 Patricia Eiko Yamakawa1*
Patricia Eiko Yamakawa1* Caio Perez Gomes2
Caio Perez Gomes2 Agatha Ribeiro Mendes2
Agatha Ribeiro Mendes2 Caio Cesar Justino de Oliveira2Florencio Porto Freitas3
Caio Cesar Justino de Oliveira2Florencio Porto Freitas3 Fabiana Bettoni4Ernande Xavier dos Santos4
Fabiana Bettoni4Ernande Xavier dos Santos4 Vinicius Campos de Molla1Matheus Vescovi Gonçalves1
Vinicius Campos de Molla1Matheus Vescovi Gonçalves1 Jessica Branquinho2Beatriz Ribeiro Nogueira2
Jessica Branquinho2Beatriz Ribeiro Nogueira2 Joao Bosco Pesquero2
Joao Bosco Pesquero2 Celso Arrais-Rodrigues1
Celso Arrais-Rodrigues1- 1Department of Hematology, Universidade Federal de São Paulo/Escola Paulista de Medicina, São Paulo, Brazil
- 2Department of Biophysics, Universidade Federal de São Paulo, São Paulo, Brazil
- 3Rudolf Virchow Zentrum (RVZ), Center for Integrative and Translational Bioimaging, University of Würzburg, Würzburg, Germany
- 4Molecular Oncology Center, Hospital Sirio Libanes, São Paulo, Brazil
Background: Paroxysmal nocturnal hemoglobinuria (PNH) is a rare clonal hematopoietic stem cell disease characterized by acquired abnormalities in the phosphatidylinositol glycan class A (PIG-A) gene.
Methods: This study analyzed PIG-A gene using polymerase chain reaction (PCR) followed by Sanger sequencing of 31 Brazilian patients with PNH, including 23 with classical PNH and 8 with subclinical PNH (aplastic anemia and a PNH clone).
Results: A diverse spectrum of acquired PIG-A variants was identified, encompassing insertions, deletions, and single-base substitutions. The majority of variants identified (17 out of 29) were deemed likely pathogenic for paroxysmal nocturnal hemoglobinuria (PNH). Six variants have undetermined significance (VUS) and six variants are probably benign. Somatic variants exhibited variability in type and location among the patients, with a predominance of small deletions and simple base changes. Notably, 41% of the variants were frameshift and 35% were missense. Among the 23 patients with hemolytic PNH, 19 had at least one detectable pathogenic variant. Subclinical PNH cases were characterized solely by polymorphisms.
Conclusion: In conclusion, the somatic variants in Brazilian PNH patients displayed variability in both site distribution and type. Contrary to mutational hotspots observed in previous studies, none were identified in this cohort. No specific correlation between the clinical characteristics of hemolytic PNH patients and their variants was found, likely due to the extensive variety of mutations.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare clonal hematopoietic stem cell disease characterized by a wide spectrum of clinical manifestations, including intravascular hemolysis, a hypercoagulable state, and bone marrow failure. Its global incidence is estimated in 1–1.5 cases per million individuals per year (1).
PNH pathogenesis involves the non-malignant clonal expansion of hematopoietic stem cells with acquired abnormalities in the phosphatidylinositol glycan class A (PIG-A) gene. PIG-A is an X-linked gene that encodes a protein required for glycosylphosphatidylinositol (GPI) anchor synthesis (2). The PIG-A gene comprises six exons and five introns spanning 17 kb (3). Exon 1 (23 bp) is a non-coding 5′-untranslated region, while exon 2 (777 bp) contains half of the coding region. Exons 3 (133 bp), 4 (133 bp), and 5 (207 bp) contain portions of the coding region, and exon 6 (2,316 bp) encompasses the remaining coding region and the 3′-untranslated region. The 5′-flanking region (583 bp) has promoter activity. PIG-A is located on the X chromosome’s short arm (Xp22.1) (2). A single inactivating mutation can produce a PNH phenotype since men have only one X chromosome, and women exhibit X chromosome inactivation (lyonization) in somatic cells, including hematopoietic stem cells (4).
Somatic PIG-A pathogenic variants in hematopoietic stem cells result in partial or complete absence of GPI-linked proteins normally present on the cell membrane. CD55 (decay-accelerating factor) and CD59 (membrane inhibitor of reactive lysis) are essential complement regulatory proteins anchored via GPI. In PNH patients, CD55 and CD59 deficiencies in the clonal blood cells lead to increased red blood cell susceptibility to complement system-mediated cell activaction, cell damage and hemolysis (5, 6).
Clinical observations highlight the heterogeneous nature of PNH patients, with variations in symptom manifestations, hemolysis intensity, and responses to eculizumab treatment. Some patients persistently experience hemolysis, including an extravascular component. A wide array of acquired PIG-A variants has been documented (7), predominantly featuring frameshift mutations (8, 9). Interestingly, these variants do not correlate with clinical factors such as anemia severity or thrombosis presence in PNH patients. This study aims to characterize the mutational profile of 31 Brazilian patients with PNH clones.
Methods
Patients harboring PNH clones, identified by flow cytometry, were selected for this study. Flow cytometry analysis was performed using monoclonal antibodies targeting GPI-linked surface antigens like CD59, CD157, and FLAER, as recommended by Borowitz et al. (10). Detection of at least 0.01% of cells lacking GPI-linked marker expression was considered a positive result. The local research ethics committee approved the study, and all patient samples and clinical information were collected following written informed consent.
The patients were classified in three groups according to International PNH Interest Group (11): group 1, classic PNH, characterized by large PNH clones and episodes of intravascular hemolysis; group 2, PNH associated with another hematologic disease, characterized by clinical/laboratory presence of hemolysis in patients who also have another concomitant diagnosis, such as aplastic anemia; and group 3, subclinical PNH, characterized by patients who have small PNH clones without clinical or laboratory manifestations of hemolysis.
Sample collection and DNA extraction
Peripheral blood samples (10 mL) were collected in EDTA-containing tubes and processed as previously described (12).
Briefly, samples underwent centrifugation to separate plasma from peripheral blood cells, followed by isolation of white blood cells (buffy coat) stored at −80°C until use.
DNA extraction from the buffy coats was performed using the QIAamp DNA Blood Mini QIAcube Kit (Qiagen, Valencia, CA, USA) on the QIAcube Connect automated nucleic acid extractor (Qiagen, Courtaboeuf, France).
