 QingHua Liu†
QingHua Liu† Yu Xiong
Yu Xiong Chenyang Xu
Chenyang Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med. , 28 January 2025
Sec. Pathology
Volume 12 - 2025 | https://doi.org/10.3389/fmed.2025.1514349
Ehlers-Danlos Syndrome (EDS) refers to a group of connective tissue disorders characterized by significant clinical and genetic variability, affecting multiple systems in the body. Classified as a rare disease, EDS includes 14 subtypes, all marked by joint hypermobility, skin extensibility, and tissue fragility. These subtypes present with a wide range of clinical manifestations and severities, including frequent joint dislocations, scoliosis, arterial dissections, and organ ruptures. Hypermobile EDS (hEDS) is the most common subtype, with newly established clinical diagnostic guidelines. In this case, a patient presented with minor hemoptysis over 8 h, and a chest CT scan revealed a massive hematoma in the left lower lung. Due to the complexity and varied presentations of EDS, misdiagnosis is common. This report shares our experience with diagnosis and treatment in this case, highlighting the importance of increasing awareness for improved survival outcomes.
Hypermobile Ehlers-Danlos Syndrome (hEDS) was classified as Type V in the EDS classification system published by the International EDS Consortium in 2017 (1). The primary diagnostic criterion for hEDS is generalized joint hypermobility, evaluated using the Beighton score (2) (Table 1), with age-adjusted cutoffs. However, a diagnosis of hEDS requires more than just generalized joint hypermobility. At least two additional criteria must be met: systemic connective tissue disease features (such as soft skin, recurrent hernias, spider-like fingers, or mitral valve prolapse), a family history of hEDS, and musculoskeletal manifestations (such as joint instability, frequent dislocations, or chronic pain). Other EDS subtypes and causes of generalized joint hypermobility must also be ruled out (Table 2). The experience in managing skeletal complications in EDS patients is limited, with almost no established guidelines Most of the literature consists of case reports or small retrospective studies (3, 4). In this case, we present a patient with hEDS who developed a hematoma in the left lung. However, the clinical presentation was only mild hemoptysis, which is previously unreported. We will provide insights on the management of this condition.
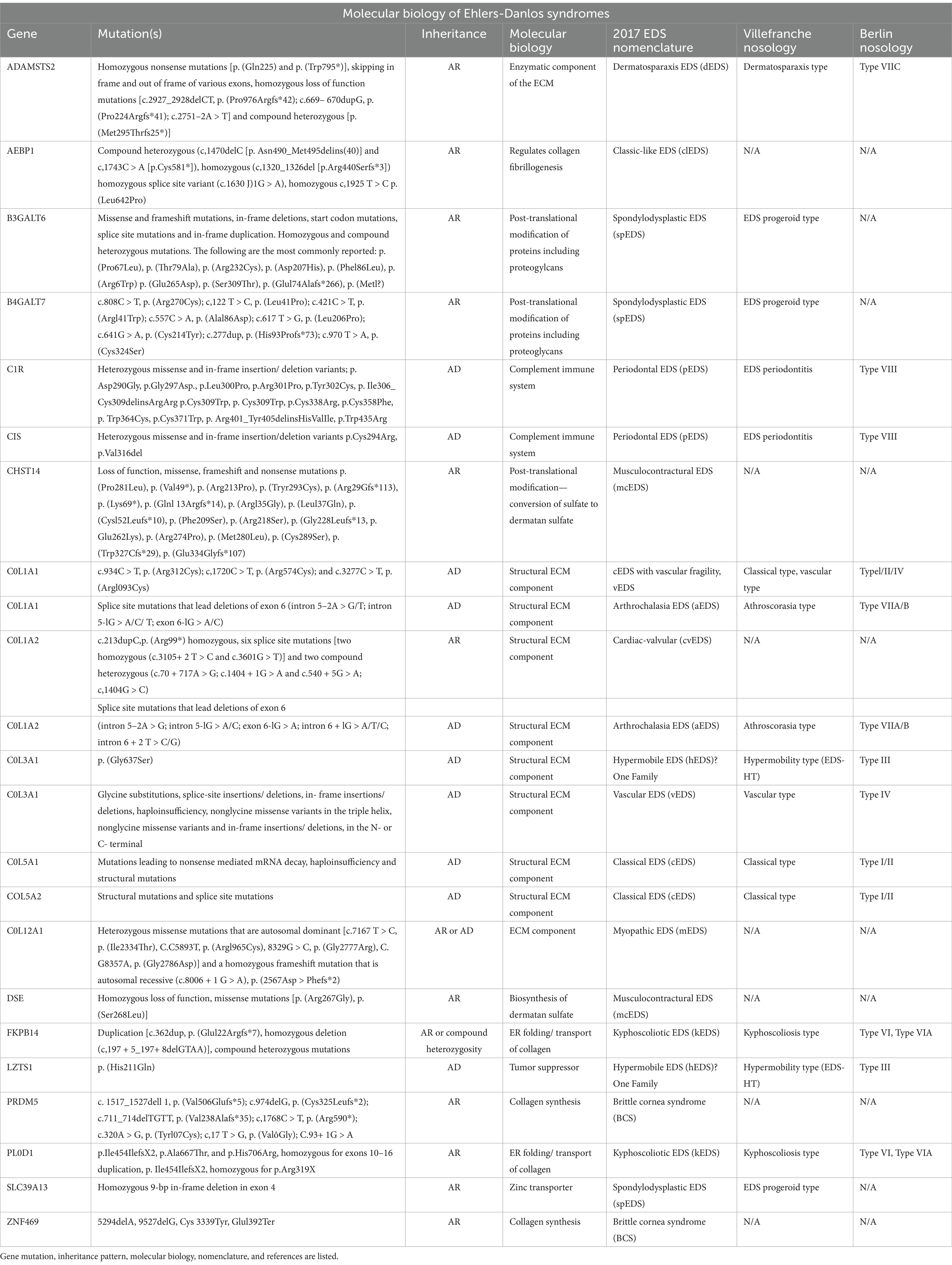
Table 1. Description of subtypes of EDS comparing 2017 nomenclature with the Villefranche and Berlin nosology.
