 Alexandra Schifferli
Alexandra Schifferli- Department of Hematology/Oncology, University Children’s Hospital Basel, Basel, Switzerland
Previous guidelines for the treatment of immune thrombocytopenia (ITP) have traditionally focused on a dichotomy between pediatric and adult ITP. Adolescents and young adults (AYAs) do not neatly fit into either the pediatric or adult ITP group. A deeper understanding of ITP’s natural history, risk factors for chronicity, and outcomes in AYAs is a crucial first step toward developing tailored treatment algorithms. Such data could form the basis for recommendations targeting this underrepresented yet clinically distinct population. Ultimately, age-adapted trials may improve long-term outcomes, reduce toxicity, and enhance quality of life for AYAs with ITP. The AYAs collaboration—drawing on data from the Pediatric and Adult Registry on Chronic ITP (PARC-ITP), Registre Midi- Pyrénéen-France (CARMEN-France) adult registry in Toulouse, and the National Prospective Cohort for Children with Chronic Autoimmune Cytopenia (OBS’CEREVANCE) in Bordeaux, France—aims to address the information gap in AYAs with ITP. To date, four analyses have been undertaken (using data from 2004 to 2021), each addressing the major clinical aspects of ITP in patients aged 12–25 years: (1) newly diagnosed ITP, (2) chronic disease, (3) refractory courses, and (4) secondary (sITP) forms.
Introduction
Previous guidelines for the treatment of immune thrombocytopenia (ITP) have traditionally focused on a dichotomy between pediatric and adult ITP. Pediatric ITP is typically acute and self-limiting, with a very low risk of life-threatening bleeding events. In contrast, adult ITP usually follows a chronic course and is associated with a higher risk of severe bleeding, particularly in individuals over 60 years of age or those with platelet counts below 20–30 × 109/L. Contributing factors to these clinical differences may include distinct immunologic triggers, comorbidities, comedications, and age-related immune senescence. Accordingly, adults with severe thrombocytopenia are generally advised to receive platelet-enhancing therapy, whereas a watch-and-wait strategy is recommended for children with minor bleeding (1, 2). These different approaches underscore the importance of age in ITP management; however, adolescents and young adults (AYAs) do not neatly fit into either the pediatric or adult ITP group. Because most large-scale ITP studies focus on children under 10 years or adults over 40 or 50, data on this intermediate age range are limited. As a result, current guidelines—largely based on preschool or older adult populations—frequently overlook AYAs, underscoring the need for a more nuanced, age-adapted approach.
AYAs are increasingly recognized as a distinct patient population with different health and psychosocial characteristics and expectations. This was first demonstrated in oncology (3, 4). This life stage is marked by significant physiological, hormonal, and social changes. AYAs are often navigating critical school or career milestones, and experiencing evolving social networks. Chronic ITP can substantially disrupt these developmental tasks (Figure 1): limitations on physical activity, frequent medical appointments, and the psychological stress of unpredictable bleeding episodes can affect educational progress and social integration. Moreover, treatment may cause side effects, adherence challenges, or be contraindicated in women who wish to become pregnant. Repeated corticosteroid use can cause weight gain, mood disturbances, acne, or reduced bone density—all of which can negatively impact self-esteem and health-related quality of life (HRQoL).
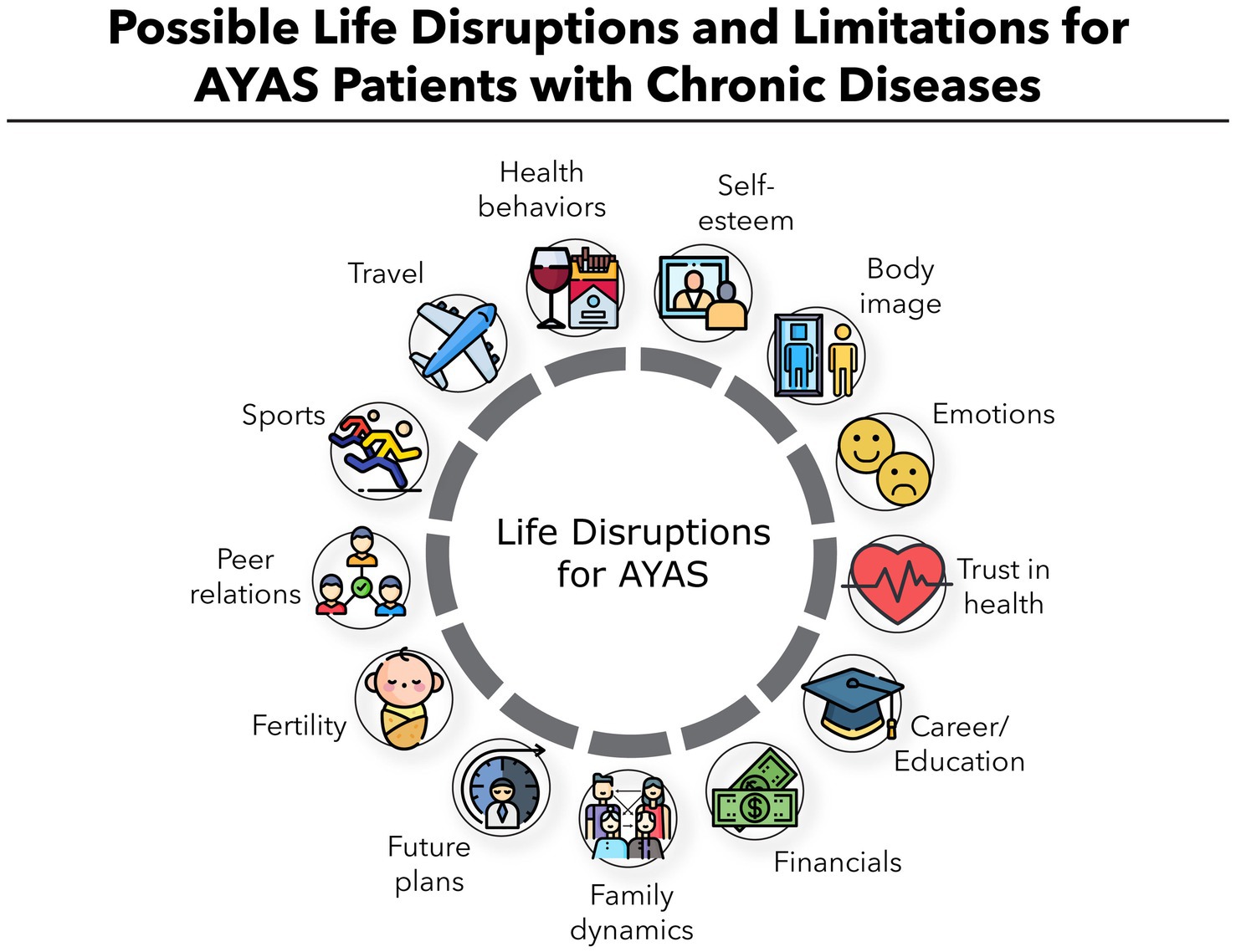
Figure 1. Possible life disruptions and limitations for AYAs patients with chronic ITP.
