 Xinlei Zhang1†
Xinlei Zhang1† Zengze Yuan
Zengze Yuan Junchao Yang
Junchao Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Med. , 20 March 2025
Sec. Pulmonary Medicine
Volume 12 - 2025 | https://doi.org/10.3389/fmed.2025.1543571
Background: Idiopathic pulmonary fibrosis (IPF) is a progressive and irreversible interstitial lung disease characterized by high mortality rates. An expanding body of evidence highlights the critical role of targeted therapies in the management of IPF. Nevertheless, there is a paucity of bibliometric studies that have comprehensively assessed this domain. This study seeks to examine global literature production and research trends related to targeted therapies for IPF.
Method: A literature search was conducted using the Web of Science Core Collection, encompassing publications from 2004 to 2024, focusing on targeted therapies for IPF. The bibliometric analysis utilized tools such as VOSviewer, CiteSpace, and the “bibliometrix” package in R.
Results: A total of 2,779 papers were included in the analysis, demonstrating a general trend of continuous growth in the number of publications over time. The United States contributed the highest number of publications, totaling 1,052, while France achieved the highest average citation rate at 75.74. The University of Michigan Medical School was the leading institution in terms of publication output, with 88 papers. Principal Investigator Naftali Kaminski was identified as the most prolific researcher in the field. The American Journal of Respiratory Cell and Molecular Biology emerged as the journal with the highest number of publications, featuring 98 articles. In recent years, the research has emerged surrounding targeted therapies for IPF, particularly focusing on agents such as TGF-β, pathogenesis, and autotaxin inhibitor.
Conclusion: In this bibliometric study, we systematically analyze research trends related to targeted therapies for IPF, elucidating recent research frontiers and emerging directions. The selected keywords-idiopathic pulmonary fibrosis, targeted therapy, bibliometric analysis, transforming growth factor β, and autotaxin inhibitor—capture the essential aspects of this research domain. This analysis serves as a reference point for future investigations into targeted therapies.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and incurable interstitial lung disease of unknown etiology, predominantly affecting middle-aged and elderly individuals, and is associated with a poor prognosis. The primary clinical manifestations include progressive dyspnea and reduced lung compliance (1). IPF is characterized by the destruction of normal lung architecture, persistent production and activation of myofibroblasts, and excessive accumulation of extracellular matrix, ultimately resulting in pulmonary fibrosis (2). The disease is estimated to affect ~3 million individuals globally, with its incidence on the rise and a high mortality rate and a median survival of 2–5 years after diagnosis (3–5). Currently, lung transplantation is regarded as the only effective treatment for IPF, but the therapy is only applicable to a small number of patients due to limitations in donor organ availability and chronic allogeneic rejection (6). The discovery of therapeutic targets highly relevant to pulmonary fibrosis and the development of effective antifibrotic therapies against these targets have been ongoing research priorities.
At present, pirfenidone and nintedanib are the only antifibrotic medications authorized for use in clinical settings (7). Pirfenidone works by decreasing the expression and activity of transforming growth factor β (TGF-β), which in turn lessens fibroblast activation and collagen production while also mitigating lung inflammation through inhibition of pro-inflammatory cytokines and chemokines. Nintedanib, a tyrosine kinase inhibitor, targets receptors associated with fibrosis (8). Although both medications help slow the decline in lung function and extend survival in individuals with IPF (9–11), they do not reverse the disease and have side effects such as gastrointestinal reactions (12). Therefore, the development of new anti-pulmonary fibrosis drugs is imperative. Over the past few years, many new therapeutic targets and drugs have emerged in clinical trials for IPF therapy. For example, dysregulation of phosphodiesterase 4B (PDE4B) can cause inflammation and fibrosis through hyperproliferation and activation. BI1015550, a novel PDE4B-selective inhibitor, has entered a phase III clinical study and prevented the decline of lung function in patients with IPF within 12 weeks (13). In all, current medications for IPF do not reverse the disease; novel targets and targeted therapeutic agents for IPF are being explored to improve efficacy, and adherence to the principles of precision medicine is urgent.
Bibliometrics is a discipline that studies the characteristics of literature and utilizes statistical methods to analyze it statistically (14). Bibliometric analysis facilitates a systematic approach to interpreting extensive volumes of unstructured data, enabling the elucidation and representation of the accumulated scientific knowledge and developmental intricacies within a well-established field. Currently, although a large number of studies have thoroughly researched and reviewed targeted therapies for IPF, there is currently no relevant literature that systematically analyzes and compares the field. This study aimed to offer a comprehensive bibliometric examination of publications and pertinent data concerning targeted therapies for IPF during the past 20 years (from January 1, 2004, to September 27, 2024) in order to evaluate the present, the current foci or hotspots and predict next research trends in the field.
In this research, the Science Citation Index Expanded (SCIE 1999–present) of Clarivate Analytics's Web of Science Core Collection (WoSCC) was utilized as the primary data source for conducting a systematic search and extracting data from literature published between 2004 and 2024. The searching strategy was formulated with reference to previous researches and the searching strategy was shown as follows: topic = (“Targeted Therapy” OR “Targeted” OR “Molecular Targeted Therapies” OR “Molecular Targeted” OR “Therapeutic Targets” OR “Targeted Drugs”) AND topic = (“IPF” OR “Idiopathic pulmonary fibrosis”). The search was carried out on September 27, 2024, covering the period from January 1, 2004, to that same date. To eliminate potential bias from daily database updates, all searches and downloads were completed within a single day. This process resulted in the retrieval of 2,913 relevant articles. After excluding 5 non-English articles and restricting the article types to theses and reviews to ensure the quality of the studies, a total of 2,779 relevant articles (2,014 theses and 765 reviews) were included, excluding meeting abstracts, editorial material, proceeding papers, early access, etc.
We conducted an analysis of the WoSCC database to evaluate bibliometric indicators such as annual publications, countries, institutions, authors, journals, citations, and keywords. Utilizing Microsoft Excel 2021, we performed quantitative analyses to determine the yearly publication totals and the average citations per paper. Additionally, we assessed both the annual and cumulative publication counts for each country, as well as the overall number of papers authored by institutions, authors, and journals, in order to evaluate the quality of the publications. To evaluate the quality of scientific information, we primarily relied on the impact factor (IF) and category data provided by the Journal Citation Reports (JCR) for the year 2023. In some cases, we also used the H index to assess the scholarly achievements of countries, institutions, journals, and researchers. The H index refers to the fact that a research researcher has published at least H papers and each paper has been cited at least H times. The H index strikes a balance between the quantity and quality of the papers (represented by the number of citations), and it is an effective measure of the scholarly impact and scientific output of researchers, countries, institutions, and journals (15). For the visualization analysis, we employed VOSviewer [version 5.8 R3; (16)], CiteSpace [version 5.8 R3; (17)], and the R package “bibliometrix” [version 3.2.1; https://www.bibliometrix.org; (18)] to extract and examine relevant information from the gathered data and to create visual representations. Specifically, VOSviewer was utilized to perform keyword co-occurrence analysis as well as co-authorship and co-citation analyses pertaining to nations, organizations, authors, and journals.
CiteSpace V (version 5.8 R3) was utilized to produce bi-graphic overlays for the journals. Additionally, the R package “bibliometrix” (version 3.2.1; https://www.bibliometrix.org) facilitated the analysis of keyword evolution and the development of global distribution networks. In the resulting visualizations, various colors indicate clusters assigned to journals through clustering methods, with each cluster representing distinct research domains. Nodes symbolize countries, institutions, authors, and similar entities, while the connections between these nodes illustrate collaborative relationships. The dimensions of each node reflect the quantity of entities it represents, whereas the thickness of the lines connecting the nodes indicates the strength of collaboration among them. Total Link Strength (TLS) quantifies the cumulative strength of co-authorships and co-citations among countries, institutions, and authors.
According to the search strategy and screening process, we collected 2,779 literatures related to targeted therapy for IPF from WOSCC database, including 2,014 papers and 765 reviews (Figure 1). As shown in Figure 2, the number of research articles on targeted therapy for IPF showed a steadily increasing trend. As of the search date, the cumulative citations of all publications amounted to 123,644, yielding an average of 44.49 citations per document. We noticed that the number of annual citations displayed a tendency of fluctuating, increasing from 2004 to 2019, whereas it showed a decline from 2019 to 2024, with a peak of 11,230 citations in 2019. Additionally, the H index also declined in a progressive manner, even though the number of annual publications continued to grow.