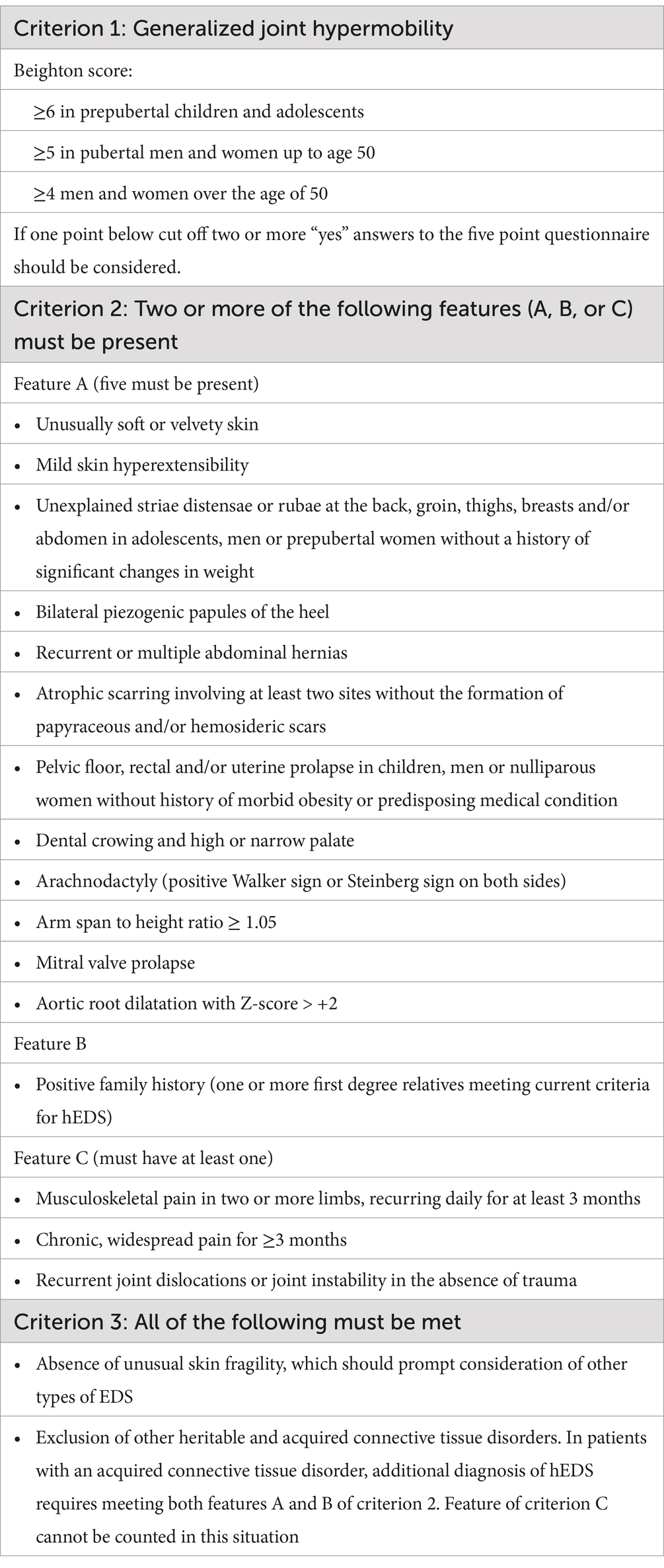
Table 2. EDS diagnostic checklist listing the three main criterion.
An 18-year-old male presented with minor hemoptysis lasting 8 h. Physical examination revealed soft, lax skin, flexible flat feet, hallux valgus, and hypertrichosis (Figures 1, 2). His medical history included spontaneous right pneumothorax, intracranial hemorrhage, spontaneous bleeding in the left calf, and chronic joint pain in the limbs. A chest CT scan revealed a hematoma in the left lower lung (Figure 3). Routine blood tests, biochemistry, and coagulation function were normal (white blood cell count: 10.32 × 10^9, neutrophils: 49.3%, lymphocytes: 29.3%, eosinophils: 8.2%; hemoglobin: 95 g/L, platelet count: 409 × 10^9), and HIV tests were negative. Due to the massive hematoma and persistent hemoptysis, a left lower lobectomy was considered. However, during surgery, the patient’s tissue displayed abnormal fragility, making hemostasis difficult, which prevented a successful lobectomy (Figure 4).