Studies indicate that patients over 10 years of age have a greater tendency to develop chronic ITP (5–7). This suggests that AYAs may exist in a “border zone” between the high self-limiting potential observed in younger children and the pronounced tendency for chronic disease seen in older adults. However, biologically, it could be hypothesized that the still-maturing immune system in AYAs retains enough plasticity to reverse abnormal immunological processes and restore self-tolerance. This might offer higher chances of long-term remission, provided therapies are tailored to foster immunomodulation. Preliminary findings suggest that early, more intensive immunomodulation might reduce the risk of ITP becoming chronic (8–11). Although such an approach has not been sufficiently evaluated in AYAs (11), it could be particularly beneficial in this age group. AYAs with ITP therefore require treatment strategies that balance the higher risk of chronicity and disease burden against the potential for overtreatment and side effects. Clinicians should also consider how best to support AYAs’ needs and minimize the adverse psychosocial effects of both the disease and its treatment.
The AYAs collaboration—drawing on data from the Pediatric and Adult Registry on Chronic ITP (PARC-ITP), the Cytopénies Auto-immunes Registre Midi-Pyrénéen-France (CARMEN-France) adult registry in Toulouse, and the National Prospective Cohort for Children with Chronic Autoimmune Cytopenia (OBS’CEREVANCE) in Bordeaux, France—aims to address the information gap in AYAs with ITP. To date, four analyses have been undertaken (using data from 2004 to 2021), each addressing the major clinical aspects of ITP in patients aged 12–25 years: (1) newly diagnosed ITP (12), (2) chronic disease (13), (3) refractory courses (14), and (4) secondary (sITP) forms (15).
A deeper understanding of ITP’s natural history, risk factors for chronicity, and outcomes in AYAs is a crucial first step toward developing tailored treatment algorithms. Such data could form the basis for recommendations targeting this underrepresented yet clinically distinct population. Ultimately, age-adapted trials may improve long-term outcomes, reduce toxicity, and enhance HRQoL for AYAs with ITP.
Discussion
The first year of ITP
In this first analysis, we included 656 AYAs (mean age 15.3 years, SD 2.5; 61% female) with newly diagnosed ITP from the PARC and CARMEN registries. Overall, the AYA cohort was homogeneous regarding clinical characteristics, with no clear rationale to separate adolescents from young adults or males from females.
The analysis revealed that AYAs exhibit clinical features overlapping both pediatric and adult forms of ITP (Table 1). Similar to younger children, AYAs typically had few comorbid conditions, frequent bleeding (82%), and very low platelet counts (median 12 × 109/L) at initial presentation. Yet, echoing the adult population, we observed a higher proportion of females (61%), a substantial likelihood (>50%) of chronic disease at 12 months, and a high use of corticosteroids throughout the one-year observation period, although dose and duration details were not captured. Possible reasons for persistent steroid use include the drug’s convenience, on-demand availability (e.g., for menstrual bleeding, preoperative treatment, rescue therapy), and limited or expensive access to alternatives [e.g., thrombopoietin receptor agonists (TPO-RAs)] during the study period.
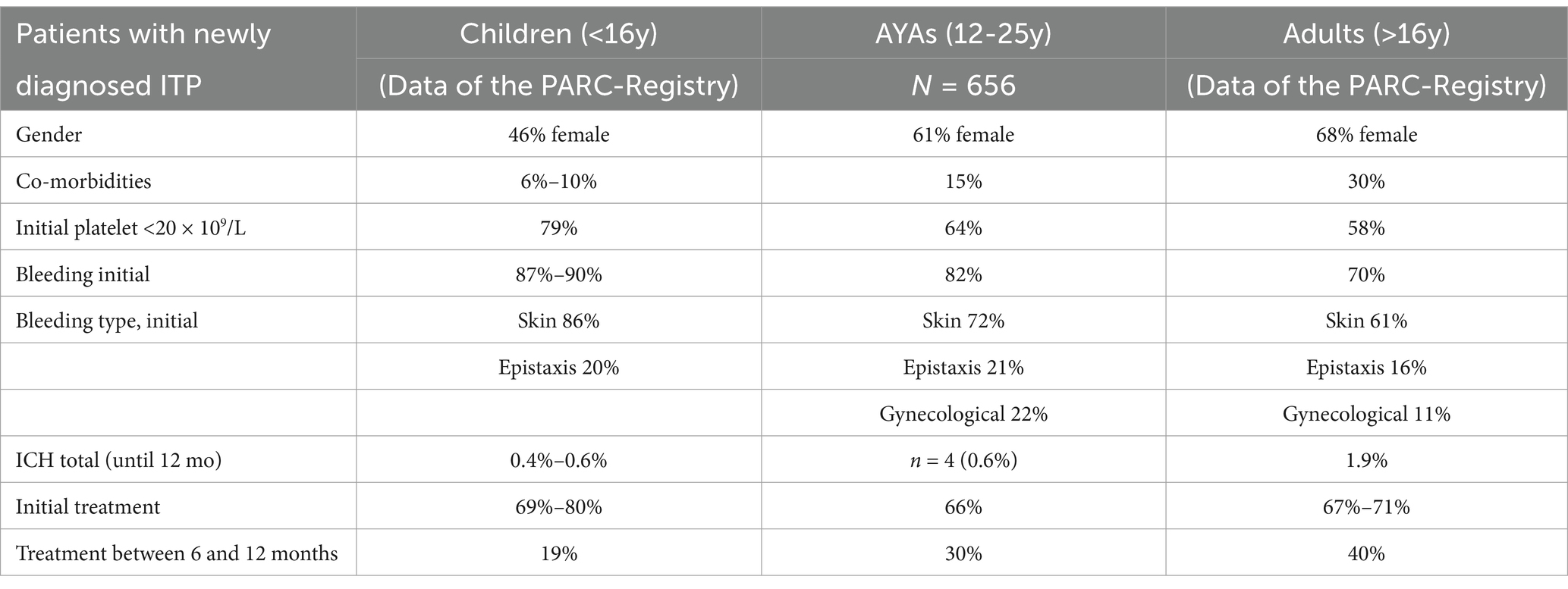
Table 1. Clinical characteristics of newly diagnosed primary ITP in different age groups [adapted and expended from Schifferli et al. (12, 23)].
