxygenation injury, oxidative stress injury, immune-inflammatory cascade reaction, mitochondrial dysfunction, cell apoptosis and necrosis, neurotransmitter disorder, and microenvironment changes (14). It is hard to interpret the pathogenesis of DEACMP from a single perspective. At present, the following statements are mainly:
The close combination of CO and hemoglobin can reduce the oxygen-carrying capacity of red blood cells and hinder oxygen release, leading to hypoxia in the body. Notably, neurons in the central nervous system are particularly susceptible to ischemia and hypoxia. Hypoxia can induce cellular injury and death in neurons, impairing neuronal function (15). In addition to neurons, CO can damage myocardial and vascular endothelial cells, leading to reduced cardiac ejection function and increased thrombosis. These injuries worsen ischemia and hypoxia in brain tissues, which may induce or aggravate DEACMP (16).
Excessive exposure to CO can lead to mitochondrial dysfunction, inhibition of mitochondrial respiration, and increased generation of reactive oxygen species (ROS). ROS include superoxide anion radical (O2.-), hydroxyl radical (.OH), hydrogen peroxide (H2O2), singlet oxygen (1O2), and nitric oxide (NO) (17, 18). High levels of ROS can elevate oxidative stress in brain cells, especially in neurons of the CNS. Central system neurons are rich in polyunsaturated fatty acids (PUFAs), which interact with ROS and can lead to lipid peroxidation, making those neurons more susceptible to the toxic effects of ROS (17). In addition, intracellular hypoxia can lead to increased xanthine oxidase (XO) to generate a large amount of O2.-, while O2.- can react with NO to form a potentially harmful oxidant peroxynitrite, exacerbating cell injury through oxidative stress (19). NO serves as a messenger molecule and may play some role in neurotransmitter release, neural development, and synaptic plasticity. However, excessive NO may be neurotoxic and significantly harm the function of nerve cells after ACOP (20–22). Moreover, it's worth noting that when hypoxic tissues receive sufficient oxygen again (such as hyperbaric oxygen therapy), high oxygen saturation facilitates ROS production, resulting in reperfusion injury and potentially exacerbating the disease. A new research reveals that ACOP induces iron accumulation in the white matter and ferrostatin-1 can reduce iron and ROS deposition to alleviate ferroptosis (23). Ferroptosis can cause lipid peroxidation and a large amount of ROS, leading to cell death. Therefore, the role of ferroptosis in the pathogenesis of DEACMP needs more attention and investagtie in the future.
ACOP can cause inflammatory reactions and immune damage in brain tissue, resulting in increased significant levels of interleukin-4 (IL-4), IL-6, IL-13, and tumor necrosis factor- α (TNF - α) in the blood or cerebrospinal fluid (CSF) of patients after ACOP (24–26, 125). In contrast, brain tissues of CO-poisoned rats has a high interferon-gamma (IFN-γ) level (27). These inflammatory factors play a crucial role in triggering the immune response. Myelin basic protein (MBP), a major myelin protein of the CNS, is synthesized by oligodendrocytes and neuromembrane cells, and its levels can reflect the extent of damage to CNS, making it represent a specific biochemical indicator of CNS damage and acute demyelination (28). Some studies have reported that MBP levels significantly increase in the CSF of ACOP patients (29, 30). CO-mediated oxidative stress induces changes in MBP and promotes the formation of MDA-MBP adducts between MBP and the product of lipid peroxidation malondialdehyde (MDA), resulting in a triggered immunological cascade. In the brain tissue of CO-poisoned rats, lymphocytes exhibited an auto-reactive proliferative response to MBP and a significantly increased number of activated microglia (31). In particular, M1 phenotype microglia can cause neuronal damage due to excessive immune responses by secreting proteolytic enzymes, reactive oxygen species (ROS), and inflammatory cytokines (32, 33).
Scholars observed neuronal apoptosis in the cerebral cortex, parahippocampal gyrus, and other brain regions, leading to the surviving rats after ACOP exhibited clinical symptoms similar to those of DEACMP patients, such as defects in cognition, learning, and memory (34, 35). When nerve cells are exposed to CO, their mitochondrial membrane potential will decline and release cytochrome c into the cytoplasm, activating the mitochondrial apoptosis pathway (36). In addition, CO can increase intracellular caspase-8 levels to activate the death receptor apoptosis pathway (37). CO-induced apoptosis may be the reason of the decline in the number of type 1 and type 2a neural precursor cells, microglial cells, and oligodendrocyte precursor cells in rats after CO exposure. Ochi et al. suggested that this change in glial cell mount can induce the decrease in hippocampal neurogenesis, leading to cognitive impairment in a rat model of delayed CO encephalopathy (38). In addition to apoptosis, tissue hypoxia would trigger autophagy (39, 40). Autophagy may act as an endogenous protective mechanism against hypoxic/ischemic injury, contributing to endogenous neuroprotective and neuro-recovery (41, 42). CO can promote autophagy in the cytoplasm of exposed cells through the increased generation of mitochondrial ROS to activate HIF-1, p53, FOXO3, and NRF2 for triggering the transcription of BNIP3 and NIX, TIGAR, LC3, and BNIP3 and p62, respectively. Low concentrations of CO-induced autophagy can inhibit cell death and inflammation, exhibiting protective actions (43–46). However, excessive autophagy could lead to the degradation of organelles by lysosomes, resulting in brain cell damage or even cell death (47, 126). Some scholars have suggested that apoptosis and autophagy can be activated by the same upstream signal and are interconnected through various crosstalk mechanisms (48, 49), leading to the coexistence of apoptosis and autophagy in the CO-exposed brain cells. Although the mechanism of transformation and coordination remains unclear, autophagy and apoptosis play crucial roles in the pathogenesis of DEACMP.
Pathological overactivation of excitatory neurotransmission can cause excitotoxicity, leading to synaptic and neuronal degeneration. Excitotoxicity is one of the primary mechanisms of cell death in the central and peripheral nervous systems. It is now regarded that the elevated Ca2+ influx induced by glutamatergic synaptic transmission facilitates the accumulation of Ca2+ inside the cell through Ca2+-permeable (CP)-AMPA receptors (AMPARs), a critical key factor in causing excitotoxicity (50). For instance, the upregulation of CP-AMPARs triggers the Ca2+ influx excitotoxic signaling pathway, resulting in mitochondrial injury, endoplasmic reticulum (ER) stress, activation of apoptotic cascades, and even alterations in the ubiquitin-proteasome system, ultimately causing neuronal cell death. In cases of brain damage caused by hypoxia and ischemia, excitotoxicity causes neuronal cell death, contributing to neurodegeneration and detrimental neurological defects (51). In addition, Ochi et al. found that the mRNA expression of nicotinic acetylcholine receptor (nAChR). Chrna3 was significantly decreased in the hippocampal tissue, while cerebellar Chrna7 expression was significantly increased in the delayed CO encephalopathy rat model, indicating that the change of nicotinic acetylcholine receptors (nAChRs) may affect the cognitive status and play a role in the pathogenesis of DEACMP (52).
So far, scholars have identified several valuable predictors for predicting DEACMP. These predictors originate from multiple research fields, including clinical characteristics, laboratory indicators, neuro-electrophysiology, imaging examination, and genetic susceptibility (Table 1). They may assist physicians in assessing the risk of developing DEACMP.