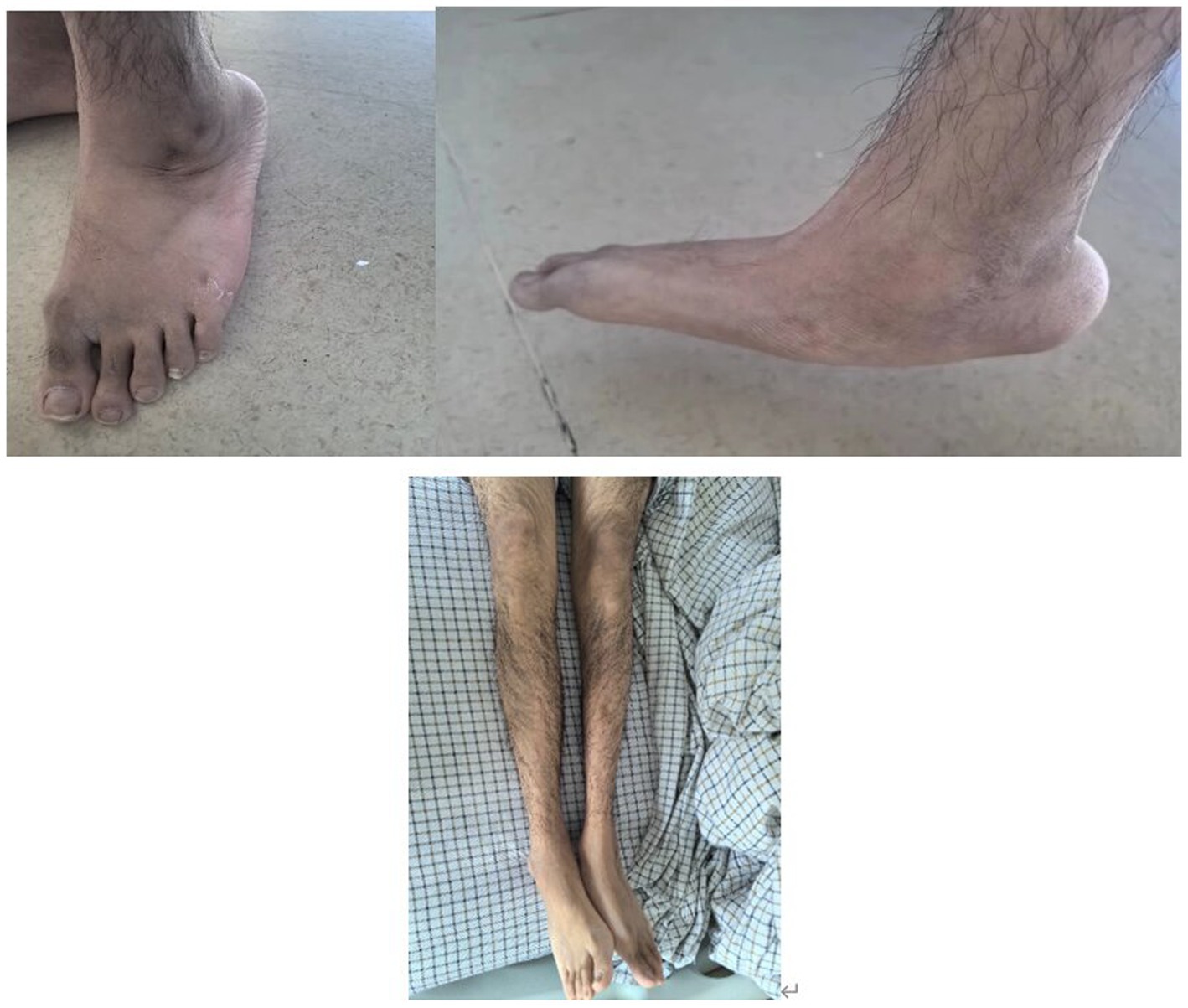
Figure 1. Increased hair on both lower extremities and flatfoot.
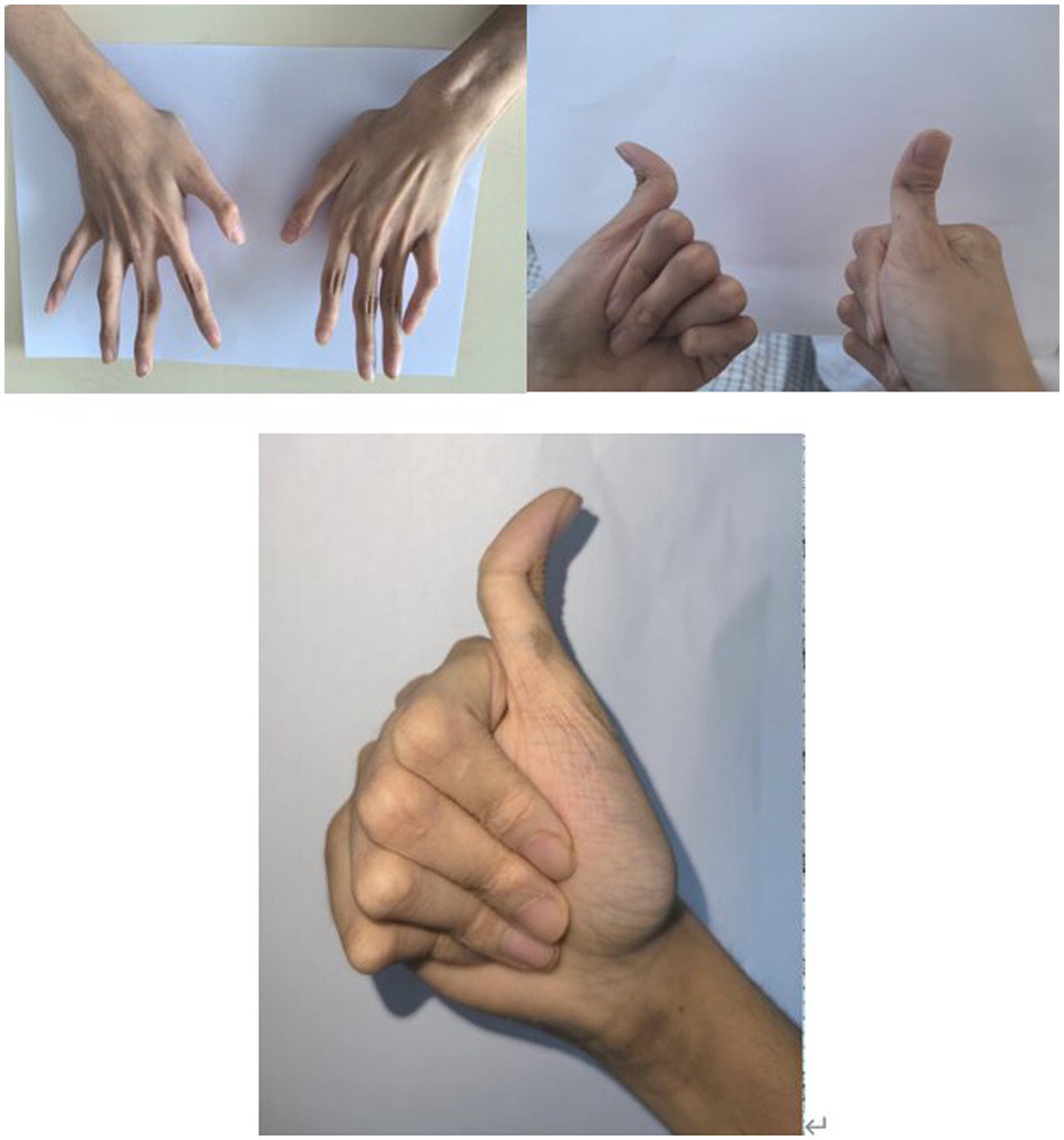
Figure 2. Joint abnormalities and Joint hyperextension.