Our data confirm that older pediatric patients (>10 years) face a higher risk of chronic disease, consistent with earlier studies, though definitions of “remission” and “chronic disease” vary in the literature. For example, the ICIS I registry reported remission rates of 72% in children aged 1–10 but only 53% in those aged 10–16 (6). Likewise, in a U.S. cohort of 10- to 18-year-olds (1976–2000), more than half developed chronic disease (16). In our cohort, adolescents aged 15–18 had a slightly higher chronic disease rate (61%) than those aged 12–15 (55%), supporting the continuum of age-dependent remission rates. There were no sex-based differences in remission rates, consistent with earlier reports (16).
Individuals entering remission had initial severe thrombocytopenia (<20 × 109/L) more frequently than those following a chronic course (69% vs. 56%). Conversely, 65% of patients with initially mild or moderate thrombocytopenia (≥20 × 109/L) developed chronic ITP, mirroring patterns previously reported in both pediatric and adult ITP. Interestingly, AYAs with severe thrombocytopenia who received upfront treatment were less likely to develop chronic disease than those managed with watch-and-wait (47% vs. 68%, p < 0.05), suggesting that early immunomodulation or immunosuppression may induce lasting responses in AYAs with “pediatric-like” acute severe ITP. Conversely, frontline therapy in with initial moderate or mild thrombocytopenia correlated with lower remission rates (26%) than a watch-and-wait approach (41%). One explanation could be that patients experiencing bleeding despite moderate thrombocytopenia may have alternative causes of thrombocytopenia (e.g., hereditary or secondary ITP), making standard therapies less effective.
Debate continues over whether aggressive intervention early in the disease can prevent chronicity. Emerging protocols using combined T- and B-cell–directed approaches, sometimes with TPO-RAs, have shown encouraging outcomes in small trials (8–11). Early rituximab in young women may also help establish prolonged immune tolerance (17). Because AYAs often exhibit a clinical course more akin to adults, they may benefit from proactive measures aimed at deeper immunological remission rather than merely controlling bleeding. However, further refinement is needed, particularly around identifying which AYAs are at the highest risk for chronic or refractory disease.
Chronic primary ITP
A total of 427 AYAs (64% female) with chronic primary ITP (pITP) from all three registries were included in this analysis. Overall, we observed steady clinical improvement up to 48 months of follow-up (FU) despite chronicity, evidenced by fewer bleeding events, higher mean platelet counts, and reduced overall treatment use. In total, 67 patients (16%) were managed with a watch-and-wait strategy until the last available FU (37 had complete data up to 48 months), while 167 (39%) received first-line therapies [corticosteroids, intravenous immunoglobulins (IVIG)] only. This finding aligns with pediatric data indicating that many children remain on first-line treatment alone—likely reflecting an on-demand approach (1, 18). In total, 188 (44%) patients received second-line drugs until the last available FU. Despite the increasing use of second-line agents over time among treated patients, the proportion receiving IVIG remained stable at about 40% across all FUs, possibly reflecting platelet fluctuations, breakthrough bleeding, or difficulties tapering treatment. Corticosteroids were administered to approximately 31–47% of treated patients, though dosage and duration were not reported. It is to note that TPO-RAs, licensed for pediatric ITP since 2016/2018, are underrepresented in our analysis, and treatment practices have evolved considerably since then.
Bleeding patterns stayed largely unchanged, with 60% of symptomatic patients presenting wet bleeding, but gynecologic bleeding rose as more girls entered puberty. Intracranial hemorrhage (ICH) was rare; six of eight events occurred before chronicity, aligning with studies indicating that ICH tends to appear early in the disease course and more frequently in older adults (≥60 years) (19–22).
Among the 427 patients with chronic ITP at 12 months, 99 (23%) achieved sustained complete remission off treatment (SCROT) during subsequent FUs, evenly spread over 3 years. SCROT was defined as platelet count >100 × 109/L without treatment for at least 12 months, independently of the previous treatment strategy. This result likely underestimates true remission rates, as those lost to FU often have milder disease courses (23). Unfortunately, variations in chronic ITP definitions, remission criteria (platelet threshold and time off treatment), and study designs complicate direct comparisons with published data (24–31). Nonetheless, in our cohort, roughly 10% of chronic ITP patients per year attained SCROT, matching some pediatric findings (31).
Identifying predictors of remission is crucial for tailoring treatment and counseling patients. Previous publications show that factors such as older age (>10 years), higher initial platelet counts, or lack of severe bleeding are associated with chronic disease risk in all age categories (6, 16, 32–34), yet evidence for late remission predictors remains limited (7, 31, 35–37). In our study, AYAs who achieved “late SCROT” (12–36 months) tended to show a more favorable disease course during the first year (excluding initial presentation) than those with “ongoing chronic disease.” Indeed, these patients presented with higher platelet counts, less bleeding, and reduced IVIG use between 6 and 12 months. This pattern was not observed in AYAs who achieved “very late SCROT” (between 36 and 48 months).
Overall, although AYAs have a higher risk of chronic disease at 12 months than younger children, their late remission rates and treatment profiles resemble pediatric patterns (Table 2). Because a notable proportion eventually achieves SCROT beyond 1 year, clinicians should consider postponing splenectomy.