Sanger sequencing
Primers for PIG-A sequencing were designed to anneal to intronic sites flanking the exonic regions, as previously described (2) (see Table 1). Polymerase chain reaction (PCR) assays utilized 25 μL reaction volumes containing genomic DNA (50 ng), 1 × Taq DNA polymerase buffer (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), 1.50 or 1.25 mM MgCl2 (see Table 1), 0.2 mM dNTPs (GE Healthcare, Little Chalfont, UK), 1 U of Taq DNA polymerase (Invitrogen), and 0.16 μM of each primer. The PCR conditions included an initial denaturation at 94°C for 5 min, followed by 35 cycles of 30 s at 94°C, 30 s at 55 or 59°C (see Table 1), and 1 min at 72°C, concluding with a final extension at 72°C for 6 min. The resulting PCR products were analyzed on 1% agarose gel and subsequently sequenced using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Thermo Fisher Scientific) on an ABI 3130xl Genetic Analyzer (Applied Biosystems).
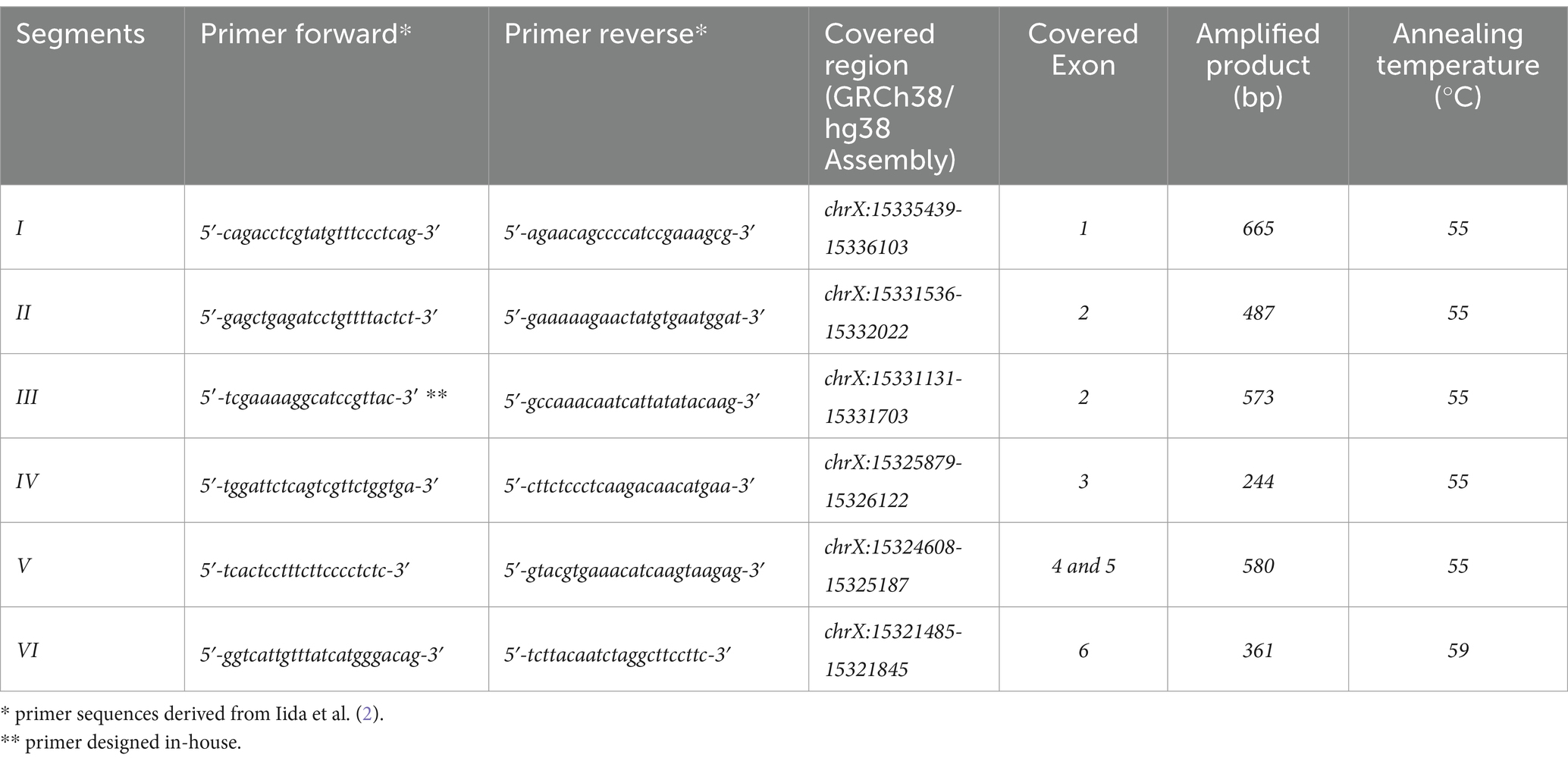
Table 1. PCR primers and conditions.
Mutation pathogenicity prediction and allelic frequency assessment
To evaluate the identified variants, we employed databases and in silico pathogenicity prediction tools in line with established literature and the parameters proposed by the American College of Medical Genetics and Genomics (ACMG) (13).
ClinVar-dbSNP (14): A public archive maintained by the National Institutes of Health that provides information on human genetic variants and their association with diseases.
REVEL (15): A tool that integrates 18 pathogenic prediction scores from 13 different tools, focusing on rare pathogenic variants. Scores above 0.5 indicate a higher likelihood of deleteriousness, with recent studies suggesting thresholds above 0.8 for deleterious variants and below 0.4 for benign ones.
PolyPhen (16): A tool that predicts the probability of a mutation being damaging by analyzing the impact of the altered amino acid on protein function.
MutationTaster (17): A tool capable of analyzing intronic, synonymous, and short indel mutations, predicting their effects. It incorporates data from sources such as 1,000 Genomes, ClinVar, and gnomAD to assess variant pathogenicity.
CADD (18): A tool that scores single nucleotide variants and small insertions or deletions based on their molecular functionality and pathogenicity, providing a probability score for deleterious effects.
gnomAD (19): A comprehensive database containing data from 76,156 whole-genome sequences and 125,748 exome sequences, used for sequence analysis.
