 Xiufang Wang1†
Xiufang Wang1† Lin Zhu
Lin Zhu Hui Huang
Hui Huang Aiping Deng
Aiping Deng Juyi Li
Juyi Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Med., 14 March 2025
Sec. Gastroenterology
Volume 12 - 2025 | https://doi.org/10.3389/fmed.2025.1527249
This article is part of the Research TopicThe Pathogenesis and Treatment Progress of Intestinal DiseasesView all 3 articles
Background: This study aimed to analyze the pathogenic variants in one family with colorectal cancer and another with endometrial cancer and provide appropriate personalized prevention strategies for carriers of these genetic mutations.
Methods: One proband with colorectal cancer and another with endometrial cancer and their family members were enrolled in this study. Whole-exome sequencing was used to identify pathogenic gene mutations in both families. We compared the structural difference between the wild-type and mutant MSH2 proteins using SWISS-MODEL and PyMOL visualization software.
Results: We identified one novel mutation (NM_000251.2:c.1486delT:p.L496*) in the MSH2 gene in Family I and a known mutation (NM_001258271.1:c.884 + 4A > G) in the MLH1 gene in Family II. The novel mutation (NM_000251.2:c.1486delT:p.L496*) caused a stop gain mutation, resulting in the absence of amino acids 496–934 in the mutant MSH2 protein. This led to the loss of Domain 5 and alterations in the sequences of Domain 3 and Domain 4 regions, resulting in premature termination of MSH2 protein coding. The known mutation (NM_001258271.1:c.884 + 4A > G) in MLH1 causes the skipping of exon 10, producing a truncated protein and undergoing nonsense-mediated decay based on literature reports. Thus, 5-fluorouracil-based adjuvant chemotherapy is not recommended for patients with lynch syndrome
Conclusion: The novel stop gain mutant (NM_000251.2:c.1486delT:p.L496*) in MSH2 is deemed pathogenic for LS, and the mutant (NM_001258271.1:c.884 + 4A > G) in MLH1 has been further confirmed to be pathogenic. These findings expand the spectrum of mismatch repair gene variations in the ethnic group Han of China and reaffirm the importance of genetic testing for LS.
Lynch syndrome (LS), also known as hereditary nonpolyposis colorectal cancer, may be the most common inherited cause of susceptibility to cancer, with an incidence rate ranging from 1 in 100 to 1 in 180 (1). Individuals with LS are prone to various cancers, the most common of which are colorectal and endometrial cancers. They are also at risk for cancers in many other organ sites, including the stomach, small intestine, ovaries, prostate, and skin (sebaceous glands) (2, 3).
LS is a hereditary, heterogeneously autosomal dominant disorder caused by pathogenic variants in mismatch repair (MMR) genes, including MSH2, MSH6, MLH1, and PMS2 (4, 5). Variations in these genes disrupt the MMR process, leading to changes in the length of DNA microsatellite repeat sequences and the emergence of microsatellite instability (MSI), which accelerates the accumulation of somatic mutations and thus, accelerates tumor formation (6). Therefore, individuals with these genetic variations are more likely to develop cancer compared to the general population and often develop cancer earlier (7).
Therefore, it is crucial to identify the mutational profile associated with LS in the Chinese population to allow the implementation of oncogenetic counseling based on genetic tests specific to this population. In this study, we report a novel MSH2 variant and a known MLH1 variant in a Chinese family with colorectal cancer and another family with endometrial cancer, respectively. This study aimed to provide genetic counseling to the family members and to develop appropriate prevention strategies and precise treatment plans for individuals carrying these mutations.
Two families (Han nationality), one affected by colorectal cancer and the other by endometrial cancer, were recruited from the Central Hospital of Wuhan. The diagnostic criteria for LS were based on a combination of the Amsterdam II criteria, clinical test reports, and detailed family pedigrees. This study was approved by the Ethics Committee of the Central Hospital of Wuhan. Informed consent was obtained from all participants involved in the study.
The proband from Family I was a 61-year-old man who had undergone a partial colectomy owing to a mass in the left colon, revealing a moderately differentiated adenocarcinoma. His father had died of oral cancer, his mother had died of rectal cancer at 65 years of age, his sister had been diagnosed with rectal cancer at 58 years of age, while his son was asymptomatic. Figure 1A shows the detailed pedigree of Family I, and Figure 1B shows the colonoscopy results of the proband in Family I.