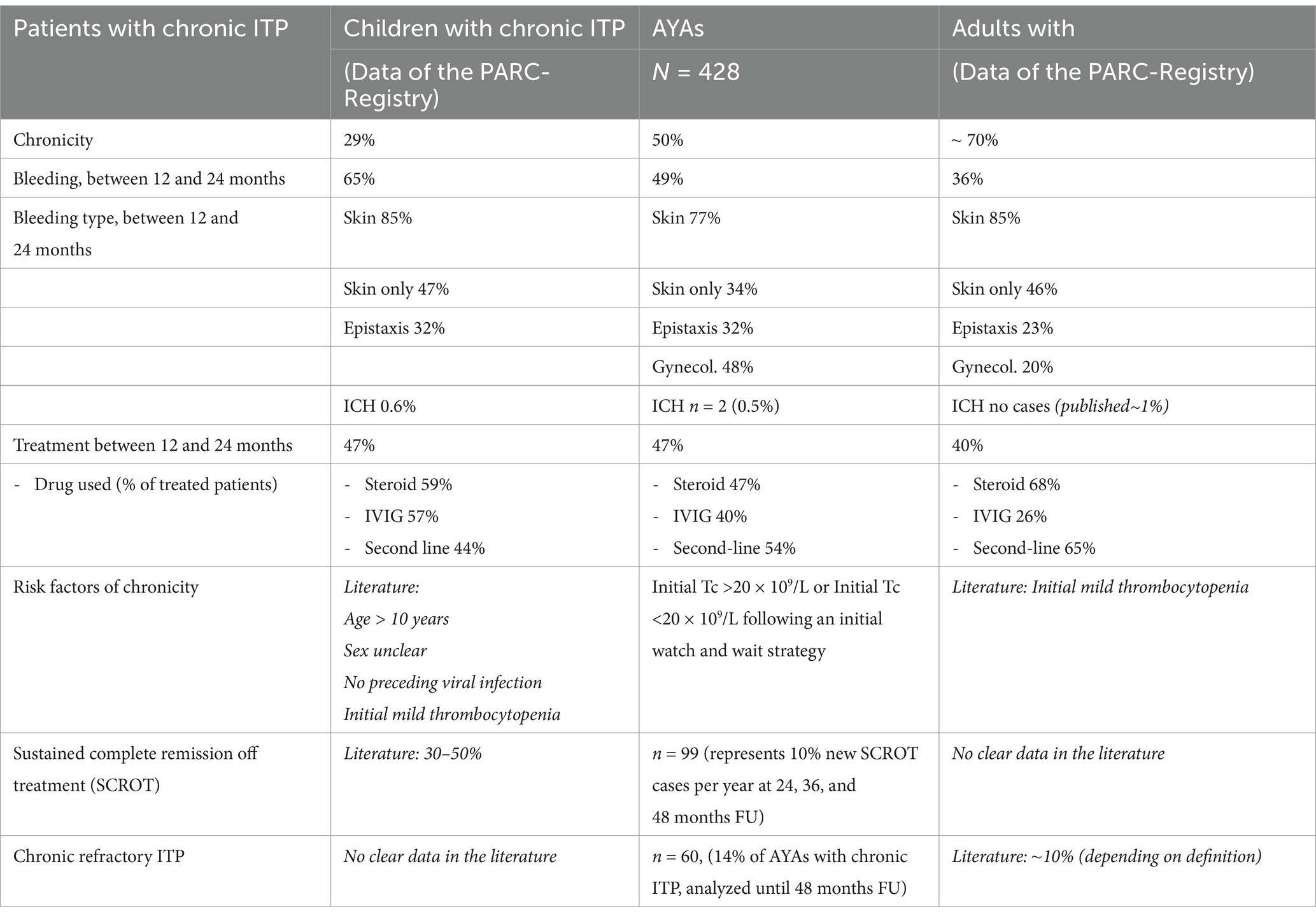
Table 2. Clinical characteristics of chronic primary ITP in different age groups [adapted and expended from Schifferli et al. (13, 23)].
Chronic refractory ITP
Only a small proportion (14%) of AYAs with chronic pITP in our cohort met our criteria for refractory disease, defined as the administration of ≥2 second-line treatments. Given that about half of AYAs develop chronic ITP, the overall proportion of newly diagnosed patients who will develop chronic refractory ITP is approximately 7%, which aligns with adult epidemiological estimates (~3%) (38). Median age at diagnosis was similar in both chronic refractory and chronic non-refractory groups, but the proportion of males was higher in the refractory group (43%) compared to the non-refractory group (35%). This difference was even greater among patients who were already refractory at 12 months (early refractory, n = 29, 48% male).
Patients with refractory disease had a notably higher clinical burden, including lower platelet counts, more frequent bleeding (especially wet bleeding), and greater ongoing treatment requirements compared to those with non-refractory ITP. In those identified as refractory early in the disease course, platelet counts and bleeding rates were even more severe at diagnosis and during the first year of FU, consistent with the literature (39). The proportion of patients with initial platelet counts <20 × 109/L was 57% in the non-refractory group, 64% in the refractory group, and 74% in the early refractory subgroup. Findings from the French CARMEN registry of adults with multirefractory ITP similarly showed very low initial platelet counts (6 × 109/L) and a high early bleeding rate (88%) (38). Likewise, a 2016 multicenter study by Mahévas et al. (40) on 37 multirefractory patients compared with a historical ITP cohort (n = 183) identified lower baseline platelet counts (median 9 × 109/L vs. 17 × 109/L) and higher bleeding/infection-related morbidity and mortality in the refractory group, although no significant gender differences emerged.
Despite the extensive use of multiline therapies at all FU points in our study, about 25% of AYAs with refractory disease continued to rely on corticosteroids at last FU (48 months), compared with only 10% of non-refractory patients. This persistent steroid use remains concerning, given that long-term or excessive steroid therapy is the most common toxicity in ITP. Corticosteroids often serve as an effective add-on or on-demand option for menstruating women, pre-procedural settings, travel, or emergencies. However, access to TPO-receptor agonists was limited in the study period, particularly for children under 16.
Overall, refractory ITP in AYAs appears to present with more severe disease features early on, implying a potentially distinct disease biology rather than just a gradual progression. However, the pathogenesis remains poorly understood and various hypotheses exists supporting progression (epitope spreading, increased drug-efflux pumps, transition from antibody- to T-cell–mediated autoimmunity, oligoclonal or monoclonal T-cell receptor expansions, expansion of long-lived plasma cells) (41–47), and supporting distinct diseases [different etiopathologies or antigen targets (48, 49), misdiagnoses like hereditary thrombocytopenia, bone marrow failure, or sITP (39, 40)]. Mahévas et al. (40) noted that 35% of multirefractory cases had sITP, versus only 9% in the control group. This heterogeneity underscores the challenges of labeling patients as “refractory” or “difficult to treat,” since they comprise a highly diverse subgroup.
Our definition of refractory ITP can also influence its comparability with other studies. Existing guidelines (50) define refractory disease as not responding to splenectomy, a criterion less applicable to adolescents, for whom splenectomy is often delayed. Although new studies have expanded the criteria, they are frequently too stringent (40), and often fail to capture the “difficult-to-treat” patients that clinicians encounter in routine practice. Our pragmatic approach—defining refractory disease as requiring ≥2 second-line therapies—focuses on a small group with hard-to-manage ITP and is easy to apply in both clinical and research settings. Strengthening the definition further would significantly reduce the number of identified patients, thereby limiting the potential for useful data analysis.
