 Karin Ashizawa1
Karin Ashizawa1 Tsuyoshi Saito1,2*Yukinori Yube3
Tsuyoshi Saito1,2*Yukinori Yube3 Shinji Mine3Tetsu Fukunaga3Cristina R. Antonescu4
Shinji Mine3Tetsu Fukunaga3Cristina R. Antonescu4 Takashi Yao1
Takashi Yao1- 1Department of Human Pathology, Graduate School of Medicine, Juntendo University, Bunkyo-ku, Japan
- 2Intractable Disease Research Center, Graduate School of Medicine, Juntendo University, Bunkyo-ku, Japan
- 3Department of Upper Gastroenterological Surgery, Juntendo University Hospital, Bunkyo-ku, Japan
- 4Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY, United States
New disease entities have been emerging based on molecular pathological findings, such as pseudoendocrine sarcoma and mesenchymal neoplasm with GLI1 gene alterations, which resemble well-differentiated neuroendocrine tumors. We report a unique case of a gastric submucosal neoplasm of approximately 1.5 cm in size with CTNNB1 mutation showing GLI1 overexpression and epithelial differentiation in a 66-year-old man. It was incidentally identified by routine health screening, and was a slowly growing tumor. Macroscopically, it was a slightly protruded tumor into the mucosa, and was primarily located from the submucosa to the muscularis propria. It was a well-defined lesion measured approximately 20 mm, and was almost stable during almost 5 years after initial identification of the tumor. Uniform round-to-epithelioid cells arranged in solid trabeculae with a microtubular/acinar appearance were seen microscopically. Occasional mitotic figures were noted, but no necrosis was observed. Immunohistochemistry (IHC) demonstrated diffuse expression of pan-cytokeratin, CD10, and CD56 without neuroendocrine markers (chromogranin A, synaptophysin, and INSM1). Molecular analysis confirmed the presence of a hot spot CTNNB1 mutation (S33C), supported by diffuse β-catenin nuclear expression by IHC. Further molecular investigations revealed the absence of GLI1 gene rearrangements, GLI1 amplification, and other fusions. Several differential diagnoses were considered; however, none adequately fit the criteria. The patient remained disease-free for 24 months postoperatively without further adjuvant therapy.
Introduction
Molecular pathological research is rapidly advancing, and new disease entities that are based on molecular pathological findings have emerged. For example, disease entities that resemble well-differentiated neuroendocrine tumors, such as pseudoendocrine sarcoma, mesenchymal neoplasms with GLI1 gene alterations described as “distinctive nested glomoid neoplasm,” gastroblastoma, and plexiform fibromyxoma have been reported (1–7). Gastroblastoma is a distinctive biphasic stomach tumor that shows epithelioid or spindle cells and harbors the MALAT::GLI1 fusion gene (5). Pseudoendocrine sarcoma is a recently recognized entity among soft tissue sarcomas, characterized by its close resemblance to well-differentiated neuroendocrine tumors, but lacking epithelial/neuroendocrine differentiation by immunohistochemistry (IHC). Additionally, nearly all cases exhibit aberrant nuclear accumulation of β-catenin caused by the secondary activating hot spot CTNNB1 mutations (1). These lesions commonly occur in deep soft tissues (1–4). These entities are morphologically similar to well-differentiated neuroendocrine tumors, which often makes their diagnosis difficult. Herein, we report a unique case of a gastric submucosal tumor that demonstrated strong expression of monoclonal antibodies directed against keratins in the absence of neuroendocrine differentiation by IHC. However, this case did not fit any of these criteria, and we report it as a gastric submucosal neoplasm with CTNNB1 mutation, showing GLI1 overexpression and epithelial differentiation.