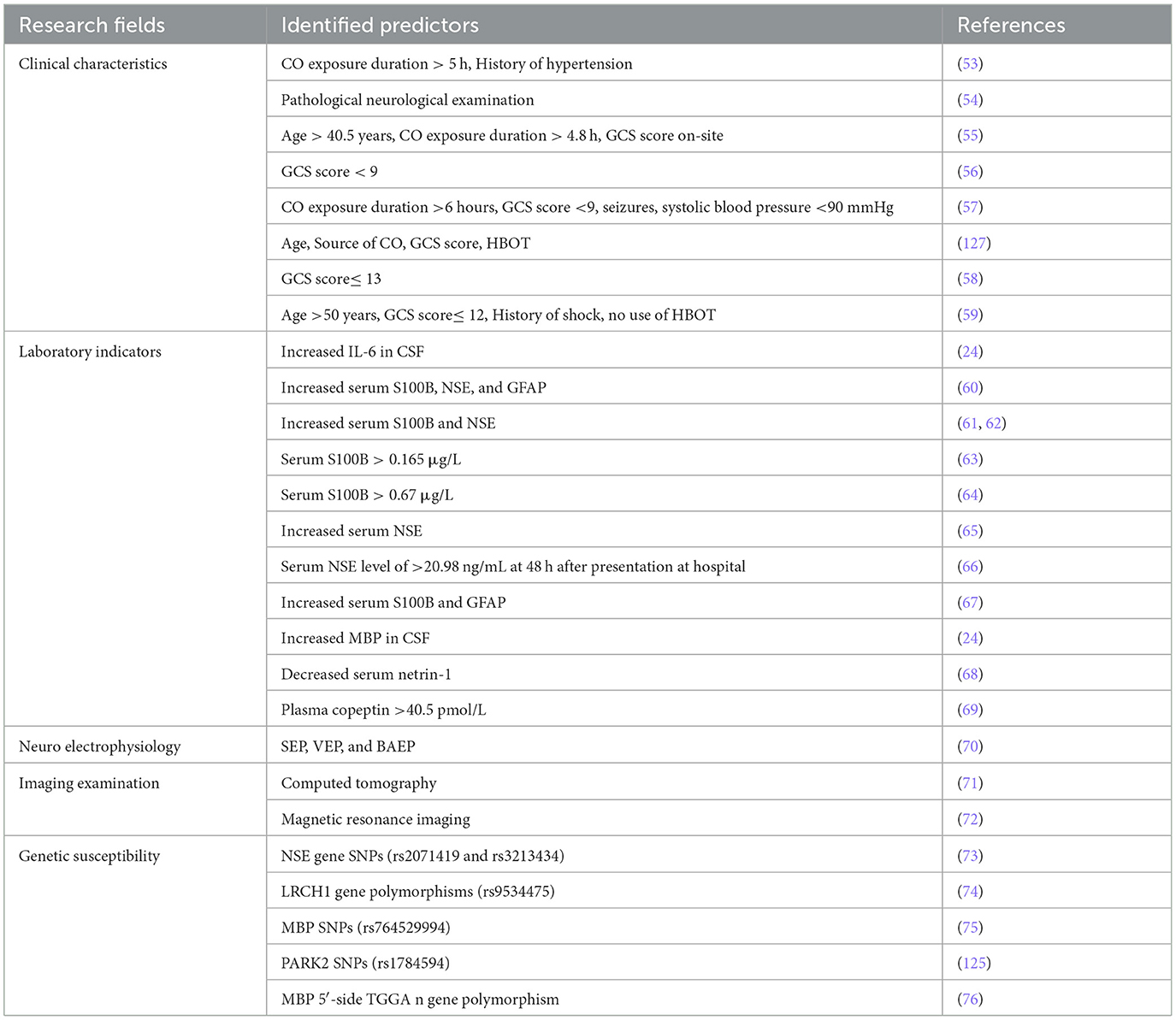
Table 1. List of the identified predictors of DEACMP.
Some retrospective analyses have identified several clinical characteristics as independent predictors of DECAMP, including age (>45 years), CO exposure duration, Glasgow coma scale (GCS) score ( ≤ 9 points), pathological neurological examination, the timing of starting hyperbaric oxygen therapy (HBOT), source of CO, and a history of hypertension and seizures [51–55; Mu et al., (127)]. Among these predictors, age, CO exposure duration, and GCS score are often mentioned in many studies. Mu et al. observed that older patients are vulnerable to DECAMP, and suggested that cardiopulmonary function declines in older patients increase the comorbidities after ACOP (127). Zhang et al. suggested that the tolerance of nerve tissue to hypoxia is weakened in the elderly population, and their gradually degraded central nervous system function will aggravate the condition of DEACMP patients (77). A longer duration of CO exposure means that inhaled CO can cause severe brain tissue hypoxia. The cerebral cortex, white matter, basal ganglia, and the globus pallidus, are the most affected areas in brain tissue. Some scholars considered that patients who are exposed to CO for >5 h have a 1.7 times higher risk of DEACMP compared to those exposed for <5 h (53). GCS score can reflect the degree of consciousness impairment in patients, especially those with traumatic brain injury (TBI). The lower scores indicate a severe coma state and a higher risk for future complications in patients. Many retrospective studies confirmed that lower GCS scores are associated with the occurrence of neurological sequelae in ACOP patients. Some scholars suggested that GCS score is a sensitive, specific, rapid, and valuable parameter in case of assessment on-site compared with that evaluated at the emergency room (55, 78). Recently, Kim et al. developed and tested a clinical scoring system (COGAS), which incorporates five factors associated with poor neurocognitive outcome: age, GCS score, serum creatine kinase (CK), hyperbaric oxygen therapy, and shock. This prediction model has demonstrated excellent discrimination performance for DECAMP and may offer significant clinical predictive value (59). However, most of the identified clinical characteristics are from retrospective studies. These studies have several limitations, such as small sample size, selection or information bias, and incomplete medical records, that can influence the sensitivity and specificity of specific clinical characteristics using logistic regression analysis, resulting in inconsistent conclusions regarding the predictive value in different studies (50, 56). Therefore, current identified clinical characteristics need validation through larger sample sizes and multicenter studies in various populations and regions.
Interleukin-6 (IL-6) is a pro-inflammatory cytokine involved in chronic inflammation and auto-immune disorders. Previous studies have confirmed significantly increased IL-6 levels in the serum and CSF of patients with ACOP. Meanwhile, the level of IL-6 in CSF may be a predictor of DECAMP rather than the serum IL-6 level (24, 26, 30). Ide et al. suggested that the changes in IL-6 levels in CSF may reflect the degree of demyelination in the cerebral white matter in the early phase of ACOP. The high IL-6 levels in CSF will return to normal, so early CSF sampling is crucial for predicting DECAMP (24). However, lumbar puncture is a relatively invasive technique for CSF sampling. Scholars need to consider its invasiveness and operational risk in the clinical application of measuring IL-6 levels in CSF.
S100B is a calcium-binding protein produced primarily by astrocytes in the CNS. A high level of S100B promotes the expression of inducible nitric oxide synthase (NOS) or pro-inflammatory cytokines, resulting in cytotoxicity to neurons in some neurodegenerative disorders (79, 80). S100B can be measured in CSF and blood and its concentration can reflect the severity of pathogenetic conditions and brain injury prognosis (60, 81). S100B in the blood of patients with ACOP also increased significantly, especially in patients with severe poisoning or suffering from DEACMP (61, 63). Previous studies have shown that S100B can reflect the early neurotoxicity of CO poisoning and is related to the degree of consciousness damage. Therefore, it can serve as a predictor of the development of DEACMP with high sensitivity and specificity (64, 82). However, some scholars suggested that the existing evidence makes it difficult to establish a clear correlation between the severity of brain disease and concentrations of S100B in CSF or serum. In addition, the extra-astrocytic and extracerebral expression of S100B involves various potential confounding factors that may induce a lack of disease specificity in predicting neurodegenerative disorders (83–85).
Neuron-specific enolase (NSE), a cell-specific isoenzyme of the glycolic enolase in nerve and neuroendocrine tissue, is involved in human glucose metabolism. NSE exhibits high activity in brain tissue and cells and is released when axons are injured. In contrast, it is lower in non-neural tissues, serum, and spinal fluid. The detection of NSE can provide a quantitative measurement of neuronal damage, reflecting the extent of primary injury to the brain and the progression of secondary damage (86). Many studies have found significantly elevated serum levels of NSE in patients after ACOP, suggesting that serum NSE could be an effective predictor of DEACMP in the early stage with better sensitivity than S100B. Notably, the detection of serum NSE enhances the prediction accuracy of initial GCS and the diagnosis accuracy for DEACMP (62, 65, 66, 87). However, scholars still are unable to confirm a clear correlation between serum NSE levels and the risk of DECAMP due to the change in serum NSE levels are also closely linked to other conditions such as neuroendocrine tumor (NET), small cell lung cancer (SCLC), melanoma and other diseases (88–90). Therefore, further large-scale studies are necessary to confirm the specificity of serum NSE as an independent factor for predicting DECAMP.
Other studies have reported several predictors for predicting DEACMP, such as glial fibrillary acid protein (GFAP), netrin-1, BMP, and plasma copeptin (67–69, 91). However, these laboratory indicators are easily influenced by the physiological and pathological conditions of the patient's body, leading to the correlation between them and DECAMP also requires further experimental discussion.
Electroencephalography (EEG) is a simple, non-invasive, and low-cost neurophysiological detection method for reflecting the functional status of the nervous system (92). Because of the persistent injury of the cerebral cortex and subcortical area, DEACMP patients' EEG may have irregular low to medium amplitude θ Wave or high amplitude δ Wave and other abnormal EEG in the pseudo-recovery period, even if they have no clinical manifestation. The abnormal rate of EEG has a positive correlation with the severity of DEACMP. By dynamically observing the EEG of patients with CO poisoning, we can gain timely insights into their brain damage and then predict whether they have DEACMP.