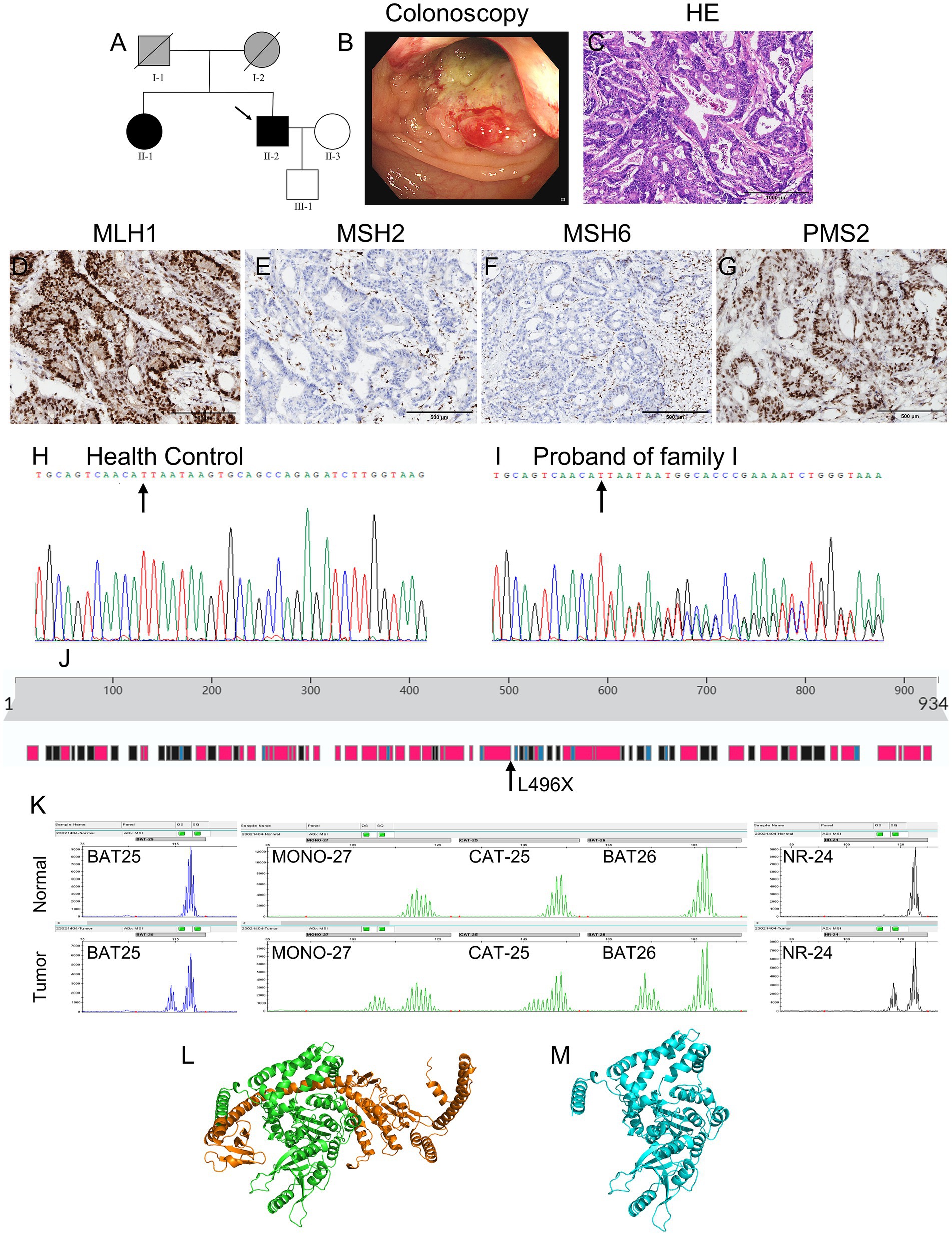
Figure 1. Clinical and basic characteristics of Family I. (A) Pedigree of family I: Circles represent females, squares represent males, a slash through the symbol indicates deceased individuals, and the arrow indicates the proband. Black symbols represent patients with LS and gray symbols represent patients with tumors that have not undergone genetic testing. (B) Colonoscopy results of the proband: findings include colon neogenesis, surface ulceration, and easy bleeding. (C) The H&E staining results of the colon tissue of the proband suggest moderately differentiated adenocarcinoma. (D–G) Figures show strong nuclear positivity for PMS2 and MLH1, partially weak nuclear positivity for MSH6, and absence of nuclear staining for MSH2. (H,I) Genetic sequencing results show that the proband carries a novel MSH2 gene mutation (NM_000251.2:c.1486delT:p.L496*), a deletion at position 1,486 in the coding region, resulting in a stop gain mutation. (J) The position of the p.L496* mutation in the secondary structures of the MSH2 protein. (K) Microsatellite instability test results. (L,M) Swiss-Prot predicts the protein structure of the wild-type and p.L496* MSH2 proteins. The orange region in the wild-type MSH2 protein model indicates the missing region of amino acids 496–934.