In conclusion, although refractory ITP in AYAs is relatively infrequent, the disease burden is high. Individuals most likely to follow a refractory course are often male, present with very low platelet counts at diagnosis, and experience repeated wet bleeding in the first year. However, these early distinctions alone do not reliably predict long-term outcomes or justify aggressive early intervention or study enrollment. Hence, new early markers of refractoriness are needed to refine patient stratification, and guide treatment strategies in AYAs.
Chronic secondary ITP
Among 481 AYAs with chronic disease from all three registries, 94 had an identifiable underlying condition (sITP), representing 20% of chronic cases and 10% of all newly diagnosed ITP. This rate lies between pediatric (3%) and adult (20%) estimates, confirming that sITP becomes more common with age. In our cohort, systemic autoimmune disorders (SAD) accounted for the largest proportion of sITP (57%), followed by Evans syndrome (ES, 20%) and primary immunodeficiencies (PID, 18%) (Figure 2). These subgroups comprise therefore 11% (SAD), 4% (ES), and 3.5% (PID) of all chronic ITP. Existing studies on sITP often mix acute and chronic patients, making direct comparisons difficult. Nonetheless, the prominence of SAD conditions—particularly systemic lupus erythematosus (SLE)—aligns with previous reports, given that about one-third of SLE patients develop thrombocytopenia, and 2–5% of adults with ITP are later diagnosed with SLE.
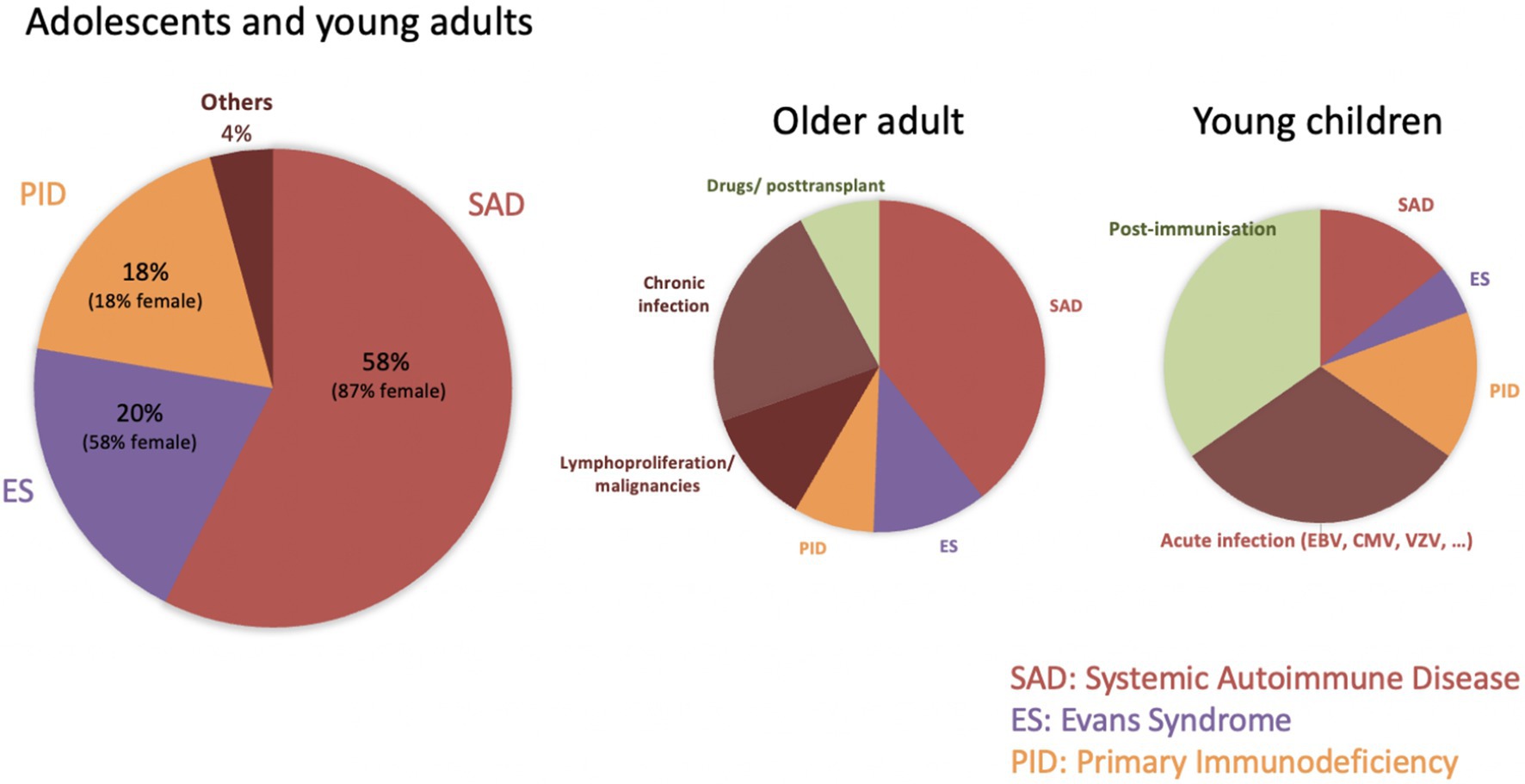
Figure 2. Causes of secondary ITP in different age groups.
Marked gender imbalances emerged among sITP subgroups, with 87% of SAD cases being female (reflecting the known female predominance in autoimmune disorders) and 82% of PID cases being male. Despite these differences, initial disease features (platelet count, bleeding incidence and types, and treatment need) were similar across the three sITP subgroups and the chronic pITP cohort.
Only 27% of sITP cases were recognized at ITP onset, whereas 30% were diagnosed more than 2 years later. Delayed diagnoses were particularly common in PID. These findings underscore the need for ongoing re-evaluation of chronic ITP, consistent with adult studies that report about 12% of presumed pITP cases are reclassified during FU (51). Current guidelines support systematic screening when new symptoms or laboratory abnormalities arise (52), and recent publications emphasize the utility of genetic testing in pediatric ITP with atypical (chronic or refractory) presentations (53, 54). Although disease-related genetic findings in ITP are rare, they can guide patient counseling and targeted therapies. Additionally, broader use of genetic diagnostics will gradually improve our understanding of immune dysregulation. In Evans syndrome, 39–65% of children exhibit PID-related gene variants, underscoring the importance of genetic counseling in multiple cytopenias (55, 56).