Brain-evoked potential (BEP) monitoring can reflect the brain function by detecting the electrical signals generated by specific stimuli (e.g., sound, light, etc.), which includes visual evoked potential (VEP), somatosensory evoked potentials (SEP), brain stem auditory evoked potentials (BAEP), etc. BAEP can sensitively evaluate the degree of brainstem functional injury. The demyelinating lesions caused by DEACMP can cause damage to brainstem function. Abnormal changes in peak and inter-peak latency of BAEP exist in patients with DEACMP. He et al. suggested that multimodality-evoked potentials based on VEP, SEP, and BAEP are sensitive indicators for evaluating brain dysfunction and predicting DEACMP in patients with ACOP (70, 93).
However, EEG or evoked potential results require expertise from professionals. Different professionals may have their perspectives on the results of multiple tests, resulting in inconsistencies in the findings. In addition, few studies have been concerned with using neurophysiological detection methods to predict DEACMP in recent years, leading to a lack of specific evaluation criteria for predicting DEACMP.
DEACMP is one uncommon subtype of acquired leukoencephalopathy with typical pathological changes, including symmetrical softening of globus pallidus, generalized or focal degeneration and necrosis of the cerebral cortex, and generalized demyelination of white matter. The low-density area of globus pallidus is the most commonly affected site of hypoxia after ACOP, especially the bilateral symmetric lesions, which are significant for diagnosing DEACMP. In addition, other affected regions, such as the hippocampus, cerebellum, and substantia nigra, also exhibit characteristic changes. Imaging examination is a crucial method for assessing the central nervous system, and scholars have found valuable information in AOCP patients' computed tomography (CT) or magnetic resonance imaging (MRI) scans.
The head computed tomography (CT) scan can reveal diffuse low-density lesions in bilateral white matter and globus pallidus, while Gray-matter—white-matter ratio (GWR) and the ratio of gray matter attenuation to white matter attenuation can be used to predict poor neurological outcomes in patients with hypoxic-ischemic encephalopathy. Wang et al. suggested that GWR-basal ganglia could serve as an indicator for predicting DEACMP with high sensitivity (93.8%) and high specificity (68.7%) (94). Du et al. have reported a brain-integrated CT score designed to identify DEACMP. The patient underwent a brain CT scan within 24 h of admission. Three experienced radiologists calculated the integrated CT score based on the characteristics of the regions of abnormal density, such as the distribution of lesions for each brain region, extent of the lesion, lesion severity, swelling of parenchyma, and others. The integrated CT score can assess the pathologic changes in the brain to semi-quantify lesion severity and may help predict the risks of developing DEACMP. Notably, the integrated CT score could be suitable for application in various medical institutions (95). However, some scholars suggest that CT's sensitivity to recognize acute ischemic lesions is relatively low, limiting its effectiveness in detecting early pathological changes of DEACMP (71).
In contrast to traditional head CT, brain MRI offers high resolution and better recognition ability for intracranial soft tissue and does not involve radiation exposure (96). Brain MRI can effectively detect the early pathological changes associated with DEACMP, such as long T1 and long T2 signals in the bilateral white matter and symmetrical globus pallidus, as well as extensive demyelination of white matter (97, 98). A recent systematic review and meta-analysis reported that brain MRI is a good predictor for DEACMP with 72% sensitivity and 80% specificity (72). Ahn et al. suggested that using MRI to detect abnormal brain damage within 72 h after ACOP patients arrive at the emergency department may assist in predicting DEACMP (71). The conventional head MRI only reflects the changes in the tissue and cell structure of the brain area and has a poor display effect of micro-structural lesions. At present, the application of diffusion-weighted imaging (DWI), diffusion tensor imaging (DTI), and diffusion kurtosis imaging (DKI) have overcome this shortcoming. DWI is a new-generation MRI based on the translational movement of water. It can detect early changes in cerebral ischemia and is sensitive to cytotoxic or vasogenic edema. The apparent diffusion coefficient (ADC) value can quantify the movement of different water molecules at the microscopic level. After ACOP, cerebral hypoxia can cause cytotoxic brain edema, which limits the movement and diffusion of water molecules, resulting in decreased ADC values and relatively increased DWI signals. Therefore, DWI can reflect the typical pathological changes of DEACMP more sensitively and earlier than conventional MRI (99). DTI can show the directionality of water molecule diffusion, and low fractional anisotropy (FA) values can sensitively reflect the slight pathologic changes from progressive demyelination in patients after ACOP, making DTI more suitable than DWI for predicting DEACMP (99). Indeed, pathological changes of DEACMP in the acute phase are too slight to observe using the conventional MRI and DWI or DTI in most cases (100), while DKI can be preferred. DKI is a straightforward extension of the DTI with greater sensitivity to detect damages to the microstructure of the brain than DWI or DTI. Even if there are no abnormal lesions in the brain white and gray matter after ACOP, the high mean kurtosis values of DKI may indicate a poor prognosis (71).
In short, brain CT and MRI all exhibited good application value in the research of DECAMP prediction (101, 102, 128). However, from the perspectives of equipment, cost, and patient compliance, medical institutions may face limitations in utilizing serial imaging examinations to predict DEACMP.
The influence of toxic substances on human DNA maps has attracted many scholars' attention. ACOP can induce acute DNA changes in the globus pallidus of the brain, which may also be genotoxic (128). Oztürk et al. suggested that the mutagenic compounds found in coal or wood smoke and ash, as well as hypoxic and reperfusion injury, may primarily contribute to the individual genotoxic of carbon monoxide exposure (103). In recent years, more and more studies have found that the polymorphism of some genes is closely related to the genetic susceptibility of DEACMP, indicating that these genes are susceptibility sites of DEACMP. Mutations in their expression could raise the risk of DEACMP and may act as potential predictors. Xu et al. conducted a screening of 6 NSE single nucleotide polymorphisms (SNPs) and compared the genotype frequencies and alleles of the 6 NSE SNPs (rs2071074, rs2071417, rs2071419, rs11064464, rs11064465, and rs3213434) by different genetic models. They found significant differences in the genotypes and allele frequencies of rs2071419 and rs3213434 in the DEACMP patients and ACOP patients, suggesting that rs2071419 and rs3213434 may be susceptible sites of DEACMP. They speculated that the C allele of the rs2071419 polymorphism and the T allele of the rs3213434 polymorphism in NSE could increase the risk of developing DEACMP, serving as potential risk factors for DEACMP (58). Gu et al. found that four leucine-rich repeats and calponin homology domain containing 1 (LRCH1) gene polymorphisms have a significant association with DEACMP, such as rs1539177 (G/A), rs17068697 (G/A), rs9534475 (A/C), and rs2236592 (T/C). Notably, the allelic A of rs9534475 polymorphism in LRCH1 may provide good early prognostic value for DEACMP (73). Zhang et al. investigated the association of MBP SNPs (rs470555, rs470724, rs4890785, rs595997, rs76452994, and rs921336) with DEACMP. They found that the MBP rs76452994 and rs921366 polymorphisms were associated with DEACMP, especially the G allele of rs76452994 and T allele of rs921336, which may serve as risk factors for DEACMP (74). Other studies have also found that the genetic susceptibility of some gene polymorphisms in DECAMP can vary between sexes. For example, Liang et al. suggested that the allelic variant of PARK2 SNPs rs1784594 is a risk factor for DECAMP, especially in females (75). In contrast, Li et al. (125) found that the allele L of MBP 5′-side TGGA n gene could increase the risk of developing DECAMP in male patients. In addition, some scholars believe that DEACMP results from the combined effects of environmental and genetic factors (104). Exploring genetic susceptibility-related gene loci offers a new perspective for predicting DECAMP. However, only focusing on the susceptibility genes of a specific ethnic group within a specific region may lead to inconsistent research results.
Given their specificity, feasibility, cost, and other considerations, the above predictive indicators for DEACMP have not yet gained expert consensus or widespread clinical use. As a result, there is still a need to explore new predictors for predicting DECAMP. The existing knowledge indicates that brain cells can secrete exosomes, which have become a hot field for evaluating brain function and diagnosing brain diseases. For example, neuronal-derived exosomes participate in the physiological and pathological processes of the brain, and changes in their content could provide valuable information for clinical diagnosis and disease evaluation of neurodegenerative diseases, as well as predict the occurrence of diseases at an early stage. Neuronal-derived exosomes could serve as potential biomarkers (105). Scholars have elucidated the process of exosome biogenesis, as illustrated in Figure 1, but the mechanisms behind the sorting of exosomal contents remain unclear. Changes in both intracellular and extracellular environments can influence the contents of exosomes and induce different biological functions. Indeed, intracellular hypoxia, ROS, and Ca2+ can significantly affect exosome biogenesis and their contents (76, 106, 107), while they play crucial roles in the pathogenesis of DEACMP.
