
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 26 March 2025
Sec. Geriatric Medicine
Volume 12 - 2025 | https://doi.org/10.3389/fmed.2025.1458281
This article is part of the Research TopicUnveiling Inflammaging – Mechanistic Insights on Aging and Related DiseasesView all 7 articles
 Senthil Kumaran Satyanarayanan1
Senthil Kumaran Satyanarayanan1 Tsz Fung Yip2
Tsz Fung Yip2 Zixu Han1
Zixu Han1 Huachen Zhu2
Huachen Zhu2 Dajiang Qin1,3,4*
Dajiang Qin1,3,4* Suki Man Yan Lee1,2,3*
Suki Man Yan Lee1,2,3*In the intricate realm of interactions between hosts and pathogens, Toll-like receptors (TLRs), which play a crucial role in the innate immune response, possess the ability to identify specific molecular signatures. This includes components originating from pathogens such as SARS-CoV-2, as well as the resulting damage-associated molecular patterns (DAMPs), the endogenous molecules released after cellular damage. A developing perspective suggests that TLRs play a central role in neuroinflammation, a fundamental factor in neurodegenerative conditions like Alzheimer’s and Parkinson’s disease (PD). This comprehensive review consolidates current research investigating the potential interplay between TLRs, their signaling mechanisms, and the processes of neurodegeneration following SARS-CoV-2 infection with an aim to elucidate the involvement of TLRs in the long-term neurological complications of COVID-19 and explore the potential of targeting TLRs as a means of implementing intervention strategies for the prevention or treatment of COVID-19-associated long-term brain outcomes.
The emergence of the Coronavirus Disease 2019 (COVID-19) pandemic, instigated by the novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) virus, has been a transformative event in the annals of global public health with a documented death of nearly 7 million (1). Beyond its immediate impact on respiratory health, the virus unleashed a series of long-term medical challenges, notably the Post-Acute Sequelae of SARS-CoV-2 infection (PASC), sometimes referred to as ‘Long COVID’, ‘Long-haul COVID’ or ‘post-COVID syndrome’, where affected individuals report persistent symptoms long after initial recovery (2–5). However, the impact of a prior SARS-CoV-2 infection on the progression of subsequent infections has been unclear (6). The prevailing clinical outcomes suggest that a combination of organ damage and systemic inflammation could be responsible for sustained health complications post-COVID-19, affecting the respiratory, cardiac, neurological, and musculoskeletal systems (2, 4, 5, 7). Among the myriads of sequelae reported, neurological manifestations have been particularly enigmatic and concerning (8–10). Acute COVID has been linked to neurodegenerative diseases like Parkinson’s disease (PD) and cognitive challenges, from ‘brain fog’ to early Alzheimer’s disease (AD) onset. Additionally, the post-COVID syndrome is associated with various psychiatric disorders, including newly diagnosed conditions like depression, anxiety, and post-traumatic stress disorder (PTSD) (3). This has prompted researchers to dive into the complex interplay between the virus, the central nervous system (CNS), and the intricate immune responses, including the role of Toll-like Receptors (TLRs) (11).
The interaction between TLRs and the CNS has gained increased importance due to the potential neurotropic properties of the SARS-CoV-2 virus, which has demonstrated a propensity for invading neural tissues (12–15). The neurodegenerative consequences observed in individuals following COVID-19 infection appear to exhibit parallels with pathways involving the activation of TLRs, a connection that may present potential therapeutic targets. Epidemiological data and clinical studies further reinforce the potential role of TLRs in COVID-19 neurological symptoms (16–18), pushing them to the forefront of therapeutic and diagnostic considerations. As the scientific community works to elucidate this intricate landscape, greater attention needs to be directed toward the repurposing of existing TLR modulators and investigating their potential to target TLR signaling as therapeutic agents for the management of PASC-related complications. In this comprehensive review, we embark on a journey to elucidate the intricate relationship between the SARS-CoV-2 virus, TLRs, and the resulting neurological sequelae. By exploring current evidence, clinical findings, and therapeutic innovations, we aim to provide a roadmap for understanding, diagnosing, and potentially treating the neurological shadows cast by the COVID-19 infection.
The emergence of SARS-CoV-2 heralded an unprecedented era in global health, largely dominated by its profound respiratory implications, with symptoms ranging from mild cough to severe respiratory distress (3, 19). Yet, as the pandemic progressed, accumulating evidence suggested that the virus has neurotropic properties, with the potential to invade and affect both the CNS and the peripheral nervous system (PNS) (20, 21). The occurrence rate of neurological symptoms following infection with SARS-CoV-2, collectively referred to as “Neuro-COVID,” varies considerably across studies and is infrequently attributed to the direct impacts of the virus (22, 23). The implications of this neurotropism have profound ramifications, as neurological manifestations have been reported in a significant proportion of COVID-19 patients, further complicating the clinical spectrum of the disease.
The SARS-CoV-2 demonstrates multiple transmission routes, prominently through respiratory fluids, underscoring its high infectiousness (24, 25). Notably, the human oral cavity and saliva have been identified as significant reservoirs for the virus (26). SARS-CoV-2 employs host entry factors such as ACE2 and TMPRSS family members (TMPRSS2 and TMPRSS4) to facilitate infection (27–29). Research has suggested that SARS-CoV-2 may travel retrogradely from peripheral regions to the CNS via olfactory sensory neurons or other neural pathways (27, 30). However, this proposed olfactory nerve route to brain infection remains controversial and lacks definitive evidence (31). Despite this, alternative pathways from the nasal cavity to the brain have been proposed, including vascular routes, CSF spaces, and the nervus terminalis system (31, 32). In humans, no longitudinal studies have mapped the timeline of neuro-invasion of SARS-CoV-2; only the end results have been documented (33, 34).
SARS-CoV-2 has been found in the olfactory epithelium, predominantly in sustentacular cells (35, 36). Studies using a rhesus monkey model have shown that SARS-CoV-2 can be transported to the CNS via the olfactory route following intranasal inoculation, inducing inflammatory cytokines and pathological lesions in the CNS (36–38). Observations in lethal COVID-19 cases indicate the virus might cross the neural-mucosal interface in the olfactory mucosa, leading to neurological diseases (36, 39). SARS-CoV-2 invades host cells primarily by binding to the ACE2 receptor on respiratory epithelial cell surfaces (25, 40, 41). Some patients have shown viral RNAs presence in the brain, particularly in the endothelial cells of the brainstem, thalamus, and hypothalamus, with occasional detection in the cerebral cortex and CSF (39, 42, 43). Other studies have reported the virus in leukocytes crossing the blood–brain barrier (BBB) or entering via endothelial cells of blood vessels, often referred to as a “Trojan horse” mechanism (44–47). Additionally, the virus can disrupt the endothelial glycocalyx and BBB integrity, facilitating its entry into the CNS (48). This disruption can lead to neurovascular dysfunction, as evidenced by increased markers of endothelial damage and coagulation abnormalities in Long COVID patients (46). Further, a novel entry route involving the CD147-spike protein has been identified, enabling the virions to enter host cells through clathrin-independent endocytosis, offering a new mechanism for SARS-CoV-2 infection and suggesting potential therapeutic strategies for COVID-19 (49). The involvement of CD147 in SARS-CoV-2 infection has also been questioned due to evidence indicating that CD147 does not have the capacity to bind to the spike protein (50).
The neurological symptoms seen in many COVID-19 patients suggest significant CNS penetrance by SARS-CoV-2 (39, 51, 52). Among the seven coronaviruses that infect humans, at least two endemic strains have demonstrated the ability to enter and persist in the CNS (39, 53, 54). The presence of SARS-CoV-2 RNA in the olfactory mucosa and neuroanatomical areas receiving olfactory tract projections implies possible neuroinvasion via axonal transport (43, 55, 56). Collectively, these findings indicate that SARS-CoV-2 can invade the CNS via the neural-mucosal interface and olfactory tract, contributing to the neurological symptoms observed in COVID-19. The exact mechanisms of SARS-CoV-2 neuroinvasion remain under investigation, with proposed routes including hematogenous spread and transneuronal dissemination through the olfactory nerve. The virus’s interaction with ACE2 receptors in various CNS cells, including neurons and glial cells, further underscores its potential to breach the BBB and infect the CNS. Understanding these pathways is crucial for developing strategies to mitigate the neurological impacts of COVID-19.
The immune response to COVID-19 and PASC is complex, involving both innate and adaptive mechanisms that contribute to the pathology and persistence of neurological symptoms (57–59). Understanding these immune reactions is critical for developing targeted therapies and improving patient outcomes. During the acute phase of COVID-19, the CNS can be affected directly by viral invasion or indirectly through systemic inflammation (60–62). SARS-CoV-2 may enter the CNS via the olfactory nerve and bloodstream or by infecting endothelial cells of the BBB, leading to neuroinflammation (43, 63). The initial innate immune response involves the activation of pattern recognition receptors (PRRs) such as TLR3, TLR7, RIG-I, and MDA5, which detect viral RNA and trigger the production of type I interferons and other pro-inflammatory cytokines (64–67). This robust immune response is intended to control viral replication but can lead to a cytokine storm characterized by elevated levels of cytokines such as interleukin (IL)-6, tumor necrosis factor (TNF)-α, and IL-1β, exacerbating inflammation and tissue damage (68–70). In severe cases, this can result in widespread neuroinflammation, microglial activation, and neuronal damage, manifesting as acute neurological symptoms like anosmia, headache, and cognitive impairment (70–72). Queiroz et al. (73) examined the involvement of the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway in COVID-19 severity and ‘Long COVID’. The research analyzed blood samples from 148 individuals, including those with acute and ‘Long COVID’. It was observed that in acute COVID-19 cases, higher expression levels of cGAS, STING, and cytokines [interferon (IFN)-α, TNF-α, IL-6] were present in patients with severe disease compared to those with non-severe manifestations. Similarly, Long COVID was associated with elevated levels of cGAS, STING, and IFN-α, indicating a persistent inflammatory response. This pathway’s activation appears to contribute to an intense systemic inflammatory state in severe COVID-19 and, after infection resolution, may induce autoinflammatory conditions in various tissues, resulting in Long COVID.
The adaptive immune system also plays a significant role in the pathogenesis of neuro-PASC (58, 59). Studies have shown that patients with neuro-PASC have elevated levels of inflammatory markers and immune cells in the CSF, including exhausted CD4+ T cells, border-associated macrophages, microglia, and granulocytes (74, 75). The presence of exhausted T cells in the CSF suggests an ongoing but ineffective immune response, potentially due to the absence of active viral replication, as most neuro-PASC CSF samples are negative for SARS-CoV-2 (2, 23, 58). Elevated levels of IL-12 in the CSF of neuro-PASC patients further indicate a skewed immune response, which may contribute to sustained neuroinflammation and tissue damage (74). There is evidence of an increased frequency of B and T cells in the CNS of neuro-PASC patients (75, 76). Some of these B cells produce IgG antibodies with anti-neural reactivity, suggesting a possible autoimmune component to the disease (75, 77). This autoimmune response may be driven by molecular mimicry, where viral antigens resemble host neural antigens, leading to cross-reactivity and autoimmunity (78). Furthermore, studies have found elevated levels of autoantibodies in patients with ‘Long COVID,’ which may contribute to persistent neurological symptoms by targeting CNS tissues and exacerbating inflammation (2).