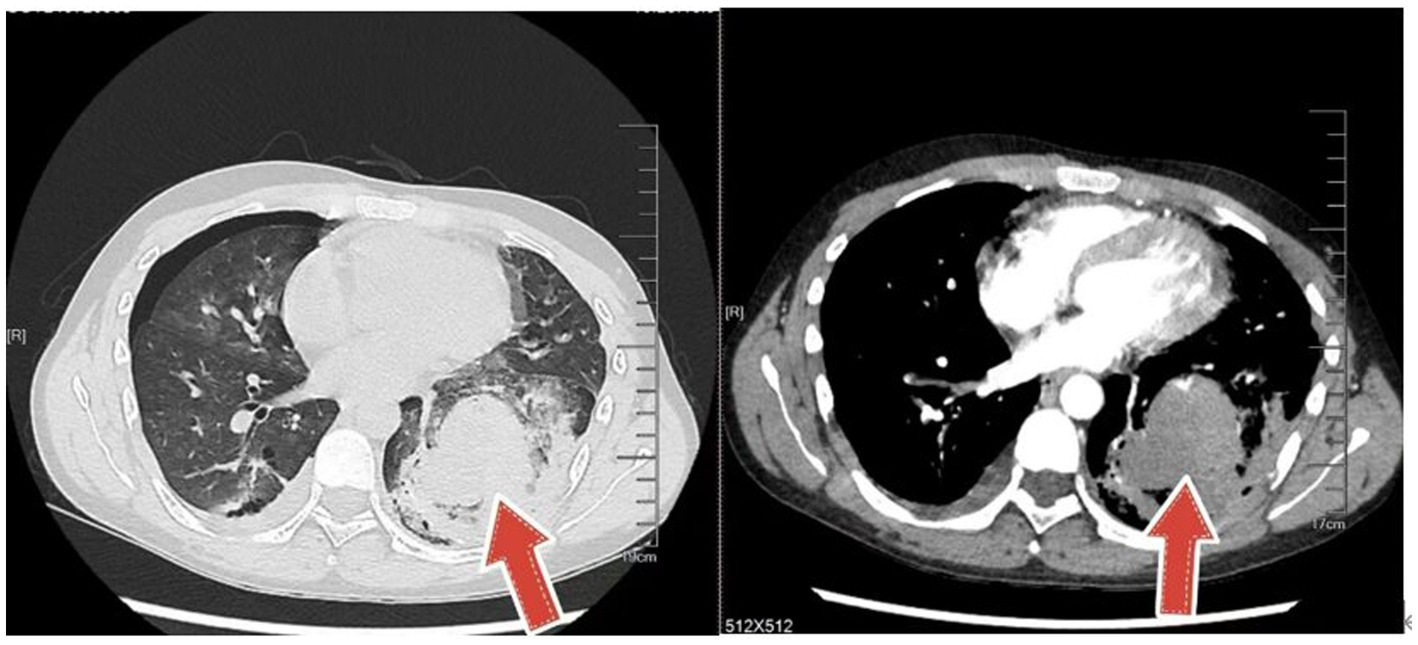
Figure 3. CT shows a massive hematoma in the left lung.
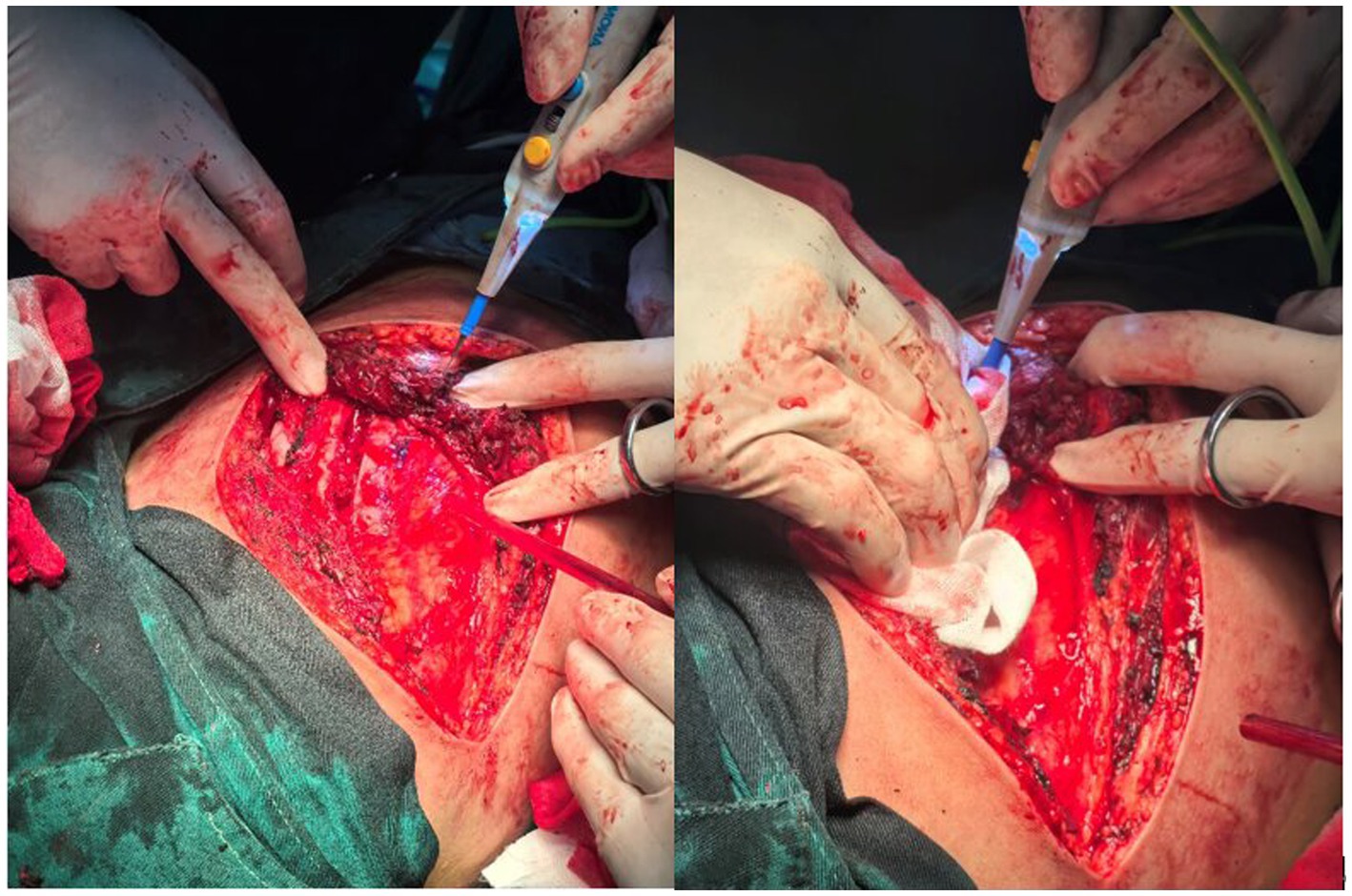
Figure 4. During the surgery, the tissues exhibited abnormal fragility and were prone to bleeding.
Postoperatively, the unusual connective tissue fragility led to further diagnostic workup, including autoimmune antibody testing, all of which returned negative. The patient’s physical appearance, with visible veins under his translucent skin, combined with a slender build (height: 159 cm, weight: 40 kg, BMI: 15.82), joint hypermobility, and elastic skin, led to a clinical suspicion of EDS. We conducted whole exome sequencing on the patient, but unfortunately, no genetic mutations were identified. However, due to financial constraints, genetic testing was not performed on the patient’s parents. However, we discovered that the patient’s grandfather had a related medical history, and the final diagnosis was hEDS associated with a large pulmonary hematoma. The patient was discharged 2 weeks after surgery. Due to the fragility of the connective tissue, further medical intervention was required. Conservative treatment was implemented, with close monitoring of the patient’s pain management. Physical therapy was provided to help improve joint stability and muscle strength, which is a key aspect of managing hypermobile Ehlers-Danlos syndrome (hEDS). We provided support for unstable joints, including finger splints and ankle stabilizing braces.The patient continued to take nonsteroidal anti-inflammatory drugs (NSAIDs) to manage postoperative chronic pain. Additionally, daily oral vitamin C (100 mg, three times a day) was recommended to enhance vascular elasticity and reduce the risk of potential bleeding.
Upon discharge, the patient was instructed to return for follow-up chest CT scans every 3 months and to avoid prolonged sun exposure. Dietary advice included consuming foods rich in vitamin C. If necessary, the patient was advised to visit the rheumatology department for evaluation of the impact of connective tissue fragility on other organ systems or the orthopedics department to assess the long-term stability and functionality of the joints.
One week after discharge, our department conducted a follow-up with the patient. The patient reported stable condition and significant pain relief compared to earlier. However, no subsequent follow-up visits or evaluations were completed. For a detailed timeline, please refer to Supplementary material.