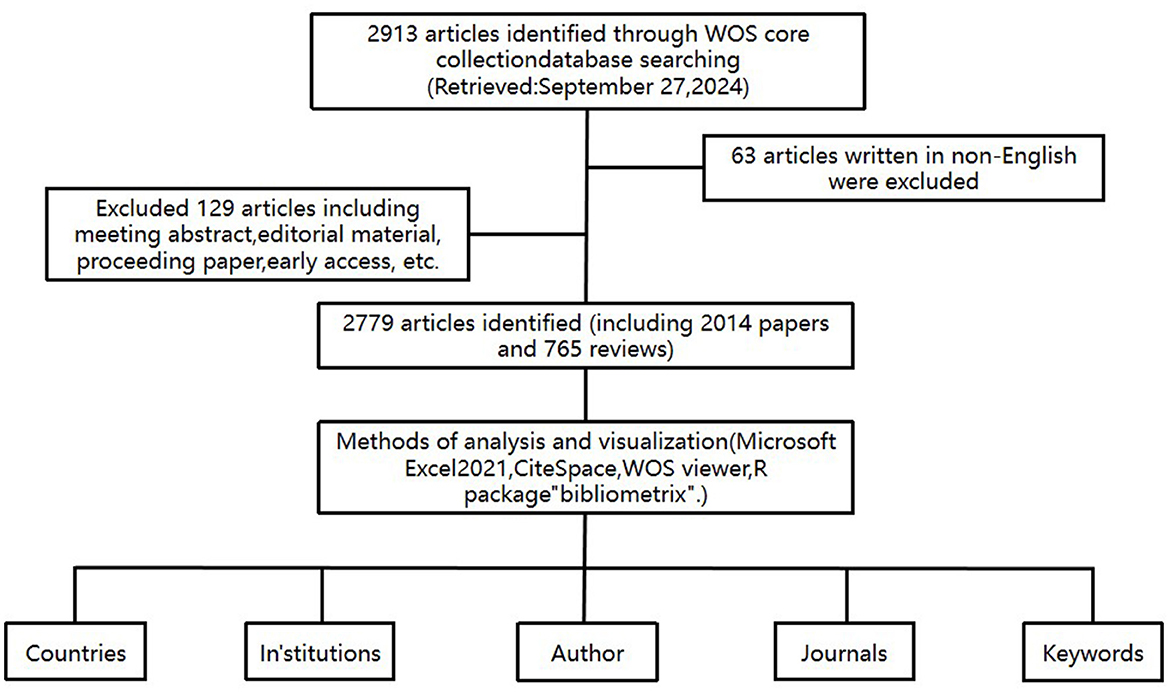
Figure 1. Flowchart of the publication's selection in the study.
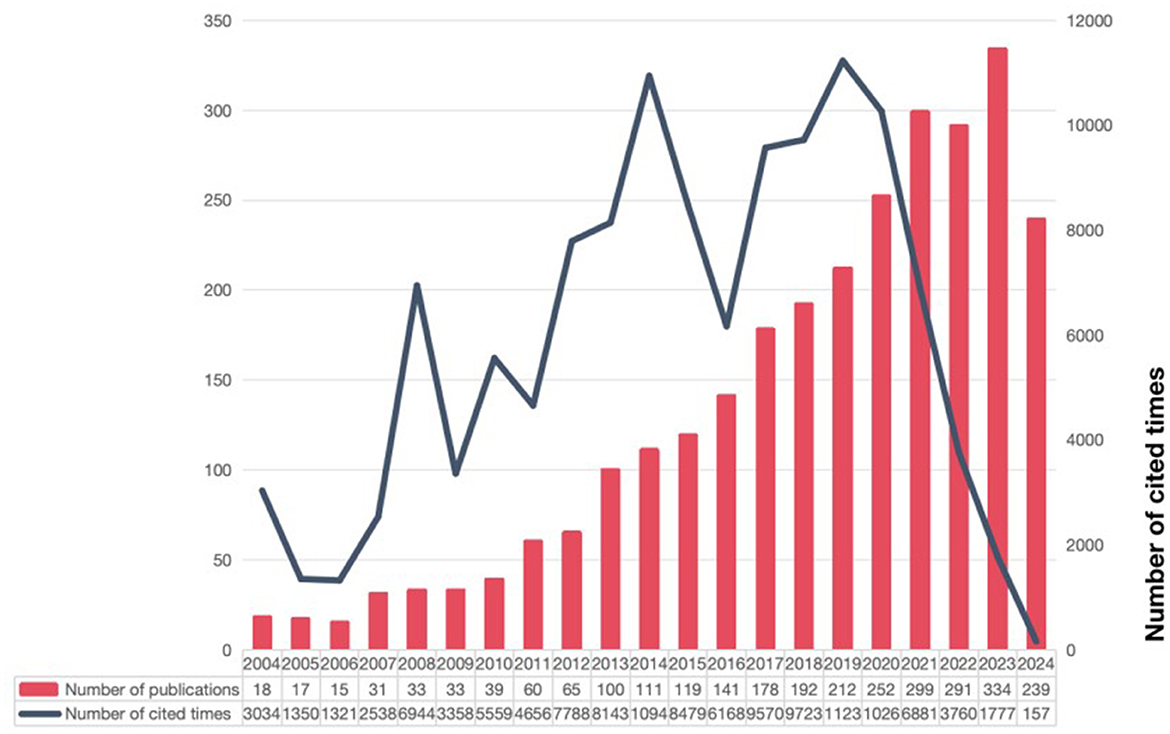
Figure 2. The number of global research publications on targeted therapy for IPF has shown a steadily increasing trend, with cumulative citations of all publications reaching 123,644 as of the search date.
Table 1 lists the top 10 productive countries on targeted therapy for IPF from 2004 to 2024. The United States possesses the most quantity of documents, including 37.86% of the total (1,052/2,779), succeeded by China (30.30%, 842/2,779) and the United Kingdom (9.43%, 262/2,779). Total citations and h-index of the United States were several times ahead of any other country, with 75,530 and 129, respectively, indicating that the United States was the most cutting-edge country in this field. Indeed, the average citation frequency reflects the quality of a paper to a large extent, and the highest average citation is in France (75.74), followed by Canada (75.61) and Germany (72.81). The average number of citations for papers in the United States, China, and the United Kingdom are 71.80, 21.48, and 68.71, respectively. China ranks second in total citations among the 10 countries, but last in the average number of citations for papers, which indicates that the quality of papers in China needs to be improved. Figure 3A illustrates the citation relationships among countries in the literature. The United States is the leading country in TLS, with a value of 13,063, followed by China at 7,818 and the United Kingdom at 4,238. Subsequently, we utilized the R package “bibliometrix” to construct a geographic distribution map based on the number of papers and collaborations in each country (Figure 3B), which represents the active cooperative relationship between different countries in this research area.
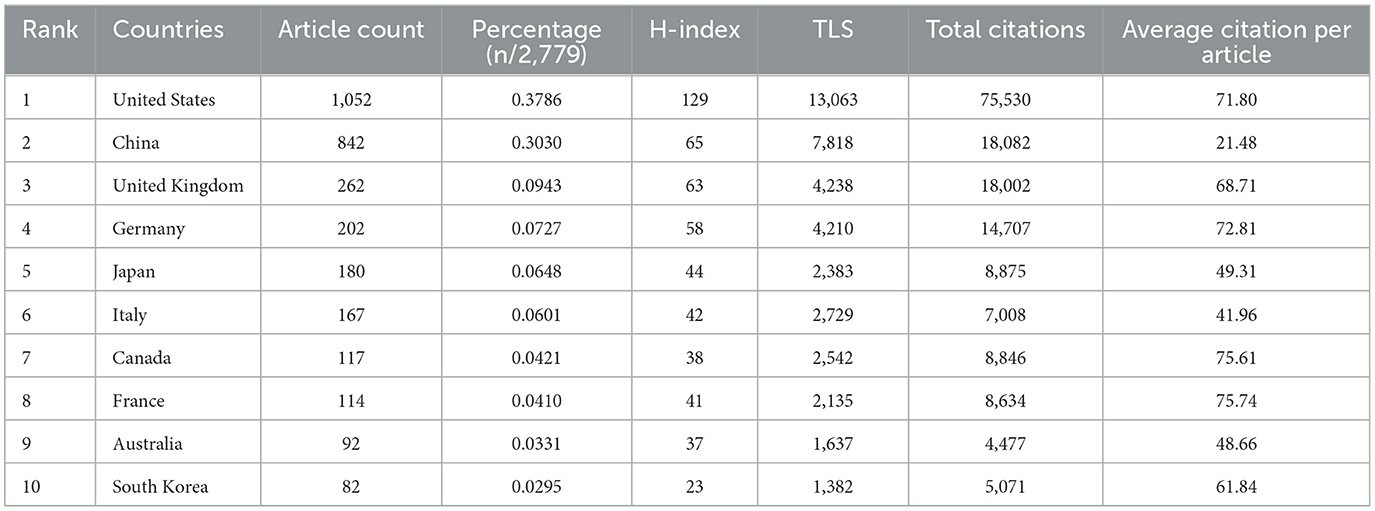
Table 1. The top 10 productive countries on targeted therapy for IPF.