The proband of Family II was a 53-year-old woman who was diagnosed with endometrioid adenocarcinoma and underwent total hysterectomy and bilateral oophorectomy. Her mother had died of gastric cancer at the age of 68 years, and her father and brother were healthy at the time of writing. Figure 2A shows the detailed pedigree of Family II.
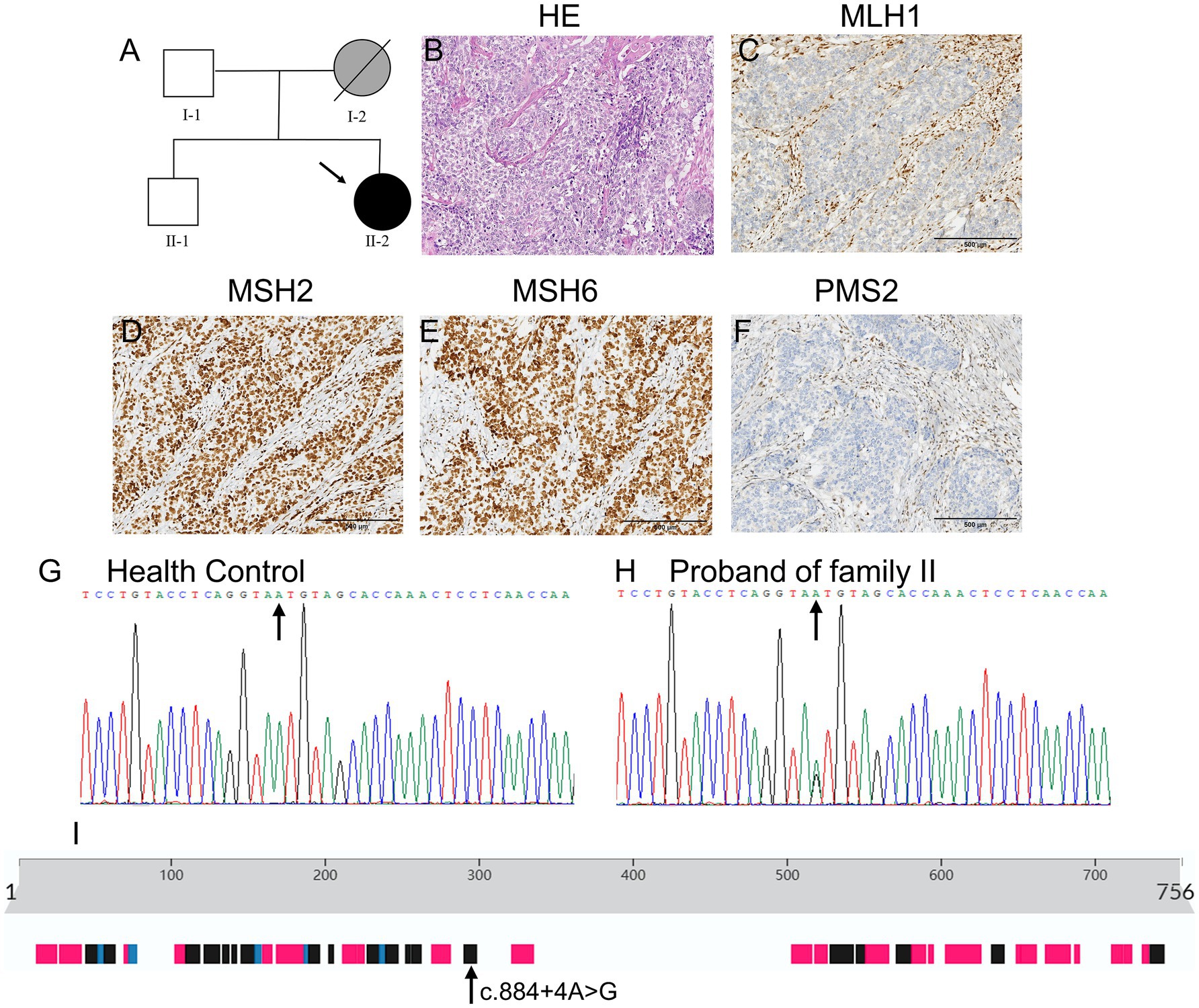
Figure 2. Clinical and basic characteristics of Family II. (A) Pedigree of family II: circles represent females, squares represent males, a slash through the symbol indicates deceased individuals, and the arrow indicates the proband. Black symbols represent patients with LS and gray symbols represent patients with tumors that have not been genetically tested. (B) The H&E staining results of the endometrial tissue of the proband suggest poorly differentiated endometrial adenocarcinoma. (C–F) Figures showed strong nuclear positivity for MSH2 and MSH6 and loss of nuclear staining for MLH1 and PMS2 in Family II. (G,H) Sanger sequencing results revealed that the proband in Family II carried a rare MLH1 gene mutation (NM_001258271.1:c.884 + 4A > G). (I) The position of the c.884 + 4A > G mutation in the secondary structures of the MLH1 protein.
Paraffin-embedded tissue sections were used for histopathological analysis (hematoxylin and eosin, H&E, Figure 1C shows the H&E staining results of Family I, and Figure 2B shows the H&E staining results of Family II) and then to detect the expression of four MMR proteins: MLH1 (clone ES05), PMS2 (clone EP51), MSH2 (clone FE11), and MSH6 (clone EP49). All four primary antibodies were purchased from Jiayuan Biomedical Engineering Co., Ltd., Wuhan, China (8).
Single and multiplex PCR reactions were conducted to amplify the five markers: BAT-25, MONO-27, CAT-25, BAT-26, and NR-24. The amplification reactions were carried out according to the manufacturer’s protocols.