Based on our observations (Figure 2), we recommend biannual assessments for AYAs with persistent or chronic ITP, comprising a thorough clinical evaluation, complete blood count with reticulocytes and blood smear review, and Coombs testing if indicated. For females, screening for autoimmune markers (e.g., ANA) and systemic autoimmune symptoms is warranted, while immunodeficiency screening (e.g., immunoglobulin levels) should be a priority for males. These measures can help detect secondary causes of ITP earlier, facilitating timely and individualized management.
Conclusion
Collectively, our four analyses demonstrate that AYAs with ITP do not fit neatly into traditional “pediatric” or “adult” categories. The initial presentation of ITP in AYAs resembles pediatric ITP (abrupt, severe, symptomatic thrombocytopenia with very low risk of ICH), but the disease course resembles adult ITP. However, AYAs have fewer comorbidities and secondary forms and may retain a greater potential for late remission compared to older adults. Although most AYAs with chronic ITP maintain relatively stable courses over time, a subset progresses to refractory or sITP, each with distinct clinical and therapeutic challenges.
With their longer life expectancies and higher HRQoL standards, AYAs require new approaches that focus on sustainable immunomodulation rather than continuous, symptom-driven management. Future studies should therefore:
1. Clearly define AYAs as a distinct patient subgroup rather than combining them with either pediatric or adult cohorts.
2. Refine treatment goals to reduce long-term chronicity, including earlier, targeted immunomodulation when appropriate.
3. Discourage extended corticosteroid use beyond the acute phase to minimize toxicity and preserve HRQoL.
4. Improve recognition and workups for secondary ITP, enabling timely, individualized care for those with underlying autoimmune disorders or immunodeficiencies.
5. Address quality of life in AYAs’ care: given the multitude of developmental and social challenges at this life stage, it is crucial to look beyond ITP symptoms and treatments alone, as the disease can negatively affect diverse aspects of their lives (Figure 1) (57–59).
Author contributions
AS: Conceptualization, Funding acquisition, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The Platelet Disease Support Association: The Barbara and Peter T. Pruitt Jr. ITP Research Award 2022 and 2023.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor TJG-L declared a past co-authorship with the author AS.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Provan, D, Arnold, DM, Bussel, JB, Chong, BH, Cooper, N, Gernsheimer, T, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. (2019) 3:3780–817. doi: 10.1182/bloodadvances.2019000812
2. Neunert, C, Terrell, DR, Arnold, DM, Buchanan, G, Cines, DB, Cooper, N, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. (2019) 3:3829–66. doi: 10.1182/bloodadvances.2019000966
3. Bibby, H, White, V, Thompson, K, and Anazodo, A. What are the unmet needs and care experiences of adolescents and young adults with cancer? A systematic review. J Adolesc Young Adult Oncol. (2017) 6:6–30. doi: 10.1089/jayao.2016.0012
4. Nass, SJ, Beaupin, LK, Demark-Wahnefried, W, Fasciano, K, Ganz, PA, Hayes-Lattin, B, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. (2015) 20:186–95. doi: 10.1634/theoncologist.2014-0265
5. Heitink-Pollé, KM, Nijsten, J, Boonacker, CW, de Haas, M, and Bruin, MC. Clinical and laboratory predictors of chronic immune thrombocytopenia in children: a systematic review and meta-analysis. Blood. (2014) 124:3295–307. doi: 10.1182/blood-2014-04-570127
6. Kühne, T, Buchanan, GR, Zimmerman, S, Michaels, LA, Kohan, R, Berchtold, W, et al. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the intercontinental childhood ITP study group. J Pediatr. (2003) 143:605–8. doi: 10.1067/S0022-3476(03)00535-3
7. Donato, H, Picón, A, Martinez, M, Rapetti, MC, Rosso, A, Gomez, S, et al. Demographic data, natural history, and prognostic factors of idiopathic thrombocytopenic purpura in children: a multicentered study from Argentina. Pediatr Blood Cancer. (2009) 52:491–6. doi: 10.1002/pbc.21872
8. Gómez Almaguer, D, Colunga Pedraza, PR, Gómez de León, A, Gutiérrez Aguirre, CH, Cantú Rodríguez, OG, and Jaime Pérez, JC. Eltrombopag, low-dose rituximab, and dexamethasone combination as frontline treatment of newly diagnosed immune thrombocytopaenia. Br J Haematol. (2019) 184:288–90. doi: 10.1111/bjh.15070
9. Zhang, L, Zhang, M, Du, X, Cheng, Y, and Cheng, G. Safety and efficacy of eltrombopag plus pulsed dexamethasone as first-line therapy for immune thrombocytopenia. Br J Haematol. (2020) 189:369–78. doi: 10.1111/bjh.16327
10. Bradbury, CA, Pell, J, Hill, Q, Bagot, C, Cooper, N, Ingram, J, et al. Mycophenolate mofetil for first-line treatment of immune thrombocytopenia. N Engl J Med. (2021) 385:885–95. doi: 10.1056/NEJMoa2100596
11. Schifferli, A, Rüfer, A, Rovo, A, Nimmerjahn, F, Cantoni, N, Holbro, A, et al. Immunomodulation with romiplostim as a second-line strategy in primary immune thrombocytopenia: the iROM study. Br J Haematol. (2023) 203:119–30. doi: 10.1111/bjh.19074
12. Schifferli, A, Moulis, G, Godeau, B, Leblanc, T, Aladjidi, N, Michel, M, et al. Adolescents and young adults with newly diagnosed primary immune thrombocytopenia. Haematologica. (2023) 108:2783–93. doi: 10.3324/haematol.2022.282524