The long-term implications of these immune reactions in the CNS are significant, with potential links to neurodegenerative diseases (79). For example, the chronic neuroinflammation observed in neuro-PASC patients may accelerate or trigger conditions like AD, especially in older individuals (80). The similarities between the neuropathological features of COVID-19 and AD, such as the involvement of the orbitofrontal cortex and parahippocampal gyrus, highlight the need for further research in this area (33, 81). Future studies should focus on elucidating the precise mechanisms of immune dysregulation in neuro-PASC and identifying biomarkers for early diagnosis and targeted therapy. Understanding the balance between protective and pathological immune responses will be crucial for developing interventions that mitigate long-term neurological sequelae while effectively controlling viral replication. Overall, the immune response in the CNS during COVID-19 and PASC involves a complex interplay of innate and adaptive mechanisms that can lead to persistent neuroinflammation and neurological symptoms. Ongoing research is essential to understand these processes fully and to develop effective treatments for those suffering from ‘Long COVID.’
The neurological manifestations directly attributed to COVID-19 encompass a broad spectrum impacting the CNS (82). The neurological manifestations of COVID-19 are diverse and can significantly impact patient outcomes (83). These manifestations can be classified into CNS symptoms like dizziness, headaches, impaired consciousness, acute cerebrovascular disease, ataxia, seizures, and PNS, including taste and smell impairments, vision disturbances, and nerve pain (21).
Among the CNS manifestations, encephalitis, an inflammation of the brain observed in COVID-19 patients, presents with symptoms such as headache, fever, confusion, seizures, and focal neurological deficits. Severe cases can lead to acute necrotizing hemorrhagic encephalopathy and rhombencephalitis, conditions confirmed by magnetic resonance imaging (MRI) showing characteristic brain changes (84–86). SARS-CoV-2 has occasionally been found in CSF, indicating direct viral invasion and immune-mediated mechanisms (2). Acute Disseminated Encephalomyelitis (ADEM), a rare inflammatory condition affecting the brain and spinal cord, has also been reported, presenting with rapid onset of neurological symptoms such as ataxia, motor deficits, and altered consciousness, often treated with immunosuppressive therapy (87, 88). COVID-19 increases the risk of cerebrovascular events, including ischemic and hemorrhagic strokes, linked to the virus-induced prothrombotic state, endothelial damage, and cytokine storm (89, 90). Strokes in COVID-19 patients, seen in 2.5 to 5% of cases, typically reveal large vessel occlusions or hemorrhagic lesions on neuroimaging (90, 91).
Among the PNS manifestations, Guillain-Barré Syndrome (GBS), characterized by acute flaccid paralysis, is associated with COVID-19, often presenting with progressive weakness and sensory disturbances following initial respiratory symptoms, likely due to immune responses misdirected at peripheral nerves (92). COVID-19 also causes cranial neuropathies, including anosmia and ageusia, resulting from viral invasion of the olfactory epithelium and spread to the olfactory bulb (93, 94). Cases of oculomotor nerve palsy and optic neuritis, causing double vision and vision loss, have been documented (95, 96). Muscle involvement ranges from mild myalgias to severe rhabdomyolysis, leading to muscle breakdown and potential kidney damage, marked by elevated creatine kinase levels (97, 98). Additionally, COVID-19 has been linked to demyelinating disorders, with MRI showing lesions and a hyperinflammatory state potentially exacerbating or triggering these conditions (99, 100). Furthermore, the post-infectious implications are only beginning to be understood. Persistent cognitive deficits, termed “brain fog,” along with fatigue and mood disorders, have been reported in PASC cases, underscoring the lasting impact of the virus on the CNS (101, 102). The neurotropic nature of SARS-CoV-2 has expanded the scope of COVID-19 beyond a respiratory illness, bringing to light the intricate and profound interplay between the virus and the nervous system. As we continue to grapple with the pandemic’s ramifications, understanding the neurological facets of COVID-19 will be pivotal in devising effective therapeutic and rehabilitative strategies.
TLRs are crucial components of the innate immune system, acting as primary detectors of pathogenic threats (103, 104). These receptors recognize conserved molecular structures known as pathogen-associated molecular patterns (PAMPs), which are essential for microbial survival but absent in host cells (105, 106). TLRs are type I transmembrane proteins with extracellular leucine-rich repeat (LRR) domains for PAMP recognition and intracellular Toll/interleukin-1 receptor (TIR) domains for initiating immune signaling pathways (105, 107). TLRs are classified into cell surface TLRs (e.g., TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10), which detect microbial membrane components, and endosomal TLRs (e.g., TLR3, TLR7, TLR8, and TLR9), which recognize microbial nucleic acids (103, 108). Upon detecting PAMPs, TLRs dimerize and recruit adaptor proteins like MyD88 and TRIF, forming complexes that activate downstream signaling pathways (107, 109). The MyD88-dependent pathway primarily induces pro-inflammatory cytokines through NF-κB and MAPKs, while the TRIF-dependent pathway promotes type I interferons and other inflammatory responses (Figure 1) (103, 110, 111). These pathways are essential for an effective immune response and linking innate and adaptive immunity.
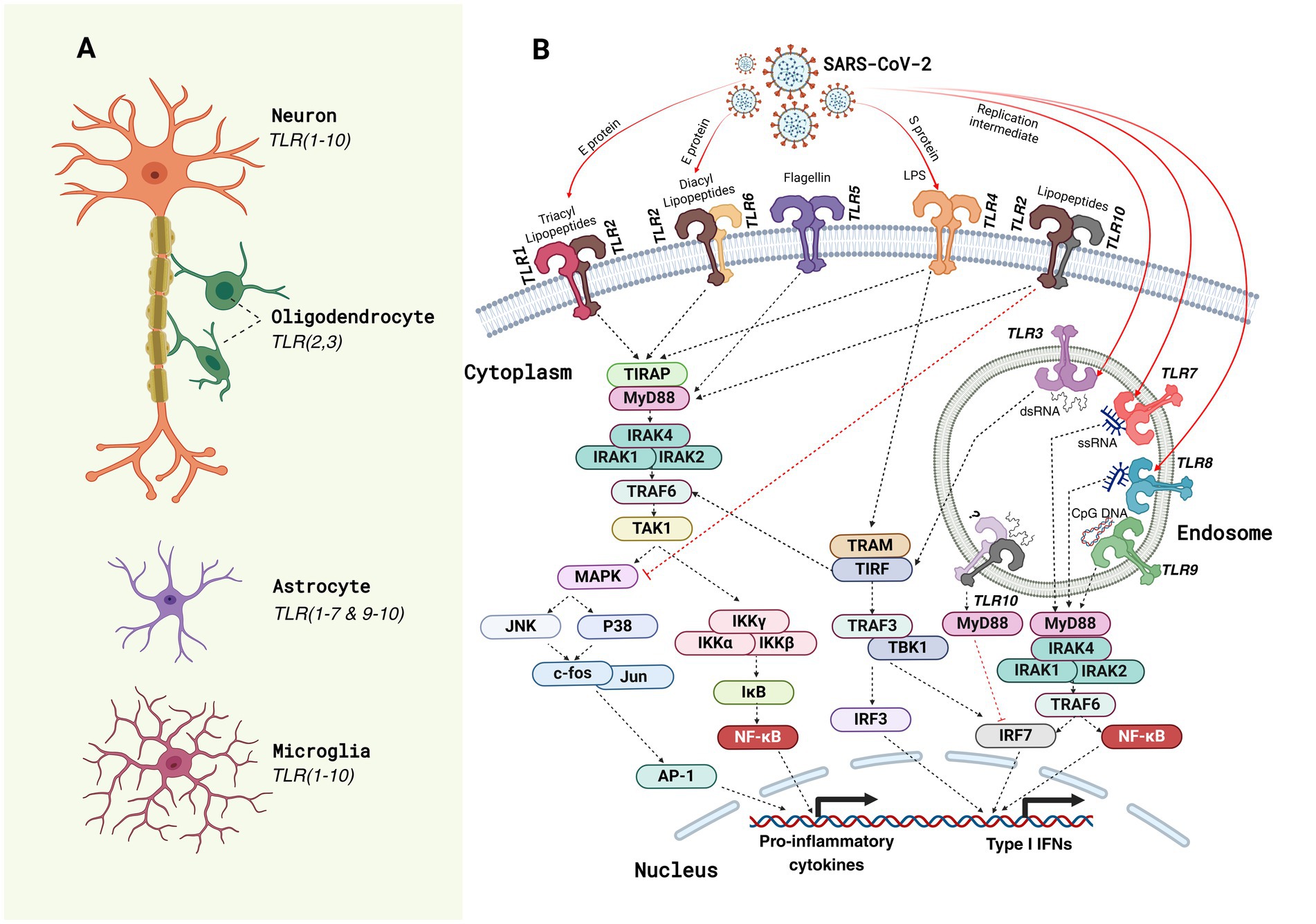
Figure 1. Toll-like receptor (TLR) expression and SARS-CoV-2-induced TLR signaling pathways in neural cells. (A) The schematic illustrates the expression profile of Toll-like receptors (TLRs) in key neural cell types of the central nervous system (CNS) in humans. Neurons predominantly express almost all TLRs (1–10); oligodendrocytes express TLR2 and TLR3; astrocytes express all TLRs except TLR8, whereas microglia, the resident immune cells of the CNS, express the broadest range of TLRs (TLR1–10) (179), underscoring their critical role in innate immune surveillance and neuroinflammatory responses. (B) TLR signaling pathways activated by SARS-CoV-2 components are depicted. The viral proteins (spike [S], envelope [E], and nucleocapsid [N]) and associated pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharides (LPS) and double-stranded RNA (dsRNA) interact with specific TLRs on the cell membrane (e.g., TLR1, TLR2, TLR4, TLR5, and TLR6) and within endosomal compartments (e.g., TLR3, TLR7, TLR8, and TLR9). These interactions activate downstream signaling cascades through adaptor proteins like myeloid differentiation primary response protein 88 (MyD88) and TIR domain-containing adapter inducing IFNβ (TRIF). Subsequent recruitment of kinases such as interleukin (IL)-1 receptor-associated kinase (IRAK)-1/4, tumor necrosis factor receptor-associated factor (TRAF6), and TANK-binding kinase 1 (TBK1) leads to the activation of transcription factors nuclear factor (NF)-κB, activator protein 1 (AP-1), and interferon regulatory factors (IRFs), which translocate to the nucleus to induce the expression of Type I interferons (IFNs) and pro-inflammatory cytokines. These signaling events mediate antiviral immunity but may contribute to the hyperinflammatory responses observed in COVID-19 neuropathology. TIR domain-containing adapter protein (TIRAP); transforming growth factor-activated kinase (TAK); mitogen-activated protein kinase (MAPK); c-Jun N-terminal kinase (JNK); inhibitor of kappa light polypeptide gene enhancer in B-cell kinase (IKK); inhibitor of nuclear factor kappa B (IĸB); Trif-related adapter molecule (TRAM); single-stranded RNA (ssRNA).