Case presentation
Clinical course
In 2017, a routine health screen incidentally revealed a submucosal gastric tumor in a 66-year-old man. The tumor located on the lesser curvature of the cardia, measuring approximately 1.5 cm, has been monitored since its discovery. Endoscopic ultrasonography (EUS) showed an almost uniform hypoechoic area contiguous to the fourth layer. There was no obvious non-echoic area or blood flow signal. While initially clinically diagnosed as leiomyoma, the possibility of a gastrointestinal stromal tumor was considered. EUS-guided fine needle aspiration (EUS-FNA) in May 2022 revealed a tumor characterized by the monotonous proliferation of epithelioid cells. IHC analysis demonstrated positive staining for CD56 but negative staining for chromogranin A, synaptophysin, and INSM-1. The MIB-1-labeling index (LI) was approximately 2%, which led to the diagnosis of a grade 1 (G1) neuroendocrine tumor, although the immunohistochemical findings were inconsistent. Upon admission, laboratory testing revealed slight abnormalities in renal function. The tumor was expected by EUS to be located in the submucosa and deeper, so the endoscopic resection was considered to be difficult. Therefore, proximal gastrectomy and lymph node dissection was performed by robotic technique in June 2022 as part of the patient’s management. The tumor was located on the lesser curvature of the cardia, allowing resection without confirmation of the tumor location during surgery, despite its small size. The operative time was 5 h and 5 min, with a blood loss of 10 mL. On 10 days after surgery, the patient developed a fever. Gastroendoscopy was performed and it revealed leakage on the left side of the esophageal margin slightly apart from the anastomotic site. Stomach tube was placed for drainage, and a central venous catheter (CV) was inserted. At 1 month after surgery, leakage improved and the drain tube was removed. However, the patient developed a fungal infection and fungal endophthalmitis associated with CV insertion. The patient received intravenous antifungal treatment for additional 1 month, then changed to oral medication. Finally, the patient discharged 2 months post-surgery.
Pathological findings of the surgically resected specimen
Macroscopically, the tumor exhibited a slight protrusion into the mucosa (Figure 1) but was primarily located from the submucosa to the muscularis propria. This well-defined lesion measured approximately 20 mm. Microscopically, the tumor was composed of uniform round-to-epithelioid cells arranged in solid trabeculae with a microtubular/acinar appearance. Tumor cells showed scant eosinophilic cytoplasm and monomorphic round-to-ovoid nuclei with fine chromatin and no pleomorphism. Occasional mitotic figures were observed (1/50 high power fields) and no necrosis was observed. In a few areas, short spindle-shaped cells arranged in tight nests were noted within the fibromyxoid stromal background (Figure 1). Totally, 31 lymph nodes were retrieved and no metastases were identified.