hEDS is a hereditary connective tissue disorder. According to Demmler et al. (5), the prevalence of hEDS and hypermobility spectrum disorders (HSD) in Wales is about 1 in 500, with 70% of diagnosed patients being female. Unlike other EDS subtypes, the genetic cause of hEDS remains elusive, complicating both research and diagnosis. Thirteen of the 14 EDS subtypes have identifiable genetic markers (6), but hEDS remains a diagnosis made based on clinical criteria (Table 3).
In hEDS, connective tissue fragility, particularly in the vascular structures, predisposes individuals to spontaneous bleeding. The underlying pathophysiology involves abnormalities in collagen synthesis, leading to weakened blood vessel walls and an increased tendency for vessels to rupture under minimal stress. This explains the patient’s intracranial hemorrhage and spontaneous bleeding in the left calf.
Pulmonary bleeding, as observed in this case, is less commonly described in hEDS but can also occur due to vascular fragility. The weakened capillaries and small blood vessels in the lungs are susceptible to rupture, especially in the context of trauma or increased intrathoracic pressure. While pulmonary hematoma is a rare complication in hEDS, it should be considered as part of the spectrum of vascular manifestations in this condition.For patients suspected of having EDS, genetic testing is recommended to confirm the diagnosis, though not all patients undergoing clinical diagnosis will have molecular diagnostic results (7). For instance, about 10% of patients clinically diagnosed with classical EDS (cEDS) may not have an identifiable genetic marker (7). Additionally, some patients with mild phenotypes of COL5A1 and COL5A2 mutations may be diagnosed with hEDS rather than cEDS. Some patients exhibit more severe phenotypes or do not completely fit the criteria for cEDS and are clinically diagnosed with vascular EDS (vEDS), dermatospraxis EDS(dEDS), or kyphoscoliotic EDS(kEDS) (8–10). Genetic testing may also reclassify patients diagnosed with EDS into other conditions like Loeys-Dietz syndrome or Cutis laxa or Multiple epiphyseal dysplasia or Hypophosphatasia (7, 11, 12). This is related not only to the strong clinical and genetic heterogeneity of EDS and its intrinsic disease characteristics, but may also be associated with inaccurate phenotype assessment or the absence of complete disease features in younger patients.