TLRs are vital in dendritic cell (DC) maturation and crucial for antigen presentation and T cell activation (103, 112). Activated DCs express higher levels of co-stimulatory molecules and MHC, enhancing their ability to initiate adaptive immune responses (113, 114). TLRs are also key regulators in macrophage function and inflammatory responses. Sanjuan et al. (115) demonstrated that TLR activation during phagocytosis mobilizes autophagy proteins to the phagosome, thereby enhancing phagosome maturation and pathogen destruction in macrophages. Recent studies underscore the critical roles of TLRs in modulating macrophage function and polarization. TLR7 agonists have been shown to repolarize M2 macrophages to the pro-inflammatory M1 phenotype, thereby enhancing phagocytosis, inflammation, and antigen presentation (116). Similarly, Vidyarthi et al. (117) elucidated the importance of TLR3 signaling in skewing M2 macrophages to the M1 subtype, both in vitro and in vivo. Further emphasizing the versatility of TLRs, Geng et al. (118) reported that TLR4 coordinates with the mechanical sensor Piezo1 to activate the CaMKII-Mst1/2 axis, driving macrophages to execute essential host defense functions. Collectively, these findings highlight the multifaceted roles of TLRs in macrophage biology, presenting them as promising therapeutic targets for enhancing immune responses.
Similarly, Veneziani et al. (119) demonstrated that TLR8 agonists effectively activate NK cells, particularly the CD56bright subset, by enhancing their proliferation, cytokine production, and cytotoxicity, which are vital in the tumor microenvironment. This activation promotes a pro-inflammatory phenotype, improving interactions between NK cells and dendritic cells (DCs) and boosting tumor antigen presentation and NK cell-mediated cytotoxicity against tumor cells. The study also highlighted that the TLR8 agonist R848 uniquely enhances CD56brightCD16− NK cell functions, especially when combined with suboptimal cytokine levels like IL-2 and IL-12, a specificity not shared by TLR3 and TLR9 agonists. This activation, exclusive to TLR8 and not TLR7, significantly increases IFN-γ and TNF-α production, suggesting TLR8 agonists as promising agents for cancer immunotherapy (119). Similarly, Noh et al. (120) reviewed the critical role of TLRs in NK cells, noting their ability to recognize pathogens and induce strong immune responses. TLR agonists enhance NK cell cytotoxicity and cytokine production, improving the efficacy of immunotherapies, including monoclonal antibody treatments for cancer.
Additionally, TLRs are expressed in non-immune cells like epithelial and endothelial cells, contributing to immune responses by producing cytokines and chemokines (121, 122). However, dysregulated TLR activation can lead to chronic inflammation and autoimmune diseases by recognizing self-derived damage-associated molecular patterns (DAMPs) (123, 124). Thus, TLR signaling must be tightly regulated to balance protective immunity and prevent pathological inflammation. Overall, TLRs are fundamental to innate immunity, recognizing PAMPs to initiate immune responses and shaping adaptive immunity. Understanding TLR signaling mechanisms is key to developing therapies for infectious, inflammatory, and autoimmune diseases.
The CNS, once considered an immune-privileged site, is now recognized as a dynamic environment where immune surveillance and responses occur (125, 126). TLRs are pivotal in this context, primarily expressed by microglia, the resident immune cells of the brain, but also found in astrocytes and other CNS cell types (127–129). Microglia express a wide array of TLRs, enabling them to detect and respond to diverse pathogens and cellular stress signals (130, 131). For instance, TLR4 recognizes lipopolysaccharides (LPS) from bacterial cell walls, while TLR3 detects viral double-stranded RNA (105, 132, 133). A significant aspect of TLR function in the CNS is their interaction with the inflammasome, particularly the NLR family pyrin domain containing 3 (NLRP3) inflammasome (134, 135). The inflammasome is a multi-protein complex that processes pro-IL-1β and pro-IL-18 into their active forms via caspase-1 (136, 137). This process requires a two-signal model: the first signal involves TLR-mediated transcription of pro-IL-1β and pro-IL-18, and the second signal, often provided by DAMPs like ATP, leads to inflammasome assembly and activation (127, 134).
Upon TLR activation, microglia release various pro-inflammatory mediators, including IL-1β, which plays a central role in the pathogenesis of numerous neurodegenerative diseases and CNS injuries (138, 139). For example, TLR4 activation in microglia can lead to significant neuroinflammation, contributing to conditions such as AD, multiple sclerosis, and stroke (128). Astrocytes also contribute to TLR-mediated responses in the CNS, albeit to a lesser extent than microglia. They express a limited repertoire of TLRs and can respond to inflammatory stimuli by producing cytokines and chemokines that influence microglial activity and overall CNS inflammation (17, 130). However, the exact role of inflammasomes in astrocytes remains less precise compared to microglia (91, 140). TLR signaling in the CNS is a double-edged sword (129, 141). While it is essential for defending against infections and maintaining homeostasis, excessive or prolonged TLR activation can lead to chronic neuroinflammation and subsequent neuronal damage (17). This is particularly relevant in neurodegenerative diseases where TLR activation can exacerbate disease progression by promoting sustained inflammation. Understanding the nuanced roles of TLRs in the CNS could pave the way for targeted therapies that modulate immune responses, aiming to protect against infections while minimizing harmful inflammation in neurodegenerative and other CNS disorders.
TLRs are key players in the pathogenesis of various neurodegenerative diseases due to their role in mediating inflammatory responses and other functions such as microglial activation and synaptic dysfunction (142–146). These receptors are expressed not only in immune cells but also in neurons, astrocytes, and microglia within the CNS, where they can recognize both PAMPs and DAMPs (130, 143).
TLRs play a significant role in the pathogenesis of AD through their involvement in neuroinflammation and the innate immune response (147). In AD, TLRs are crucial in recognizing amyloid-beta (Aβ) plaques and tau protein tangles, which are hallmarks of the disease (148). For instance, TLR4 has been implicated in recognizing Aβ plaques (148–150). Upon activation, TLR4 triggers signaling pathways that lead to the production of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6, contributing to neuroinflammation and neuronal damage (107, 151). This chronic inflammation can exacerbate the pathology of AD, suggesting that TLRs are significant contributors to the disease’s progression (152). Moreover, TLR2 and TLR9 also play roles in AD by enhancing the clearance of Aβ and modulating neuroinflammatory pathways (153).
A detailed examination of TLRs in AD draws on multiple studies and offers a comprehensive understanding of their contributions to the disease (Table 1). TLR4 is one of the most studied TLRs in relation to AD (153–157). They are localized on the surface of microglia, the resident immune cells in the brain, and are responsible for recognizing LPS and Aβ peptides, both associated with inflammatory responses in AD (139, 157). Research has shown that genetic polymorphisms in TLR4 can influence AD occurrence. Specifically, the Asp299Gly polymorphism of TLR4 has been associated with a decreased risk of late-onset AD (LOAD). This polymorphism attenuates inflammatory response due to reduced receptor signaling (155). Further, in this study, which involved the Italian population (277 LOAD patients), the frequency of the minor TLR4 299Gly allele was significantly higher in controls (300 cognitively healthy controls) compared to LOAD patients, and this variant was suggested to be protective toward the development of LOAD (155). Studies have demonstrated that TLR4 expression is significantly upregulated in the brains of AD patients (158, 159). This upregulation correlates with the presence of Aβ plaques and neurofibrillary tangles, the pathological hallmarks of AD. Further, the studies suggest that variations in TLR4 can affect its expression and function, thereby modulating the risk of developing AD (154). For instance, Zhang et al. (153) found increased expressions of TLR4 on peripheral blood mononuclear cells from AD patients, indicating a systemic inflammatory response in addition to the local brain inflammation. TLR2, like TLR4, is involved in recognizing Aβ and plays a role in mediating inflammatory responses in AD (153). Increased expression of TLR2 has been observed in microglia surrounding Aβ plaques. The activation of TLR2 by Aβ leads to the production of pro-inflammatory cytokines, contributing to the neuroinflammatory milieu characteristic of AD (153).
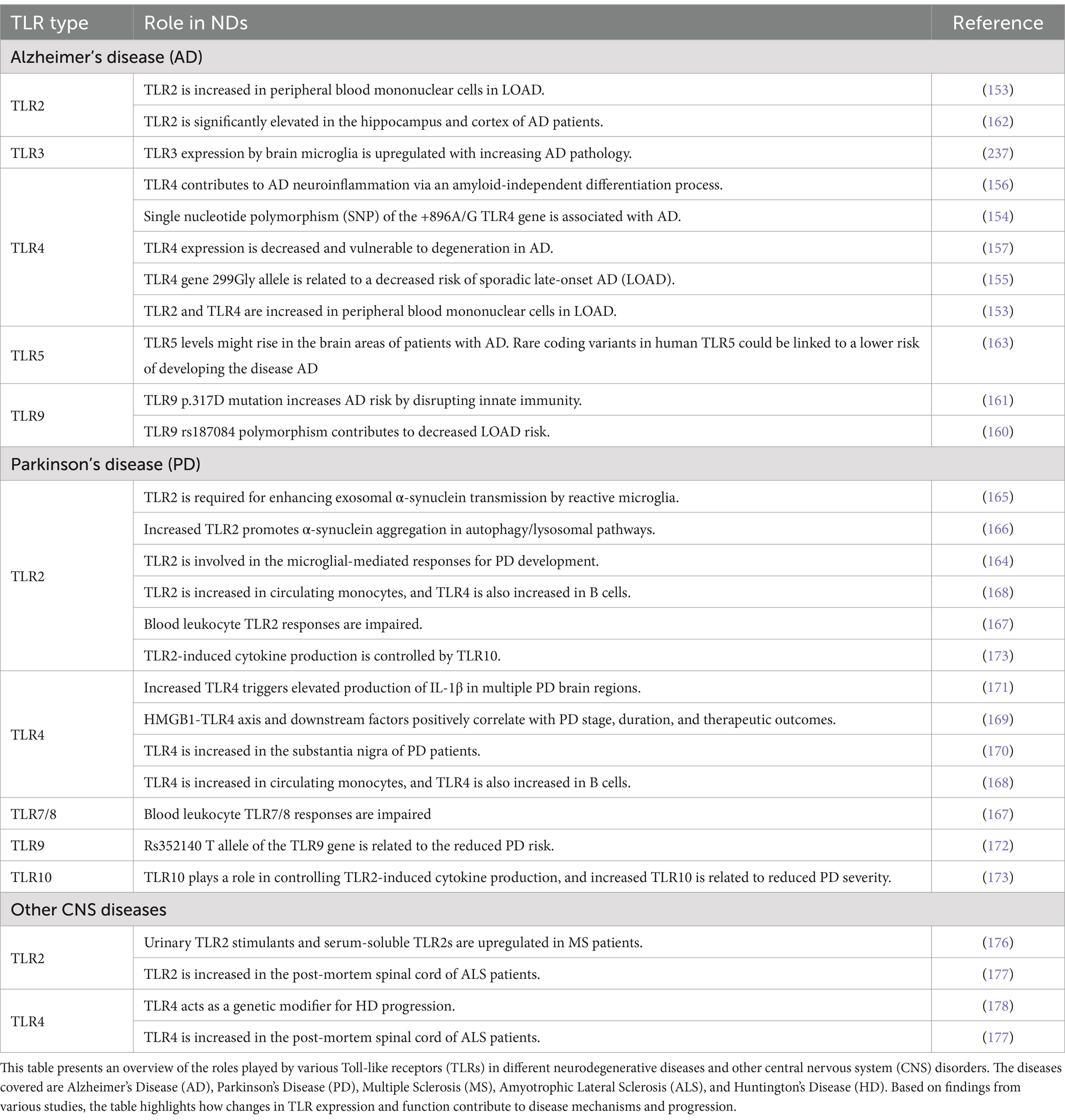
Table 1. Summary of TLR involvement in neurodegenerative diseases and other CNS disorders.