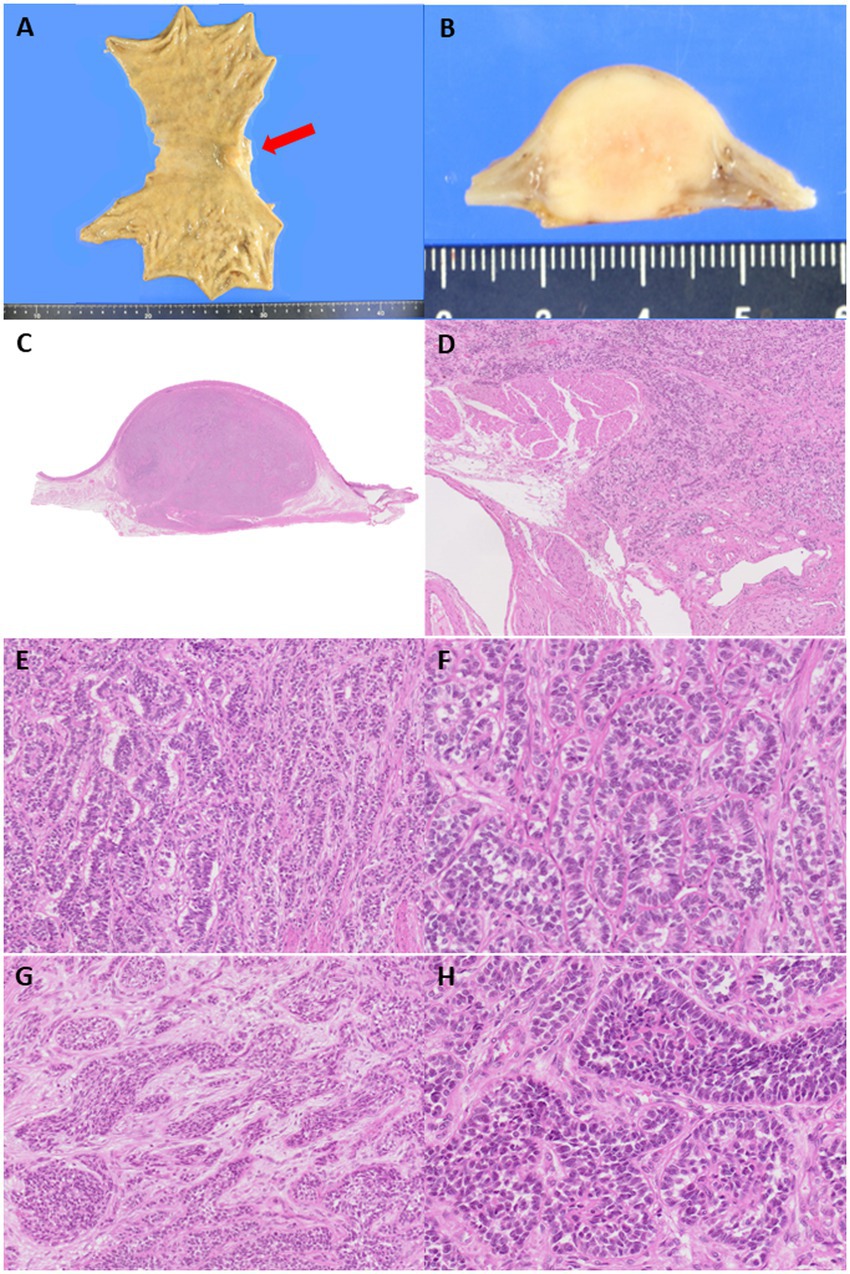
Figure 1. (A) A submucosal-centered tumor with slight protrusion toward the mucosal aspect is identified at the cardia and lesser curvature (red arrow). (B) The cut surface of the tumor shows a well-demarcated, white-yellow solid lesion measuring 20 mm in the largest dimension. (C) The tumor extends from the submucosa to the muscularis propria. (D,E) Tumor cells are arranged in microacinar and trabecular architectural arrangements without necrosis, reminiscent of a well-differentiated neuroendocrine tumor. The tumor mainly appeared well-demarcated but irregularly infiltrated the surrounding tissue in a few areas. (F) Tumor cells are monomorphic and exhibited scant, pale eosinophilic cytoplasm, round nuclei, and speckled chromatin. Occasional rosette-like structural formations are observed, similar to those observed in well-differentiated neuroendocrine tumors. (G) In a few areas, short, spindle-shaped cells arranged in tight nests are observed within the fibromyxoid stroma. (H) Tumor cells form solid sheets with small amounts of hyalinized stromal bodies.
IHC analysis revealed that the tumor cells were diffusely positive for vimentin, CD56, CD10, CAM5.2, AE1/AE3, estrogen receptor (ER), progesterone receptor (PgR) and androgen receptor (AR), STAT6 (cytoplasmic), and focally positive for S-100, E-cadherin, cyclinD1 and ATRX, whereas they were negative for chromogranin A, synaptophysin, INSM-1, smooth muscle antibody, c-kit, calretinin, epithelial membrane antigen, inhibin, SALL4, AFP, Glypican-3, p53, p40, CD34, bcl-10, SF-1, PAX-8, CDX2, CD99, MelanA, CEA, CA19-9, and CA125 (Figure 2). As this immune profile was deemed nondiagnostic for neuroendocrine neoplasms, further molecular studies were performed. As one of the primary differential diagnoses was gastroblastoma, RT-PCR for MALAT1::GLI1 fusion was performed, which yielded a negative result. Furthermore, DDIT3/GLI1 FISH analysis was negative for GLI1 gene rearrangements, and targeted RNA sequencing using the Illumina TruSight panel (500 genes) was negative for gene fusion candidates. RNA sequencing was predominantly performed to identify gene fusions rather than mutations, and coincidentally, manual inspection revealed a 10-fold increase in the mRNA expression of STAT6 and GLI1; however, no gene amplification was detected by FISH. Additional IHC showed focal and weak staining for GLI1 of uncertain significance.