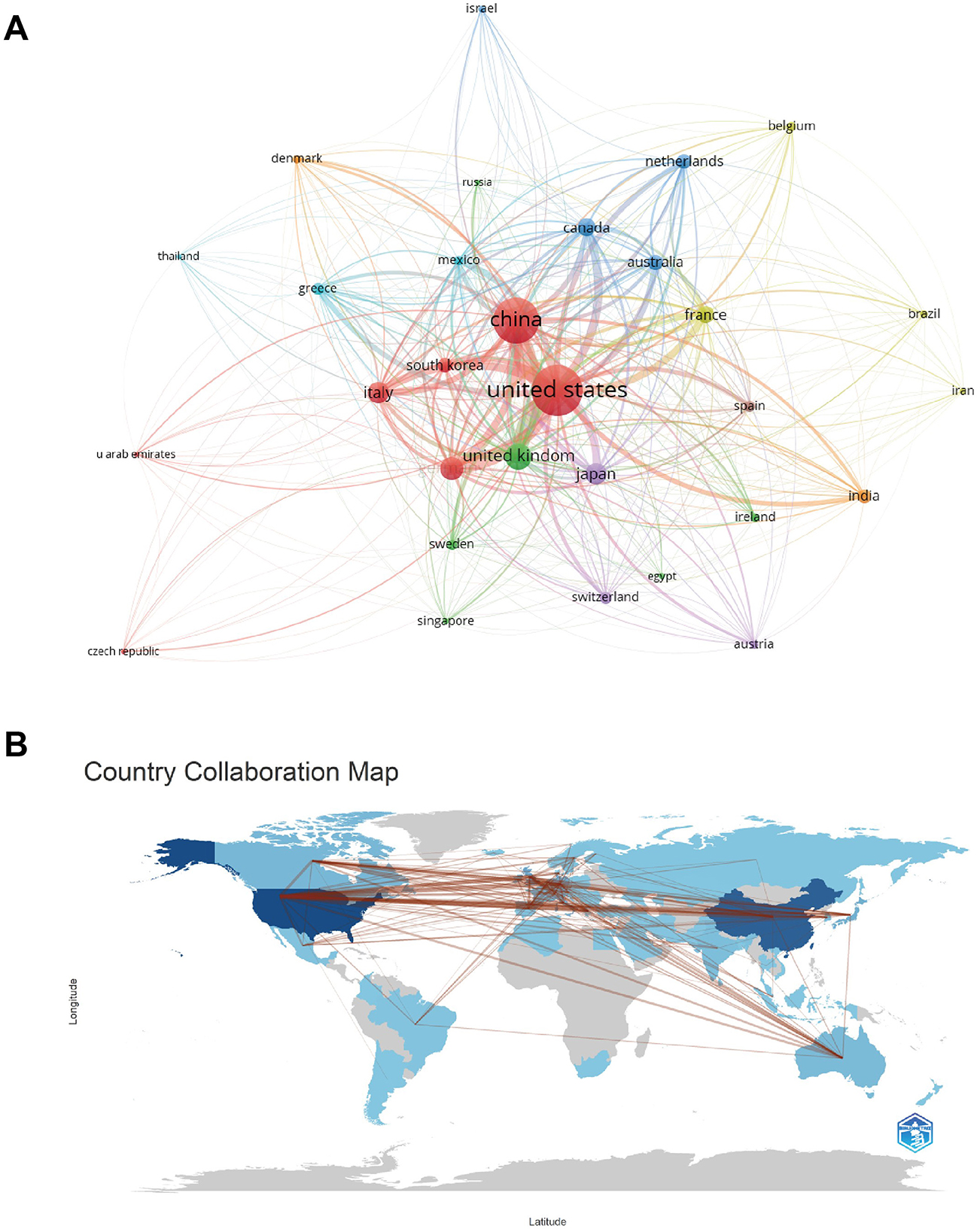
Figure 3. (A) A national citation visualization network map among countries, generated using VOSviewer version 5.8 R3, illustrates the citation relationships in the literature. (B) A cross-country/region collaboration visualization map highlights the active cooperative relationships between different countries in this research field.
Table 2 presents the 10 leading institutions ranked by their publication volume, and the majority of these organizations are based in the United States. The University of Michigan Medical School tops the list with the highest number of published articles, followed closely by the University of Pittsburgh and the University of Alabama at Birmingham. Additionally, the University of Michigan Medical School boasts the highest H index at 43, while Imperial College London has achieved the highest average citation count per article, with 123.60 citations.
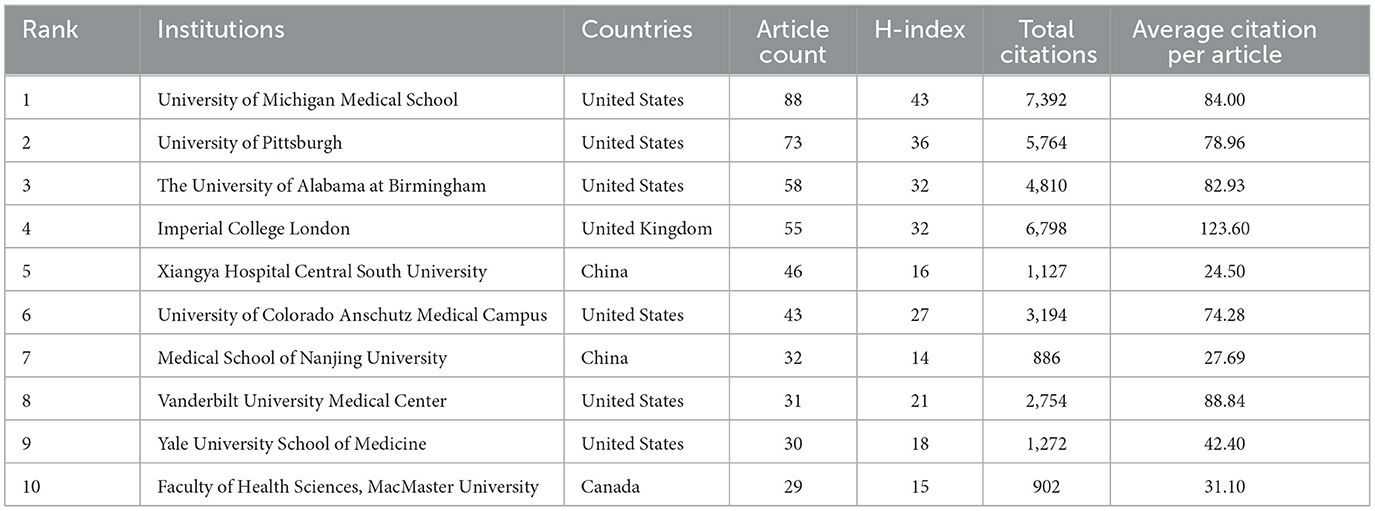
Table 2. The top 10 productive institutions ranked by the numbers of publications.
Close inter-agency collaboration has led to the involvement of more organizations in this area. Figure 4A summarized 150 entries and 1,636 links, with the 147 entries color-coded into eight clusters. Institutions within each cluster are intricately interconnected. Figure 4B presents a citation analysis network diagram including 130 items and 6,828 linkages. The University of Michigan possesses the highest TLS at 1,119, underscoring its substantial connectedness inside the network. The above results suggested the complex partnerships and citations between global institutions.

Figure 4. (A) A cross-institutional collaboration visualization network map, generated using VOSviewer version 5.8 R3, illustrates the intricate interconnections within each cluster. (B) An institutional citation analysis network diagram reveals the complex partnerships and citation relationships among global institutions.
Table 3 lists the top 10 most productive authors, and these authors were mostly from the United States. Notably, Kaminski, Naftali is the author with the most publications, with 38 articles and an H index of 27. While Paul W. published fewer papers, his total citations were as high as 5,404 and is the author with the highest number of citations per article on average with 245.64 citations. Note that both individuals were from the United States. Figure 5A visualizes the map of author co-authorship analysis generated by VOSviewer. It is worth noting that Crestani, Bruno, a prolific figure in the field, has a much wider network of collaborators. Figure 5B illustrates a graphical representation of the authors' collaborative citation network, including 106 entries, 6 clusters, and 3,411 links. The authors with the highest TLSs include Noble, Paul W. (TLS = 998), Thannickal, victor j. (TLS = 808), and Selman, moises (TLS = 753).