Recent research highlights the potential involvement of TLR9 in AD pathogenesis, particularly its role in the immune system’s response to Aβ deposits. TLR9 is known for recognizing unmethylated CpG DNA from pathogens, triggering an immune response. Studies have suggested TLR9 signaling may be implicated in Aβ clearance, an AD hallmark (160). In a study focusing on a large Han Chinese population, a significant association was found between a polymorphism in the TLR9 gene (rs187084) and a decreased risk of LOAD. This polymorphism was linked to higher expression levels of TLR9 in peripheral blood monocytes, suggesting that enhanced TLR9 activity might facilitate the clearance of Aβ, thus reducing AD risk (160). Moreover, research involving a Flanders-Belgian family identified a novel variant (p.E317D) in TLR9 that co-segregates with early-onset AD (EOAD) in an autosomal dominant manner. This variant caused a 50% reduction in TLR9 activation in an NF-κB luciferase assay, indicating a loss-of-function mutation. The resulting cytokine profile from human peripheral blood mononuclear cells (PBMCs) upon TLR9 activation showed an anti-inflammatory response that promoted phagocytosis of Aβ42 oligomers in human iPSC-derived microglia. This finding underscores the protective role of TLR9 signaling in AD pathogenesis and suggests that loss-of-function mutations may disrupt peripheral-central immune crosstalk, leading to increased neuroinflammation and pathogenic protein aggregates in AD (161). TLR3, another member of the TLR family, is also implicated in the pathogenesis of AD through its role in recognizing double-stranded RNA and activating immune responses. Specifically, TLR3-induced neuroinflammation can contribute to neuronal damage and the progression of AD pathology (153). For instance, TLR3 signaling has been shown to upregulate the expression of genes that modulate the production and clearance of Aβ, suggesting a potential mechanism by which TLR3 contributes to AD progression (153). The involvement of TLRs in AD extends beyond genetic associations. Mechanistically, TLR activation triggers downstream signaling pathways, such as NF-κB, which promotes the transcription of pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6. The role of TLR4 in mediating these responses has been particularly well-documented, with studies showing that TLR4-deficient mice exhibit reduced microglial activation and cytokine production in response to Aβ (157). Research findings also suggest a role for TLR2 in AD pathogenesis (153). TLR2 has been shown to be upregulated in the prefrontal cortex and hippocampus of AD patients and 5XFAD mouse models (162). Furthermore, TLR5 has also emerged as a potential modulator of AD pathology. Genetic analyses conducted in human AD brains have suggested that rare protein-coding variants in TLR5 may be associated with a reduced risk of AD (163).
In PD, the misfolded protein alpha-synuclein (α-syn) accumulates and activates TLR2 and TLR4 in microglia. This activation leads to the production of inflammatory mediators and reactive oxygen species (ROS), which contribute to the neurodegenerative process. Studies have shown that TLR2 expression is significantly increased in the substantia nigra and anterior cingulate cortex of PD patients, correlating with disease severity (164). This upregulation correlates with the accumulation of α-syn, a protein that aggregates and forms Lewy bodies, characteristic of PD (165). Experimental models have demonstrated that TLR2 deficiency can reduce alpha-synuclein accumulation and neuroinflammation, highlighting its role in PD pathogenesis (143). TLR2 is found in microglia, neurons, and within Lewy bodies, indicating its widespread involvement in the pathogenesis of PD (166). In addition to its expression in the CNS, TLR2 is also present in peripheral immune cells. Blood monocytes from PD patients exhibit altered responses to TLR2 activation, with decreased production of cytokines such as TNF-α, IL-1β, IL-6, and IL-10 compared to controls. This impaired cytokine response suggests a dysregulation of TLR2 and TLR7/8-mediated immune functions in PD (167). Further, TLR2 and TLR4 are also significantly involved in PD, with increased expression in the blood and brain leading to microglial activation, inflammatory cytokine production, and neurodegeneration, indicating potential therapeutic targets to mitigate inflammation and slow disease progression (168).
TLR4 plays a pivotal role in the pathogenesis of PD by mediating inflammatory responses and contributing to neurodegeneration. Yang et al. (169) demonstrated that TLR4 expression is significantly elevated in the serum of PD patients compared to healthy controls, and this elevation correlates with disease progression, duration, and poor responses to drug treatments, highlighting TLR4’s critical involvement in PD pathology. This finding underscores the systemic nature of TLR4-related inflammation and its potential as a biomarker for disease severity (169). Shin et al. (170) expanded on this by showing that prothrombin kringle-2 (pKr-2), a non-toxic fragment of prothrombin, induces TLR4 expression in microglia within the substantia nigra of PD patients. The study suggests that inhibiting pKr-2-induced TLR4 activation could be a protective strategy against dopaminergic neuron loss in PD (170). Further supporting these findings, Kouli et al. (171) reported that TLR4 activation by α-syn aggregates significantly contributes to the chronic neuroinflammatory environment observed in PD. They noted increased TLR4 expression in both the CNS and peripheral immune cells of PD patients, emphasizing its role in exacerbating neurodegeneration. The study by Miri et al. (172) found that the rs352140T polymorphism in the TLR9 gene acts as a protective factor against PD in the northern Iranian population. This single-nucleotide polymorphism (SNP) was significantly associated with a reduced risk of PD, suggesting that individuals carrying the rs352140T allele have a lower likelihood of developing the disease. Additionally, the study highlighted that the TLR9 rs352140T allele could be considered a potential prognostic marker and therapeutic target for PD, emphasizing the importance of genetic factors in modulating immune responses and disease susceptibility (172). The study by da Rocha Sobrinho et al. found that TLR10 plays a crucial role in controlling TLR2-induced cytokine production in monocytes from PD patients. The elevated expression of TLR10 on monocytes was associated with reduced production of pro-inflammatory cytokines, suggesting a protective mechanism against PD progression (173).
Multiple sclerosis (MS) is characterized by chronic inflammation and demyelination in the CNS. TLR2 and TLR4 activation in response to myelin debris and other DAMPs leads to pro-inflammatory cytokines and chemokines, promoting immune cell infiltration and further demyelination. The complex role of TLRs in MS is evidenced by varying effects of TLR deficiencies, which can either ameliorate or exacerbate the disease in experimental models (143). TLR3 and TLR9 have also been implicated in MS, with their activation contributing to neuroinflammation and neurodegeneration (174, 175). A recent study found that soluble TLR2 (sTLR2) levels were significantly elevated in the serum of MS patients during both relapse and remission compared to healthy controls, suggesting sTLR2 as a potential biomarker for MS (176). Further, TLR2 stimulants were significantly higher in the urine of MS patients than in healthy controls. This finding indicates that MS patients are exposed to higher levels of TLR2 ligands, possibly contributing to disease activity and progression (176).
Amyotrophic lateral sclerosis (ALS) involves selective motor neuron degeneration, and TLRs have been implicated in its pathogenesis. TLR2 and TLR4 levels are elevated in the spinal cord of ALS patients, particularly in microglia and astrocytes. These receptors mediate neuroinflammatory responses that exacerbate motor neuron death. In mouse models of ALS, the deletion of TLR4 has been shown to improve survival and reduce neurological deficits, underscoring the detrimental role of TLR-mediated inflammation in ALS (143). A research study findings demonstrate heightened expression of TLR2, TLR4, the receptor for advanced glycation end products (RAGE), and high mobility group box 1 (HMGB1) in reactive glia in the ALS spinal cord, suggesting the activation of TLR/RAGE signaling pathways resulting in the injury of motor neurons (177). Vuono et al. (178) observed a significant increase in the number of cells expressing TLR4 in the striatum of postmortem brain samples from Huntington’s disease (HD) patients compared to controls. This finding suggests that TLR4-mediated inflammatory responses are involved in HD pathogenesis. The study also identified that specific TLR4 SNPs were associated with changes in motor progression in HD patients. Specifically, the rs1927911 and rs1927914 polymorphisms were linked to a faster rate of motor decline, indicating that genetic variations in TLR4 may influence the clinical progression of HD (178).
Across these diseases, TLRs function as critical modulators of neuroinflammation and neuronal degeneration. Their activation often results in the production of pro-inflammatory cytokines and reactive oxygen species, contributing to a chronic inflammatory environment that accelerates disease progression. However, the precise role of each TLR may vary, as TLR-mediated signaling can have context-dependent effects in different disease states, potentially resulting in either detrimental or beneficial outcomes. In AD, TLR4 and TLR2 play pivotal roles in identifying and eliminating Aβ plaques and tau tangles. On the other hand, these receptors have the capacity to initiate chronic inflammation, which aggravates the progression of the disease. In a similar manner, in PD, the activation of TLR2 and TLR4 by α-synuclein aggregates prompts microglial activation and oxidative stress, thereby expediting neurodegeneration. Whereas, TLRs such as TLR9 and TLR10 function as protective factors by mitigating the production of pro-inflammatory cytokines. However, in MS, TLR9 as well as other TLRs such as TLR2, TLR3, TLR4 mediate inflammatory cascades linked to demyelination. Additionally, TLR-mediated signaling pathways are implicated in ALS and HD, highlighting their systemic contributions to disease-specific inflammatory processes and progression. Understanding the distinct roles of TLRs in these diseases offers valuable insights into the complex interplay between the immune system and neurodegeneration, providing a foundation for the development of targeted therapies aimed at modulating TLR signaling to mitigate neuroinflammation and slow disease progression. Modulating TLR activity could help balance the beneficial and detrimental effects of inflammation. For example, TLR4 inhibitors might reduce neuroinflammation in AD and PD, while strategies to enhance TLR-mediated clearance of protein aggregates could be beneficial in early disease stages. Nevertheless, therapeutic interventions must be meticulously tailored to prevent undesired suppression of protective immune responses (179).