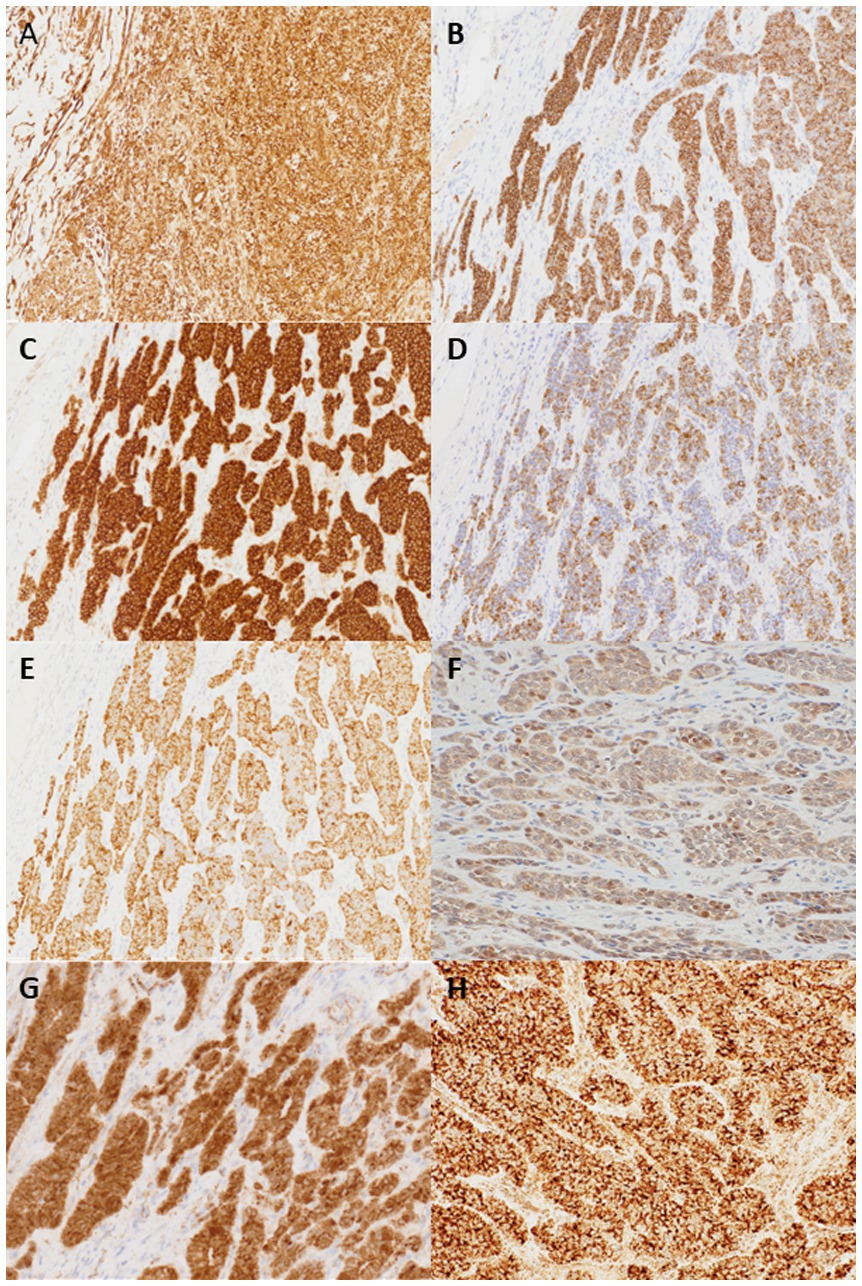
Figure 2. Tumor cells are positive for vimentin (A), CD56 (B), CD10 (C), CAM5.2 (D), AE1/3 (E). Focal and weak nuclear staining for GLI1 (F) and cytoplasmic and partial nuclear staining for β-catenin (G) are also noted. STAT6 IHC shows cytoplasmic staining but not nuclear staining (H) (×200).
The next possibility to be considered was a pseudoendocrine sarcoma. β-catenin immunostaining was performed, which showed aberrant nuclear expression (Figure 2). This result was further confirmed by Sanger sequencing, which showed a hot spot CTNNB1 mutation (S33C) (Figure 3). Consistent with these results was the tumor content of 80% in this case, which equated to an allele frequency of the CTNNB1 mutation (S33C) of 40%. Therefore, the CTNNB1 mutation was deemed to be clonal.