Genomic DNA was extracted from the peripheral blood of the proband from Family I, and the tumor tissue from paraffin-embedded sections of the proband from Family II. Target regions were captured using the SureSelect Human All Exon V6 (Agilent) hybridization capture kit. High-throughput sequencing was employed using the Illumina NovaSeq (sequencing read length: 2 × 150 bp) to analyze the genomic DNA sequences (8–10). The UCSC hg19 and NCBI build 37 were used as reference genomes for WES.
The results of high-throughput sequencing were validated using Sanger sequencing. The polymerase chain reaction (PCR) conditions were as follows: an initial predenaturation step at 95°C for 5 min, followed by denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and finally, extension at 72°C for 10 min. The PCR primers used were as follows: For Family I, the forward primer was 3′-ATAACTATTACAAGTTTTGCACA-5′ and the reverse primer was 3′-GTGACATTTAAAATAGGGCT-5′. For Family II, the forward primer was 3′-GTGACCTCACCCCTCAGGAC-5′ and the reverse primer was 3′-ACATCCTTTTGCCAGTGGTG-5′.
To construct the three-dimensional spatial structure of MSH2, SWISS-MODEL1 was used, and the structural differences between the wild-type and mutant MSH2 proteins were compared using the visualization software PyMOL.2
Figures 1D–G show strong nuclear positivity for MLH1 (Figure 1D) and PMS2 (Figure 1G), weak nuclear positivity for MSH6 (Figure 1F), and an absence of nuclear staining for MSH2 (Figure 1E) in Family I. Figures 2C–F show strong nuclear positivity for MSH2 (Figure 2D) and MSH6 (Figure 2E) and absence of nuclear staining for MLH1 (Figure 2C) and PMS2 (Figure 2F) in Family II. The absence of MSH2, MLH1, and PMS2 expression in these patients suggests the possibility of a germline mutation in the MMR genes, indicating a high risk for LS.