TLRs play significant roles in recognizing pathogens and initiating immune responses (104). Several studies have investigated the involvement of TLRs in neuroinflammation induced by SARS-CoV-2 infection, providing insights into their mechanisms and potential therapeutic targets (180–183). SARS-CoV-2, the virus responsible for COVID-19, interacts with various TLRs to trigger immune responses (182). TLR2, for instance, has been identified as a receptor that recognizes the envelope (E) protein of SARS-CoV-2 (184). This interaction leads to the activation of downstream signaling pathways, resulting in the production of pro-inflammatory cytokines (141). TLR4 is also implicated in SARS-CoV-2 infection, where it recognizes viral components and contributes to the inflammatory response (185). Both TLR2 and TLR4 play pivotal roles in mediating neuroinflammation during SARS-CoV-2 infection (183, 186). TLR2, expressed on microglia and neurons, is activated by SARS-CoV-2, leading to the release of inflammatory cytokines and the promotion of neuroinflammation. This process is suggested to exacerbate neurodegenerative conditions such as AD and PD by increasing Aβ and α-syn aggregation (185). TLRs’ involvement in SARS-CoV-2 infection is also linked to their effects on the BBB. The inflammation induced by TLR activation can compromise the integrity of the BBB, allowing SARS-CoV-2 and immune cells to infiltrate the CNS. This breach is a critical step in the pathogenesis of COVID-19-related neurological symptoms, including encephalitis, cerebrovascular events, and neurodegeneration (141). Given their central role in mediating neuroinflammation, TLRs present potential therapeutic targets for mitigating SARS-CoV-2-induced neurological damage. Modulating TLR activity, either through agonists or antagonists, could help regulate the immune response and reduce the excessive inflammation that contributes to CNS pathology. This approach holds promise for preventing or treating the neuroinflammatory consequences of COVID-19.
Evidence from recent research provides comprehensive insights into the role of various TLRs in COVID-19 infection and severity, highlighting the intricate interplay between genetic variations, immune responses, and clinical outcomes (Table 2). Zheng et al. (187) demonstrated that TLR2 recognizes the SARS-CoV-2 E protein, initiating inflammatory signaling pathways and cytokine production, essential for the inflammatory response during β-coronavirus infections. Inhibition of TLR2 signaling significantly reduces key inflammatory cytokines and chemokines, such as IL-6, CXCL10, and MCP-1, and improves survival rates in SARS-CoV-2-infected mice, suggesting a potential therapeutic strategy to mitigate severe COVID-19 pathology. Building on this, Nayak et al. (188) reveal the minor deletion allele of the TLR2 rs111200466 polymorphism is inversely correlated with susceptibility to SARS-CoV-2 infection and mortality. This indicates that TLR2 genetic variants, specifically the rs111200466 polymorphism, may influence COVID-19 incidence and severity, warranting further validation in diverse populations. Alhabibi et al. (189) highlight TLR4’s critical role in the inflammatory response to SARS-CoV-2, with the minor alleles 299Gly (G) and 399Ile (T) significantly associated with severe COVID-19 and increased IL-6 levels, contributing to cytokine storm and higher mortality rates. They also identify TLR2 and TLR9 variants linked to increased risk and severity of COVID-19, specifically the TLR2 rs5743708 (G/A) and TLR9 rs5743836 (C/C) genotypes, which correlate with pro-inflammatory responses and disease progression.
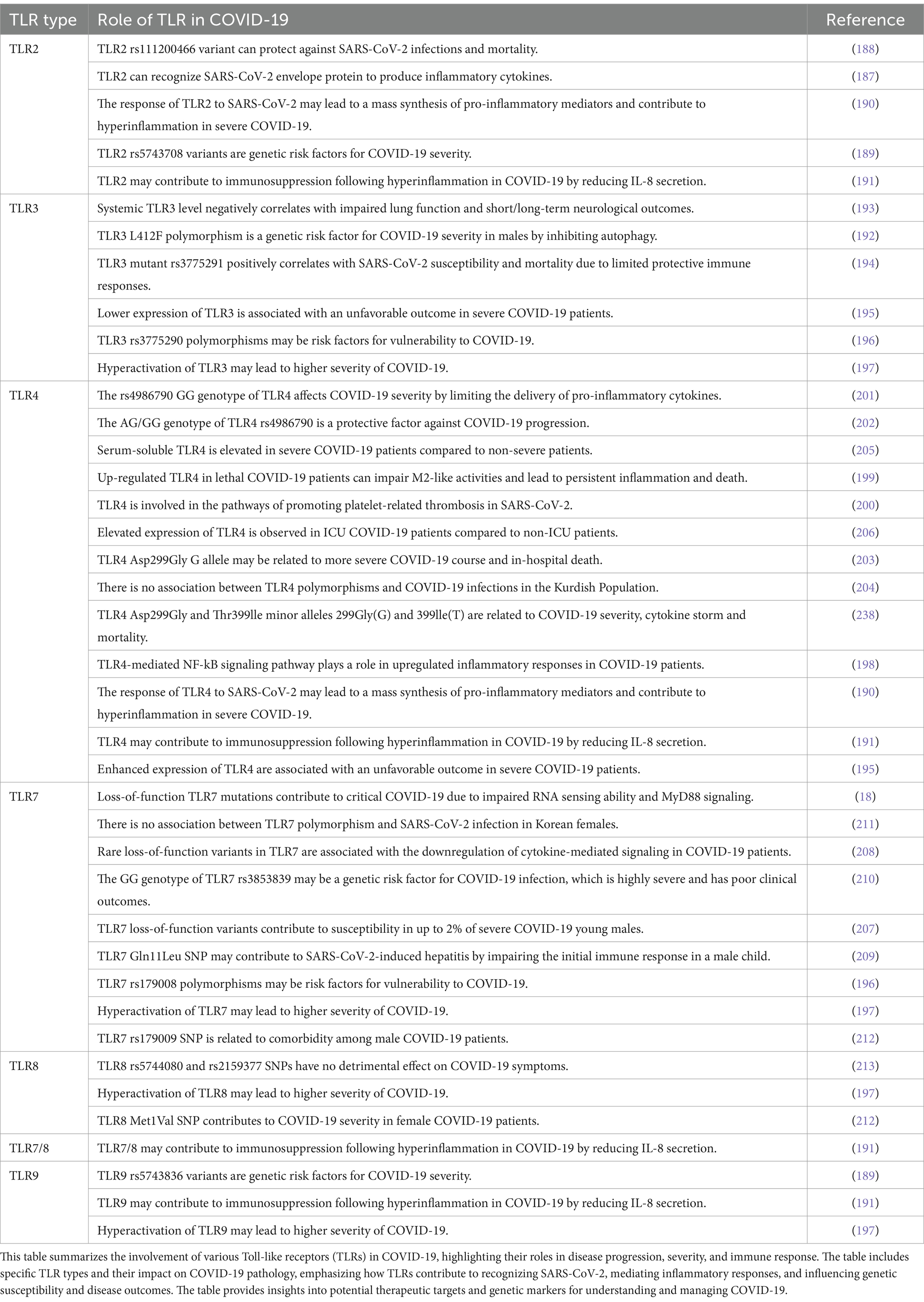
Table 2. Roles of toll-like receptors (TLRs) in COVID-19 pathogenesis.
Sahanic et al. (190) further underscore TLR4’s importance by showing that SARS-CoV-2 activates the TLR4/MyD88 pathway in human macrophages, triggering strong pro-inflammatory responses linked to severe COVID-19. Their study suggests that TLR4 blockade can significantly reduce exaggerated inflammatory responses in macrophages infected with various SARS-CoV-2 variants, presenting a potential therapeutic target for severe COVID-19. Carreto-Binaghi et al. (191) find that IL-8 secretion via TLR2, TLR4, TLR7/8, and TLR9 receptors in blood cells from COVID-19 patients is significantly reduced at admission, indicating an impaired initial immune response. However, receptor functionality improves after 2 weeks of hospitalization, signaling recovery in the patient’s immune response. Additionally, compromised NOD2 receptor functionality at the onset of COVID-19 contributes to lower IL-8 levels, suggesting this impairment in innate immune receptors plays a role in the early-stage immunosuppression observed in severe COVID-19, which is later restored with continued hospitalization.
Several studies have collectively emphasized the significant role of TLR3 and related TLR polymorphisms in modulating the immune response to SARS-CoV-2, influencing COVID-19 severity and outcomes. Croci et al. (192) highlight the L412F polymorphism in TLR3 as a significant indicator of severe COVID-19, particularly in males. This genetic variant disrupts autophagy, impairing the innate immune response and exacerbating disease severity. Patients with this polymorphism exhibit reduced TNF production and a higher incidence of autoimmune conditions, underscoring the critical role of autophagy and immune regulation in the progression of severe COVID-19. Supporting these findings, Lieberum et al. (193) report that systemic TLR3 expression in blood negatively correlates with impaired lung function and neurological outcomes in severe COVID-19. Higher TLR3 levels are associated with less severe disease progression and better recovery. This study identifies TLR3 expression in blood, rather than bronchoalveolar lavage (BAL), as a stronger predictor of disease severity and long-term outcomes, highlighting its crucial role in the systemic immune response during severe COVID-19. In line with this, Dhangadamajhi and Rout (194) demonstrate that the TLR3 functional variant rs3775291 significantly increases susceptibility to and mortality from COVID-19. This variant impairs TLR3 expression and its ability to recognize SARS-CoV-2 dsRNA, leading to inadequate immune responses and higher disease severity and death rates across diverse populations. Further elucidating TLR3’s role, Menezes et al. (195) discovered that lower TLR3 expression in peripheral blood is associated with unfavorable outcomes in severe COVID-19 patients. This reduced expression correlates with a decreased interferon-gamma response, indicating a compromised antiviral defense mechanism in critically ill patients. Conversely, enhanced TLR4 expression in severe COVID-19 patients may reflect a compensatory mechanism for the impaired TLR3 response but also contribute to the heightened inflammatory state characteristic of severe COVID-19.