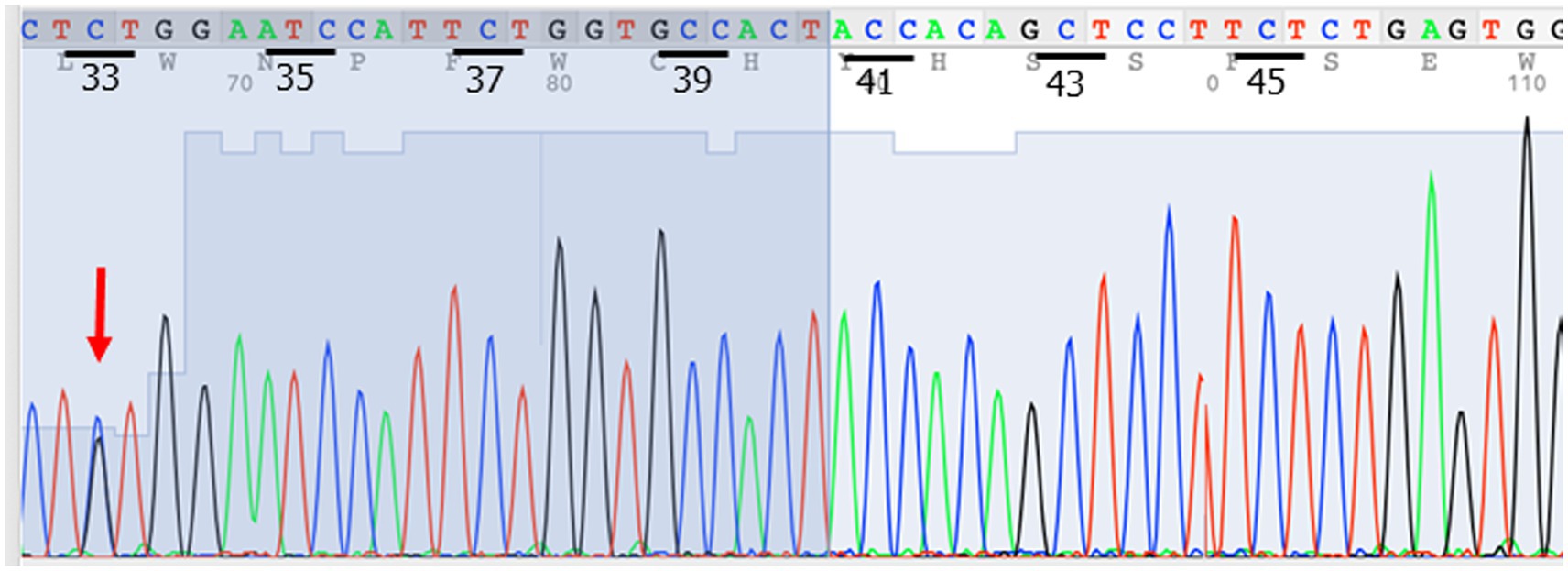
Figure 3. Sanger sequencing confirmed the CTNNB1 mutation (S33C) (red arrow).
Discussion
Gastric mesenchymal tumors are rare, with gastrointestinal stromal tumors, schwannomas, and leiomyomas being the most common. Based on the histological features observed in the initial biopsy, a diagnosis of a well-differentiated neuroendocrine tumor was considered; however, the IHC findings were not fully supportive. Gastric neuroendocrine neoplasms typically exhibit positive immunohistochemical staining for chromogranin A, synaptophysin, and INSM-1. In contrast, IHC findings suggested the possibility of gastroblastoma (5). However, gastroblastomas, similar to synovial sarcomas, usually exhibit a biphasic pattern characterized by a mixture of spindle cells and nests of epithelioid cells with abundant eosinophilic cytoplasm. These features were not observed in the present case. In addition, RT-PCR, FISH, and RNA sequencing failed to identify the presence of GLI1 gene rearrangements, MALAT1::GLI1 fusion, PTCH1::GLI1 fusion, or an EWSR1-CTBP1, recently identified in a patient with Wiskott-Aldrich syndrome (6, 8). Therefore, a diagnosis of gastroblastoma was excluded. The next possibility was a pseudoendocrine sarcoma. CTNNB1 mutation and nuclear expression for β-catenin IHC are consistent with pseudoendocrine sarcoma; however, these sarcomas occur commonly in the truncal locations such as the soft tissues of the vertebral body region and posterior head (1), so the site, in this case, did not fit pseudoendocrine sarcoma. In addition, short spindle cells arranged in tight nests were observed within a fibromyxoid stromal background that did not fit pseudoendocrine sarcoma. The case also showed focal and weak staining expression of GLI1 on IHC and monoclonal antibodies directed against keratins, such as CAM5.2 and AE1/AE3, although prior studies have not reported any positive staining for monoclonal antibodies directed against keratins and GLI1 on IHC (1, 2, 9). Based on these findings, the diagnosis of pseudoendocrine sarcoma was excluded.