Allelic frequencies were assessed using the 1,000 Genomes Project database, which includes over 88 million variants, covering more than 99% of SNPs with a frequency greater than 1% across diverse ancestries (20).
Results
A total of 31 patients were included in this study. The median age was 36 years (range 19–72 years), with 55% being male. Twenty patients had classical PNH with large PNH clones (Group 1), three had classical PNH with another hematologic disease (Group 2), and eight had aplastic anemia with small PNH clones (Group 3). The clinical characteristics and laboratory features of the patients are summarized in Tables 2, 3. Among these, 16 (52%) from Groups 1 and 2 experienced hemoglobinuria, and 11 (35%) reported abdominal pain. Twenty-five patients (81%) required transfusion. Eight patients (26%) experienced thrombotic events; seven venous (22%) and one arterial (3%), Including five intra-abdominal venous thromboses (16%), with two cases (6%) being Budd-Chiari syndrome. The median hemoglobin level at sample collection was 9.1 g/dL (range 4.7–14.4 g/dL). The median lactate dehydrogenase (LDH) level was 1,455 U/L (range 159–3,989 U/L, upper limit 480 U/L). Median PNH clone sizes were 79% in monocytes (range 0.03–99.9%), 70% in granulocytes (range 0.01–99.9%), and 37% in red blood cells (range 0.01–95%).
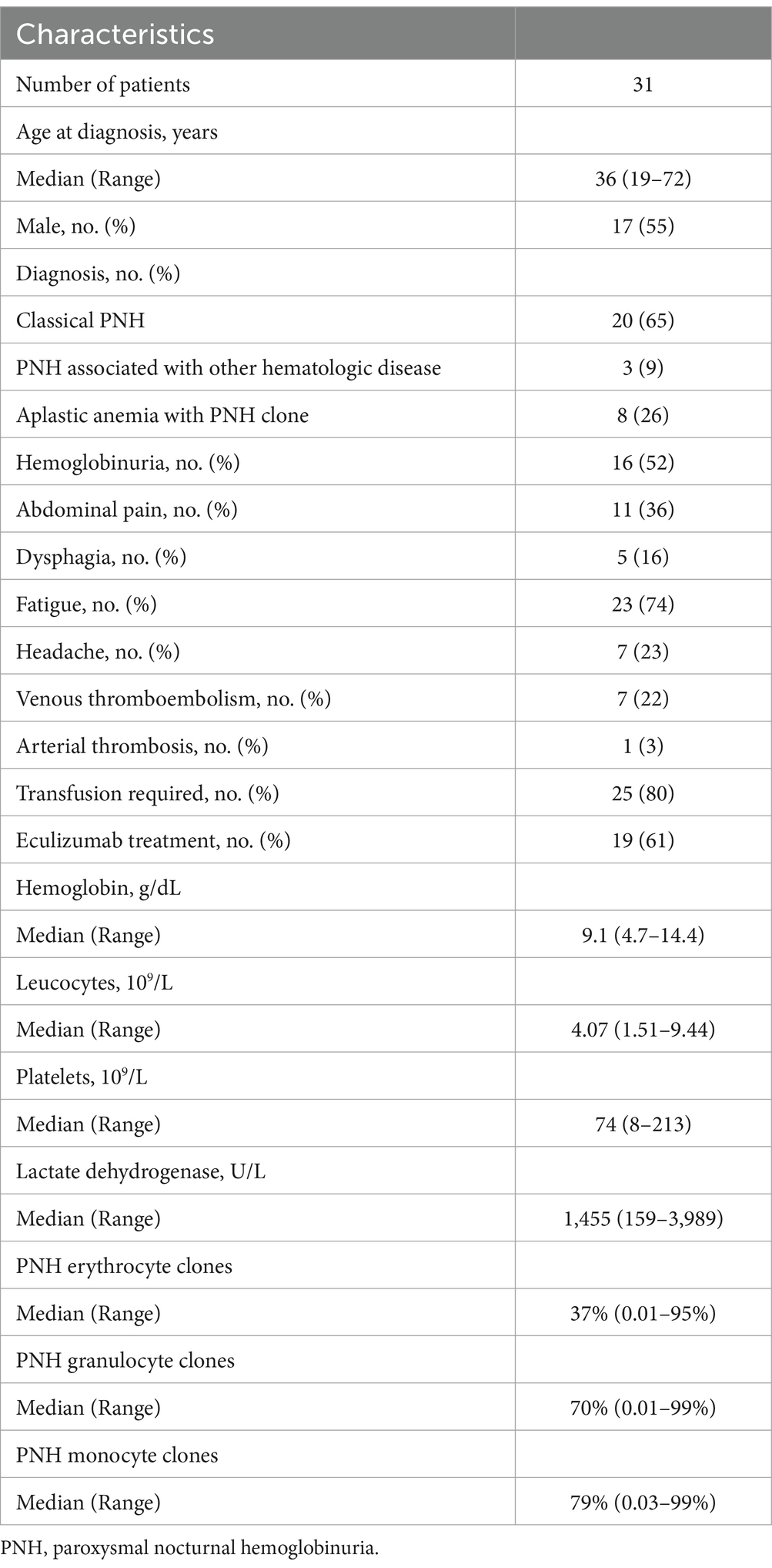
Table 2. Patient and disease characteristics.