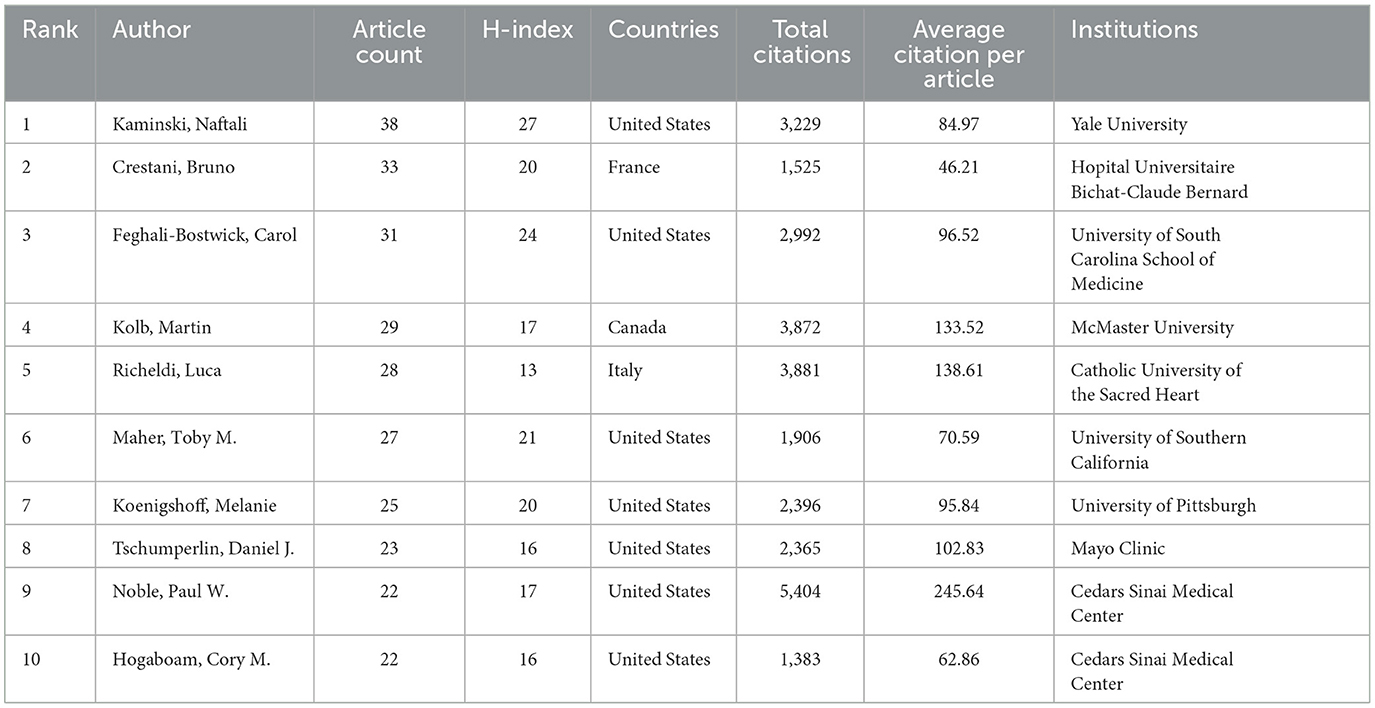
Table 3. The top 10 most productive authors on targeted therapy for IPF.
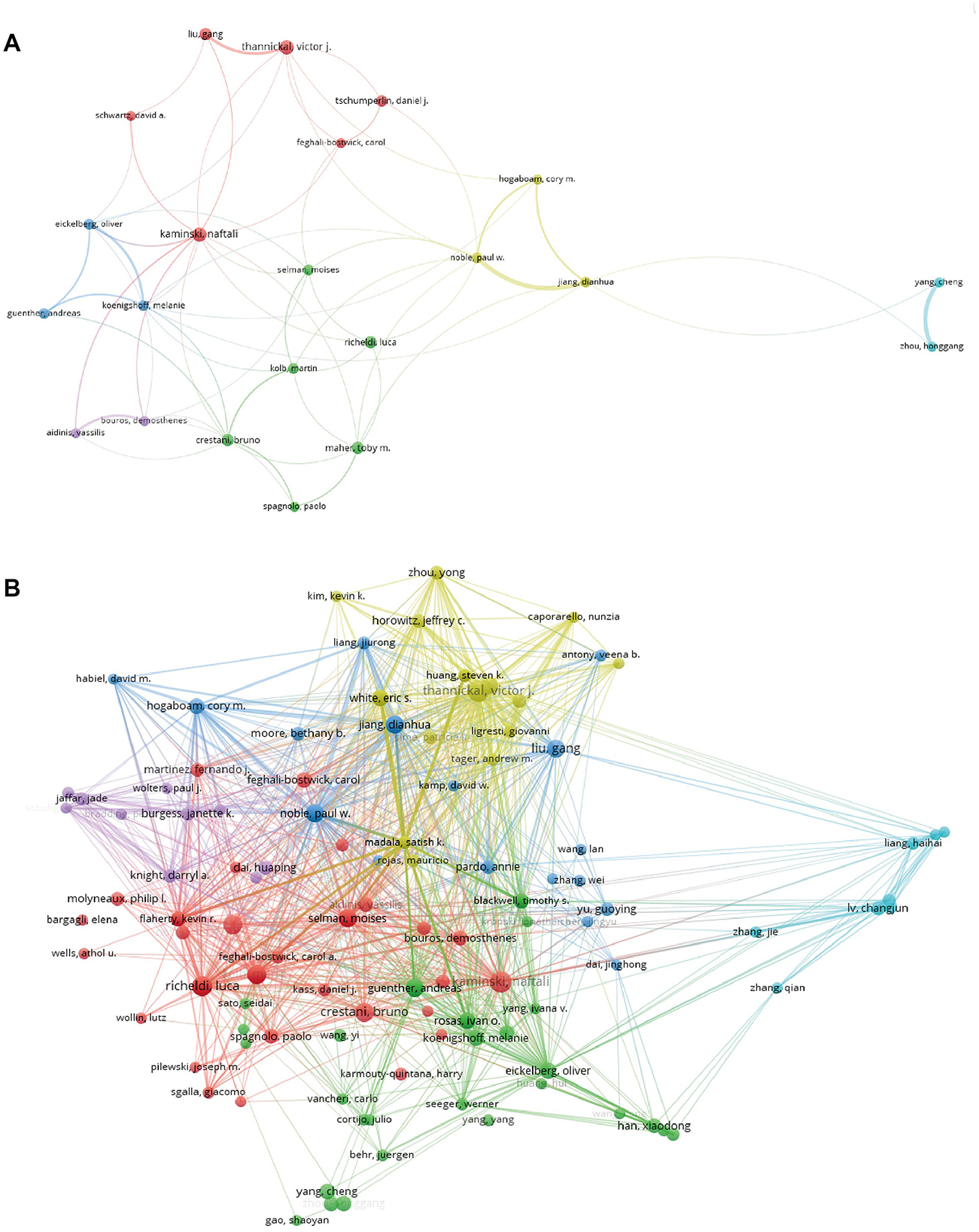
Figure 5. (A) The co-authorship analysis network visualization map, generated using VOSviewer version 5.8 R3. (B) A graphical representation of the authors' collaborative citation network, created using VOSviewer version 5.8 R3.
Table 4 lists the top 10 journals that have published the highest number of articles in this research area. As shown in the table, as of September 2024, the top 10 journals have published a total of 567 articles, representing 20.40% of the included literature. American Journal of Respiratory Cell and Molecular Biology (IF 2023 = 5.9) had the highest number of articles with 98, followed by International Journal of Molecular Sciences (IF 2023 = 4.9) and American Journal of Molecular Sciences (IF 2023 = 4.9) and American Journal of Physiology-Lung Cellular and Molecular Physiology (IF 2023 = 3.6). Among the top 10 journals, 4 originate from the United States, 3 from the United Kingdom, and 3 from Switzerland. The American Journal of Respiratory and Critical Care Medicine possesses the greatest H-index (43), total citations (7,184), and impact factor (IF 2023 = 19.3).

Table 4. The top 10 journals related to the research of IPF targeted therapy ranked by publication number.
Figure 6 visualizes the linkage of related journals by overlaying a double figure. The left side of the figure illustrates the domain of the administering literature (applied research), while the right side depicts the domain of the cited literature (basic research). Various colors denote distinct citation trajectories, which subsequently illustrate the causal linkages among the citations. Three main citation paths are identified in this viewable view, including one green path and two yellow paths. The yellow routes signify that the provided citations predominantly pertain to the domains of molecules, biology, and immunology, whereas the cited references primarily relate to the subjects of molecules, biology, genetics, health, nursing, and medicine. The green path signifies that the sizing literature is predominantly disseminated throughout the domains of medicine, medical, and clinical fields, whereas the cited literature is primarily concentrated in the areas of molecular, biology, and genetics. Furthermore, the elliptical curves on the periphery illustrate the extent of effect of the referenced material in the domain.
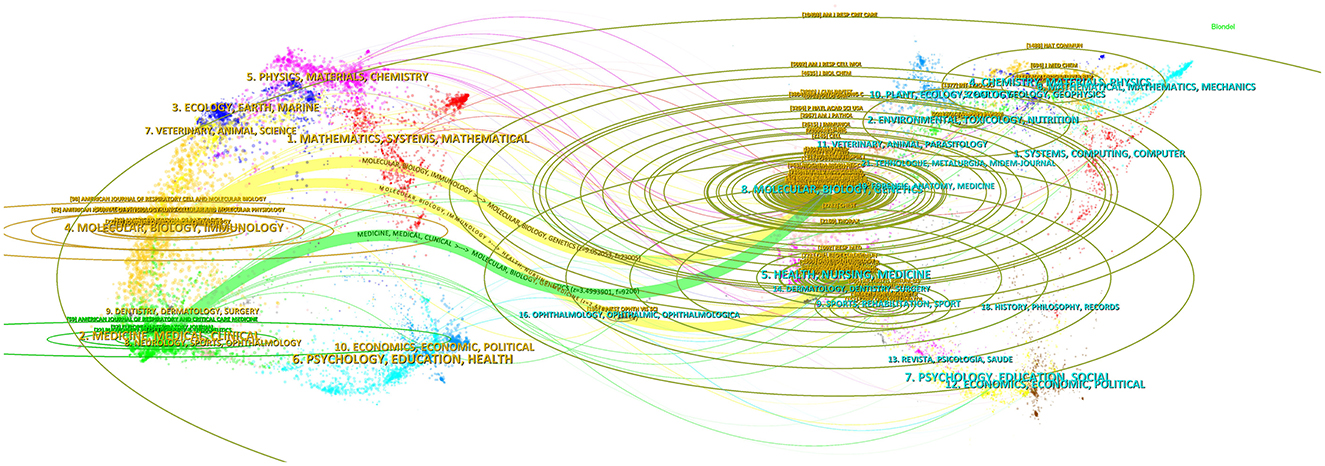
Figure 6. A dual-map overlap of journals on IPF targeted therapy (drawn by CiteSpace V version5.8 R3). The left side of the figure illustrates the domain of the administering literature (applied research), while the right side depicts the domain of the cited literature (basic research). Various colors denote distinct citation trajectories, which subsequently illustrate the causal linkages among the citations.