13. Schifferli, A, Le Gavrian, G, Aladjidi, N, Moulis, G, Godeau, B, Leblanc, T, et al. Sustained remission at long term follow-up in adolescents and young adults with chronic primary immune thrombocytopenia. Blood Adv. (2024) 8:6183–94. doi: 10.1182/bloodadvances.2024014381
14. Schifferli, A, Le Gavrian, G, Aladjidi, N, Moulis, G, Godeau, B, and Kühne, T. Chronic refractory immune thrombocytopenia in adolescents and young adults. Br J Haematol. (2023) 203:36–42. doi: 10.1111/bjh.19081
15. Schifferli, A, Aladjidi, N, Moulis, G, Gisin, M, Leverger, G, Heritier, S, et al. Adolescents and young adults with secondary chronic immune thrombocytopenia: a Switzerland/France collaboration project. Blood. (2024) 144:548. doi: 10.1182/blood-2024-200034
16. Lowe, EJ, and Buchanan, GR. Idiopathic thrombocytopenic purpura diagnosed during the second decade of life. J Pediatr. (2002) 141:253–8. doi: 10.1067/mpd.2002.125909
17. Bussel, JB, Lee, CS, Seery, C, Imahiyerobo, AA, Thompson, MV, Catellier, D, et al. Rituximab and three dexamethasone cycles provide responses similar to splenectomy in women and those with immune thrombocytopenia of less than two years duration. Haematologica. (2014) 99:1264–71. doi: 10.3324/haematol.2013.103291
18. Ducassou, S, Gourdonneau, A, Fernandes, H, Leverger, G, Pasquet, M, Fouyssac, F, et al. Second-line treatment trends and long-term outcomes of 392 children with chronic immune thrombocytopenic purpura: the French experience over the past 25 years. Br J Haematol. (2020) 189:931–42. doi: 10.1111/bjh.16448
19. Hato, T, Shimada, N, Kurata, Y, Kuwana, M, Fujimura, K, Kashiwagi, H, et al. Risk factors for skin, mucosal, and organ bleeding in adults with primary ITP: a nationwide study in Japan. Blood Adv. (2020) 4:1648–55. doi: 10.1182/bloodadvances.2020001446
20. Tsuda, H, Tsuji, T, Tsuji, M, and Yamasaki, H. Life-threatening bleeding episodes in primary immune thrombocytopenia: a single-center retrospective study of 169 inpatients. Ann Hematol. (2017) 96:1915–20. doi: 10.1007/s00277-017-3095-6
21. Psaila, B, Petrovic, A, Page, LK, Menell, J, Schonholz, M, and Bussel, JB. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood. (2009) 114:4777–83. doi: 10.1182/blood-2009-04-215525
22. Butros, LJ, and Bussel, JB. Intracranial hemorrhage in immune thrombocytopenic purpura: a retrospective analysis. J Pediatr Hematol Oncol. (2003) 25:660–4. doi: 10.1097/00043426-200308000-00017
23. Schifferli, A, Holbro, A, Chitlur, M, Coslovsky, M, Imbach, P, Donato, H, et al. A comparative prospective observational study of children and adults with immune thrombocytopenia: 2-year follow-up. Am J Hematol. (2018) 93:751–9. doi: 10.1002/ajh.25086
24. Rosthøj, S, Rajantie, J, Treutiger, I, Zeller, B, Tedgård, U, Henter, JI, et al. Duration and morbidity of chronic immune thrombocytopenic purpura in children: five-year follow-up of a Nordic cohort. Acta Paediatr. (2012) 101:761–6. doi: 10.1111/j.1651-2227.2012.02671.x
25. Imbach, P, Kühne, T, Müller, D, Berchtold, W, Zimmerman, S, Elalfy, M, et al. Childhood ITP: 12 months follow-up data from the prospective registry I of the intercontinental childhood ITP study group (ICIS). Pediatr Blood Cancer. (2006) 46:351–6. doi: 10.1002/pbc.20453
26. Medeiros, D, and Buchanan, GR. Current controversies in the management of idiopathic thrombocytopenic purpura during childhood. Pediatr Clin N Am. (1996) 43:757–72. doi: 10.1016/S0031-3955(05)70431-4
27. Blanchette, VS, and Price, V. Childhood chronic immune thrombocytopenic Purpura: unresolved issues. J Pediatr Hematol Oncol. (2003) 25:S28–33. doi: 10.1097/00043426-200312001-00007
28. Shim, YJ, Kim, UH, Suh, JK, and Lee, KS. Natural course of childhood chronic immune thrombocytopenia using the revised terminology and definitions of the international working group: a single center experience. Blood Res. (2014) 49:187–91. doi: 10.5045/br.2014.49.3.187
29. Kim, CY, Lee, EH, and Yoon, HS. High remission rate of chronic immune thrombocytopenia in children: result of 20-year follow-up. Yonsei Med J. (2016) 57:127–31. doi: 10.3349/ymj.2016.57.1.127
30. Donato, H, Picón, A, Rapetti, MC, Rosso, A, Schvartzman, G, Drozdowski, C, et al. Splenectomy and spontaneous remission in children with chronic idiopathic thrombocytopenic purpura. Pediatr Blood Cancer. (2006) 47:737–9. doi: 10.1002/pbc.20982
31. Jayabose, S, Levendoglu-Tugal, O, Ozkaynkak, MF, Visintainer, P, and Sandoval, C. Long-term outcome of chronic idiopathic thrombocytopenic purpura in children. J Pediatr Hematol Oncol. (2004) 26:724–6. doi: 10.1097/00043426-200411000-00007
32. Bennett, CM, Neunert, C, Grace, RF, Buchanan, G, Imbach, P, Vesely, SK, et al. Predictors of remission in children with newly diagnosed immune thrombocytopenia: data from the intercontinental cooperative ITP study group registry II participants. Pediatr Blood Cancer. (2018) 65:e26736. doi: 10.1002/pbc.26736
33. Grimaldi-Bensouda, L, Nordon, C, Leblanc, T, Abenhaim, L, Allali, S, Armari-Alla, C, et al. Childhood immune thrombocytopenia: a nationwide cohort study on condition management and outcomes. Pediatr Blood Cancer. (2017) 64:e26389. doi: 10.1002/pbc.26389
34. Tamminga, RYJ, Berchtold, W, Bruin, MCA, Buchanan, GR, and Kuehne, T. Less chronic ITP after IVIG for acute childhood ITP? A matched pairs analysis from registry I of the intercontinental ITP study group (ICIS). Blood. (2007) 110:3917–7. doi: 10.1182/blood.V110.11.3917.3917
35. Bansal, D, Bhamare, TA, Trehan, A, Ahluwalia, J, Varma, N, and Marwaha, RK. Outcome of chronic idiopathic thrombocytopenic purpura in children: chronic ITP in children. Pediatr Blood Cancer. (2010) 54:403–7. doi: 10.1002/pbc.22346