In Family I, as shown in Figure 1K, all markers (BAT25, MONO-27, CAT-25, BAT26, and NR-24) showed a leftward shift in the cancerous tissues compared to the normal tissues, indicating MSI in the proband’s tumor tissue.
Table 1 shows the detailed information of WES for the proband (II-2) in Family I. A novel mutation (NM_000251.2:c.1486delT:p.L496*) in the MSH2 gene was identified (Figure 1I, the mutant position indicated by the arrow, and Figure 1H as the wild type control) in the proband (II-2). This mutation involved the deletion of thymine (T) at position 1,486 in the coding region of the MSH2 gene, resulting in a stop gain mutation. The codon TTA encoding leucine was shifted to a stop codon TAA, leading to premature termination of the MSH2 protein translation and loss of protein integrity and function. This mutation was pathogenic for the proband and has not been previously reported.

Table 1. Whole-exome sequencing detail of the proband in family I and II.
Sanger sequencing showed that the germline mutation in MSH2 was also found in the proband’s sister (II-1), who had been diagnosed with rectal cancer at 58 years of age, but not in the proband’s son who was in good health (III-1). Given that the proband’s mother (I-2) had died of rectal cancer, we speculated that the pathogenic mutation observed in the proband was inherited from his mother.
Table 1 shows the detailed information from WES of the proband (II-2) in Family II. A heterozygous mutation (NM_001258271.1:c.884 + 4A > G) was identified in MLH1 (Figure 2H, the mutant position indicated by the arrow, and Figure 2G as the wild type control) in the proband in Family II (II-2). This mutation causes the skipping of exon 10, producing a truncated protein, and is recognized as a pathogenic mutation (rs267607777) (11). Given that the proband’s mother (I-2) had died of gastric cancer at the age of 68 years, we speculated that this pathogenic mutation was inherited from her.
The three-dimensional spatial structures of the MSH2 proteins, both wild-type and the p. L496* MSH2, are shown in Figure 1L (wild-type) and Figure 1M (mutant type). The c.1486del mutation leads to the substitution of leucine at position 496 with a stop codon, resulting in the absence of amino acids 496–934 in the mutant MSH2 protein model, which leads to the loss of Domain 5 in the MSH2 protein and alterations in the sequences of Domain 3 and Domain 4 regions (12). The orange region in the wild-type MSH2 protein model is the missing region of amino acids 496–934.
In this study, two significant mutations were identified in two typical families with LS: one novel mutation in the MSH2 gene (NM_000251.2:c.1486delT:p.L496*), which was not recorded in any database including GnomAD; and a previously known mutation in the MLH1 gene (NM_001258271.1:c.884 + 4A > G), which was very rare and the mutation frequency was not recorded in any database including GnomAD. According to the American College of Medical Genetics and Genomics classification criteria, these two mutants were considered pathogenic for LS, with the MSH2 mutant (NM_000251.2:c.1486delT:p.L496*) meeting the criteria for PVS1, PM2, PM6, and PP4, and the MLH1 mutant (NM_001258271.1:c.884 + 4A > G) being confirmed as pathogenic.
Consistent with previous studies, immunohistochemical analysis of MMR proteins can guide the analysis of mutations in MMR genes. MLH1 combines with PMS2 to form a heterodimer, and the combined deletion of MLH1 and PMS2 is a characteristic of patients with MLH1 mutations, while the deletion of PMS2 protein in tumor tissue indicates a germline mutation in PMS2 (13). Similarly, loss of nuclear staining in MSH2 and MSH6 proteins indicates mutants in MSH2 and MSH6, respectively (13). Our data supported these findings, with immunohistochemistry results for the proband in Family I revealing MSH2 as the pathogenic gene and those for the proband in Family II revealing MLH1.