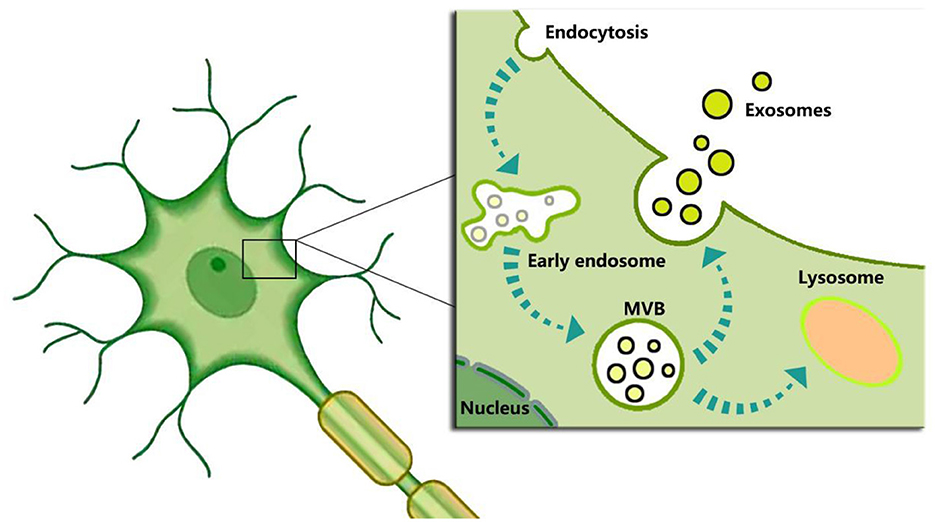
Figure 1. Schematic representation of exosome biogenesis. In short, exosome biogenesis begins with the inward budding of the plasma membrane to form endosomes. Then endosomes encapsulate nucleic acids, proteins, and other bioactive molecules to form intraluminal vesicles (ILVs) and multivesicular bodies (MVBs). Finally, the MVBs not fused by lysosomes are released into the extracellular environment to form “exosomes” after the fusion with the plasma membrane.
Hypoxia promotes the release of exosomes and changes the composition of the exosomal contents, especially miRNAs (108). Chiang et al. harvested the exosomes from the rat cortical primary neuronal cells under normoxic or oxygen and glucose deprivation (OGD) conditions. They compared the exosomal miRNA expression levels in normoxic and OGD conditions using next-generation sequencing. They found that 45 exosomal miRNAs were significantly different in their expression levels. The expression levels of 18 exosomal miRNAs were significantly higher, but those in 27 exosomal miRNAs were lower than those of normoxic, respectively. These exosomal miRNAs may play a role in cellular survival or death processes, as well as neuronal signaling (109). In the upregulated exosomal miRNAs, miRNA-34a-5p and miR-10a-5p have caught our attention. Previous studies have found that increased miRNA-34a facilitates neuronal injury and death in ischemic brain injury (110), while miR-10a induces apoptosis and inflammatory responses in neurons by inhibiting the PI3K/Akt/mTOR pathway (111, 112). In contrast, in the downregulated exosomal miRNAs, miR-25-5p and miR-532-3p have caught our attention, due to their neuroprotective effects in ischemic brain injury (113, 127). CO can cause cerebral hypoxia after prolonged exposure, resulting in affecting the sorting mechanisms of exosomal miRNAs, and the above changed exosomal miRNA profiles may reflect the risk of DECAMP.
Oxidative stress influences exosome biogenesis through different pathways, showing both prompt or inhibit effects on the release of exosomes (114, 115). On one hand, ROS inhibits the release of calcium ions from lysosomes through the transient receptor potential mucin 1 (TRPML1) channel and blocks the fusion between multivesicular bodies (MVBs) and lysosomes (114). ROS also affects the function of normal lysosomes to form nonfunctional lysosomes by inhibiting the activity of mTOR, resulting in reduced degradation of MVBs (116), further upregulating exosomal release. On the other hand, ROS enhances the biogenesis of autophagosomes, and MVBs can be selectively degraded by autophagosomes, further downregulating exosomal release (117). In simple terms, low oxidative stress restrains MVBs from being degraded in lysosomes, thereby prompting the release of exosomes. In contrast, high oxidative stress promotes the degradation of MVBs by activating cellular autophagy, thereby inhibiting the release of exosomes (118). Indeed, these effect of oxidative stress also depend on donor cell type and extracellular stimulus conditions. In addition, oxidative stress may affect the sorting mechanisms of exosomal content, leading to changes in exosomal miRNA profiles. In turn, exosomal miRNAs can regulate various genes associated with oxidative stress. This crosstalk between exosomal miRNAs and oxidative stress plays a significant role in the pathophysiological process of neurodegenerative diseases (105). Several exosomal miRNAs have been associated with neurodegenerative disorders, such as exosomal miRNA-34a. It is well known that CO can promote the production of mitochondrial ROS, leading to intracellular oxidative stress in brain cells, that can influence the release and the molecular cargo of brain cell-derived exosomes after ACOP. Special exosomal miRNAs could be involved in the development of DEACMP and their changes in quantity can provide dynamic information for predicting DEACMP.
Intracellular Ca2+ also regulates exosome biogenesis, and increasing intracellular Ca2+ stimulates the release of exosomes (106). Hettiarachchi et al. found that CO stimulates the generation of NO and ROS, leading to the formation of peroxynitrite in brain tissue, aggravating oxidative stress, and disrupting neuronal Ca2+ homeostasis (119), which may inhibit the release of exosomes. Some scholars suggested that exosomes play a role as the bridge between the inflammasome and autophagy activation under CO exposure conditions (120).
Therefore, gaining an in-depth and comprehensive understanding of the role of exosomes in ACOP can clarify the pathogenesis of DECAMP and provide theoretical support for the research of exosomes as biomarkers to predict DECAMP. To date, there are few reports focused on the relationship between exosomes and DECAMP, as well as the potential use of exosomes as clinical indicators for DEACMP. This scarcity may stem from the blood or cerebrospinal fluid samples containing exosomes derived from various tissues or cells except brain cells. Existing isolation and purification methods of exosomes (e.g., ultracentrifugation, exosome isolation reagent, ultrafiltration, immunoprecipitation/affinity capture, etc.) do not effectively isolate large quantities of pure and specific brain cell-derived exosomes from in blood or cerebrospinal fluid samples. Hence, it is not easy to identify the exosomes needed to predict DECAMP under the current research conditions.
DEACMP is a serious complication of ACOP that often shows poor therapeutic responses to current clinical treatment methods. Evaluating the risk of the development of DEACMP becomes very important. So far, some valuable predictors have emerged in clinical characteristics, laboratory indicators, neuroelectrophysiology, imaging examination, and genetic susceptibility. Recent research has confirmed that imaging examination (High cranial DWI signal within 24 h) and clinical characteristics (duration of CO >5.5 h) can serve as independent predictors of DEACMP (121). Some scholars highlighted the importance of the period of inability to walk in the acute stage and suggested that the clinical score of peak CK (U/L) and 40 × WALK (time for which walking was impossible during the acute stage of intoxication, days) is an early predictor for predicting DEACMP with 100% sensitivity and 82% specificity (122). More independent factors will also emerge in the future. Given the pathogenesis of DEACMP is diverse and complex, we need some new insights to explore the independent predictors. The studies on the unique role of exosomes in neurodegenerative diseases may offer new candidates to us. First, changes in both intracellular and extracellular environments of brain tissue after ACOP will influence the biogenesis and selection of contents in brain cell-derived exosomes, and exosomal contents may play roles in the pathogenesis of DEACMP, particularly miRNA. Identifying specific exosomal miRNA can reflect the development process of DEACMP. Second, brain cell-derived exosomes can cross the blood-brain barrier (BBB) and present in cerebrospinal fluid and blood circulation, facilitating us to collect them for serial determination about their amount and cargo. At last, CSF exosomal miRNAs can reflect brain pathophysiology while plasma exosomal miRNAs are not as easily degraded (123), which provides the samples to detect specific miRNA sequences using the ddPCR-based method. Based on this, exosomes have a potential advantage over currently identified predictors. Developing an effective isolation and purification method for wanted exosomes is a significant challenge in the current research on identifying specific exosomes for predicting DECAMP. Encouragingly, scholars have found that neuronal-derived exosome membranes are rich in the neuronal cell adhesion molecule and the L1 cell adhesion molecule (L1CAM). They utilized immunoprecipitation with biotinylated antibodies against neuronal surface markers to isolate neuronal-derived exosomes. This isolation method for separating will provide specific exosomes needed for DECAMP research (115, 124). With advancements in technology and increased research investment, we are optimistic that scholars will gain deeper insights into the role of exosomes in DECAMP and develop exosome-based detection methods for predicting DECAMP.