36. Tamary, H, Kaplinsky, C, Levy, I, Cohen, IJ, Yaniv, I, Stark, B, et al. Chronic childhood idiopathic thrombocytopenia purpura: long-term follow-up. Acta Paediatr. (1994) 83:931–4. doi: 10.1111/j.1651-2227.1994.tb13175.x
37. Calleja Gero, ML, Sevilla, J, and Madero, L. What is the prognosis of chronic immune thrombocytopenia? An Pediatr (Barc). (2011) 74:317–23. doi: 10.1016/j.anpedi.2010.09.035
38. Moulis, G, Rueter, M, Mahevas, M, Viallard, JF, Comont, T, Chèze, S, et al. Multirefractory primary immune thrombocytopenia in adults: prevalence and burden. Results from the CARMEN-France registry. Blood. (2022) 140:5557–8. doi: 10.1182/blood-2022-157723
39. Miltiadous, O, Hou, M, and Bussel, JB. Identifying and treating refractory ITP: difficulty in diagnosis and role of combination treatment. Blood. (2020) 135:472–90. doi: 10.1182/blood.2019003599
40. Mahévas, M, Gerfaud-Valentin, M, Moulis, G, Terriou, L, Audia, S, Guenin, S, et al. Characteristics, outcome, and response to therapy of multirefractory chronic immune thrombocytopenia. Blood. (2016) 128:1625–30. doi: 10.1182/blood-2016-03-704734
41. Cines, DB, and Blanchette, VS. Immune thrombocytopenic purpura. N Engl J Med. (2002) 346:995–1008. doi: 10.1056/NEJMra010501
42. Levy, AS, Cunningham-Rundles, S, Mazza, B, Simm, M, Gorlick, R, and Bussel, J. High P-glycoprotein-mediated export observed in patients with a history of idiopathic thrombocytopenic purpura. Br J Haematol. (2002) 118:836–8. doi: 10.1046/j.1365-2141.2002.03709.x
43. Chapin, J, Lee, CS, Zhang, H, Zehnder, JL, and Bussel, JB. Gender and duration of disease differentiate responses to rituximab-dexamethasone therapy in adults with immune thrombocytopenia. Am J Hematol. (2016) 91:907–11. doi: 10.1002/ajh.24434
44. Stasi, R, Cooper, N, Del Poeta, G, Stipa, E, Laura Evangelista, M, Abruzzese, E, et al. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood. (2008) 112:1147–50. doi: 10.1182/blood-2007-12-129262
45. Zhang, H, Zhang, BM, Guo, X, Xu, L, You, X, West, RB, et al. Blood transcriptome and clonal T cell correlates of response and nonresponse to eltrombopag therapy in a cohort of patients with chronic immune thrombocytopenia. Haematologica. (2020) 105:e129–32. doi: 10.3324/haematol.2019.226688
46. Fogarty, PF, Rick, ME, Zeng, W, Risitano, AM, Dunbar, CE, and Bussel, JB. T cell receptor VB repertoire diversity in patients with immune thrombocytopenia following splenectomy. Clin Exp Immunol. (2003) 133:461–6. doi: 10.1046/j.1365-2249.2003.02239.x
47. Mahévas, M, Patin, P, Huetz, F, Descatoire, M, Cagnard, N, Bole-Feysot, C, et al. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. J Clin Invest. (2013) 123:432–42. doi: 10.1172/JCI65689
48. Zeng, Q, Zhu, L, Tao, L, Bao, J, Yang, M, Simpson, EK, et al. Relative efficacy of steroid therapy in immune thrombocytopenia mediated by anti-platelet GPIIbIIIa versus GPIbα antibodies. Am J Hematol. (2012) 87:206–8. doi: 10.1002/ajh.22211
49. Webster, ML, Sayeh, E, Crow, M, Chen, P, Nieswandt, B, Freedman, J, et al. Relative efficacy of intravenous immunoglobulin G in ameliorating thrombocytopenia induced by antiplatelet GPIIbIIIa versus GPIbalpha antibodies. Blood. (2006) 108:943–6. doi: 10.1182/blood-2005-06-009761
50. Rodeghiero, F, Stasi, R, Gernsheimer, T, Michel, M, Provan, D, Arnold, DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. (2009) 113:2386–93. doi: 10.1182/blood-2008-07-162503
51. Arnold, DM, Nazy, I, Clare, R, Jaffer, AM, Aubie, B, Li, N, et al. Misdiagnosis of primary immune thrombocytopenia and frequency of bleeding: lessons from the McMaster ITP registry. Blood Adv. (2017) 1:2414–20. doi: 10.1182/bloodadvances.2017010942
52. Matzdorff, A, Alesci, SR, Gebhart, J, Holzhauer, S, Hütter-Krönke, ML, Kühne, T, et al. Expert report on immune thrombocytopenia: current diagnostics and treatment-recommendations from an expert group from Austria, Germany, and Switzerland. Oncol Res Treat. (2023) 46:5–44. doi: 10.1159/000529662
53. Rotz, SJ, Ware, RE, and Kumar, A. Diagnosis and management of chronic and refractory immune cytopenias in children, adolescents, and young adults. Pediatr Blood Cancer. (2018) 65:e27260. doi: 10.1002/pbc.27260
54. Zhao, S, Ma, J, Zhu, X, Zhang, J, and Wu, R. Chronic refractory immune thrombocytopenia is associated with variants in immune genes. Clin Appl Thromb Hemost. (2021) 27:10760296211059813. doi: 10.1177/10760296211059813
55. Aladjidi, N, Pincez, T, Rieux-Laucat, F, and Nugent, D. Paediatric-onset Evans syndrome: breaking away from refractory immune thrombocytopenia. Br J Haematol. (2023) 203:28–35. doi: 10.1111/bjh.19073
56. Hadjadj, J, Aladjidi, N, Fernandes, H, Leverger, G, Magérus-Chatinet, A, Mazerolles, F, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. (2019) 134:9–21. doi: 10.1182/blood-2018-11-887141
57. Phillips, RH. Adolescents and chronic illness. Available online at: https://pdsa.org/images/AdolescentsChronicIllness.pdf
58. Platelet Disorder Support Association. ITP in teens—frequently asked questions. Available online at: https://pdsa.org/images/stories/pdf/itp-in-teens-engl.pdf
Keywords: ITP, AYAs, secondary ITP, chronic ITP, sustained remission
Citation: Schifferli A (2025) Immune thrombocytopenia in adolescents and young adults. Front. Med. 12:1553936. doi: 10.3389/fmed.2025.1553936
Edited by:
Tomás José González-López, Burgos University Hospital, SpainReviewed by:
Neelufar Mozaffarian, Ouro Medicines, United StatesCopyright © 2025 Schifferli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandra Schifferli, YWxleGFuZHJhLnNjaGlmZmVybGlAdWtiYi5jaA==