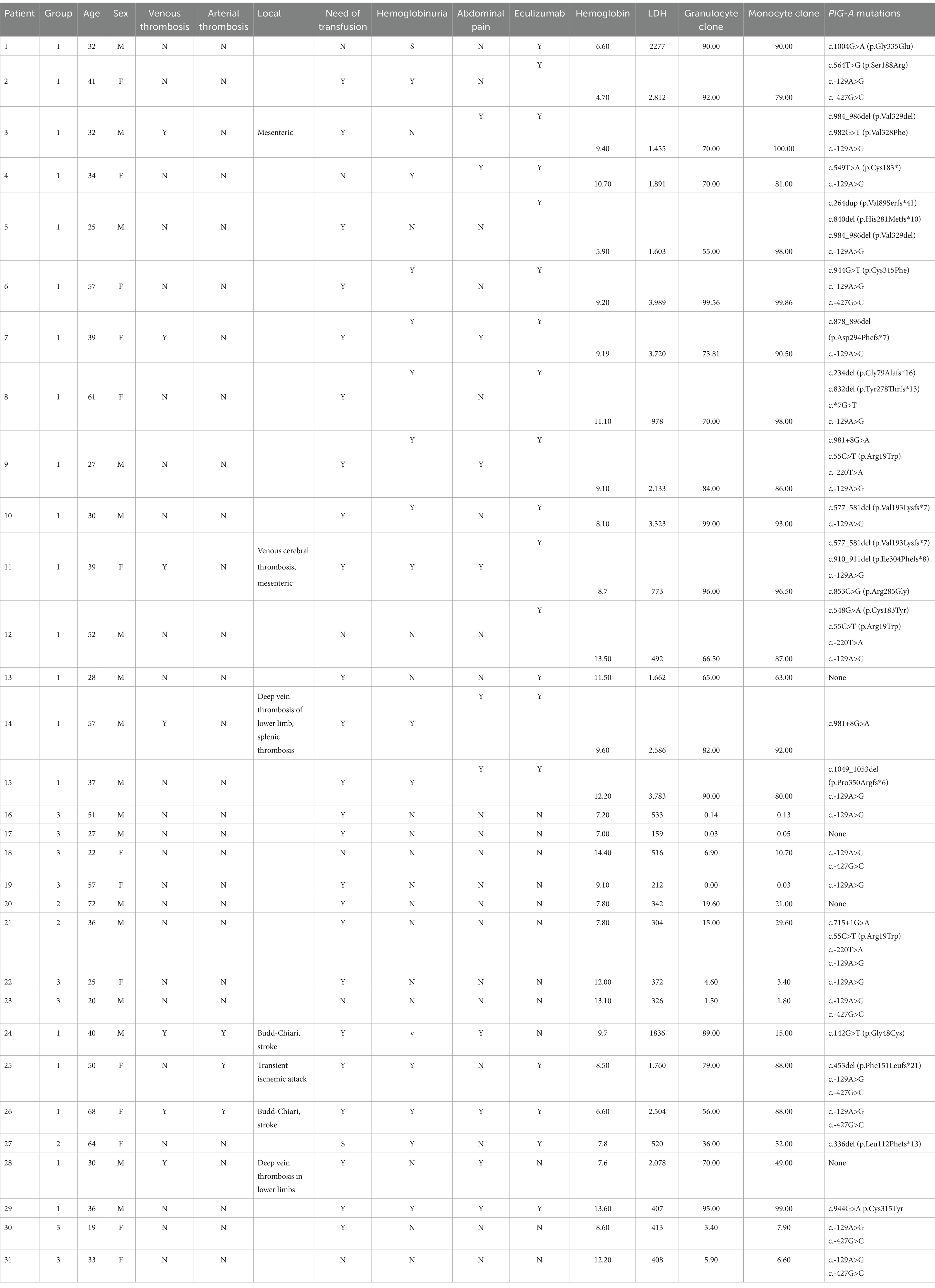
Table 3. Clinical characteristics and PIG-A mutations in PNH patients.
We detected 29 distinct variants in 27 of the 31 patients (refer to Table 4). One mutation had been previously identified: c.55C > T (p.Arg19Trp), a benign polymorphism in exon 2 already reported (Nafa et al., Endo et al.). A c.*7G > T mutation found in the 1,000 Genomes project and two SNPs, c.—427G > C and c.-129A > G, were observed in the general population and are cataloged in the 1,000 Genomes databases. Twenty-five newly identified variants had no prior descriptions in the literature.
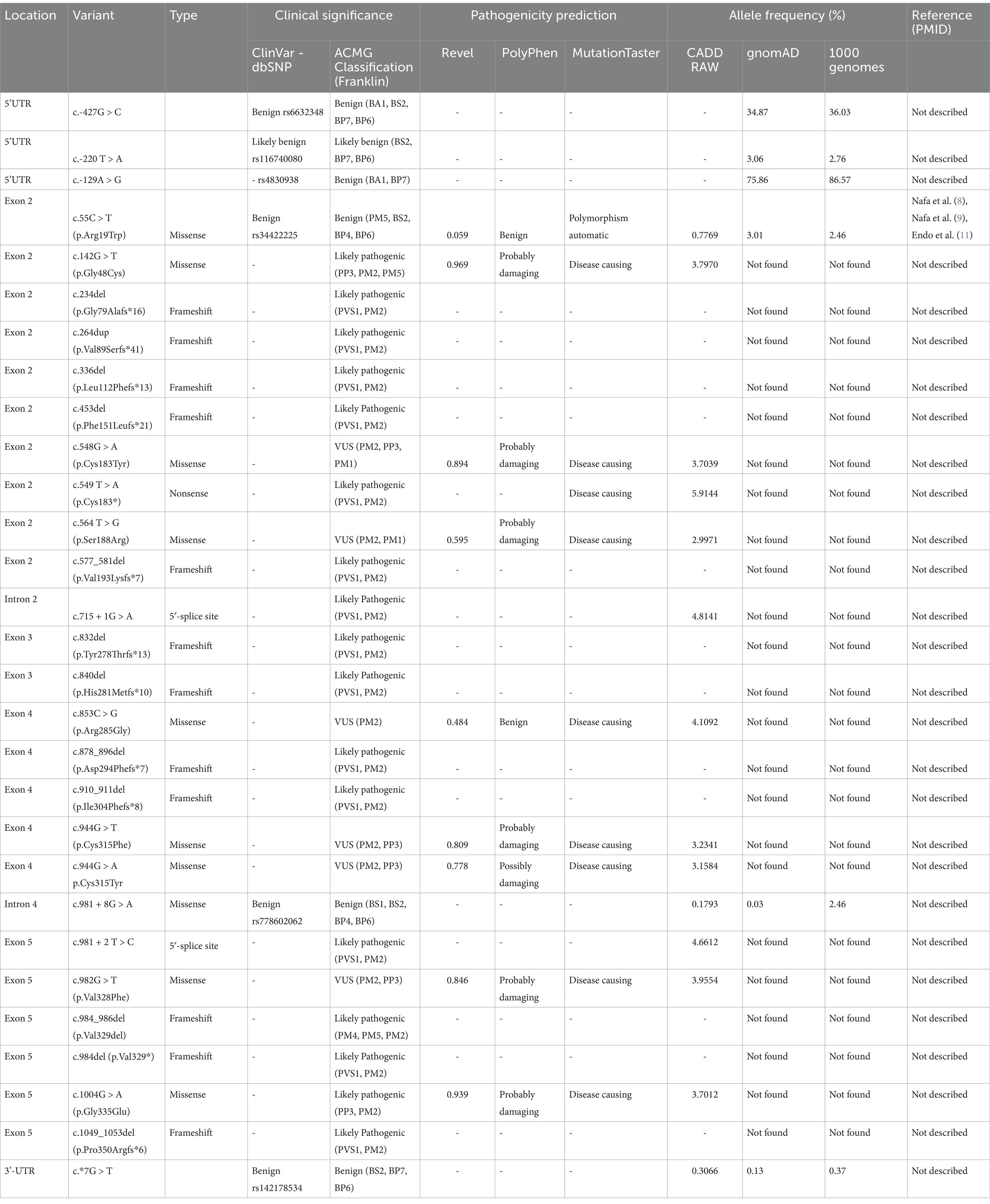
Table 4. Mutations detected in this study.