The proband in Family I showed high MSI (MSI-H). Patients with stage II colon cancer with MSI-H have a better prognosis but do not benefit from single-agent adjuvant chemotherapy with fluorouracil. Patients with colorectal cancer and MSI-H should be further tested for MMR gene mutations to confirm a diagnosis of LS. Figure 1J displays the secondary structure of the wild-type MSH2 protein, which has 934 amino acids. A variation in the MSH2 gene (NM_000251.2:c.1486delT:p.L496*) leads to a stop gain mutation in the MSH2 protein. This mutation changes the codon TTA, which encodes leucine, to a stop codon TAA, altering the tertiary structure of the protein and eventually leading to the formation of a non-functional protein. The characteristics of the disrupted MSH2 protein may include reduced MMR activity, defective interactions with MSH6, and loss of protein expression. Given the pattern of morbidity in the family, this MSH2 gene (NM_000251.2:c.1486delT:p.L496*) mutation was considered to be pathogenic. Therefore, the offspring of individual II-1 in Family I should undergo genetic testing. If they carry the pathogenic mutation MSH2 (NM_000251.2:c.1486delT:p.L496*), they should undergo regular physical examinations, including gastrointestinal and gynecological endoscopy, for early diagnosis and treatment.
Furthermore, we identified a known mutation (NM_001258271.1:c.884 + 4A > G) in the MLH1 gene that affects a donor splice site in intron 10 (Figure 2I, the mutant position in secondary structure of the wild-type MLH1 protein indicated by the arrow). Many similar MLH1 gene variations, such as c.1668–2 A > G (14) and c.790 + 1 G > A (15), have been reported as pathogenic mutations of LS. The known mutation (NM_001258271.1:c.884 + 4A > G) in MLH1 causes skipping of exon 10, producing a truncated protein (11). This protein is subject to nonsense-mediated decay (16), reinforcing the classification of this mutation as pathogenic. Hence, the offspring of individual II-2 in Family II should undergo genetic testing. If they carry the pathogenic MLH1 mutation (NM_001258271.1:c.884 + 4A > G), they should undergo regular physical examinations, including gastrointestinal and gynecological endoscopy, for early diagnosis and treatment.
In 2018, Patrick summarized the strategies for surveillance, prevention, and precision medicine for managing LS-associated colorectal cancer (CRC) (17, 18). Carriers of LS should undergo colonoscopy every 1–2 years. Carriers of pathogenic MLH1, MSH2, and MSH6 mutations can reduce their risk of developing LS-associated endometrial and ovarian cancers through preventive hysterectomy with salpingo-oophorectomy. In addition, aspirin intake reduces the risk of CRC and all LS-associated cancers. Postoperative adjuvant therapy for advanced CRC often entails the administration of 5-fluorouracil (5-FU) as a standalone treatment or in conjunction with other drugs (19). However, patients with CRC exhibiting MSI show limited response to 5-FU-based adjuvant chemotherapy. As a result, the use of 5-FU is not recommended for patients with MSI who require chemotherapy. Tumors characterized by deficient MMR pathways present ideal targets for immunotherapy, specifically, immune checkpoint inhibitors (20) targeting cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed cell death 1 (PD-1), and programmed cell apoptosis ligand 1 (PD-L1), which negatively regulate T cell activation. Such patients are likely to benefit from anti-PD-1/PD-L1/CTLA-4 treatments (21–23). Ongoing trials are evaluating the effectiveness of combining chemotherapy and immunotherapy, immunotherapy and targeted therapy, and the role of immunotherapy in the adjuvant setting for patients with LS (22, 24, 25).
This study sheds light on the characteristics of LS in the Chinese population, which is of great significance for disease diagnosis, prevention, and precise treatment. The highest risk of LS-associated cancers has been linked to mutations in MLH1 or MSH2 (26). Previous studies have found that carriers of MSH6 gene mutations are more likely to develop endometrial cancer, while carriers of MSH2 gene mutations are more likely to develop extracolonic tumors or various other tumors (8). Therefore, research on the association between genotype and phenotype is crucial to provide more precise treatment strategies for patients with LS.
In summary, the study identified a novel pathogenic mutation (NM_000251.2:c.1486delT:p.L496*) in the MSH2 gene and confirmed a known mutation in the MLH1 gene (NM_001258271.1:c.884 + 4A > G) in two unrelated Chinese families with LS. The findings broaden our understanding of the spectrum of MMR gene mutations in China and reaffirm the importance of genetic testing for LS. Although the results of this study suggest that the novel MSH2 mutation (NM_000251.2:c.1486delT:p.L496*) is pathogenic, further research is required to investigate its underlying pathogenic mechanisms. Genetic diagnosis, regular follow-ups, and individualized treatment should be provided to cancer-afflicted families with evidence of compromised MMR gene function.