YW: Writing – original draft. ZZ: Writing – original draft. DZ: Writing – original draft, Writing – review & editing. YJ: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the grants from Research Project of the Science and Technology Department of Sichuan province (2021YJ0217) and Chengdu Medical Research Project (2023203).
The authors express their sincere thanks to Mr. Chongzhi Liu, Mr. Yi Yuan, and Ms. Limei Wang for their helpful assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gozubuyuk AA, Dag H, Kacar A, Karakurt Y, Arica V. Epidemiology, pathophysiology, clinical evaluation, and treatment of carbon monoxide poisoning in child, infant, and fetus. North Clin Istanb. (2017) 4:100–7. doi: 10.14744/nci.2017.49368
2. Mattiuzzi C, Lippi G. Worldwide epidemiology of carbon monoxide poisoning. Hum Exp Toxicol. (2020) 39:387–92. doi: 10.1177/0960327119891214
3. Al-Matrouk A, Al-Hemoud A, Al-Hasan M, Alabouh Y, Dashti A, Bojbarah H. Carbon Monoxide poisoning in Kuwait: a five-year, retrospective, epidemiological study. Int J Environ Res Public Health. (2021) 18:8854. doi: 10.3390/ijerph18168854
4. Zhang L, Wu D, Xu M, Bian Y, Wang Y, Gao G, et al. Acute carbon monoxide poisoning in Shandong, China: an observational study. Chin Med J (Engl). (2022) 135:1539–44. doi: 10.1097/CM9.0000000000001942
5. Xu X, Zhang H, Wang K, Tu T, Jiang Y. Protective effect of edaravone against carbon monoxide induced apoptosis in rat primary cultured astrocytes. Biochem Res Int. (2017) 2017:5839762. doi: 10.1155/2017/5839762
6. Xu XM, Luo H, Rong BB, Zheng XM, Wang FT, Zhang SJ, et al. Management of delayed encephalopathy after CO poisoning: an evidence-based narrative review. Medicine (Baltimore). (2019) 98:e18199. doi: 10.1097/MD.0000000000018199
7. Li, C., Liang, M. L., and Zhang, X. G. (2022). Research progress on the mechanisms of delayed encephalopathy in acute carbon monoxide poisoning. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 40, 543-546. Chinese.
8. Shankar GM, Balaj L, Stott SL, Nahed B, Carter BS. Liquid biopsy for brain tumors. Expert Rev Mol Diagn. (2017) 17:943–7. doi: 10.1080/14737159.2017.1374854
9. Lee EC, Ha TW, Lee DH, Hong DY, Park SW, Lee JY, et al. Utility of exosomes in ischemic and hemorrhagic stroke diagnosis and treatment. Int J Mol Sci. (2022) 23:8367. doi: 10.3390/ijms23158367
10. Tao H, Gao B. Exosomes for neurodegenerative diseases: diagnosis and targeted therapy. J Neurol. (2024) 271:3050–62. doi: 10.1007/s00415-024-12329-w
11. Meldolesi J. Extracellular vesicles (exosomes and ectosomes) play key roles in the pathology of brain diseases. Mol Biomed. (2021) 2:18. doi: 10.1186/s43556-021-00040-5
12. Winston CN, Goetzl EJ, Akers JC, Carter BS, Rockenstein EM, Galasko D, et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer's Dement. (2016) 3:63–72. doi: 10.1016/j.dadm.2016.04.001
13. Jia L, Qiu Q, Zhang H, Chu L, Du Y, Zhang J, et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. (2019) 15:1071–80. doi: 10.1016/j.jalz.2019.05.002
14. Huang YQ, Peng ZR, Huang FL, Yang AL. Mechanism of delayed encephalopathy after acute carbon monoxide poisoning. Neural Regen Res. (2020) 15:2286–95. doi: 10.4103/1673-5374.284995
15. Corcoran A, O'Connor JJ. Hypoxia-inducible factor signalling mechanisms in the central nervous system. Acta Physiol (Oxf). (2013) 208:298–310. doi: 10.1111/apha.12117
16. Satran D, Henry CR, Adkinson C, Nicholson CI, Bracha Y, Henry TD. Cardiovascular manifestations of moderate to severe carbon monoxide poisoning. J Am Coll Cardiol. (2005) 45:1513–6. doi: 10.1016/j.jacc.2005.01.044
17. Akyol S, Erdogan S, Idiz N, Celik S, Kaya M, Ucar F, et al. The role of reactive oxygen species and oxidative stress in carbon monoxide toxicity: an in-depth analysis. Redox Rep. (2014) 19:180–9. doi: 10.1179/1351000214Y.0000000094
18. Stucki D, Stahl W. Carbon monoxide - beyond toxicity? Toxicol Lett. (2020) 333:251–60. doi: 10.1016/j.toxlet.2020.08.010
19. Akyol S, Yuksel S, Pehlivan S, Erdemli HK, Gulec MA, Adam B, et al. Possible role of antioxidants and nitric oxide inhibitors against carbon monoxide poisoning: Having a clear conscience because of their potential benefits. Med Hypotheses. (2016) 92:3–6. doi: 10.1016/j.mehy.2016.04.015
20. Yun HY, Dawson VL, Dawson TM. Neurobiology of nitric oxide. Crit Rev Neurobiol. (1996) 10:291–316. doi: 10.1615/CritRevNeurobiol.v10.i3-4.20
21. Thom SR, Fisher D, Zhang J, Bhopale VM, Cameron B, Buerk DG. Neuronal nitric oxide synthase and N-methyl-D-aspartate neurons in experimental carbon monoxide poisoning. Toxicol Appl Pharmacol. (2004) 194:280–95. doi: 10.1016/j.taap.2003.09.017
22. Ivanova VO, Balaban PM, Bal NV. Modulation of AMPA receptors by nitric oxide in nerve cells. Int J Mol Sci. (2020) 21:981. doi: 10.3390/ijms21030981
23. Wang S, Xiong B, Tian Y, Hu Q, Jiang X, Zhang J, et al. Targeting ferroptosis promotes functional recovery by mitigating white matter injury following acute carbon monoxide poisoning. Mol Neurobiol. (2024) 61:1157–74. doi: 10.1007/s12035-023-03603-5
24. Ide T, Kamijo Y. The early elevation of interleukin 6 concentration in cerebrospinal fluid and delayed encephalopathy of carbon monoxide poisoning. Am J Emerg Med. (2009) 27:992–6. doi: 10.1016/j.ajem.2008.07.009
25. Thom SR, Bhopale VM, Milovanova TM, Hardy KR, Logue CJ, Lambert DS, et al. Plasma biomarkers in carbon monoxide poisoning. Clin Toxicol (Phila). (2010) 48:47–56. doi: 10.3109/15563650903468209
26. Akcali G, Uzun G, Arziman I, Aydin I, Yildiz S. The relationship between intoxication severity and blood interleukin 6, interleukin 10 and CRP levels in carbon monoxide-poisoned patients. Undersea Hyperbaric Med. (2018) 45:646–52. doi: 10.22462/11.12.2018.4
27. Wang W, Li J, Chang Y, Xie X, Ren J, Wang X, et al. Effects of immune reaction in rats after acute carbon monoxide poisoning. Undersea Hyperb Med. (2011) 38:239–46.