Predominantly, point mutations were observed, including 16 variants involving a simple exchange of base pairs, one insertion, 10 small deletion mutations (ranging from one to five base pairs), and one large deletion (19 base pairs). The alterations were categorized as follows: 12 frameshift (41%), 10 missense (35%), two splice site (7%), and one nonsense variant (3%). Four mutations (14%) were found in non-coding regions.
Variants in the PIG-A gene were distributed throughout the gene: three in the 5′ region, 10 in exon 2, two in exon 3, five in exon 4, five in exon 5, one in intron 2, two in intron 4, and one in the 3′ region (Figure 1).

Figure 1. Representation of the location of mutations found in the PIG-A gene.
Four patients showed no detectable variants: two with hemolytic PNH (group 1), one with aplastic anemia and PNH clone (group 2) and one with subclinical PNH (group 3).
In silico analysis
The impact of variants found in these patients was assessed using software tools outlined in the methods section. The majority of variants identified (17 out of 29) were deemed likely pathogenic for paroxysmal nocturnal hemoglobinuria (PNH).
Six variants have undetermined significance (VUS). c.548G > A, c 564 T > G, c.944G > T, c.944G > A, and c.982G > T were predicted as probably pathogenic by PolyPhen and Mutation taster, and have Revel scores above 0.5, indicating a higher likelihood of deleteriousness. Inconsistent scoring was noted for the c.853C > G (p.Arg285Gly) variant; MutationTaster rated it as likely pathogenic, whereas PolyPhen-2 assessed it as benign.
Intronic variants were evaluated. The c.981 + 8G > A mutation was found to have no impact on splicing, while c.715 + 1G > A and c.981 + 2 T > C were predicted to potentially affect splicing, suggesting they may have pathological consequences.
Two variants, c.*7G > T and c.55C > T, were categorized as benign, with the latter also described as a polymorphism in multiple studies.
Three variants in the 5′ regulatory region—c.-220 T > A and two polymorphisms, c.-427G > C and c.-129A > G (both with high frequencies in the general population as described in Table 4) — were classified as having undetermined effects, suggesting they may exhibit benign behavior.
Out of the 23 patients with hemolytic PNH, 19 presented with at least one variant assessed as pathogenic. Notably, Patient 9, despite harboring variants generally regarded as benign or of unknown significance, such as c.55C > T (p.Arg19Trp), c.-129A > G, c.-220 T > A, and a c.981 + 8G > A intronic variant (which according to ACMG classification does not affect splicing), displayed classical PNH with significant hemolysis and a large PNH clone presence (99%). Similarly, Patient 26, diagnosed with classical PNH, had only the SNPs c.-427G > C and c.-129A > G in the 5′ regulatory region, which are not known to have clinical impact. In addition, two patients with hemolytic PNH did not show any variant in the PIG-A gene.
A significant proportion of patients (15 out of 23, or 65%) with hemolytic PNH possessed multiple variants concurrently, with the predominant ones being potentially pathogenic in conjunction with a polymorphism. Specifically, five patients carried more than one variant likely to be pathogenic. No clear correlation was observed between the clinical presentation and the type or quantity of variants identified.
Among the variants, three were confirmed as polymorphisms. The c.55C > T (p.Arg19Trp) polymorphism was detected in three individuals within the hemolytic PNH subgroup. Two SNPs, c.-427G > C and c.-129A > G, were frequently observed; c.-129A > G was present in 22 of the 31 patients (approximately 70%), while c.-427G > C was found in eight patients (around 26%). These polymorphisms have high frequency related in population, approximately 86 and 36%, respectively, in 1000 Genomes database, and were the only genetic variants identified in seven of eight patients from Group 3, who had subclinical PNH manifestations.
Discussion
To the best of our knowledge, this study represents the first comprehensive evaluation of PNH cases using PIG-A gene sequencing in Brazil. We identified 29 somatic variants in 27 out of 31 patients. Among these, only one variant was previously characterized in the literature. Three patients had the c.55C > T variant, initially described by Iida et al. (2). The majority of variants identified (17 out of 29) were deemed likely pathogenic for paroxysmal nocturnal hemoglobinuria (PNH). Six variants have undetermined significance (VUS) and six variants are probably benign.
Since Miyata et al.’s landmark discovery in 1993 (21), which linked PNH pathogenesis to variants in the X-linked PIG-A gene, over a hundred variants have been documented worldwide (9, 22–28). In a smaller cohort, De Carvalho et al. (29) described three Brazilian patients with classical PNH. They used conformation-sensitive gel electrophoresis followed by direct sequencing to identify three distinct variants.
The spectrum of PIG-A variants is broad, predominantly composed of single-nucleotide substitutions, small insertions, and deletions, with large gene deletions being relatively rare. Frameshift mutations often result in premature stop codons, potentially leading to truncated proteins with compromised or lost function. PIG-A variants include nonsense, splice-site variants affecting mRNA processing, and missense that result in single amino acid changes. Some missense variants may confer partial protein functionality, explaining the partial deficiency of GPI-linked proteins in the PNH type II phenotype, while others may lead to a complete loss of function. Our findings resonate with existing literature, wherein frameshift and missense variants are reported frequently; in our study, these accounted for 41 and 35%, respectively. Notably, missense variants with widespread effects are usually situated in pivotal regions; here, four out of nine missense mutations were located in exon 2, which is consistent with previous suggestions that this exon is a critical site for function-affecting variants.