Additionally, Alseoudy et al. (196) report that TLR3 and TLR7 polymorphisms are linked to increased prevalence of COVID-19 pneumonia. Specifically, the TLR3 rs3775290 and TLR7 rs179008 polymorphisms significantly increase the risk of severe COVID-19 symptoms, suggesting their potential role in disease progression. However, these polymorphisms did not significantly correlate with COVID-19 pneumonia outcomes. Other factors such as male sex, low SPO2 levels, high INR, high LDH, and lymphopenia were identified as independent predictors of mortality, highlighting the complex interplay of genetic and clinical factors in disease prognosis. Bagheri-Hosseinabadi et al. (197) find that mRNA expression levels of TLR3, TLR7, TLR8, and TLR9 are significantly upregulated in nasopharyngeal epithelial cells of COVID-19 patients compared to controls. This upregulation correlates with disease severity and clinical markers of inflammation, suggesting that these TLRs play a critical role in COVID-19 pathogenesis and could serve as potential biomarkers for predicting disease severity. Collectively, these studies underscore the importance of TLR3 and other TLR polymorphisms in SARS-CoV-2 recognition and the immune response, highlighting their potential as therapeutic targets.
Several studies have demonstrated that TLR4 plays a critical role in the immune response to SARS-CoV-2 infection, with significant implications for disease severity and potential therapeutic strategies. TLR4-mediated signaling molecules are significantly upregulated in the PBMCs of COVID-19 patients compared to healthy controls, correlating with the role of S100A9, a TLR4 ligand, as a potential biomarker for severe COVID-19 due to its inverse correlation with serum albumin levels (198). In damaged pneumocytes and lung macrophages of patients who succumbed to COVID-19, TLR4 expression is significantly increased, suggesting a pivotal role in the hyper-inflammatory response observed in lethal cases of the disease (199). This inflammatory environment leads to a pathological shift in macrophage populations, with an upregulation of pro-inflammatory TLR4(+) macrophages and a depletion of GAL-3(+) macrophages, crucial for resolving inflammation and promoting tissue repair.
Furthermore, TLR4 plays a crucial role in platelet activation and subsequent thrombus formation in SARS-CoV-2 infection. The interaction of the SARS-CoV-2 spike protein with TLR4 induces oxidative stress and platelet activation, pivotal in COVID-19-associated thrombosis. Inhibition of TLR4 or its oxidative stress pathways significantly reduces platelet activation and thrombus growth, suggesting promising therapeutic strategies (200). Additionally, TLR4 activation leads to increased production of pro-inflammatory cytokines such as IL-6 and IL-1β, contributing to the severity of COVID-19. Inhibition of TLR4 significantly reduces the expression of these cytokines, highlighting the therapeutic potential of targeting TLR4 to manage severe COVID-19 cases (201).
Genetic studies have revealed that specific TLR4 polymorphisms correlate with COVID-19 severity. The TLR4 rs4986790 AG/GG genotype acts as a protective factor against severe COVID-19 outcomes, significantly reducing the risk of hospitalization, intensive care, or death. Patients with this genotype exhibit lower inflammatory markers, such as IL-6 and procalcitonin, associated with a more favorable prognosis (202). In contrast, the TLR4 (Asp299Gly and Thr399Ile) polymorphisms are significantly associated with an increased risk of severe COVID-19 and elevated levels of IL-6, indicating a heightened inflammatory response and severe disease progression (203). However, in the Kurdish population, no significant association was found between these polymorphisms and COVID-19 infection, suggesting that these SNPs may not influence COVID-19 susceptibility in this cohort (204). TLR4 also emerges as a potential biomarker and therapeutic target for COVID-19. Serum levels of soluble TLR4 (sTLR4) and soluble CD14 (sCD14) are significantly elevated in patients with severe COVID-19 compared to non-severe cases, indicating that sTLR4 and sCD14 could serve as promising markers for assessing clinical severity and potential therapeutic targets for managing severe COVID-19 (205). Moreover, TLR4 mRNA expression is significantly elevated in ICU COVID-19 patients compared to non-ICU patients, correlating with disease severity. High levels of inflammatory chemokines, such as IFN-β and CXCL13, present in ICU patients suggest these molecules could serve as biomarkers for severe COVID-19 and potential targets for immunotherapeutic strategies (206).
Research on TLR7 reveals its significant role in influencing COVID-19 severity, particularly among males. Fallerini et al. (207) discovered that rare loss-of-function variants in TLR7 are significantly linked to severe COVID-19 in males. These genetic alterations hinder the TLR7 signaling pathway, leading to diminished expression of type I and II interferon-related genes, crucial for antiviral defenses. The study found that 2.1% of severely affected males carried these TLR7 variants, while none of the asymptomatic individuals did, highlighting TLR7’s role in susceptibility to severe COVID-19 among young male patients. Mantovani et al. (208) identified similar rare loss-of-function variants in X-chromosomal TLR7 in young men with severe COVID-19, associated with impaired TLR7 signaling and reduced type I and II interferon responses. This genetic impairment led to significant downregulation of cytokine-mediated signaling, contributing to severe disease progression. Functional characterization of newly identified TLR7 variants in severely affected male patients demonstrated decreased mRNA levels in IFNα, IFNγ, RSAD2, ACOD1, IFIT2, and CXCL10 genes, revealing profound impairment of the TLR7 pathway.
Pessoa et al. (209) reported the first known case of SARS-CoV-2-induced hepatitis in a male child with the TLR7 Gln11Leu rs179008 polymorphism, suggesting this genetic variation may impair the initial immune response against the virus. This indicates that polymorphisms in the TLR7 gene, such as Gln11Leu rs179008, can negatively impact the innate immune response, leading to higher susceptibility to severe outcomes in viral infections like COVID-19. Naushad et al. (18) demonstrated that men with loss-of-function mutations in TLR7 have a significantly increased risk of severe COVID-19 due to disrupted viral RNA sensing and impaired MyD88 signaling. These findings highlight TLR7’s critical role in the innate immune response to SARS-CoV-2 and suggest that TLR7 agonists could potentially restore antiviral responses in individuals with these genetic variants. Interestingly, specific hypofunctional and neutral variants of TLR7 still maintain their ability to form the TLR7-MyD88-TIRAP complex and respond to TLR7 agonists, indicating potential for targeted therapies using TLR7 agonists to enhance immune responses in COVID-19 patients with specific TLR7 mutations.
El-Hefnawy et al. (210) found that the GG genotype of the TLR7 SNP (rs3853839) is significantly more prevalent in COVID-19 patients, suggesting a genetic predisposition to severe disease outcomes. They also observed that TLR7 mRNA expression levels were substantially higher in COVID-19 patients, particularly those with the GG genotype, correlating with increased disease severity and poorer clinical outcomes. In contrast, Zayed et al. (211) found no significant differences in the genotype or allele frequencies of the TLR7 rs864058 polymorphism between female COVID-19 patients and healthy controls, indicating this specific genetic variation in TLR7 is not associated with COVID-19 susceptibility in the Korean female population. This research suggests that while TLR7 plays a critical role in the innate immune response to SARS-CoV-2, the low-frequency TLR7 rs864058 polymorphism does not contribute to variations in disease severity or susceptibility, emphasizing the need to explore other genetic factors and TLR variants that might influence COVID-19 outcomes.
Research on the genetic factors influencing COVID-19 severity has highlighted the roles of TLR7 and TLR8 single nucleotide polymorphisms (SNPs). Bagci et al. (212) identified that the TLR8 Met1Val SNP is significantly associated with increased COVID-19 severity in female patients, particularly those admitted to the ICU. In male patients, the TLR7 rs179009 SNP A allele was more prevalent among individuals with comorbidities, suggesting its role in exacerbating disease severity under these conditions. Conversely, Mahallawi and Suliman (213) found that specific SNPs in TLR8, namely rs5744080 and rs2159377, did not affect the severity of COVID-19 symptoms in the Saudi population. Their findings suggest that the innate immune response, once activated, does not depend on the affinity level of the TLR8 receptor for identifying SARS-CoV-2 glycoprotein structures. DNA sequencing revealed that TLR8 is highly conserved among the Saudi population, with an average sequence homology of 99.63%, indicating minimal variation. This conservation implies that TLR8 mutations do not significantly impact the receptor’s function or the severity of COVID-19 symptoms in this population.
The collective findings from these studies illustrate the complex role of TLRs in COVID-19. TLR4 is consistently implicated in severe inflammatory and thrombotic responses, while TLR3 and TLR7 genetic variants are linked to differential disease outcomes and immune responses. These insights highlight the potential for targeted therapies, such as TLR agonists and inhibitors, to modulate immune responses and improve clinical outcomes in COVID-19 patients. Understanding the genetic and molecular mechanisms underlying TLR-mediated immune responses can guide the development of personalized treatments and preventive strategies for COVID-19 and other viral infections.
Furthermore, all mentioned studies were conducted to elucidate the role of TLRs in the neuroinflammation and neuroinvasion associated with SARS-CoV-2; however, they lack conclusive experimental evidence in humans to substantiate these mechanisms. Addressing this limitation necessitates conducting human studies, such as postmortem analyses of brain tissues from COVID-19 patients, alongside advanced transcriptomic and proteomic investigations, to verify TLR-mediated pathways. Longitudinal cohort studies across diverse populations are imperative to validate the influence of TLR polymorphisms on disease severity and long-term neurological outcomes. While TLR activation is implicated in the disruption of the BBB, additional studies utilizing ex vivo BBB models or imaging techniques are required to elucidate these interactions in humans. Although TLR-targeted therapies exhibit promise in preclinical investigations, robust clinical trials are essential to assess their safety and efficacy in alleviating neurological complications. Moreover, there is a pressing need for research to delineate specific cytokine profiles associated with TLR activation in neuroinflammation, correlating these profiles with cerebrospinal fluid and serum markers in affected patients. The interplay between TLRs and other innate immune receptors, such as NOD-like receptors, remains another underexplored domain, potentially uncovering synergistic mechanisms in SARS-CoV-2-induced inflammation. Limited data exist regarding the long-term cognitive and neurological ramifications of TLR-mediated neuroinflammation, thus necessitating neuropsychological assessments and imaging within long-COVID cohorts. Besides, the molecular underpinnings of sex-specific differences in TLR-related immune responses warrant further examination. Addressing these research gaps through multidisciplinary approaches will enhance the scientific framework for comprehending TLR-mediated mechanisms in SARS-CoV-2 infection and will inform the development of personalized therapeutic and preventive strategies.
Considering the putative shared mechanisms by which TLRs contribute to the pathogenesis of neurodegenerative diseases and the host immune response to SARS-CoV-2 infection suggest potential synergistic pathways that may underlie the development of neurodegenerative phenotypes observed in a subset of Long COVID patients. Persistent neuroinflammation is a hallmark of ‘Long COVID’ (214), and TLRs are crucial in mediating this inflammatory response (67). Activation of TLRs, particularly TLR2 and TLR4, by SARS-CoV-2 infection or the resulting inflammatory mediators, can lead to sustained microglial activation and the release of pro-inflammatory cytokines in the CNS (183). This chronic neuroinflammation can contribute to neuronal dysfunction and degeneration. Studies have documented that amyloid precursor protein (APP) facilitates SARS-CoV-2 virus entry into cells and enhances Aβ-associated pathology (215). In some cases of Long COVID, the potential for SARS-CoV-2 infection to induce the aggregation of certain proteins, such as Aβ, has been observed (216–219). These protein aggregates can act as endogenous ligands for TLRs, further activating inflammatory pathways and exacerbating neurodegeneration (220). The interplay between protein aggregation and TLR-mediated signaling may play a role in developing neurodegenerative phenotypes in Long COVID (221). Further, TLR activation has been linked to synaptic dysfunction and impairment of synaptic plasticity (222), which are early events in the pathogenesis of neurodegenerative diseases. The dysregulation of TLR signaling observed in Long COVID may contribute to the loss of synaptic connections and the impairment of neuronal communication, leading to cognitive impairment and other neurological manifestations.