28. Deber CM, Reynolds SJ. Central nervous system myelin: structure, function, and pathology. Clin Biochem. (1991) 24:113–34. doi: 10.1016/0009-9120(91)90421-A
29. Kuroda H, Fujihara K, Kushimoto S, Aoki M. Novel clinical grading of delayed neurologic sequelae after carbon monoxide poisoning and factors associated with outcome. Neurotoxicology. (2015) 48:35–43. doi: 10.1016/j.neuro.2015.03.002
30. Omi T. Cerebrospinal fluid biomarkers for monitoring delayed neurologic sequelae after carbon monoxide poisoning. Neurol India. (2022) 70:1668–9. doi: 10.4103/0028-3886.355093
31. Thom SR, Bhopale VM, Fisher D, Zhang J, Gimotty P. Delayed neuropathology after carbon monoxide poisoning is immune-mediated. Proc Natl Acad Sci U S A. (2004) 101:13660–5. doi: 10.1073/pnas.0405642101
32. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. (2016) 53:1181–94. doi: 10.1007/s12035-014-9070-5
33. Wu Z, Yu J, Zhu A, Nakanishi H. Nutrients, microglia aging, and brain aging. Oxid Med Cell Longev. (2016) 2016:7498528. doi: 10.1155/2016/7498528
34. Piantadosi CA, Zhang J, Levin ED, Folz RJ, Schmechel DE. Apoptosis and delayed neuronal damage after carbon monoxide poisoning in the rat. Exp Neurol. (1997) 147:103–14. doi: 10.1006/exnr.1997.6584
35. Guan L, Wen T, Zhang Y, Wang X, Zhao J. Induction of heme oxygenase-1 with hemin attenuates hippocampal injury in rats after acute carbon monoxide poisoning. Toxicology. (2009) 262:146–52. doi: 10.1016/j.tox.2009.06.001
36. Tofighi R, Tillmark N, Daré E, Aberg AM, Larsson JE, Ceccatelli S. Hypoxia-independent apoptosis in neural cells exposed to carbon monoxide in vitro. Brain Res. (2006) 1098:1–8. doi: 10.1016/j.brainres.2006.04.095
37. Jurič DM, Finderle Ž, Šuput D, Brvar M. The effectiveness of oxygen therapy in carbon monoxide poisoning is pressure- and time-dependent: a study on cultured astrocytes. Toxicol Lett. (2015) 233:16–23. doi: 10.1016/j.toxlet.2015.01.004
38. Ochi S, Sekiya K, Abe N, Funahashi Y, Kumon H, Yoshino Y, et al. Neural precursor cells are decreased in the hippocampus of the delayed carbon monoxide encephalopathy rat model. Sci Rep. (2021) 11:6244. doi: 10.1038/s41598-021-85860-9
39. Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med. (2012) 25:30–4. doi: 10.3109/14767058.2012.663176
40. Chun Y, Kim J. AMPK-mTOR signaling and cellular adaptations in hypoxia. Int J Mol Sci. (2021) 22:9765. doi: 10.3390/ijms22189765
41. Liu X, Tian F, Wang S, Wang F, Xiong L. Astrocyte autophagy flux protects neurons against oxygen-glucose deprivation and ischemic/reperfusion injury. Rejuvenation Res. (2018) 21:405–15. doi: 10.1089/rej.2017.1999
42. Lu N, Li X, Tan R, An J, Cai Z, Hu X, et al. HIF-1α/Beclin1-mediated autophagy is involved in neuroprotection induced by hypoxic preconditioning. J Mol Neurosci. (2018) 66:238–50. doi: 10.1007/s12031-018-1162-7
43. Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim HP, Choi AM, et al. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol. (2011) 45:867–73. doi: 10.1165/rcmb.2010-0352OC
44. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. (2015) 35:615–21. doi: 10.1007/s10571-015-0166-x
45. Kim DS, Song L, Wang J, Wu H, Gou W, Cui W, et al. Carbon monoxide inhibits islet apoptosis via induction of autophagy. Antioxid Redox Signal. (2018) 28:1309–22. doi: 10.1089/ars.2016.6979
46. Figueiredo-Pereira C, Villarejo-Zori B, Cipriano PC, Tavares D, Ramírez-Pardo I, Boya P, et al. Carbon monoxide stimulates both mitophagy and mitochondrial biogenesis to mediate protection against oxidative stress in astrocytes. Mol Neurobiol. (2023) 60:851–63. doi: 10.1007/s12035-022-03108-7
47. Corti O, Blomgren K, Poletti A, Beart PM. Autophagy in neurodegeneration: new insights underpinning therapy for neurological diseases. J Neurochem. (2020) 154:354–71. doi: 10.1111/jnc.15002
48. Cao W, Li J, Yang K, Cao D. An overview of autophagy: mechanism, regulation and research progress. Bull Cancer. (2021) 108:304–22. doi: 10.1016/j.bulcan.2020.11.004
49. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. (2007) 8:741–52. doi: 10.1038/nrm2239
50. Guo C, Ma YY. Calcium permeable-AMPA receptors and excitotoxicity in neurological disorders. Front Neural Circuits. (2021) 15:711564. doi: 10.3389/fncir.2021.711564
51. Greco P, Nencini G, Piva I, Scioscia M, Volta CA, Spadaro S, et al. Pathophysiology of hypoxic-ischemic encephalopathy: a review of the past and a view on the future. Acta Neurol Belg120. (2020) 277–88. doi: 10.1007/s13760-020-01308-3
52. Ochi S, Abe M, Li C, Mori Y, Ishimaru T, Yoshino Y, et al. The nicotinic cholinergic system is affected in rats with delayed carbon monoxide encephalopathy. Neurosci Lett. (2014) 569:33–7. doi: 10.1016/j.neulet.2014.03.054
53. Huang CC, Lee JC, Lin KC, Lin HJ, Su SB, Hsu CC, et al. Exposure duration and history of hypertension predicted neurological sequelae in patients with carbon monoxide poisoning. Epidemiology. (2019) 30:S76–81. doi: 10.1097/EDE.0000000000001000
54. Sari Dogan F, Güneysel Ö, Gökdag E, Güneş M, Sümen SG. Demographic characteristics and delayed neurological sequelae risk factors in carbon monoxide poisoning. Am J Emerg Med. (2020) 38:2552–6. doi: 10.1016/j.ajem.2019.12.037
55. Zhang Y, Lu Q, Jia J, Xiang D, Xi Y. Multicenter retrospective analysis of the risk factors for delayed neurological sequelae after acute carbon monoxide poisoning. Am J Emerg Med. (2021) 46:165–9. doi: 10.1016/j.ajem.2020.06.090
56. Han S, Choi S, Nah S, Lee SU, Cho YS, Kim GW, et al. Cox regression model of prognostic factors for delayed neuropsychiatric sequelae in patients with acute carbon monoxide poisoning: a prospective observational study. Neurotoxicology. (2021) 82:63–8. doi: 10.1016/j.neuro.2020.11.006
57. Pepe G, Castelli M, Nazerian P, Vanni S, Del Panta M, Gambassi F, et al. Delayed neuropsychological sequelae after carbon monoxide poisoning: predictive risk factors in the Emergency Department. A retrospective study. Scand J Trauma Resuscit Emerg Med. (2011) 19:16. doi: 10.1186/1757-7241-19-16
58. Xu P, Wang Y, Cao L, Huang W, Zhang J, Gao X, et al. Glasgow Coma Scale is a better delayed neurological sequelae risk factor than neurological examination abnormalities in carbon monoxide poisoning. Am J Emerg Med. (2020) 38:2468–9. doi: 10.1016/j.ajem.2020.02.047
59. Kim SH, Lee Y, Kang S, Paik JH, Kim H, Cha YS. Derivation and validation of a score for predicting poor neurocognitive outcomes in acute carbon monoxide poisoning. JAMA Netw Open. (2022) 5:e2210552. doi: 10.1001/jamanetworkopen.2022.10552
60. Akdemir HU, Yardan T, Kati C, Duran L, Alacam H, Yavuz Y, et al. The role of S100B protein, neuron-specific enolase, and glial fibrillary acidic protein in the evaluation of hypoxic brain injury in acute carbon monoxide poisoning. Hum Exp Toxicol. (2014) 33:1113–20. doi: 10.1177/0960327114521049
61. Zhang L, Zhao J, Hao Q, Xu X, Han H, Li J. Serum NSE and S100B protein levels for evaluating the impaired consciousness in patients with acute carbon monoxide poisoning. Medicine (Baltimore). (2021) 100:e26458. doi: 10.1097/MD.0000000000026458