Variants in the PIG-A gene are distributed across the coding region, occurring at random with no significant hotspots, except for a specific sequence in exon 2 reported by some studies (22). The majority of variants were identified in exon 2, likely due to its size and coding significance within the PIG-A gene.
In our study, 19 out of 23 patients with hemolytic PNH exhibited more than one PIG-A variant, suggesting a possible oligoclonal nature of the disease, as previously proposed (8, 30). This theory is supported by the finding that multiple hematopoietic stem cell lineages can acquire distinct PIG-A variants and propagate independently. Genotypic mosaicism within T-cell clones, as demonstrated by Endo et al. (30), underlines the phenotypic diversity observed in PNH. This genotypic diversity within PNH may be due to a selective autoimmune pressure, a hypothesis not yet conclusively proven (31, 32).
Interestingly, patients with concomitant variants did not exhibit clinical differences compared to those with a single mutation. This observation reinforces the idea that PNH is a complex disorder with a multifactorial pathogenesis. In cases of subclinical PNH, no pathogenic variants were identified; only polymorphisms with a high incidence in the general population were present. This finding, coupled with the absence of a sorting method for GPI-deficient leukocytes, suggests that these polymorphisms are likely benign.
The absence of detectable PIG-A variants in four patients, including one with subclinical PNH and three with hemolytic PNH, suggests that mutations may be located in regions not examined by our study, such as non-coding intronic regions. It’s noteworthy that other genes involved in GPI-anchor biosynthesis, like PIG-T and PIG-M, have been implicated in GPI deficiency, but pathogenic variants in these genes are rare and typically associated with autosomal chromosomes, requiring biallelic mutations to manifest (33, 34). These cases often present with a broader clinical spectrum, including developmental delay and epilepsy, which differs from the hemolysis predominant in PNH (35).
The relationship between aplastic anemia and PNH clones, as well as the role of immune selection, provides an interesting angle on the survival advantage of PNH clones (36, 37). This is highlighted by the varying mutation profiles in patients with aplastic anemia and myelodysplastic syndrome compared to those with classical PNH, as reported by Okamoto et al. (38). The diversity of PIG-A variants and their association with different disease phenotypes underscore the complexity of PNH pathogenesis.
Furthermore, the detection of somatic PIG-A variants in individuals with normal hematopoiesis and their prevalence in healthy individuals indicate that these mutations alone are not sufficient for clonal expansion (39–41). The existence of additional genetic abnormalities in PNH patients, such as variants in genes like TET2 and JAK2, suggests a multistep clonal evolution that may contribute to the expansion of PNH clones (42, 43). Additionally, the expression of HMGA2, an architectural transcription factor, has been found to be abnormally high in some PNH patients, potentially contributing to clonal expansion (44).
In terms of clinical manifestations, our study did not reveal a clear correlation between the mutational profile and the severity of hemolytic PNH. This may be due to the wide range of mutations observed. The study also reflects on additional genetic factors, such as BMPR2 and THBD, that may contribute to the risk of thrombosis in PNH patients (45).
In a comprehensive analysis by Chen et al. (46), the clinical implications of variants were elucidated through whole-exome sequencing of genes commonly mutated in PNH, including PIG-A, BCORL1, RUNX1T1, MAP3K4, CSMD1, NOTCH1, FANCD2, PEG3, DIS3, and SETBP1. They found correlations between specific variants and clinical features: the RUNX1T1 mutation was linked to larger PNH clones, higher levels of unconjugated bilirubin, and lower hemoglobin levels; the BCORL1 mutation tended to occur in younger patients; the SRRD mutation was tied to visceral thrombosis; and the EGR4 mutation was associated with myocardial infarction. This emphasizes the potential for targeted mutation analysis to predict clinical outcomes and tailor prophylaxis in PNH patients.
To summarize, the genetic landscape of PNH in Brazilian patients is diverse, with a variety of variant sites and types, and no significant mutational hotspots have been identified. The variants observed were predominantly small deletions and single-nucleotide alterations. Pathogenicity prediction tools suggested that a significant proportion of these variants might play a pathogenic role in the disease: 17 variants were likely pathogenic and five of the six VUS described were probably damaging according to the tools of pathogenicity prediction. In the cohort with hemolytic PNH, a majority (19 out of 23) had variants deemed pathogenic. Interestingly, only polymorphisms, which are common in the general population and likely benign, were detected in patients with subclinical PNH.
The study did not establish a direct correlation between the clinical presentation of hemolytic PNH and specific variants, which may be due to the heterogeneity of the genetic alterations observed. This finding suggests that PNH severity and clinical manifestations are influenced by a complex interplay of factors beyond the PIG-A gene variants alone.
To enhance our understanding of PNH’s heterogeneity, further research involving larger cohorts, including patients from different geographical regions, and a broader analysis of genetic variants, is essential. Such investigations may uncover additional variants implicated in PNH, offering insights into the pathogenesis of the disease and potentially informing the development of personalized therapeutic approaches.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The studies involving humans were approved by Comite de ética do hospital Sirio Libanes- São Paulo, Brazil. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
PE: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft. CP: Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Validation, Writing – original draft. AR: Data curation, Formal analysis, Methodology, Software, Writing – original draft. CC: Data curation, Formal analysis, Writing – review & editing. FP: Conceptualization, Investigation, Methodology, Supervision, Writing – original draft. FB: Conceptualization, Investigation, Methodology, Supervision, Writing – original draft. EX: Methodology, Project administration, Writing – original draft. VC: Formal analysis, Validation, Writing – review & editing. MV: Investigation, Supervision, Validation, Writing – review & editing. JB: Data curation, Methodology, Writing – review & editing. BR: Formal analysis, Validation, Writing – review & editing. JB: Data curation, Supervision, Validation, Writing – review & editing. CA-R: Conceptualization, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hill, A, DeZern, AE, Kinoshita, T, and Brodsky, RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. (2017) 3:17028. doi: 10.1038/nrdp.2017.28
2. Iida, Y, Takeda, J, Miyata, T, Inoue, N, Nishimura, J, Kitani, T, et al. Characterization of genomic PIG-A gene: a gene for glycosylphosphatidylinositol-anchor biosynthesis and paroxysmal nocturnal hemoglobinuria. Blood. (1994) 83:3126–31. doi: 10.1182/blood.V83.11.3126.3126
3. Rotoli, B, and Boccuni, P. The PIG-A gene somatic mutation responsible for paroxysmal nocturnal hemoglobinuria. Haematologica. (1995) 80:539–45.