However, despite the importance of TLRs, there is a significant dearth of comprehensive studies on the role of TLRs in post-COVID-19 conditions, underscoring the need for further investigation in this area. A study by Noor Eddin et al. identified a novel hemizygous loss-of-function variant in the TLR7 gene in a pediatric patient who experienced severe neurological deterioration following COVID-19 infection. This finding suggests a critical role for TLR7 in mediating immune response and neurological outcomes post-infection, emphasizing the importance of genetic screening for TLR7 variants in predicting COVID-19 severity and long-term effects (223). Complementing this, a study by Fontes-Dantas et al. (16) highlighted the role of TLR4 in mediating long-term cognitive dysfunction, a significant symptom of post-COVID-19 syndrome. The study demonstrated that infusion of the SARS-CoV-2 Spike protein into the brains of mice induced late cognitive dysfunctions, marked by neuroinflammation and synapse loss. This cognitive impairment was mediated via TLR4 signaling, with genetic or pharmacological blockage of TLR4 protecting against synapse elimination and memory dysfunction. Moreover, the study identified that in a cohort of COVID-19 patients, those with the TLR4-2604G > A GG genotype (rs10759931) were associated with poorer cognitive outcomes, underscoring TLR4’s pivotal role in long-term neurological effects post-COVID-19.
Collectively, these studies underscore the significant role of TLRs, especially TLR4 and TLR7, in the pathogenesis of post-COVID-19 conditions. They highlight the potential mechanisms these receptors contribute to prolonged inflammation and tissue damage, leading to the diverse and persistent symptoms observed in ‘Long COVID’. The findings suggest that targeting these receptors could offer therapeutic potential in mitigating the long-term effects of COVID-19. However, the current body of research remains limited, necessitating further studies to comprehensively understand the molecular underpinnings and develop targeted treatments for post-COVID-19 syndrome.
The aftermath of COVID-19, often termed post-COVID-19 syndrome or Long COVID, has prompted a deeper exploration into novel therapeutic strategies. Targeting Toll-like receptors (TLRs) and their signaling pathways emerges as a promising approach due to the crucial role these receptors play in the innate immune response and inflammation.
Several studies emphasize the significance of TLR4 in COVID-19 pathogenesis. TLR4 recognizes viral components, triggering a cascade that results in the production of pro-inflammatory cytokines such as IL-6 and TNF-α, which are pivotal in the cytokine storm observed in severe COVID-19 cases (224). Thus, therapeutic agents that inhibit these TLR signaling might potentially reduce chronic inflammation and modulate immune responses, thereby preventing tissue damage and promoting recovery. Another promising strategy involves using nucleic acid-based therapeutics, such as small interfering RNAs (siRNAs) and antisense oligonucleotides (ASOs), which can specifically target and downregulate the expression of TLRs or their signaling components. These therapies might offer a high degree of specificity and can be designed to silence genes involved in aberrant TLR signaling, thereby mitigating the detrimental effects of chronic inflammation. One study discusses using BZL-sRNA-20, an oligonucleotide targeting TLR4, which has shown efficacy in reducing acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) in mice models. This suggests that targeting TLR4 could potentially mitigate severe respiratory complications associated with COVID-19 (225).
Furthermore, modulation of TLR signaling using synthetic ligands or agonists may help recalibrate the immune response, promoting a more balanced and controlled inflammatory environment. Agonists of these receptors can enhance antiviral responses, whereas antagonists might be used to suppress excessive inflammation. For instance, the antagonist M5049 (enpatoran), which targets TLR7 and TLR8, is currently under clinical evaluation and has shown promise in reducing inflammatory responses in COVID-19 patients (226, 227). A study by Proud et al. investigated the prophylactic use of the TLR2/6 agonist INNA-051 in a ferret model to reduce viral shedding of SARS-CoV-2. Prophylactic intranasal administration of INNA-051 markedly decreased viral RNA levels in the nose and throat of infected ferrets by up to 96%, highlighting its potential to limit person-to-person transmission of COVID-19. These findings strongly support the further development of TLR2/6 agonists as a therapeutic approach to activate innate immune responses at mucosal surfaces, thereby providing a rapid and effective defense against SARS-CoV-2 (228). The conjugation of the TLR1/2 agonist Pam3CSK4 to the SARS-CoV-2 receptor-binding domain (RBD) significantly boosted both the antibody and cellular immune responses against RBD. This approach effectively inhibited RBD-ACE2 binding and provided protection against SARS-CoV-2 and its variants, underscoring the potential of TLR1/2 agonists in enhancing vaccine efficacy (229). PUL-042, a combination therapeutic currently undergoing clinical trials, comprises the TLR2/6 ligand Pam2CSK4 and the TLR9 ligand ODN M362. It is being evaluated for its efficacy as a prophylactic agent in reducing the infection rate and progression of COVID-19 (230, 231). Various TLR modulators are undergoing clinical trials to assess their efficacy in treating COVID-19. For instance, TAK-242 (resatorvid), a TLR4 antagonist, has shown the potential to reduce the inflammatory response in preclinical models (190, 232). Further, in a study investigating the link between TLR7 loss-of-function (LOF) mutations and the severity of COVID-19, researchers discovered that TLR7 agonists can effectively bind to hypofunctional and neutral TLR7 variants. These agonists stimulate TLR7-MyD88-TIRAP-mediated signaling, thereby restoring antiviral responses. Agonists capable of binding to most TLR7 variants have the potential to serve as effective adjuvants in vaccines (18).
Similarly, other TLR-targeted therapies, including agonists and antagonists, are being explored for their ability to modulate the immune response and mitigate the severity of COVID-19. A recent study has also highlighted the development of inulin acetate-based nanoparticles (InAc-NPs) as a TLR4 agonist for intranasal vaccination, which significantly enhances both systemic and mucosal immune responses, indicated by elevated IgG1, IgG2a, and sIgA levels. InAc-NPs effectively activate TLR4 on macrophages, leading to a robust immune response and demonstrating their potential as a novel adjuvant for mucosal vaccines (233). Likewise, recent research successfully developed a SARS-CoV-2 spike protein subunit vaccine incorporating a dual TLR ligand liposome adjuvant, which demonstrated high efficacy in mice by inducing potent systemic neutralizing antibodies and significant levels of anti-spike IgA. The vaccine’s efficacy was further evidenced by the complete protection of mice from a lethal viral challenge, highlighting its potential for robust protective immunity against COVID-19 (234). A recent study found that stimulating PBMCs from moderate COVID-19 patients with a TLR8 agonist and the spike protein (SP) of SARS-CoV-2 significantly increased the frequency of CD3 + IFN-β + T cells and upregulated IFN-β gene expression compared to healthy controls. Notably, the TLR8 agonist induced the highest frequency of IFN-β-producing T cells in moderate patients, highlighting its potential to enhance antiviral immune responses in COVID-19 (235).
Targeting TLRs offers a dual benefit: enhancing antiviral responses during the acute phase of infection and mitigating chronic inflammation associated with post-COVID-19 syndrome. Modulating TLR pathways could address the prolonged immune activation and inflammation seen in long COVID-19, thereby reducing symptoms and improving patient outcomes. The strategic targeting of TLRs and their signaling pathways presents a viable therapeutic avenue for managing both acute and long-term complications of COVID-19. By fine-tuning the immune response, it is possible to enhance antiviral defenses while mitigating the harmful effects of hyperinflammation. As research progresses, TLR-targeted therapies could become integral components of the therapeutic arsenal against COVID-19 and its lingering effects. No regulatory TLR drugs have been approved for clinical use due to insufficient studies on their efficacy and safety and the lack of standard control treatments for COVID-19 (236). Additionally, the variability in COVID-19 severity among patients complicates the understanding of the disease.
Despite the promising potential of therapies targeting TLRs in addressing COVID-19 and its associated complications, it is imperative to recognize several limitations and potential adverse effects. A predominant concern pertains to the risk of off-target effects, wherein the therapeutic modulation of TLR signaling may unintentionally exacerbate systemic inflammation. For example, excessive activation of TLRs by agonists could enhance the production of pro-inflammatory cytokines, potentially resulting in cytokine storm-like conditions in susceptible patients. Conversely, the inhibition of TLRs may diminish critical antiviral defenses, thereby increasing the risk of secondary infections. Furthermore, these interventions might disrupt the delicate equilibrium between immune activation and regulation, culminating in immune dysregulation and possible autoimmune reactions. Prolonged suppression of TLR pathways could impair the immune system’s ability to resolve infections or inflammation, while overstimulation may contribute to persistent inflammatory signaling and autoimmune pathologies. Another substantial challenge resides in the variability of patient responses, influenced by factors such as genetic polymorphisms, comorbidities, and the severity of the disease. This variability complicates the formulation of standardized therapeutic approaches and underscores the necessity for personalized strategies. Finally, the absence of extensive clinical trials and regulatory approvals for TLR-targeted pharmaceuticals in COVID-19 treatment emphasizes the urgent need for further research to ascertain their safety, efficacy, and long-term effects in the management of this disease and its prolonged sequelae.
The COVID-19 pandemic has led to significant public health concerns regarding the long-term neurological impacts of the disease. This review examines the role of TLRs in mediating the immune response to SARS-CoV-2. The possible intricate interactions between TLRs and post-COVID-19 neurodegenerative processes highlight the crucial role these receptors play in long-term neurological consequences. The evidence demonstrates that TLRs are essential mediators of the immune response to SARS-CoV-2, contributing to both the immediate and long-term effects of infection. Specifically, certain TLRs have been identified as critical players in the inflammatory response, potentially exacerbating neuroinflammation and driving the development of neurodegenerative diseases, such as AD and PD, in individuals who have recovered from acute COVID-19. Furthermore, genetic polymorphisms in TLRs have been linked to variations in COVID-19 severity and the persistence of neurological symptoms. These findings suggest that individuals with specific genetic profiles may be more susceptible to severe outcomes and long-term complications, highlighting the need for personalized approaches in managing post-COVID-19 conditions.