The presence of the CTNNB1 mutation and the morphological similarity in this case may raise the possibility of a solid pseudopapillary neoplasm or Sertoli-like neoplasm, such as a Sertoli cell tumor of the testis. However, ectopic pancreatic tissue was not observed in the present case. Furthermore, the absence of a history of gonadal systemic tumors and no abnormalities in the gonads on PET-CT ruled out this possibility. However, the overexpression of hormone receptors such as ER, PgR, and AR in this case remains puzzling.
The possibility of a mesenchymal neoplasm with GLI1 gene alterations was raised because of GLI1 mRNA and protein overexpression (7). Recent studies have highlighted the morphological similarities between well-differentiated neuroendocrine tumors and soft tissue sarcomas with GLI1 gene alterations described as “distinctive nested glomoid neoplasm” (1, 7, 10, 11). However, it was excluded because of the absence of GLI1 fusion and the presence of the CTNNB1 mutation. Furthermore, irregular fibrous septa, lobular growth patterns, a prominent capillary network, or lobules protruding into vascular lumina beneath intact endothelium are frequently seen in distinctive nested glomoid neoplasms, but were not observed in this case. A study by Parrack et al. reported that GLI1 IHC was highly sensitive (91.3%) and specific (98.0%) for mesenchymal tumors with driver GLI1 alterations among morphological mimics (9). However, in this case, GLI1 IHC was focally and weakly positive rather than diffusely and strongly positive, which does not positively support a GLI1-altered mesenchymal neoplasm.
Furthermore, the strong and diffuse cytokeratin expression in this case seemed unusual for a GLI1-altered mesenchymal neoplasm (12). Since this case does not fit the disease concepts reported to date, we report it as “gastric submucosal neoplasm with CTNNB1 mutation showing GLI1 overexpression and epithelial differentiation.”
Regarding the prognosis of this case, the patient was followed up for almost 5 years without any treatment before undergoing surgery, during which the tumor size remained relatively stable. The MIB-1 proliferation index in the surgical specimen was approximately 5%, and the patient survived for 2 years without any evidence of recurrence or metastasis after surgery. Gastroendoscopy has been performed every year after surgery, and no new lesions are observed. These outcomes show the slow growth and low-grade nature of this tumor. This case report provides new insights into the clinicopathological characteristics of this disease.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Research Ethics Committee, Faculty of Medicine, Juntendo University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
KA: Writing – original draft, Writing – review & editing. TS: Writing – original draft, Writing – review & editing, Data curation, Funding acquisition, Investigation, Supervision. YY: Investigation, Writing – review & editing, Resources. SM: Investigation, Writing – review & editing, Resources. TF: Investigation, Writing – review & editing, Resources. CA: Investigation, Writing – review & editing, Data curation. TY: Writing – review & editing, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported in part by grants from the Grant-in-Aid for Scientific Research (KAKENHI) program of the Japan Society for the Promotion of Science (JSPS) (grant number 20K07415 to TS).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Papke, DJ Jr, Dickson, BC, Sholl, L, and Fletcher, CDM. Pseudoendocrine sarcoma: Clinicopathologic analysis of 23 cases of a distinctive soft tissue neoplasm with metastatic potential, recurrent CTNNB1 mutations, and a predilection for truncal locations. Am J Surg Pathol. (2022) 46:33–43. doi: 10.1097/PAS.0000000000001751