The data presented in the study are deposited in the following repository: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1226963.
The studies involving humans were approved by the Ethics Committee of the Central Hospital of Wuhan. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
XW: Validation, Visualization, Writing – review & editing. HN: Data curation, Resources, Writing – review & editing. LZ: Investigation, Software, Supervision, Writing – original draft, Writing – review & editing. HH: Investigation, Methodology, Resources, Writing – review & editing. AD: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing – review & editing. JH: Project administration, Resources, Supervision, Writing – review & editing. WC: Validation, Data curation, Writing – review & editing. JL: Data curation, Investigation, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This project was founded by the Wuhan Municipal Health Commission (No. WX18M02), the Horizontal Subject of the Hospital of Wuhan (No. 2023-024) and the Central Guiding Local Science and Technology Development Special Project (No. 2022BGE272).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sinicrope, FA. Lynch syndrome-associated colorectal cancer. N Engl J Med. (2018) 379:764–73. doi: 10.1056/NEJMcp1714533
2. Møller, P, Seppälä, T, Bernstein, I, Holinski-Feder, E, Sala, P, Evans, DG, et al. Cancer incidence and survival in lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective lynch syndrome database. Gut. (2017) 66:464–72. doi: 10.1136/gutjnl-2015-309675
3. Møller, P, Seppälä, TT, Bernstein, I, Holinski-Feder, E, Sala, P, Gareth Evans, D, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the prospective lynch syndrome database. Gut. (2018) 67:1306–16. doi: 10.1136/gutjnl-2017-314057
4. Chen, W, Swanson, BJ, and Frankel, WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. (2017) 12:24. doi: 10.1186/s13000-017-0613-8
5. Cini, G, Carnevali, I, Quaia, M, Chiaravalli, AM, Sala, P, Giacomini, E, et al. Concomitant mutation and epimutation of the MLH1 gene in a lynch syndrome family. Carcinogenesis. (2015) 36:452–8. doi: 10.1093/carcin/bgv015
6. Cohen, SA, and Leininger, A. The genetic basis of lynch syndrome and its implications for clinical practice and risk management. Appl Clin Genet. (2014) 7:147–58. doi: 10.2147/TACG.S51483
7. Akizawa, Y, Yamamoto, T, Tamura, K, Kanno, T, Takahashi, N, Ohki, T, et al. A novel MLH1 mutation in a Japanese family with lynch syndrome associated with small bowel cancer. Hum Genome Var. (2018) 5:13. doi: 10.1038/s41439-018-0013-y
8. Liu, Y, Wang, M, Chen, Q, Zheng, Q, Li, G, Cheng, Q, et al. A novel heterozygous large deletion of MSH6 gene in a Chinese family with lynch syndrome. Gene. (2019) 704:103–12. doi: 10.1016/j.gene.2019.04.011
9. Wang, Y, Sun, Y, Liu, M, Zhang, X, and Jiang, T. Functional characterization of Argininosuccinate Lyase gene variants by Mini-gene splicing assay. Front Genet. (2019) 10:436. doi: 10.3389/fgene.2019.00436
10. Fu, F, Tao, X, Jiang, Z, Gao, Z, Zhao, Y, Li, Y, et al. Identification of germline mutations in east-Asian young never-smokers with lung adenocarcinoma by whole-exome sequencing. Phenomics. (2023) 3:182–9. doi: 10.1007/s43657-022-00062-1
11. Rahner, N, Friedrichs, N, Wehner, M, Steinke, V, Aretz, S, Friedl, W, et al. Nine novel pathogenic germline mutations in MLH1, MSH2, MSH6 and PMS2 in families with lynch syndrome. Acta Oncol (Stockholm, Sweden). (2007) 46:763–9. doi: 10.1080/02841860701230217
12. Warren, JJ, Pohlhaus, TJ, Changela, A, Iyer, RR, Modrich, PL, and Beese, LS. Structure of the human Mut Salpha DNA lesion recognition complex. Mol Cell. (2007) 26:579–92. doi: 10.1016/j.molcel.2007.04.018