4. Nishimura, J, Murakami, Y, and Kinoshita, T. Paroxysmal nocturnal hemoglobinuria: an acquired genetic disease. Am J Hematol. (1999) 62:175–82. doi: 10.1002/(SICI)1096-8652(199911)62:3<175::AID-AJH7>3.0.CO;2-8
5. Brodsky, RA. Paroxysmal nocturnal hemoglobinuria. Blood. (2014) 124:2804–11. doi: 10.1182/blood-2014-02-522128
6. Boccuni, P, Del Vecchio, L, Di Noto, R, and Rotoli, B. Glycosyl phosphatidylinositol (GPI)-anchored molecules and the pathogenesis of paroxysmal nocturnal hemoglobinuria. Crit Rev Oncol Hematol. (2000) 33:25–43. doi: 10.1016/S1040-8428(99)00052-9
7. Bessler, M, Mason, PJ, Hillmen, P, and Luzzatto, L. Mutations in the PIG-A gene causing partial deficiency of GPI-linked surface proteins (PNH II) in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. (1994) 87:863–6. doi: 10.1111/j.1365-2141.1994.tb06754.x
8. Nafa, K, Mason, P, Hillmen, P, Luzzatto, L, and Bessler, M. Mutations in the PIG-A gene causing paroxysmal nocturnal hemoglobinuria are mainly of the frameshift type. Blood. (1995) 86:4650–5. doi: 10.1182/blood.V86.12.4650.bloodjournal86124650
9. Nafa, K, Bessler, M, Castro-Malaspina, H, Jhanwar, S, and Luzzatto, L. The spectrum of somatic mutations in the PIG-A gene in paroxysmal nocturnal hemoglobinuria includes large deletions and small duplications. Blood Cells Mol Dis. (1998) 24:370–84. doi: 10.1006/bcmd.1998.0203
10. Borowitz, MJ, Craig, FE, Digiuseppe, JA, Illingworth, AJ, Rosse, W, Sutherland, DR, et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom. (2010) 78B:211–30. doi: 10.1002/cyto.b.20525
11. Parker, CJ. Update on the diagnosis and management of paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program. (2016) 2016:208–16. doi: 10.1101/pdb.rec079152
12. RBC Lysis Buffer. Red blood cell lysis solution. Cold Spring Harb Protoc. (2015) 2015:pdb.rec079152
13. Richard, S, Aziz, N, Bale, S, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
14. Landrum, MJ, Lee, JM, Benson, M, Brown, GR, Chao, C, Chitipiralla, S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. (2018) 46:D1062–7. doi: 10.1093/nar/gkx1153
15. Ioannidis, NM, Rothstein, JH, Pejaver, V, Middha, S, Mc Donnel, SK, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
16. Adzhubei, I, Jordan, DM, and Sunyaev, SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. (2013) 7:20. doi: 10.1002/0471142905.hg0720s76
17. Schwarz, JM, Cooper, DN, Schuelke, M, and Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
18. Kircher, M, Witten, DM, Jain, P, O’Roak, BJ, Cooper, GM, and Shendure, J. A general gramework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
19. Karczewski, KJ, Francioli, L, Tiao, G, Cummings, BB, Alfoldi, J, Wang, Q, et al. The mutational constraint spectrum quantified from variation in 141456 humans. Nature. (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
20. Auton, A, Ld, B, Rm, D, Ep, G, Hm, K, et al. A global reference for human genetic variation. Nature. (2015) 526:68–74. doi: 10.1038/nature15393
21. Miyata, T, Takeda, J, Iida, Y, Yamada, N, Inoue, N, Takahashi, M, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. (1993) 259:1318–20. doi: 10.1126/science.7680492
22. Mortazavi, Y, Merk, B, McIntosh, J, Marsh, JC, Schrezenmeier, H, Rutherford, TR, et al. The spectrum of PIG-A gene mutations in aplastic anemia/paroxysmal nocturnal hemoglobinuria (AA/PNH): a high incidence of multiple mutations and evidence of a mutational hot spot. Blood. (2003) 101:2833–41. doi: 10.1182/blood-2002-07-2095
23. Yamada, N, Miyata, T, Maeda, K, Kitani, T, Takeda, J, and Kinoshita, T. Somatic mutations of the PIG-A gene found in Japanese patients with paroxysmal nocturnal hemoglobinuria. Blood. (1995) 85:885–92. doi: 10.1182/blood.V85.4.885.bloodjournal854885
24. Pavlu, J, Mortazavi, Y, Tooze, J, Marsh, JC, Gordon-Smith, EC, and Rutherford, TR. Paroxysmal nocturnal haemoglobinuria due to an 88 bp direct tandem repeat insertion in the PIG-A gene. Br J Haematol. (1997) 98:289–91. doi: 10.1046/j.1365-2141.1997.2343051.x
25. Bessler, M, Mason, PJ, Hillmen, P, Miyata, T, Yamada, N, Takeda, J, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. (1994) 13:110–7. doi: 10.1002/j.1460-2075.1994.tb06240.x
26. Araten, DJ, Nafa, K, Pakdeesuwan, K, and Luzzatto, L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Med Sci. (1999) 96:5209–14. doi: 10.1073/pnas.96.9.5209
27. Yoon, JH, Cho, HI, Park, SS, Chang, YH, and Kim, BK. Mutation analysis of the PIG-A gene in Korean patients with paroxysmal nocturnal haemoglobinuria. J Clin Pathol. (2002) 55:410–3. doi: 10.1136/jcp.55.6.410
28. Pramoonjago, P, Pakdeesuwan, K, Siripanyaphinyo, U, Chinprasertsuk, S, Kinoshita, T, and Wanachiwanawin, W. Genotypic, immunophenotypic and clinical features of Thai patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. (1999) 105:497–504. doi: 10.1111/j.1365-2141.1999.01325.x