Table 5 lists the top 10 most cited articles in the field. It is worth noting that Nature is an important source of significant contributions to the field, with 40% of the top 10 most cited articles coming from Nature and its subpublications, respectively. All of the references in the top 10 were cited a total of 670 times or more, underscoring their far-reaching influence. The article by Wynn (43), published in the Journal of Pathology, was the most cited, with 3,214 citations. Figure 7 illustrates our investigation of citation burstiness utilizing CiteSpace V (version 5.8 R3), revealing a total of 25 papers exhibiting the highest citation burstiness. The literature featuring citation bursts initially emerged in 2008, with its inception traced back to a publication published in 2006. Seventy-six percent of these references underwent a citation surge from 2010 to 2020. The latest instance of a citation burst occurred in 2022 and continues to persist.
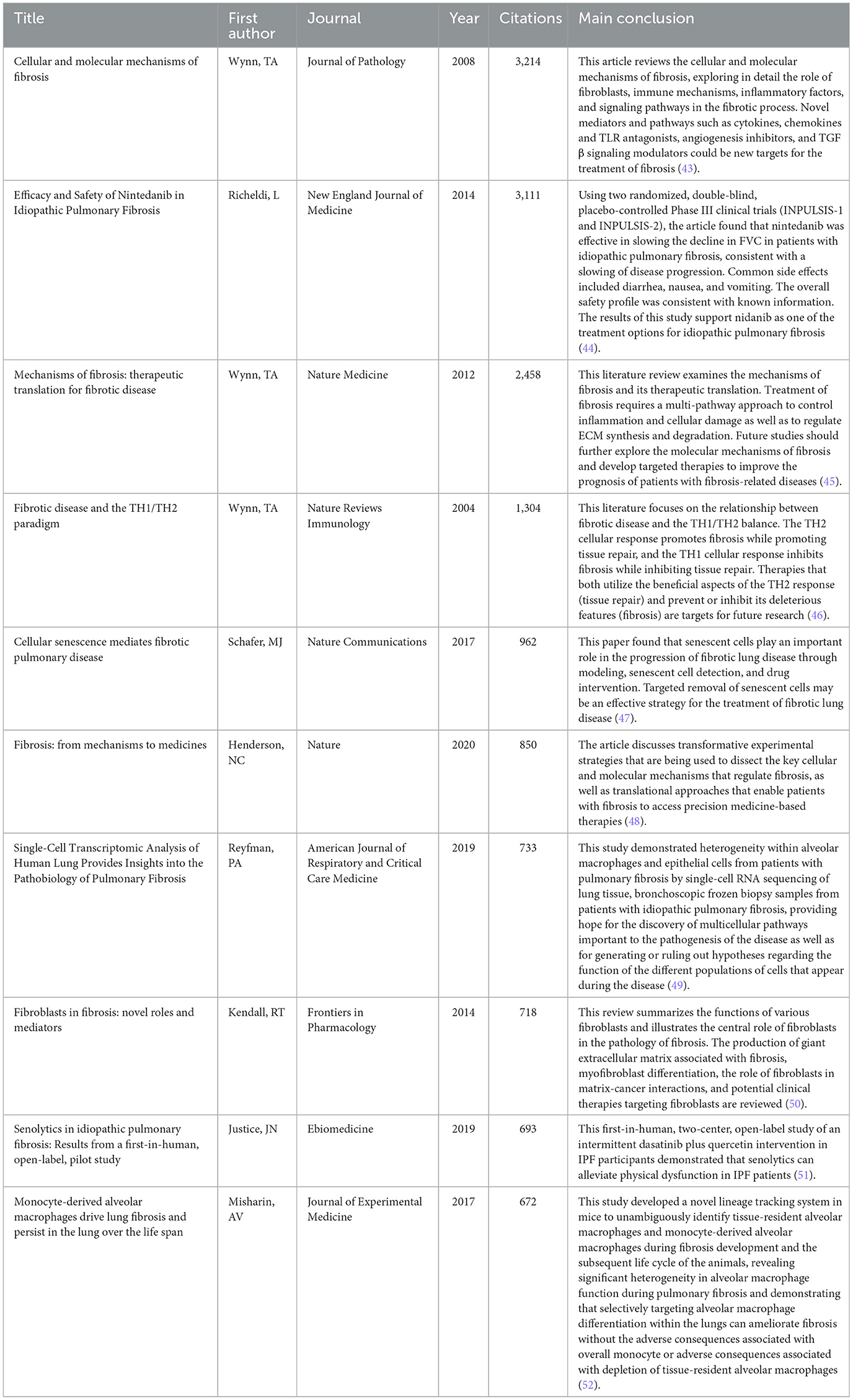
Table 5. The top 10 related articles with the most citations concerning the IPF targeted therapy.
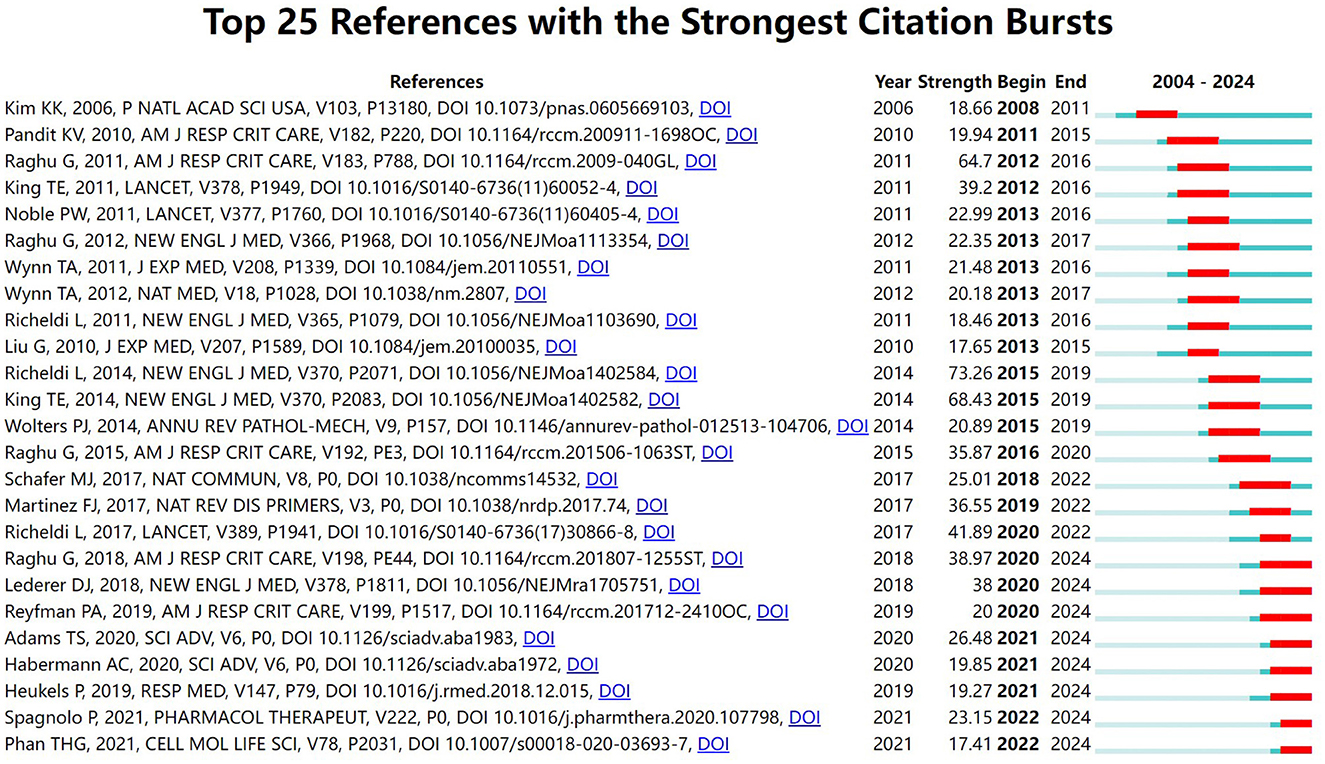
Figure 7. Top 25 references with the strongest citation bursts on IPF targeted therapy (The green line segment represents the time interval, and the red line segment represents the active time).