13. Hendriks, YM, Jagmohan-Changur, S, van der Klift, HM, Morreau, H, van Puijenbroek, M, Tops, C, et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (lynch syndrome). Gastroenterology. (2006) 130:312–22. doi: 10.1053/j.gastro.2005.10.052
14. Li, J, Zhu, L, Li, Y, Huang, H, Huang, K, and Deng, A. Two novel and one known pathogenic germline mutations in MMRs in Chinese families with lynch syndrome. Genes Dis. (2022) 9:292–5. doi: 10.1016/j.gendis.2021.10.006
15. Auclair, J, Busine, MP, Navarro, C, Ruano, E, Montmain, G, Desseigne, F, et al. Systematic mRNA analysis for the effect of MLH1 and MSH2 missense and silent mutations on aberrant splicing. Hum Mutat. (2006) 27:145–54. doi: 10.1002/humu.20280
16. Rossi, BM, Palmero, EI, López-Kostner, F, Sarroca, C, Vaccaro, CA, Spirandelli, F, et al. A survey of the clinicopathological and molecular characteristics of patients with suspected lynch syndrome in Latin America. BMC Cancer. (2017) 17:623. doi: 10.1186/s12885-017-3599-4
17. Boland, PM, Yurgelun, MB, and Boland, CR. Recent progress in lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J Clin. (2018) 68:217–31. doi: 10.3322/caac.21448
18. Rohani, S, Suhada, N, Mohammad, Z, and Azhar, S. Traditional herbal medicine as adjunctive therapy for colorectal cancer: a scoping review. Trad Med Res. (2022) 7:15–8. doi: 10.53388/TMR20220127260
19. Aggarwal, N, Quaglia, A, McPhail, MJW, and Monahan, KJ. Systematic review and meta-analysis of tumour microsatellite-instability status as a predictor of response to fluorouracil-based adjuvant chemotherapy in colorectal cancer. Int J Color Dis. (2022) 37:35–46. doi: 10.1007/s00384-021-04046-x
20. Ferriss, JS, and Williams-Brown, MY. Immunotherapy: checkpoint inhibitors in lynch-associated gynecologic cancers. Curr Treat Options in Oncol. (2019) 20:75. doi: 10.1007/s11864-019-0676-8
21. Diaz, LA Jr, and Le, DT. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 373:1979. doi: 10.1056/NEJMc1510353
22. Le, DT, Durham, JN, Smith, KN, Wang, H, Bartlett, BR, Aulakh, LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. doi: 10.1126/science.aan6733
23. Dedeurwaerdere, F, Claes, KB, Van Dorpe, J, Rottiers, I, Van der Meulen, J, Breyne, J, et al. Comparison of microsatellite instability detection by immunohistochemistry and molecular techniques in colorectal and endometrial cancer. Sci Rep. (2021) 11:12880. doi: 10.1038/s41598-021-91974-x
24. Le, DT, Uram, JN, Wang, H, Bartlett, BR, Kemberling, H, Eyring, AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
25. Zhang, L, Jiang, B, Zhu, N, Tao, M, Jun, Y, Chen, X, et al. Mitotic checkpoint kinase Mps 1/TTK predicts prognosis of colon cancer patients and regulates tumor proliferation and differentiation via PKCα/ERK1/2 and PI3K/Akt pathway. Med Oncol (Northwood, London, England). (2019) 37:5. doi: 10.1007/s12032-019-1320-y
Keywords: lynch syndrome, mismatch repair gene, whole-exome-sequencing, Sanger sequencing, genetic counseling
Citation: Wang X, Ni H, Zhu L, Huang H, Deng A, Hu J, Cai W and Li J (2025) Analyzing pathogenic variants in mismatch repair genes: personalized prevention strategies for lynch syndrome in Chinese families. Front. Med. 12:1527249. doi: 10.3389/fmed.2025.1527249
Edited by:
Jianbing Zhang, Dalian Medical University, ChinaReviewed by:
Zheng Jin Tu, Cleveland Clinic, United StatesCopyright © 2025 Wang, Ni, Zhu, Huang, Deng, Hu, Cai and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aiping Deng, ZGFweXhiQDE2My5jb20=; Jifa Hu, amlmYWh1QHNpbmEuY29t; Wei Cai, NzYzMDg1MDFAcXEuY29t; Juyi Li, bGp5d3hmMTEwQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.