The complexity of EDS diagnosis, stemming from clinical and genetic heterogeneity, can lead to uncertainties for clinicians. Negative or ambiguous genetic results do not rule out EDS, which underscores the need for regular re-evaluation of genetic data using advanced methods like RNA sequencing. Studies have shown that regularly re-analyzing genetic data can significantly increase diagnostic accuracy due to evolving knowledge of gene-disease associations and improved phenotypic assessments (13–15).
Pain is the most common symptom in hEDS, and nearly all hEDS patients experience pain, although its severity and frequency can vary widely. Patients with joint hypermobility in EDS may progress from hypermobility to chronic, progressive pain, leading to reduced mobility, decreased muscle mass, proprioceptive dysfunction, and conditions like arthritis. Other common skeletal manifestations include scoliosis (kyphoscoliosis), clubfoot, pectus excavatum or carinatum, osteopenia or osteoporosis, and congenital hip dislocation (16).
One study (17) suggests a multidisciplinary approach for hEDS patients, incorporating medication for pain management, physical therapy, occupational therapy, and psychological support. Medications primarily focus on symptom relief. For example, over-the-counter NSAIDs and analgesics may help alleviate pain and discomfort frequently experienced by EDS patients. However, given that EDS patients often have gastrointestinal issues, medication selection and usage should be guided by a gastroenterologist (18).
Physical therapy serves as a crucial conservative treatment method. It can help enhance joint stability, improve muscle strength, and manage pain.
hEDS is often linked with autonomic dysfunction, and when it impacts the cardiovascular system, it manifests as orthostatic intolerance, including Postural Orthostatic Tachycardia Syndrome (POTS). POTS is defined as an increase in heart rate of at least 30 beats per minute or a heart rate exceeding 120 beats per minute within 10 min of standing, without orthostatic hypotension. However, in this case, POTS symptoms were not observed. Some studies report that the prevalence of hEDS among POTS patients ranges from 15 to 22% (19, 20).
Regarding POTS management (21, 22), early exercise training and increased salt and fluid intake are recommended to expand blood volume. Other non-pharmacological strategies include elevating the head of the bed during sleep, using lower limb compression garments or abdominal binders to reduce venous pooling while standing, and physical maneuvers such as squeezing a rubber ball, crossing legs, muscle pumping, squatting, or negative pressure breathing to prevent orthostatic intolerance and manage acute clinical symptoms in POTS patients. Autonomic dysfunction may also present as gastrointestinal motility disorders, bladder dysfunction, skin discoloration, or abnormal sweating (23).
In terms of the skin, many types of EDS can exhibit skin hyperextensibility, but it is usually less pronounced than in hEDS or cEDS, along with a reduction in dermal thickness and increased skin fragility (24, 25). Patients may experience wound healing defects, potentially leading to atrophic scar formation (25–27). Increased capillary fragility can result in frequent bruising and delayed healing. Oral or intravenous vitamin C can help improve vascular fragility and reduce bruising, but the dose should not exceed 500 mg/day (27, 28). The patient was instructed to take Vitamin C orally, three times a day, 100 mg per dose, upon discharge.
hEDS is frequently associated with symptoms such as fatigue, urticaria, flushing, angioedema, rhinitis, and diarrhea, potentially linked to mast cell activation disorder (MCAD) in hEDS (29). Treatment for MCAD involves avoiding triggers such as certain foods, medications, and temperature changes. Symptomatic treatment includes antihistamines for allergic symptoms, mast cell stabilizers like ketotifen to reduce the release of mediators from mast cells, corticosteroids for acute or severe symptoms to suppress inflammation, leukotriene receptor antagonists like montelukast to alleviate inflammation, NSAIDs for pain and inflammation control, and immunosuppressants in refractory cases (30). Regular monitoring of mast cell mediator levels is also necessary to assess treatment efficacy.
Psychological and mental health management is crucial, not only in hEDS but across all EDS types. Psychological issues such as depression, anxiety, attention deficit hyperactivity disorder (ADHD), and post-traumatic stress disorder (PTSD) are frequently observed in EDS patients (31). Pain is a prominent and pervasive symptom that troubles EDS patients. Research indicates (32, 33) that psychological health is closely associated with pain in EDS, revealing a significant positive correlation between pain intensity and the severity of depressive symptoms. Furthermore, psychological and emotional issues may exacerbate the perception of pain. In this patient, the ongoing involvement of multiple disciplines and frequent hospital visits led to a loss of confidence in the treatment. This required us to provide clear explanations about the condition and offer emotional support.
Physical therapy and medication can improve the patient’s physical condition, while cognitive-behavioral therapy and other psychological interventions can enhance self-efficacy and emotional well-being, helping to reduce fear associated with movement and pain during treatment.
According to the literature (34), due to the increased vascular and tissue fragility in EDS patients, numerous surgical complications can occur, such as spontaneous expansion of surgical incisions and rupture of deep tissues upon contact with the scalpel. Therefore, extreme caution is required when handling tissues, and the use of skin retractors should be minimized. If the patient has significant vascular fragility, adequate hemostasis through electrocautery should be performed during surgery, and the use of vascular clamps should be avoided. Because the tensile strength of the patient’s blood vessels is poor, arterial repairs can be challenging. Hemostasis can be achieved through gauze compression and intermittent horizontal mattress suturing.When closing the incision, skin closure should be performed in two layers, with minimal tension, sufficient suture material, deep sutures, and three-dimensional support. The sutures should be placed at a proper distance from the incision to avoid cutting through fragile tissues, and skin clamps should not be used. Finally, the duration for retaining the sutures should be twice the usual recommended time to avoid wound dehiscence (35).