We meticulously extracted keywords from the titles and abstracts of 2,779 articles included in our analysis. Subsequently, we employed the VOS browser (version 5.8 R3) for both visualization and examination, concentrating on keywords that appeared more than 50 times. As illustrated in Figure 8A, a comprehensive cluster analysis followed, leading to the identification of 79 frequently occurring keywords. These keywords were categorized into distinct clusters, each illustrated by a unique color (clusters 1–4 are depicted in green, red, yellow, and blue). The relationships among these keywords created a network consisting of 2,671 connections.

Figure 8. (A) The keyword co-occurrence network visualization map, generated using VOSviewer version 5.8 R3, categorizes keywords into distinct clusters, each represented by a unique color (clusters 1–4 are shown in green, red, yellow, and blue). The interconnections among these keywords form a network comprising 2,671 links. (B) A comprehensive thematic analysis of keyword trends was conducted using the R package “bibliometrix.” The results reveal a significant evolution in research themes over time, and suggest that these keywords likely represent emerging research focal points within the field of targeted therapies for IPF.
To refine our analysis, we conducted a thorough thematic examination of keyword trends utilizing the R package “bibliometrix.” The findings of this analysis indicate a notable evolution in themes over the years (Figure 8B). From 2009, prevailing research keywords included growth factor β, anti-interferon gamma-1b, factor alpha, and peripheral blood fibrocytes. Following 2016, there was a substantial rise in topics concerning fibroblasts, nintedanib, and pirfenidone. Furthermore, the terms “optimization,” “resolution,” and “autotaxin inhibitors” have experienced a significant increase in usage over the past 1–4 years. This upward trend indicates that these keywords likely highlight current research focal points within the domain of targeted therapies for IPF.
In this study, bibliometric techniques were utilized to perform an extensive analysis of the global literature concerning targeted therapy for IPF spanning the period from 2004 to 2024. The results reveal a consistent upward trend in the volume of publications over the years, a pattern projected to persist into the future. From 2004 to 2019, the total number of citations exhibited a fluctuating yet generally upward trajectory. However, post-2019, there was a notable decline in both the aggregate citation count and the H-index of articles within this field. This downturn may suggest a potential decline in the impact or influence of research in this area. While it is essential to consider potential factors such as citation lag, there is an urgent necessity to identify novel research focal points in this domain to rejuvenate and enhance the quality of findings within related disciplines.
A comprehensive analysis of global academic publication trends indicates that the United States contributes most significantly to the field. The United States not only produces the highest volume of publications but also excels in metrics such as the H Index, Total Link Strength (TLS), and Total Citation Count. These indicators underscore the superior quality and extensive impact of the United States' scholarly output. China and the United Kingdom closely follow. Despite China securing the second position in terms of the number of publications, total link strength (TLS), and total citations, the average citation count per paper remains relatively low. This suggests that Chinese scholars should prioritize enhancing the quality of their academic publications to increase the international impact of their research.
Among the top 10 research institutions, the United States comprises 66% of this group based on the volume of published papers, underscoring its dominance in the relevant research field. China follows with 12%. However, it is important to recognize the achievements and status of other developing research institutions, as they play a significant role in enhancing the academic influence of their nations. Acknowledging the contributions of these emerging institutions is vital for improving the scholarly reputation of their respective countries.
The correlation analysis of highly productive scholars reveals that 7 of the top 10 scholars are from the United States, identified as having the highest H-index, total citations of their articles, and average citations per article. Notable authors include Kaminski, Naftali from Yale University, Crestani, Bruno from Hopital Universitaire Bichat-Claude Bernard, and Feghali-Bostwick, Carol from the University of South Carolina School of Medicine, who are recognized as major contributors to the field. A visual comparison and analysis of co-authored publications provide insights into existing collaborations and help identify significant or prospective collaborators in the area. Furthermore, the present research depicts co-citation networks among authors, indicating that those sharing the same color are engaged in similar research domains. The size of each node corresponds to the influence and prominence of the respective researcher within their discipline. Notably, Crestani, Bruno and Richeldi, Luca exhibit extensive collaboration networks, while Noble, Paul W. has the most robust co-citation connections. These researchers play a crucial role in the study of targeted therapy for IPF, and their teams are expected to make substantial contributions to high-impact publications in related areas.
In the area of targeted therapy for IPF, leading publications like the American Journal of Respiratory and Critical Care Medicine, European Respiratory Journal, American Journal of Respiratory Cell and Molecular Biology, and Frontiers in Immunology have established themselves as key academic resources. These results offer essential insights for researchers, encouraging them to target these esteemed journals for their manuscript submissions and inspiring the dissemination of their research outcomes within these influential platforms. Importantly, among the top 10 journals in this field, 2 possess IF exceeding 10.0: the American Journal of Respiratory and Critical Care Medicine (IF 2023: 19.3) and the European Respiratory Journal (IF 2023: 16.6). Furthermore, there are two additional journals within this top tier that have IF ratings between 5.0 and 10.0, specifically the American Journal of Respiratory Cell and Molecular Biology (IF 2023: 5.9) and Frontiers in Immunology (IF 2023: 5.7). Overall, the pursuit of publication in high-impact journals within the domain of targeted therapy for IPF continues to present significant challenges.
In the domain of targeted therapy for idiopathic pulmonary fibrosis (IPF), prominent academic journals, including the American Journal of Respiratory and Critical Care Medicine, the European Respiratory Journal, the American Journal of Respiratory Cell and Molecular Biology, and Frontiers in Immunology, have established themselves as crucial scholarly resources. These journals offer invaluable insights to researchers, encouraging the submission of manuscripts to these esteemed platforms and promoting the dissemination of research findings within influential academic communities. Notably, among the top 10 journals in this field, 2 have impact factors (IF) exceeding 10.0: the American Journal of Respiratory and Critical Care Medicine (IF 2023: 19.3) and the European Respiratory Journal (IF 2023: 16.6). Additionally, there are two other journals within this elite tier that possess IF ratings between 5.0 and 10.0, specifically the American Journal of Respiratory Cell and Molecular Biology (IF 2023: 5.9) and Frontiers in Immunology (IF 2023: 5.7). Overall, the pursuit of publication in high-impact journals within the domain of targeted therapy for IPF continues to present significant challenges.
“Highly cited references” denote studies that have been frequently cited within a specified period. This metric indicates the research interest and dynamics surrounding these papers in the field of targeted therapies for idiopathic pulmonary fibrosis (IPF), highlighting their substantial impact on the scientific community during this timeframe. The initial surge in citations occurred in 2008, following a seminal publication by Kim et al. (19). This study provided evidence that alveolar epithelial cells serve as progenitors for fibroblasts in vivo, underscoring the significant regulatory function of the transient extracellular matrix in the transdifferentiation of epithelial cells during fibrogenesis. This conclusion was reached through experiments involving genetically engineered mice that express β-galactosidase exclusively in lung epithelial cells, which were subsequently monitored within a well-established pulmonary fibrosis model. The increasing number of citations further underscores the burgeoning scientific interest in developing targeted therapies for IPF.
The synthesis of pertinent keywords is crucial for efficiently understanding targeted therapies for idiopathic pulmonary fibrosis (IPF). By employing cluster analysis and thematic trend analysis of these keywords, we identified that research on targeted therapies for IPF is focused on several key domains. In the context of IPF, persistent harmful stimuli, coupled with the compromised function of the alveolar epithelium, disrupt the cytokine equilibrium in lung tissue. This modified microenvironment is marked by increased concentrations of pro-fibrotic substances, including platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and TGF-β (20). Since 2009, scholars have focused on TGF-β, which is a crucial factor in the pathogenesis for IPF. Following injury to epithelial cells, TGF-β acts as a significant pro-fibrotic agent that promotes the progression of pulmonary fibrosis (21). Its multifaceted roles are vital in the proliferation and differentiation of both epithelial cells and fibroblasts, while also stimulating the formation of myofibroblasts. This leads to an increased production of extracellular matrix (ECM), and the uncontrolled accumulation of ECM contributes to pulmonary sclerosis and impaired gas exchange, ultimately resulting in a decline in lung function in patients with IPF (22). Furthermore, TGF-β is implicated in promoting epithelial-mesenchymal transition (EMT), enhancing apoptosis and migration of epithelial cells, and inducing the synthesis of connective tissue growth factor (CTGF) along with other relevant mediators (23, 24). Therefore, TGF-β is a key target of IPF, and many targeted drugs achieve antifibrotic effects by inhibiting TGF-β signaling.
Currently, only pirfenidone and nintedanib are approved antifibrotic drugs for clinical use. Pirfenidone inhibits TGF-β signaling, thereby enhancing the efficacy of transplanted progenitor cells (25). Although the precise mechanism of action of nintedanib remains unclear, its effects on TGF-β signaling and atypical autophagy have been demonstrated (26). Furthermore, nintedanib effectively blocks the activation of key receptors, including the PDGF receptor, fibroblast growth factor receptor, vascular endothelial growth factor receptor, and Src-family kinases, all of which are implicated in the pathogenesis of IPF (27). In addition to nintedanib and pirfenidone, which have been approved for clinical use, recent advances have emerged in the development of drugs targeting novel mechanisms that inhibit TGF-β signaling. Integrins are known to facilitate the activation of TGF-β, with avβ6 integrins being among the most extensively studied potential therapeutic targets in IPF. However, a phase II clinical trial of an anti-integrin drug, BG00001, revealed no significant change in forced vital capacity (FVC) between the treatment group and the placebo group, and a subset of patients even experienced acute exacerbations (28). Besides avβ6, several other integrins, such as avβ1 and avβ3, also regulate TGF-β activity and promote fibroblast proliferation and growth, indicating that they may serve as important targets for future research.