62. Yardan T, Cevik Y, Donderici O, Kavalci C, Yilmaz FM, Yilmaz G, et al. Elevated serum S100B protein and neuron-specific enolase levels in carbon monoxide poisoning. Am J Emerg Med. (2009) 27:838–42. doi: 10.1016/j.ajem.2008.04.016
63. Park E, Ahn J, Min YG, Jung YS, Kim K, Lee J, et al. The usefulness of the serum s100b protein for predicting delayed neurological sequelae in acute carbon monoxide poisoning. Clin Toxicol (Phila). (2012) 50:183–8. doi: 10.3109/15563650.2012.658918
64. Liu H, Zhang Y, Ren YB, Kang J, Xing J, Qi QH, et al. Serum S100B level may be correlated with carbon monoxide poisoning. Int Immunopharmacol. (2015) 27:69–75. doi: 10.1016/j.intimp.2015.04.027
65. Cha YS, Kim H, Do HH, Kim HI, Kim OH, Cha KC, et al. Serum neuron-specific enolase as an early predictor of delayed neuropsychiatric sequelae in patients with acute carbon monoxide poisoning. Hum Exp Toxicol. (2018) 37:240–6. doi: 10.1177/0960327117698544
66. Nah S, Choi S, Kim GW, Moon JE, Lee YH, Han S. Prediction of delayed neuropsychiatric sequelae after carbon monoxide poisoning via serial determination of serum neuron-specific enolase levels. Hum Exp Toxicol. (2021) 40:S339–46. doi: 10.1177/09603271211043475
67. Di C, Zeng Y, Mao J, Shen Z, Gu W. Dynamic changes and clinical significance of serum S100B protein and glial fibrillary acidic protein in patients with delayed encephalopathy after acute carbon monoxide poisoning. Pak J Med Sci. (2018) 34:945–9. doi: 10.12669/pjms.344.15363
68. Kokulu K, Mutlu H, Sert ET. Serum netrin-1 levels at presentation and delayed neurological sequelae in unintentional carbon monoxide poisoning. Clin Toxicol (Phila). (2020) 58:1313–9. doi: 10.1080/15563650.2020.1743302
69. Pang L, Wang HL, Wang ZH, Wu Y, Dong N, Xu DH, et al. Plasma copeptin as a predictor of intoxication severity and delayed neurological sequelae in acute carbon monoxide poisoning. Peptides. (2014) 59:89–93. doi: 10.1016/j.peptides.2014.07.007
70. He F, Liu X, Yang S, Zhang S, Xu G, Fang G, et al. Evaluation of brain function in acute carbon monoxide poisoning with multimodality evoked potentials. Environ Res. (1993) 60:213–26. doi: 10.1006/enrs.1993.1029
71. Ahn C, Oh J, Kim CW, Lee H, Lim TH, Kang H. Early neuroimaging and delayed neurological sequelae in carbon monoxide poisoning: a systematic review and meta-analysis. Sci Rep. (2022) 12:3529. doi: 10.1038/s41598-022-07191-7
72. Feng SY. Magnetic resonance imaging for predicting delayed neurologic sequelae caused by carbon monoxide poisoning: a systematic review and meta-analysis. Medicine (Baltimore). (2022) 101:e31981. doi: 10.1097/MD.0000000000031981
73. Gu J, Zeng J, Wang X, Gu X, Zhang X, Zhang P, et al. LRCH1 polymorphisms linked to delayed encephalopathy after acute carbon monoxide poisoning identified by GWAS analysis followed by Sequenom MassARRAY® validation. BMC Med Genet. (2019) 20:197. doi: 10.1186/s12881-019-0931-7
74. Zhang F, Zeng J, Zhang X, Gu J, Han Y, Zhang P, et al. Investigation of the relationship between MBP gene polymorphisms and delayed encephalopathy after acute carbon monoxide poisoning. Neurotoxicology. (2023) 94:217–22. doi: 10.1016/j.neuro.2022.12.002
75. Liang F, Li W, Zhang P, Zhang Y, Gu J, Wang X, et al. A PARK2 polymorphism associated with delayed neuropsychological sequelae after carbon monoxide poisoning. BMC Med Genet. (2013) 14:99. doi: 10.1186/1471-2350-14-99
76. Li G, Huang D, Li N, Ritter JK, Li PL. Regulation of TRPML1 channel activity and inflammatory exosome release by endogenously produced reactive oxygen species in mouse podocytes. Redox Biol. (2021) 43:102013. doi: 10.1016/j.redox.2021.102013
77. Zhang Y, Bai Y, Feng T, Li Y, Zhang H, Wang T. Establishment and application of severity assessment system for patients with delayed encephalopathy caused by carbon monoxide poisoning. Am J Transl Res. (2023) 15:6558–64.
78. Xu L, Liu X, Zhao J, Zeng J, Gu J, Zhang X, et al. Association between neuron-specific enolase gene polymorphism and delayed encephalopathy after acute carbon monoxide poisoning. Behav Neurol. (2020) 2020:8819210. doi: 10.1155/2020/8819210
79. Michetti F, Corvino V, Geloso MC, Lattanzi W, Bernardini C, Serpero L, et al. The S100B protein in biological fluids: more than a lifelong biomarker of brain distress. J Neurochem. (2012) 120:644–59. doi: 10.1111/j.1471-4159.2011.07612.x
80. Astrand R, Undén J, Romner B. Clinical use of the calcium-binding S100B protein. Methods Mol Biol. (2013) 963:373–84. doi: 10.1007/978-1-62703-230-8_23
81. Yuan XS, Bian XX. S100B protein and its clinical effect on craniocerebral injury. Chin J Traumatol. (2008) 11:54–7. doi: 10.1016/S1008-1275(08)60012-7
82. Hafez AS, El-Sarnagawy GN. S-100β in predicting the need of hyperbaric oxygen in CO-induced delayed neurological sequels. Hum Exp Toxicol. (2020) 39:614–23. doi: 10.1177/0960327119897104
83. Rasmussen LS, Poulsen MG, Christiansen M, Jansen EC. Biochemical markers for brain damage after carbon monoxide poisoning. Acta Anaesthesiol Scand. (2004) 48:469–73. doi: 10.1111/j.1399-6576.2004.00362.x
84. Steiner J, Bogerts B, Schroeter ML, Bernstein HG. S100B protein in neurodegenerative disorders. Clin Chem Lab Med. (2011) 49:409–24. doi: 10.1515/CCLM.2011.083
85. Akelma AZ, Celik A, Ozdemir O, Kavak Akelma F, Abaci A, Razi CH, et al. Neuron-specific enolase and S100B protein in children with carbon monoxide poisoning: children are not just small adults. Am J Emerg Med. (2013) 31:524–8. doi: 10.1016/j.ajem.2012.10.009
86. Cheng F, Yuan Q, Yang J, Wang W, Liu H. The prognostic value of serum neuron-specific enolase in traumatic brain injury: systematic review and meta-analysis. PLoS ONE. (2014) 9:e106680. doi: 10.1371/journal.pone.0106680
87. Abdel Salam ME, Elawady EH, Khater AS, Eweda SA, Abd El Moneam MH. Neuropsychiatric sequelae of acute carbon monoxide poisoning: the predictive role of neuron specific enolase and glial fibrillary acidic protein. Neurotoxicology. (2021) 85:115–20. doi: 10.1016/j.neuro.2021.05.003
88. Cakir Z, Aslan S, Umudum Z, Acemoglu H, Akoz A, Turkyilmaz S, et al. S-100beta and neuron-specific enolase levels in carbon monoxide-related brain injury. Am J Emerg Med. (2010) 28:61–7. doi: 10.1016/j.ajem.2008.10.032
89. Isgrò MA, Bottoni P, Scatena R. Neuron-specific enolase as a biomarker: biochemical and clinical aspects. Adv Exp Med Biol. (2015) 867:125–43. doi: 10.1007/978-94-017-7215-0_9
90. Moon JM, Chun BJ, Lee SD, Jung EJ. Serum neuron-specific enolase levels at presentation and long-term neurological sequelae after acute charcoal burning-induced carbon monoxide poisoning. Clin Toxicol (Phila). (2018) 56:751–8. doi: 10.1080/15563650.2017.1415347
91. Ide T, Kamijo Y. Myelin basic protein in cerebrospinal fluid: a predictive marker of delayed encephalopathy from carbon monoxide poisoning. Am J Emerg Med. (2008) 26:908–12. doi: 10.1016/j.ajem.2007.11.021
92. Müller-Putz GR. Electroencephalography. Handb Clin Neurol. (2020) 168:249–62. doi: 10.1016/B978-0-444-63934-9.00018-4
93. Emerson TS, Keiler J. Pattern shift visual evoked potential screening for HBO2 in mild-to-moderate carbon monoxide poisoning. Undersea Hyperb Med. (1998) 25:27–32.