In summary, we have depicted a case of hypermobile Ehlers-Danlos Syndrome (hEDS) presenting as mild hemoptysis, a clinical manifestation not previously documented. This case highlights the importance of considering hEDS in patients with joint deformities and pain. hEDS is a hereditary connective tissue disorder primarily diagnosed through clinical features rather than genetic markers. The patient exhibited tissue fragility during treatment, complicating the surgical process and emphasizing the complexity of the condition. Pain is the most common symptom in hEDS, requiring multidisciplinary management that includes medication, physical therapy, and psychological support. hEDS is also associated with autonomic dysfunction and mental health issues, necessitating psychological interventions to improve quality of life. This case serves as a reminder for clinicians to consider hEDS when encountering joint deformities and unexplained bleeding.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Ethics Committee of Ganzhou People’s Hospital (Jiangxi, China). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
QL: Writing – original draft, Writing – review & editing. GZ: Writing – original draft, Writing – review & editing. YX: Writing – original draft, Writing – review & editing. CX: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Ganzhou City Science and Technology Planning Project (GZ2024ZSF059).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1514349/full#supplementary-material
1. Malfait, F, Francomano, C, Byers, P, Belmont, J, Berglund, B, Black, J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. (2017) 175:8–26. doi: 10.1002/ajmg.c.31552
2. Høiseth, TCJ, Tsykunova, G, Bruserud, Ø, and Tvedt, THA. The Beighton scoring system. Tidsskrift for den Norske laegeforening: tidsskrift for praktisk medicin, ny raekke. (2020) 140. doi: 10.4045/tidsskr.19.0487
3. Ibrahim, R, Hamadah, O, Abdul-Hak, M, Alshawa, A, and Alouda, MA. Cleidocranial dysplasia with hypermobile Ehlers-Danlos syndrome: a case report. Radiol Case Reports. (2023) 18:289–94. doi: 10.1016/j.radcr.2022.10.038
4. Gullapalli, PA, and Javed, S. Multidisciplinary chronic pain management strategies in patients with Ehlers-Danlos syndromes. Pain Manag. (2023) 13:5–14. doi: 10.2217/pmt-2022-0050
5. Demmler, JC, Atkinson, MD, Reinhold, EJ, Choy, E, Lyons, RA, and Brophy, ST. Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: a national electronic cohort study and case-control comparison. BMJ Open. (2019) 9:e031365. doi: 10.1136/bmjopen-2019-031365
6. Malfait, F, Castori, M, Francomano, CA, Giunta, C, Kosho, T, and Byers, PH. The Ehlers-Danlos syndromes. Nat Rev Dis Prim. (2020) 6:64. doi: 10.1038/s41572-020-0194-9
7. Damseh, N, Dupuis, L, O'Connor, C, Oh, RY, Wang, YW, Stavropoulos, DJ, et al. Diagnostic outcomes for molecular genetic testing in children with suspected Ehlers-Danlos syndrome. Am J Med Genet A. (2022) 188:1376–83. doi: 10.1002/ajmg.a.62672
8. Colman, M, Syx, D, De Wandele, I, Dhooge, T, Symoens, S, and Malfait, F. Clinical and molecular characteristics of 168 probands and 65 relatives with a clinical presentation of classical Ehlers-Danlos syndrome. Hum Mutat. (2021) 42:1294–306. doi: 10.1002/humu.24258
9. Lavanya, K, Mahtani, K, Abbott, J, Jain, A, Selvam, P, Atwal, H, et al. A patient with a novel pathogenic variant in COL5A1 exhibiting prominent vascular and cardiac features. Am J Med Genet A. (2022) 188:2192–7. doi: 10.1002/ajmg.a.62745
10. Colombi, M, Dordoni, C, Cinquina, V, Venturini, M, and Ritelli, M. A classical Ehlers-Danlos syndrome family with incomplete presentation diagnosed by molecular testing. Eur J Med Genet. (2018) 61:17–20. doi: 10.1016/j.ejmg.2017.10.005
11. Greally, MT, Kalis, NN, Agab, W, Ardati, K, Giurgea, S, Kornak, U, et al. Autosomal recessive cutis laxa type 2A (ARCL2A) mimicking Ehlers-Danlos syndrome by its dermatological manifestations: report of three affected patients. Am J Med Genet A. (2014) 164:1245–53. doi: 10.1002/ajmg.a.36411
12. Vandersteen, AM, Weerakkody, RA, Parry, DA, Kanonidou, C, Toddie-Moore, DJ, Vandrovcova, J, et al. Genetic complexity of diagnostically unresolved Ehlers-Danlos syndrome. J Med Genet. (2024) 61:232–8. doi: 10.1136/jmg-2023-109329
13. Liu, P, Meng, L, Normand, EA, Xia, F, Song, X, Ghazi, A, et al. Reanalysis of clinical exome sequencing data. N Engl J Med. (2019) 380:2478–80. doi: 10.1056/NEJMc1812033
14. Chi, CS, Tsai, CR, and Lee, HF. Resolving unsolved whole-genome sequencing data in paediatric neurological disorders: a cohort study. Arch Dis Child. (2024) 109:730–5. doi: 10.1136/archdischild-2024-326985
15. Surl, D, Won, D, Lee, ST, Lee, CS, Lee, J, Lim, HT, et al. Clinician-driven reanalysis of exome sequencing data from patients with inherited retinal diseases. JAMA Netw Open. (2024) 7:e2414198. doi: 10.1001/jamanetworkopen.2024.14198
16. Yonko, EA, LoTurco, HM, Carter, EM, and Raggio, CL. Orthopedic considerations and surgical outcomes in Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. (2021) 187:458–65. doi: 10.1002/ajmg.c.31958