Pentraxin 2 inhibits the production of TGF-β, likely through the inhibition of monocyte-to-macrophage differentiation, which subsequently reduces the levels of TGF-β. Zinpentraxin alfa (rhPTX-2) is a recombinant form of human pentraxin-2. In a phase II trial (NCT02550873), rhPTX-2 demonstrated significant efficacy in slowing the decline of forced vital capacity (FVC) and the 6-min walk distance [6 MWD; (29)]. However, in the 52-week phase 3 randomized controlled trial (RCT) STARSCAPE, no significant difference was observed between rhPTX-2 and placebo in patients with IPF (30). Galectin-3 (Gal-3) is a lectin that binds to β-galactosides and is significantly elevated in various fibrotic disorders. This β-galactoside-binding lectin promotes the development of fibrosis by modulating the expression of TGF-β receptors. A phase I/IIa trial demonstrated that the expression of Gal-3 was reduced in individuals receiving TD139, a galectin-3 inhibitor, compared to control subjects. The findings from this study suggest that changes in serum biomarkers, including platelet-derived growth factor-BB, fibrinogen activator inhibitor-1, CCL18, and YKL-40 in patients with idiopathic pulmonary fibrosis (IPF), are associated with the downregulation of Gal-3 (31).
In addition to TGF-β, CTGF, interleukin 13 (IL-13), and FGF are all involved in the fibrotic process associated with IPF. FG-3019 (pamrevlumab) is a human monoclonal antibody that inhibits CTGF. The phase II PRAISE trial (NCT01890265) indicated that FG-3019 slowed the decline in forced vital capacity (FVC) and improved diffusing capacity of the lungs for carbon monoxide (32). However, no evidence supporting the antifibrotic effectiveness of FG-3019 was obtained in the subsequent phase III trial. IL-13 activates fibroblasts, promotes extracellular matrix synthesis, and induces myofibroblast transformation and epithelial cell apoptosis through both TGF-β-dependent and non-dependent pathways (33, 34). Nevertheless, in a phase II trial, lebrikizumab, a monoclonal antibody targeting soluble IL-13, did not achieve the anticipated slowing of FVC decline (34). FGF signaling plays a significant role in the pathogenesis of IPF. This signaling pathway is activated through a group of cell surface receptors known as fibroblast growth factor receptors (FGFRs). Notably, FGF-2 expression is elevated in lung tissues affected by IPF, and it is essential for epithelial repair following bleomycin-induced injury in mice, although it does not directly participate in the fibrotic process (35). Similarly, FGF-1 levels are also increased in IPF lung tissue, where it inhibits transforming growth factor-beta 1 (TGF-β1)-stimulated myofibroblast differentiation and epithelial-mesenchymal transition (EMT), thereby functioning as an antifibrotic agent (36). Therefore, therapeutic strategies that target FGF signaling may prove to be effective in mitigating fibrosis.
Additionally, autotaxin inhibitors have emerged as a prominent trend in 2023. Lysophosphatidic acid plays a significant role in mediating epithelial cell apoptosis and fibroblast recruitment during pulmonary fibrosis in Hoyles et al. (36) and Funke et al. (37). Autotaxin, an enzyme responsible for the production of lysophosphatidic acid (38), is upregulated in patients with IPF, making it a potential target for novel therapeutic approaches. Ziritaxet, a new Autotaxin inhibitor, demonstrated encouraging results by reducing plasma lysophosphatidic acid concentrations in a Phase IIa study involving 23 patients with IPF (39). However, in the subsequent Phase III trial, Ziritaxet did not lead to improved clinical outcomes compared to placebo (40). IPF pathogenesis can be driven by lysophosphatidic acid (LPA), which signals through six LPA receptors (LPA1–6). Among these, LPA1 signaling plays a critical role in the development of fibrotic diseases. Admilparant (BMS-986278), an LPA receptor antagonist, has been evaluated in a large phase 2 trial. Phase 3 trials, ALOFT-IPF and ALOFT-PPF, comparing admilparant to placebo in patients with IPF and PPF, respectively, have been initiated (41). Additionally, bexotegrast (PLN-74809), a dual-selective inhibitor of avβ6 and avβ1 integrins, is expressed at low levels in normal lung tissue but upregulated in IPF patients. It may block TGF-β activation during fibrogenesis. In the INTEGRIS-IPF phase 2 trial (42), IPF patients were randomized to receive various doses of bexotegrast or placebo, showing favorable safety and tolerability up to 12 weeks. A phase 2b/3 program (BEACON-IPF) is underway to further assess its efficacy, tolerability, and safety in IPF patients (41).
In conclusion, IPF is a progressive and irreversible lung disease with an etiology that remains incompletely understood. Consequently, further research into the pathogenesis of IPF is imperative. As our understanding of the disease's pathogenesis advances, it will be possible to develop novel targeted therapies based on these research findings, which can then be progressively introduced into clinical trials. The initial exploration of IPF's pathophysiology reveals numerous potential therapeutic targets, including αvβ6 integrin, Pentraxin 2, Galectin-3, connective tissue growth factor (CTGF), interleukin-13 (IL-13), fibroblast growth factor (FGF), and autotaxin. Notably, we identify significant shortcomings in recent phase III clinical trials, wherein certain drugs demonstrated efficacy in preclinical and phase II trials but failed to achieve desired outcomes in phase III trials. Future research should prioritize the identification of novel therapeutic targets and expedite their progression to phase III clinical trials.
Our study provides a systematic overview of the global research advancements in targeted therapy for IPF over the past two decades. Nevertheless, it is important to acknowledge certain limitations. First, this review is limited to literature published in English, which may lead to the exclusion of important studies available in other languages. Second, data collection was conducted solely through the WOSCC database, potentially missing significant research accessible in other databases, such as PubMed and Embase. Third, bibliometric analyses typically depend on bibliographic indexes, which may not provide a comprehensive view of new publications when faced with imperfect or insufficient indexes. Fourthly, due to the continuous updating of database, recently published high-quality clinical studies may be underestimated for their unsatisfactory citations. Finally, we acknowledge that this study did not analyze the funding information associated with the publications included, and we plan to address this aspect in our future research to provide a more comprehensive understanding of the funding landscape related to this field.
In conclusion, the exploration of targeted therapies for IPF is undergoing significant development, as evidenced by the growing annual number of pertinent publications. Notably, the United States leads in both the quantity and quality of research, exerting substantial influence on the trajectory of this field. Previous studies have delineated various therapeutic mechanisms and their associated targets. Currently, the focus has shifted toward phase III clinical trials, which are critical for the validation of these targeted therapies. It is expected that concepts, such as “optimization,” “resolution,” and “autotaxin inhibitors” will become increasingly prominent in future research endeavors.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
XZ: Data curation, Investigation, Methodology, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing. ZY: Formal analysis, Investigation, Methodology, Validation, Writing – original draft. XS: Conceptualization, Project administration, Supervision, Writing – review & editing. JY: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the Data Center of Management Science, National Natural Science Foundation of China – Peking University (grant number: 81774231 and 82174300), and the “Pioneer” and “Leading Goose” R&D Program of Zheiiang (grant number: 2025C02196).