29. De Carvalho, RF, Arruda, VR, Saad, ST, and Costa, FF. Detection of somatic mutations of the PIG-A gene in Brazilian patients with paroxysmal nocturnal hemoglobinuria. Braz J Med Biol Res. (2001) 34:763–6. doi: 10.1590/S0100-879X2001000600010
30. Endo, M, Ware, RE, Vreeke, TM, Singh, SP, Howard, TA, Tomita, A, et al. Molecular basis of the heterogeneity of expression of glycosyl phosphatidylinositol anchored proteins in paroxysmal nocturnal hemoglobinuria. Blood. (1996) 87:2546–57. doi: 10.1182/blood.V87.6.2546.bloodjournal8762546
31. Araten, DJ, and Luzzatto, L. The mutation rate in PIG-A is normal in patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood. (2006) 108:734–6. doi: 10.1182/blood-2006-01-0256
32. Clemente, MJ, Przychodzen, B, Hirsch, CM, Nagata, Y, Bat, T, Wlodarski, MW, et al. Clonal PIG-A mosaicism and dynamics in paroxysmal nocturnal hemoglobinuria. Leukemia. (2018) 32:2507–11. doi: 10.1038/s41375-018-0138-5
33. Krawitz, PM, Höchsmann, B, Murakami, Y, Teubner, B, Krüger, U, Klopocki, E, et al. A case of paroxysmal nocturnal hemoglobinuria caused by a germline mutation and a somatic mutation in PIGT. Blood. (2013) 122:1312–5. doi: 10.1182/blood-2013-01-481499
34. Almeida, AM, Murakami, Y, Layton, DM, Hillmen, P, Sellick, GS, Maeda, Y, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med. (2006) 12:846–51. doi: 10.1038/nm1410
35. Luzzatto, L. PNH from mutations of another PIG gene. Blood. (2013) 122:1099–100. doi: 10.1182/blood-2013-06-508556
36. Luzzatto, L. Paroxysmal nocturnal hemoglobinuria: an acquired X-linked genetic disease with somatic-cell mosaicism. Curr Opin Genet Dev. (2006) 16:317–22. doi: 10.1016/j.gde.2006.04.015
37. Savage, WJ, Barber, JP, Mukhina, GL, Hu, R, Chen, G, Matsui, W, et al. Glycosylphosphatidylinositol-anchored protein deficiency confers resistance to apoptosis in PNH. Exp Hematol. (2009) 37:42–51.e1. doi: 10.1016/j.exphem.2008.09.002
38. Okamoto, M, Shichishima, T, Noji, H, Ikeda, K, Nakamura, A, Akutsu, K, et al. High frequency of several PIG-A mutations in patients with aplastic anemia and myelodysplastic syndrome. Leukemia. (2006) 20:627–34. doi: 10.1038/sj.leu.2404135
39. Hu, R, Mukhina, GL, Piantadosi, S, Barber, JP, Jones, RJ, and Brodsky, RA. PIG-A mutations in normal hematopoiesis. Blood. (2005) 105:3848–54. doi: 10.1182/blood-2004-04-1472
40. Brodsky, RA, and Hu, R. PIG-A mutations in paroxysmal nocturnal hemoglobinuria and in normal hematopoiesis. Leuk Lymphoma. (2006) 47:1215–21. doi: 10.1080/10428190600555520
41. Araten, DJ, Bessler, M, McKenzie, S, Castro-Malaspina, H, Childs, BH, Boulad, F, et al. Dynamics of hematopoiesis in paroxysmal nocturnal hemoglobinuria (PNH): no evidence for intrinsic growth advantage of PNH clones. Leukemia. (2002) 16:2243–8. doi: 10.1038/sj.leu.2402694
42. Sugimori, C, Padron, E, Caceres, G, Shain, K, Sokol, L, Zhang, L, et al. Paroxysmal nocturnal hemoglobinuria and concurrent JAK2V617F mutation. Blood Cancer J. (2012) 2:e63. doi: 10.1038/bcj.2012.7
43. Shen, W, Clemente, MJ, Hosono, N, Yoshida, K, Przychodzen, B, Yoshizato, T, et al. Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin Invest. (2014) 124:4529–38. doi: 10.1172/JCI74747
44. Murakami, Y, Inoue, N, Shichishima, T, Ohta, R, Noji, H, Maeda, Y, et al. Deregulated expression of HMGA2 is implicated in clonal expansion of PIG-A deficient cells in paroxysmal nocturnal haemoglobinuria. Br J Haematol. (2012) 156:383–7. doi: 10.1111/j.1365-2141.2011.08914.x
45. Li, L, Wang, H, Liu, H, Liu, Z, Li, L, Ding, K, et al. Gene mutations associated with thrombosis detected by whole-exome sequencing in paroxysmal nocturnal hemoglobinuria. Int J Lab Hematol. (2019) 41:424–32. doi: 10.1111/ijlh.13018
Keywords: paroxysmal nocturnal hemoglobinuria, PIG-A mutation, hemolytic anemia, PIG-A gene, pathogenicity
Citation: Eiko Yamakawa P, Perez Gomes C, Ribeiro Mendes A, Cesar Justino de Oliveira C, Porto Freitas F, Bettoni F, Xavier dos Santos E, Campos de Molla V, Vescovi Gonçalves M, Branquinho J, Ribeiro Nogueira B, Bosco Pesquero J and Arrais-Rodrigues C (2025) Somatic mutations in Brazilian patients with paroxysmal nocturnal hemoglobinuria: a comprehensive analysis. Front. Med. 12:1472186. doi: 10.3389/fmed.2025.1472186
Edited by:
Alberto Lazarowski, University of Buenos Aires, ArgentinaReviewed by:
Patricia Esperon, Universidad de la República, UruguayAndres Brodsky, Hospital de Clínicas José de San Martín, Argentina
Copyright © 2025 Eiko Yamakawa, Perez Gomes, Ribeiro Mendes, Cesar Justino de Oliveira, Porto Freitas, Bettoni, Xavier dos Santos, Campos de Molla, Vescovi Gonçalves, Branquinho, Ribeiro Nogueira, Bosco Pesquero and Arrais-Rodrigues. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia Eiko Yamakawa, cGF0cmljaWFfeWFtYWthd2FAeWFob28uY29tLmJy