17. Sulli, A, Talarico, R, Scirè, CA, Avcin, T, Castori, M, Ferraris, A, et al. Ehlers-Danlos syndromes: state of the art on clinical practice guidelines. RMD Open. (2018) 4:e000790. doi: 10.1136/rmdopen-2018-000790
18. Chopra, P, Tinkle, B, Hamonet, C, Brock, I, Gompel, A, Bulbena, A, et al. Pain management in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. (2017) 175:212–9. doi: 10.1002/ajmg.c.31554
19. Deb, A, Morgenshtern, K, Culbertson, CJ, Wang, LB, and Hohler, AD. A survey-based analysis of symptoms in patients with postural orthostatic tachycardia syndrome. Proc (Baylor Univ Med Cent). (2015) 28:157–9. doi: 10.1080/08998280.2015.11929217
20. Wallman, D, Weinberg, J, and Hohler, AD. Ehlers-Danlos syndrome and postural tachycardia syndrome: a relationship study. J Neurol Sci. (2014) 340:99–102. doi: 10.1016/j.jns.2014.03.002
21. Fu, Q, and Levine, BD. Exercise and non-pharmacological treatment of POTS. Auton Neurosci. (2018) 215:20–7. doi: 10.1016/j.autneu.2018.07.001
22. Fedorowski, A. Postural orthostatic tachycardia syndrome: clinical presentation, aetiology and management. J Intern Med. (2019) 285:352–66. doi: 10.1111/joim.12852
23. Tai, FWD, Palsson, OS, Lam, CY, Whitehead, WE, Sperber, AD, Tornblom, H, et al. Functional gastrointestinal disorders are increased in joint hypermobility-related disorders with concomitant postural orthostatic tachycardia syndrome. Neurogastroenterol Motil. (2020) 32:e13975. doi: 10.1111/nmo.13975
24. Castori, M. Ehlers-danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatology. (2012) 2012:751768:1–22. doi: 10.5402/2012/751768
25. Catala-Pétavy, C, Machet, L, Georgesco, G, Pétavy, F, Maruani, A, and Vaillant, L. Contribution of skin biometrology to the diagnosis of the Ehlers-Danlos syndrome in a prospective series of 41 patients. Skin Res Technol: Official J Int Society Bioeng Skin (ISBS) [and] Int Society Digital Imag Skin (ISDIS) [and] Int Society Skin Imag (ISSI). (2009) 15:412–7. doi: 10.1111/j.1600-0846.2009.00379.x
26. Eisenbeiss, C, Martinez, A, Hagedorn-Greiwe, M, Reinhardt, DP, Bätge, B, and Brinckmann, J. Reduced skin thickness: a new minor diagnostic criterion for the classical and hypermobility types of Ehlers-Danlos syndrome. Br J Dermatol. (2003) 149:850–2. doi: 10.1046/j.1365-2133.2003.05439.x
27. Tinkle, B, Castori, M, Berglund, B, Cohen, H, Grahame, R, Kazkaz, H, et al. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome type III and Ehlers-Danlos syndrome hypermobility type): clinical description and natural history. Am J Med Genet C Semin Med Genet. (2017) 175:48–69. doi: 10.1002/ajmg.c.31538
28. Engelbert, RH, Juul-Kristensen, B, Pacey, V, de Wandele, I, Smeenk, S, Woinarosky, N, et al. The evidence-based rationale for physical therapy treatment of children, adolescents, and adults diagnosed with joint hypermobility syndrome/hypermobile Ehlers Danlos syndrome. Am J Med Genet C Semin Med Genet. (2017) 175:158–67. doi: 10.1002/ajmg.c.31545
29. Seneviratne, SL, Maitland, A, and Afrin, L. Mast cell disorders in Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. (2017) 175:226–36. doi: 10.1002/ajmg.c.31555
30. Monaco, A, Choi, D, Uzun, S, Maitland, A, and Riley, B. Association of mast-cell-related conditions with hypermobile syndromes: a review of the literature. Immunol Res. (2022) 70:419–31. doi: 10.1007/s12026-022-09280-1
31. Hershenfeld, SA, Wasim, S, McNiven, V, Parikh, M, Majewski, P, Faghfoury, H, et al. Psychiatric disorders in Ehlers-Danlos syndrome are frequent, diverse and strongly associated with pain. Rheumatol Int. (2016) 36:341–8. doi: 10.1007/s00296-015-3375-1
32. Palomo-Toucedo, IC, Leon-Larios, F, Reina-Bueno, M, Vázquez-Bautista, MDC, Munuera-Martínez, PV, and Domínguez-Maldonado, G. Psychosocial influence of Ehlers-Danlos syndrome in daily life of patients: a qualitative study. Int J Environ Res Public Health. (2020) 17. doi: 10.3390/ijerph17176425
33. Niermeyer, M, Ball, D, Green, M, Jensen, B, Pace, L, Shingleton, R, et al. Interoceptive attention regulation in Ehlers-Danlos syndromes: associations between pain and psychiatric symptom severity. Transl Behav Med. (2021) 11:1923–30. doi: 10.1093/tbm/ibab049
34. Ryan, N, Walkden, G, and Akbar, S. Some wounds are hard to heal: an interesting presentation of Ehlers-Danlos syndrome. J Wound Care. (2012) 21:223–6. doi: 10.12968/jowc.2012.21.5.223
Keywords: pulmonary hematoma, case report, hypermobile Ehlers-Danlos syndrome (hEDS), hemoptysis, thoracic surgery
Citation: Liu Q, Zeng G, Xiong Y and Xu C (2025) A case of massive hematoma: reflections on hypermobile Ehlers-Danlos syndrome. Front. Med. 12:1514349. doi: 10.3389/fmed.2025.1514349
Edited by:
Dimitrios Liakopoulos, General Hospital Nice Piraeus Saint Panteleimon, GreeceReviewed by:
George Galyfos, National and Kapodistrian University of Athens, GreeceCopyright © 2025 Liu, Zeng, Xiong and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Xiong, MTMxNjMxNzMxNUBxcS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.