The authors acknowledged the developers of R software and the relevant R packages for their contributions to the tools used in the analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official Ats/Ers/Jrs/Alat clinical practice guideline. Am J Respir Crit Care Med. (2022) 205:e18–47. doi: 10.1164/rccm.202202-0399ST
2. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. (2017) 389:1941–52. doi: 10.1016/S0140-6736(17)30866-8
3. Martinez FJ, Chisholm A, Collard HR, Flaherty KR, Myers J, Raghu G, et al. The diagnosis of idiopathic pulmonary fibrosis: current and future approaches. Lancet Respir Med. (2017) 5:61–71. doi: 10.1016/S2213-2600(16)30325-3
4. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. (2015) 46:795–806. doi: 10.1183/09031936.00185114
5. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An Official Ats/Ers/Jrs/Alat clinical practice guideline. Am J Respir Crit Care Med. (2018) 198:e44–68. doi: 10.1164/rccm.201807-1255ST
6. Bos S, Vos R, Van Raemdonck DE, Verleden GM. Survival in adult lung transplantation: where are we in 2020? Curr Opin Organ Transplant. (2020) 25:268–73. doi: 10.1097/MOT.0000000000000753
7. Kolilekas L, Papiris S, Bouros D. Existing and emerging treatments for idiopathic pulmonary fibrosis. Expert Rev Respir Med. (2019) 13:229–39. doi: 10.1080/17476348.2019.1568244
8. Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. (2014) 349:209–20. doi: 10.1124/jpet.113.208223
9. Pardo A, Selman M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. (2016) 13(Suppl. 5):S417–21. doi: 10.1513/AnnalsATS.201605-341AW
10. Carlos WG, Strek ME, Wang TS, Patel H, Raghu G, Wilson KC, et al. Treatment of idiopathic pulmonary fibrosis. Ann Am Thorac Soc. (2016) 13:115–7. doi: 10.1513/AnnalsATS.201510-713CME
11. Petnak T, Lertjitbanjong P, Thongprayoon C, Moua T. Impact of antifibrotic therapy on mortality and acute exacerbation in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Chest. (2021) 160:1751–63. doi: 10.1016/j.chest.2021.06.049
12. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (capacity): two randomised trials. Lancet. (2011) 377:1760–9. doi: 10.1016/S0140-6736(11)60405-4
13. Richeldi L, Azuma A, Cottin V, Hesslinger C, Stowasser S, Valenzuela C, et al. Trial of a preferential phosphodiesterase 4b inhibitor for idiopathic pulmonary fibrosis. N Engl J Med. (2022) 386:2178–87. doi: 10.1056/NEJMoa2201737
14. Kim YM, Delen D. Medical informatics research trend analysis: a text mining approach. Health Inform J. (2018) 24:432–52. doi: 10.1177/1460458216678443
15. Yuan Z, Zhang W, Jin Z, Wang Y, Lin Z, Xie Z, et al. Global research trends in precision-targeted therapies for systemic lupus erythematosus (2003-2023): a bibliographic study. Heliyon. (2024) 10:e33350. doi: 10.1016/j.heliyon.2024.e33350
16. Yeung AWK, Tzvetkov NT, Balacheva AA, Georgieva MG, Gan RY, Jozwik A, et al. Lignans: quantitative analysis of the research literature. Front Pharmacol. (2020) 11:37. doi: 10.3389/fphar.2020.00037
17. Synnestvedt MB, Chen C, Holmes JH. Citespace II: visualization and knowledge discovery in bibliographic databases. AMIA Annu Symp Proc. (2005) 2005:724–8.
18. Li C, Ojeda-Thies C, Renz N, Margaryan D, Perka C, Trampuz A. The global state of clinical research and trends in periprosthetic joint infection: a bibliometric analysis. Int J Infect Dis. (2020) 96:696–709. doi: 10.1016/j.ijid.2020.05.014
19. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. (2006) 103:13180–5. doi: 10.1073/pnas.0605669103
20. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. (2005) 166:1321–32. doi: 10.1016/S0002-9440(10)62351-6
21. Fernandez IE, Eickelberg O. The impact of tgf-beta on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc. (2012) 9:111–6. doi: 10.1513/pats.201203-023AW
22. Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An official Ats/Ers/Jrs/Alat clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. (2015) 192:e3–19. doi: 10.1164/rccm.201506-1063ST
23. Walters GI. Occupational exposures and idiopathic pulmonary fibrosis. Curr Opin Allergy Clin Immunol. (2020) 20:103–11. doi: 10.1097/ACI.0000000000000610
24. Kropski JA, Blackwell TS. Progress in understanding and treating idiopathic pulmonary fibrosis. Annu Rev Med. (2019) 70:211–24. doi: 10.1146/annurev-med-041317-102715
25. Ma Q, Ma Y, Dai X, Ren T, Fu Y, Liu W, et al. Regeneration of functional alveoli by adult human SOX9(+) airway basal cell transplantation. Prot Cell. (2018) 9:267–82. doi: 10.1007/s13238-018-0506-y
26. Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, et al. Novel mechanisms for the antifibrotic action of nintedanib. Am J Respir Cell Mol Biol. (2016) 54:51–9. doi: 10.1165/rcmb.2014-0445OC
27. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. (2015) 45:1434–45. doi: 10.1183/09031936.00174914
28. Raghu G, Mouded M, Chambers DC, Martinez FJ, Richeldi L, Lancaster LH, et al. A phase iib randomized clinical study of an anti-alpha(V)beta(6) monoclonal antibody in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2022) 206:1128–39. doi: 10.1164/rccm.202112-2824OC
29. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. JAMA. (2018) 319:2299–307. doi: 10.1001/jama.2018.6129
30. Richeldi L, Schiffman C, Behr J, Inoue Y, Corte TJ, Cottin V, et al. Zinpentraxin alfa for idiopathic pulmonary fibrosis: the randomized phase III starscape trial. Am J Respir Crit Care Med. (2024) 209:1132–40. doi: 10.1164/rccm.202401-0116OC
31. Hirani N, MacKinnon AC, Nicol L, Ford P, Schambye H, Pedersen A, et al. Target inhibition of galectin-3 by inhaled Td139 in patients with idiopathic pulmonary fibrosis. Eur Respir J. (2021) 57:2002559. doi: 10.1183/13993003.02559-2020
32. Richeldi L, Fernandez Perez ER, Costabel U, Albera C, Lederer DJ, Flaherty KR, et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (praise): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. (2020) 8:25–33. doi: 10.1016/S2213-2600(19)30262-0
33. Ballester B, Milara J, Cortijo J. Idiopathic pulmonary fibrosis and lung cancer: mechanisms and molecular targets. Int J Mol Sci. (2019) 20:593. doi: 10.3390/ijms20030593
34. Maher TM, Costabel U, Glassberg MK, Kondoh Y, Ogura T, Scholand MB, et al. Phase 2 trial to assess lebrikizumab in patients with idiopathic pulmonary fibrosis. Eur Respir J. (2021) 57:1902442. doi: 10.1183/13993003.02442-2019
35. Guzy RD, Stoilov I, Elton TJ, Mecham RP, Ornitz DM. Fibroblast growth factor 2 is required for epithelial recovery, but not for pulmonary fibrosis, in response to bleomycin. Am J Respir Cell Mol Biol. (2015) 52:116–28. doi: 10.1165/rcmb.2014-0184OC
36. Hoyles RK, Derrett-Smith EC, Khan K, Shiwen X, Howat SL, Wells AU, et al. An essential role for resident fibroblasts in experimental lung fibrosis is defined by lineage-specific deletion of high-affinity type II transforming growth factor beta receptor. Am J Respir Crit Care Med. (2011) 183:249–61. doi: 10.1164/rccm.201002-0279OC
37. Funke M, Zhao Z, Xu Y, Chun J, Tager AM. The lysophosphatidic acid receptor Lpa1 promotes epithelial cell apoptosis after lung injury. Am J Respir Cell Mol Biol. (2012) 46:355–64. doi: 10.1165/rcmb.2010-0155OC
38. Kraljic K, Jelic D, Ziher D, Cvrtila A, Dragojevic S, Sinkovic V, et al. Benzoxaboroles-novel autotaxin inhibitors. Molecules. (2019) 24:3419. doi: 10.3390/molecules24193419
39. Maher TM, van der Aar EM, Van de Steen O, Allamassey L, Desrivot J, Dupont S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir Med. (2018) 6:627–35. doi: 10.1016/S2213-2600(18)30181-4
40. Maher TM, Ford P, Brown KK, Costabel U, Cottin V, Danoff SK, et al. Ziritaxestat, a novel autotaxin inhibitor, and lung function in idiopathic pulmonary fibrosis: the Isabela 1 and 2 randomized clinical trials. JAMA. (2023) 329:1567–78. doi: 10.1001/jama.2023.5355
41. Cottin V, Valenzuela C. Evidence from recent clinical trials in fibrotic interstitial lung diseases. Curr Opin Pulm Med. (2024) 30:484–93. doi: 10.1097/MCP.0000000000001089
42. Lancaster L, Cottin V, Ramaswamy M, Wuyts WA, Jenkins RG, Scholand MB, et al. Bexotegrast in patients with idiopathic pulmonary fibrosis: the INTEGRIS-IPF clinical trial. Am J Respir Crit Care Med. (2024) 210:424–34. doi: 10.1164/rccm.202403-0636OC
43. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.2277
44. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2071–82. doi: 10.1056/NEJMoa1402584
45. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18:1028–40. doi: 10.1038/nm.2807
46. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. (2004) 4:583–94. doi: 10.1038/nri1412
47. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:14532. doi: 10.1038/ncomms14532
48. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
49. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. (2019) 199:1517–36. doi: 10.1164/rccm.201712-2410OC
50. Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. (2014) 5:123. doi: 10.3389/fphar.2014.00123
51. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
Keywords: idiopathic pulmonary fibrosis, targeted therapy, bibliometric analysis, transforming growth factor β, autotaxin inhibitor
Citation: Zhang X, Yuan Z, Shi X and Yang J (2025) Targeted therapy for idiopathic pulmonary fibrosis: a bibliometric analysis of 2004–2024. Front. Med. 12:1543571. doi: 10.3389/fmed.2025.1543571
Received: 11 December 2024; Accepted: 03 March 2025;
Published: 20 March 2025.
Edited by:
Stephen C. Land, University of Dundee, United KingdomReviewed by:
Guojun Tong, Huzhou Central Hospital, ChinaCopyright © 2025 Zhang, Yuan, Shi and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junchao Yang, eWFuZ2p1bmNoYW96akB6Y211LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.