94. Wang SL, Ma MM, Lv GW, Zhang M, Du YS, Zhang SL, et al. Predictive value of gray-matter-white-matter ratio on brain computed tomography for delayed encephalopathy after acute carbon monoxide poisoning: a retrospective cohort study. Biomed Res Int. (2021) 2021:5511290. doi: 10.1155/2021/5511290
95. Du X, Gu H, Hao F, Gao L, Wang J, Sun C, et al. Utility of brain CT for predicting delayed encephalopathy after acute carbon monoxide poisoning. Exp Ther Med. (2019) 17:2682–8. doi: 10.3892/etm.2019.7233
96. Biegger P, Ladd ME, Komljenovic D. Multifunctional magnetic resonance imaging probes. Recent Results Cancer Res. (2020) 216:189–226. doi: 10.1007/978-3-030-42618-7_6
97. Kim YS, Cha YS, Kim MS, Kim HJ, Lee YS, Youk H, et al. The usefulness of diffusion-weighted magnetic resonance imaging performed in the acute phase as an early predictor of delayed neuropsychiatric sequelae in acute carbon monoxide poisoning. Hum Exp Toxicol. (2018) 37:587–95. doi: 10.1177/0960327117722821
98. Wu J, Peng T, Xu Z, Yang X, Zhao L, Yang H, et al. Evaluation of changes in magnetic resonance diffusion tensor imaging after treatment of delayed encephalopathy due to carbon monoxide poisoning. J Integr Neurosci. (2019) 18:475–9. doi: 10.31083/j.jin.2019.04.160
99. Kim DM, Lee IH, Park JY, Hwang SB, Yoo DS, Song CJ. Acute carbon monoxide poisoning: MR imaging findings with clinical correlation. Diagn Interv Imaging. (2017) 98:299–306. doi: 10.1016/j.diii.2016.10.004
100. Beppu T. The role of MR imaging in assessment of brain damage from carbon monoxide poisoning: a review of the literature. Am J Neuroradiol. (2014) 35:625–31. doi: 10.3174/ajnr.A3489
101. Tsai PH, Chou MC, Chiang SW, Chung HW, Liu HS, Kao HW, et al. Early white matter injuries in patients with acute carbon monoxide intoxication: a tract-specific diffusion kurtosis imaging study and STROBE compliant article. Medicine. (2017) 96:e5982. doi: 10.1097/MD.0000000000005982
102. Zhang Y, Wang T, Lei J, Guo S, Wang S, Gu Y, et al. Cerebral damage after carbon monoxide poisoning: a longitudinal diffusional kurtosis imaging study. AJNR Am J Neuroradiol. (2019) 40:1630–7. doi: 10.3174/ajnr.A6201
103. Oztürk S, Vatansever S, Cefle K, Palanduz S, Güler K, Erten N, et al. Acute wood or coal exposure with carbon monoxide intoxication induces sister chromatid exchange. J Toxicol Clin Toxicol. (2002) 40:115–20. doi: 10.1081/CLT-120004398
104. Li W, Zhang Y, Gu R, Zhang P, Liang F, Gu J, et al. DNA pooling base genome-wide association study identifies variants at NRXN3 associated with delayed encephalopathy after acute carbon monoxide poisoning. PLoS ONE. (2013) 8:e79159. doi: 10.1371/journal.pone.0079159
105. Wang X, Zhou Y, Gao Q, Ping D, Wang Y, Wu W, et al. The role of exosomal microRNAs and oxidative stress in neurodegenerative diseases. Oxid Med Cell Longev. (2020) 2020:3232869. doi: 10.1155/2020/3232869
106. Savina A, Furlán M, Vidal M, Colombo MI. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem. (2003) 278:20083–90. doi: 10.1074/jbc.M301642200
107. Jiang H, Zhao H, Zhang M, He Y, Li X, Xu Y, et al. Hypoxia induced changes of exosome cargo and subsequent biological effects. Front Immunol. (2022) 13:824188. doi: 10.3389/fimmu.2022.824188
108. Bister N, Pistono C, Huremagic B, Jolkkonen J, Giugno R, Malm T. Hypoxia and extracellular vesicles: a review on methods, vesicular cargo and functions. J Extracell Vesicles. (2020) 10:e12002. doi: 10.1002/jev2.12002
109. Chiang CS, Fu SJ, Hsu CL, Jeng CJ, Tang CY, Huang YS, et al. Neuronal exosomes secreted under oxygen-glucose deprivation/reperfusion presenting differentially expressed miRNAs and affecting neuronal survival and neurite outgrowth. Neuromolecular Med. (2021) 23:404–15. doi: 10.1007/s12017-020-08641-z
110. Chua CEL, Tang BL. miR-34a in neurophysiology and neuropathology. J Mol Neurosci. (2019) 67:235–46. doi: 10.1007/s12031-018-1231-y
111. Wang J, Wang A, He H, She X, He Y, Li S, et al. Trametenolic acid B protects against cerebral ischemia and reperfusion injury through modulation of microRNA-10a and PI3K/Akt/mTOR signaling pathways. Biomed Pharmacother. (2019) 112:108692. doi: 10.1016/j.biopha.2019.108692
112. Lu Y, Wang W, Ma Y, Fan Z, Xiong L, Zhao J, et al. miR-10a induces inflammatory responses in epileptic hippocampal neurons of rats via PI3K/Akt/mTOR signaling pathway. Neuroreport. (2023) 34:526–34. doi: 10.1097/WNR.0000000000001920
113. Zhou X, Qiao B. Inhibition of HDAC3 and ATXN3 by miR-25 prevents neuronal loss and ameliorates neurological recovery in cerebral stroke experimental rats. J Physiol Biochem. (2022) 78:139–49. doi: 10.1007/s13105-021-00848-3
114. Chiaradia E, Tancini B, Emiliani C, Delo F, Pellegrino RM, Tognoloni A, et al. Extracellular vesicles under oxidative stress conditions: biological properties and physiological roles. Cells. (2021) 10:1763. doi: 10.3390/cells10071763
115. Chen Z, Chang F, Yao L, Yuan F, Hong J, Wu D, et al. Clinical significance of the cognition-related pathogenic proteins in plasma neuronal-derived exosomes among normal cognitive adults over 45 years old with olfactory dysfunction. Eur Arch Otorhinolaryngol. (2022) 279:3467–76. doi: 10.1007/s00405-021-07143-3
116. Zheng K, Ma J, Wang Y, He Z, Deng K. Sulforaphane inhibits autophagy and induces exosome-mediated paracrine senescence via regulating mTOR/TFE3. Mol Nutr Food Res. (2020) 64:e1901231. doi: 10.1002/mnfr.201901231
117. Zhu L, Zang J, Liu B, Yu G, Hao L, Liu L, et al. Oxidative stress-induced RAC autophagy can improve the HUVEC functions by releasing exosomes. J Cell Physiol. (2020) 235:7392–409. doi: 10.1002/jcp.29641
118. Zhang W, Liu R, Chen Y, Wang M, Du J. Crosstalk between oxidative stress and exosomes. Oxid Med Cell Longev. (2022) 2022:3553617. doi: 10.1155/2022/3553617
119. Hettiarachchi NT, Boyle JP, Bauer CC, Dallas ML, Pearson HA, Hara S, et al. Peroxynitrite mediates disruption of Ca2+ homeostasis by carbon monoxide via Ca2+ ATPase degradation. Antioxid Redox Signal. (2012) 17:744–55. doi: 10.1089/ars.2011.4398
120. Chen RJ, Lee YH, Chen TH, Chen YY, Yeh YL, Chang CP, et al. Carbon monoxide-triggered health effects: the important role of the inflammasome and its possible crosstalk with autophagy and exosomes. Arch Toxicol. (2021) 95:1141–59. doi: 10.1007/s00204-021-02976-7
121. Gao X, Wei W, Yang GD. Clinical factors for delayed neuropsychiatric sequelae from acute carbon monoxide poisoning: a retrospective study. Front Med. (2024) 11:1333197. doi: 10.3389/fmed.2024.1333197
122. Suzuki Y. Risk factors for delayed encephalopathy following carbon monoxide poisoning: Importance of the period of inability to walk in the acute stage. PLoS ONE. (2021) 16:e0249395. doi: 10.1371/journal.pone.0249395
123. Li DB, Liu JL, Wang W, Li RY, Yu DJ, et al. Plasma Exosomal miR-422a and miR-125b-2-3p Serve as Biomarkers for Ischemic Stroke. Curr Neurovasc Res. (2017) 14:330–7. doi: 10.2174/1567202614666171005153434
124. Eren E, Leoutsakos JM, Troncoso J, Lyketsos CG, Oh ES, Kapogiannis D. Neuronal-derived EV biomarkers track cognitive decline in Alzheimer's disease. Cells. (2022) 11:436. doi: 10.3390/cells11030436
125. Li SG, Li WQ, Wang JK, Zhang HY, Li W, Zhang P, et al. Association of the genes for tumor necrosis factor-α and myelin basic protein with delayed encephalopathy after acute carbon monoxide poisoning. Genet Mol Res. (2012) 11:4479–86. doi: 10.4238/2012.December.19.1
126. Xu Y, Tian Y, Tian Y, Li X, Zhao P. Autophagy activation involved in hypoxic-ischemic brain injury induces cognitive and memory impairment in neonatal rats. J Neurochem. (2016) 139:795–805. doi: 10.1111/jnc.13851
127. Mu J, Cheng X, Zhong S, Chen X, Zhao C. Neuroprotective effects of miR-532-5p against ischemic stroke. Metab Brain Dis. (2020) 35:753–63. doi: 10.1007/s11011-020-00544-z
Keywords: carbon monoxide poisoning, DEACMP, predictors, exosomes, miRNAs
Citation: Wang Y, Zhou Z, Zhang D and Jiang Y (2025) Predictors of delayed encephalopathy after acute carbon monoxide poisoning: a literature review. Front. Med. 12:1559264. doi: 10.3389/fmed.2025.1559264
Received: 12 January 2025; Accepted: 11 March 2025;
Published: 26 March 2025.
Edited by:
Luca Zazzeron, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Enrico M. Camporesi, USF Health, United StatesCopyright © 2025 Wang, Zhou, Zhang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dailiang Zhang, MjcyMDkzMzUwQHFxLmNvbQ==; Yuan Jiang, ODU3NDE5MjBAcXEuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.