2. Vizcaino, MA, Folpe, AL, Huffman, H, Panchal, RR, Nielsen, GP, Kipp, BR, et al. Pseudoendocrine sarcoma: clinicopathologic, molecular, and epigenetic features of one case. Virchows Arch. (2023) 483:899–904. doi: 10.1007/s00428-023-03695-3
3. Bellan, E, Zanco, F, Baciorri, F, Toffolatti, L, Dei Tos, AP, and Sbaraglia, M. Case report: pseudoendocrine sarcoma, a clinicopathologic report of a newly described soft tissue neoplasm. Virchows Arch. (2023) 482:1057–63. doi: 10.1007/s00428-022-03476-4
4. Moran, JMT, Hung, YP, Selig, MK, and Nielsen, GP. Meningioma-like ultrastructural features of Pseudoendocrine sarcoma. Am J Surg Pathol. (2022) 46:1014–6. doi: 10.1097/pas.0000000000001890
5. Graham, RP, Nair, AA, Davila, JI, Jin, L, Jen, J, Sukov, WR, et al. Gastroblastoma harbors a recurrent somatic MALAT1-GLI1 fusion gene. Mod Pathol. (2017) 30:1443–52. doi: 10.1038/modpathol.2017.68
6. Koo, SC, LaHaye, S, Kovari, BP, Schieffer, KM, Ranalli, MA, Aldrink, JH, et al. Gastroblastoma with a novel EWSR1-CTBP1 fusion presenting in adolescence. Genes Chromosomes Cancer. (2021) 60:640–6. doi: 10.1002/gcc.22973
7. Papke, DJ Jr, Dickson, BC, Oliveira, AM, et al. Distinctive nested Glomoid neoplasm: Clinicopathologic analysis of 20 cases of a mesenchymal neoplasm with frequent GLI1 alterations and indolent behavior. Am J Surg Pathol. (2023) 47:12–24. doi: 10.1097/pas.0000000000001979
8. Chen, C, Lu, J, and Wu, H. Case report: submucosal gastroblastoma with a novel PTCH1::GLI2 gene fusion in a 58-year-old man. Front Oncol. (2022) 12:935914. doi: 10.3389/fonc.2022.935914
9. Parrack, PH, Mariño-Enríquez, A, Fletcher, CDM, Hornick, JL, and Papke, DJ. GLI1 immunohistochemistry distinguishes mesenchymal neoplasms with GLI1 alterations from morphologic mimics. Am J Surg Pathol. (2023) 47:453–60. doi: 10.1097/pas.0000000000002018
10. Antonescu, CR, Agaram, NP, Sung, YS, Zhang, L, Swanson, D, and Dickson, BC. A distinct malignant epithelioid neoplasm with GLI1 gene rearrangements, frequent S100 protein expression, and metastatic potential: expanding the Spectrum of pathologic entities with ACTB/MALAT1/PTCH1-GLI1 fusions. Am J Surg Pathol. (2018) 42:553–60. doi: 10.1097/pas.0000000000001010
11. Agaram, NP, Zhang, L, Sung, YS, Singer, S, Stevens, T, Prieto-Granada, CN, et al. GLI1-amplifications expand the spectrum of soft tissue neoplasms defined by GLI1 gene fusions. Mod Pathol. (2019) 32:1617–26. doi: 10.1038/s41379-019-0293-x
12. Saoud, C, Agaimy, A, Dermawan, JK, Chen, JF, Rosenblum, MK, Dickson, BC, et al. A comprehensive Clinicopathologic and molecular reappraisal of GLI1-altered mesenchymal tumors with pooled outcome analysis showing poor survival in GLI1- amplified versus GLI1-rearranged tumors. Am J Surg Pathol. (2024) 48:1302–17. doi: 10.1097/pas.0000000000002272
Keywords: pseudoendocrine sarcoma, stomach, CTNNB1, cytokeratin, epithelial, neuroendocrine, GLI1
Citation: Ashizawa K, Saito T, Yube Y, Mine S, Fukunaga T, Antonescu CR and Yao T (2025) Case Report: Gastric submucosal neoplasm with CTNNB1 mutation showing GLI1 overexpression and epithelial differentiation. Front. Med. 12:1526614. doi: 10.3389/fmed.2025.1526614
Edited by:
Luigi Tornillo, University of Basel, SwitzerlandReviewed by:
Shabnam Momtahen, Dartmouth College, United StatesRahul Gupta, Synergy Institute of Medical Sciences, India
Qi-Xing Gong, First Affiliated Hospital of Nanjing Medical University, China
Copyright © 2025 Ashizawa, Saito, Yube, Mine, Fukunaga, Antonescu and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tsuyoshi Saito, dHlzYWl0b3VAanVudGVuZG8uYWMuanA=