Given the pivotal role of TLRs in post-COVID-19 neurodegenerative disorders, future research should focus on several key areas to improve understanding and therapeutic strategies. Developing specific inhibitors or modulators of TLR signaling pathways offers a promising therapeutic avenue, with potential drugs that selectively inhibit TLR activation from reducing chronic inflammation and preventing neurodegenerative disease progression in post-COVID-19 patients. Genetic screening for TLR polymorphisms could inform personalized medicine approaches, tailoring interventions based on individual risk profiles. Longitudinal studies are essential to understand the temporal relationship between TLR activation, neuroinflammation, and neurodegenerative symptom onset. Further mechanistic studies using advanced imaging, animal models, and in vitro systems are needed to elucidate how TLR signaling contributes to CNS pathology. Additionally, exploring immunomodulatory therapies that balance protective and pathological aspects of TLR activation could offer dual benefits in managing post-COVID-19 neurological outcomes. Integrating these findings into public health strategies is crucial for monitoring and managing neurological health in COVID-19 survivors, thereby addressing the long-term impact of the pandemic.
SS: Conceptualization, Writing – original draft, Writing – review & editing. TY: Writing – review & editing. ZH: Writing – review & editing. HZ: Writing – review & editing. DQ: Writing – review & editing. SL: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a grant from the Health@InnoHK Program to the Centre for Regenerative Medicine and Health. The authors were supported by the following grants: General Research Fund 17116018 from the Hong Kong Research Grant Council, and T11-712/19-N and T11-705/21-N from the RGC Theme-based Research Scheme.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. WHO. (2023). Who coronavirus (Covid-19) dashboard. World Healh Organisation. Availableonline at: https://covid19.who.int/ [Accessed 02 June 2023 2023].
2. Davis, HE, Mccorkell, L, Vogel, JM, and Topol, EJ. Long Covid: major findings, mechanisms and recommendations. Nat Rev Microbiol. (2023) 21:133–46. doi: 10.1038/s41579-022-00846-2
3. Mehandru, S, and Merad, M. Pathological sequelae of long-haul Covid. Nat Immunol. (2022) 23:194–202. doi: 10.1038/s41590-021-01104-y
4. Nalbandian, A, Sehgal, K, Gupta, A, Madhavan, MV, Mcgroder, C, Stevens, JS, et al. Post-acute Covid-19 syndrome. Nat Med. (2021) 27:601–15. doi: 10.1038/s41591-021-01283-z
5. Su, S, Zhao, Y, Zeng, N, Liu, X, Zheng, Y, Sun, J, et al. Epidemiology, clinical presentation, pathophysiology, and management of long Covid: an update. Mol Psychiatry. (2023) 28:4056–69. doi: 10.1038/s41380-023-02171-3
6. Kissler, SM, Hay, JA, Fauver, JR, Mack, C, Tai, CG, Anderson, DJ, et al. Viral kinetics of sequential Sars-CoV-2 infections. Nat Commun. (2023) 14:6206. doi: 10.1038/s41467-023-41941-z
7. Chilunga, FP, Appelman, B, Van Vugt, M, Kalverda, K, Smeele, P, Van Es, J, et al. Differences in incidence, nature of symptoms, and duration of long Covid among hospitalised migrant and non-migrant patients in the Netherlands: a retrospective cohort study. Lancet Regional Health –Europe. (2023) 29:100630. doi: 10.1016/j.lanepe.2023.100630
8. Ferini-Strambi, L, and Salsone, M. Covid-19 and neurological disorders: are neurodegenerative or neuroimmunological diseases more vulnerable? J Neurol. (2021) 268:409–19. doi: 10.1007/s00415-020-10070-8
9. Heneka, MT, Golenbock, D, Latz, E, Morgan, D, and Brown, R. Immediate and long-term consequences of Covid-19 infections for the development of neurological disease. Alzheimers Res Ther. (2020) 12:69. doi: 10.1186/s13195-020-00640-3
10. Taquet, M, Luciano, S, Geddes, JR, and Harrison, PJ. Bidirectional associations between Covid-19 and psychiatric disorder: retrospective cohort studies of 62 354 Covid-19 cases in the Usa. Lancet Psychiatry. (2021) 8:130–40. doi: 10.1016/S2215-0366(20)30462-4
11. Larenas-Linnemann, D, Rodríguez-Pérez, N, Arias-Cruz, A, Blandón-Vijil, MV, Del Río-Navarro, BE, Estrada-Cardona, A, et al. Enhancing innate immunity against virus in times of Covid-19: trying to untangle facts from fictions. World Allergy Organ J. (2020) 13:100476. doi: 10.1016/j.waojou.2020.100476
12. Baig, AM, Khaleeq, A, Ali, U, and Syeda, H. Evidence of the Covid-19 virus targeting the Cns: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. (2020) 11:995–8. doi: 10.1021/acschemneuro.0c00122
13. Ghaderi, S, Olfati, M, Ghaderi, M, Hadizadeh, H, Yazdanpanah, G, Khodadadi, Z, et al. Neurological manifestation in Covid-19 disease with neuroimaging studies. Am J Neurodegener Dis. (2023) 12:42–84.
14. Iacono, S, Schirò, G, Davì, C, Mastrilli, S, Abbott, M, Guajana, F, et al. Covid-19 and neurological disorders: what might connect Parkinson's disease to Sars-CoV-2 infection. Front Neurol. (2023) 14:1172416. doi: 10.3389/fneur.2023.1172416
15. Paterson, RW, Brown, RL, Benjamin, L, Nortley, R, Wiethoff, S, Bharucha, T, et al. The emerging spectrum of Covid-19 neurology: clinical, radiological and laboratory findings. Brain. (2020) 143:3104–20. doi: 10.1093/brain/awaa240
16. Fontes-Dantas, FL, Fernandes, GG, Gutman, EG, De Lima, EV, Antonio, LS, Hammerle, MB, et al. Sars-CoV-2 spike protein induces Tlr4-mediated long-term cognitive dysfunction recapitulating post-Covid-19 syndrome in mice. Cell Rep. (2023) 42:112189. doi: 10.1016/j.celrep.2023.112189
17. Li, L, Acioglu, C, Heary, RF, and Elkabes, S. Role of astroglial toll-like receptors (Tlrs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav Immun. (2021) 91:740–55. doi: 10.1016/j.bbi.2020.10.007
18. Naushad, SM, Mandadapu, G, Ramaiah, MJ, Almajhdi, FN, and Hussain, T. The role of Tlr7 agonists in modulating Covid-19 severity in subjects with loss-of-function Tlr7 variants. Sci Rep. (2023) 13:13078. doi: 10.1038/s41598-023-40114-8
19. Huang, C, Wang, Y, Li, X, Ren, L, Zhao, J, Hu, Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
20. Bark, L, Larsson, I-M, Wallin, E, Simrén, J, Zetterberg, H, Lipcsey, M, et al. Central nervous system biomarkers Gfap and NfL associate with post-acute cognitive impairment and fatigue following critical Covid-19. Sci Rep. (2023) 13:13144. doi: 10.1038/s41598-023-39698-y
21. Mao, L, Jin, H, Wang, M, Hu, Y, Chen, S, He, Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. (2020) 77:683–90. doi: 10.1001/jamaneurol.2020.1127
22. Etter, MM, Martins, TA, Kulsvehagen, L, Pössnecker, E, Duchemin, W, Hogan, S, et al. Severe neuro-Covid is associated with peripheral immune signatures, autoimmunity and neurodegeneration: a prospective cross-sectional study. Nat Commun. (2022) 13:6777. doi: 10.1038/s41467-022-34068-0
23. Heming, M, Li, X, Räuber, S, Mausberg, AK, Börsch, AL, Hartlehnert, M, et al. Neurological manifestations of Covid-19 feature T cell exhaustion and dedifferentiated monocytes in cerebrospinal fluid. Immunity. (2021) 54:164–175.e6. doi: 10.1016/j.immuni.2020.12.011
24. Da Silva, SJR, Do Nascimento, JCF, Germano Mendes, RP, Guarines, KM, Targino Alves Da Silva, C, Da Silva, PG, et al. Two years into the Covid-19 pandemic: lessons learned. Acs Infectious Dis. (2022) 8:1758–814. doi: 10.1021/acsinfecdis.2c00204
25. Jackson, CB, Farzan, M, Chen, B, and Choe, H. Mechanisms of Sars-CoV-2 entry into cells. Nat Rev Mol Cell Biol. (2022) 23:3–20. doi: 10.1038/s41580-021-00418-x
26. Hoffmann, D. The role of the oral cavity in Sars-CoV-2- and other viral infections. Clin Oral Investig. (2023) 27:15–22. doi: 10.1007/s00784-023-05078-z
27. Behboudi, E, Nooreddin Faraji, S, Daryabor, G, Mohammad Ali Hashemi, S, Asadi, M, Edalat, F, et al. Sars-CoV-2 mechanisms of cell tropism in various organs considering host factors. Heliyon. (2024) 10:e26577. doi: 10.1016/j.heliyon.2024.e26577
28. Hoffmann, M, Kleine-Weber, H, Schroeder, S, Krüger, N, Herrler, T, Erichsen, S, et al. Sars-CoV-2 cell entry depends on Ace2 and Tmprss2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–280.e8. doi: 10.1016/j.cell.2020.02.052
29. Wruck, W, and Adjaye, J. Sars-CoV-2 receptor Ace2 is co-expressed with genes related to transmembrane serine proteases, viral entry, immunity and cellular stress. Sci Rep. (2020) 10:21415. doi: 10.1038/s41598-020-78402-2
30. De Melo, GD, Perraud, V, Alvarez, F, Vieites-Prado, A, Kim, S, Kergoat, L, et al. Neuroinvasion and anosmia are independent phenomena upon infection with Sars-CoV-2 and its variants. Nat Commun. (2023) 14:4485. doi: 10.1038/s41467-023-40228-7
31. Butowt, R, Meunier, N, Bryche, B, and Von Bartheld, CS. The olfactory nerve is not a likely route to brain infection in Covid-19: a critical review of data from humans and animal models. Acta Neuropathol. (2021) 141:809–22. doi: 10.1007/s00401-021-02314-2
32. Bilinska, K, Von Bartheld, CS, and Butowt, R. Expression of the Ace2 virus entry protein in the Nervus terminalis reveals the potential for an alternative route to brain infection in Covid-19. Front Cell Neurosci. (2021) 15:674123. doi: 10.3389/fncel.2021.674123
33. Douaud, G, Lee, S, Alfaro-Almagro, F, Arthofer, C, Wang, C, Mccarthy, P, et al. Sars-CoV-2 is associated with changes in brain structure in Uk biobank. Nature. (2022) 604:697–707. doi: 10.1038/s41586-022-04569-5
34. Serrano, GE, Walker, JE, Arce, R, Glass, MJ, Vargas, D, Sue, LI, et al. Mapping of Sars-CoV-2 brain invasion and histopathology in Covid-19 disease. medRxiv. (2021). doi: 10.1101/2021.02.15.21251511
35. Verma, AK, Zheng, J, Meyerholz, DK, and Perlman, S. Sars-CoV-2 infection of sustentacular cells disrupts olfactory signaling pathways. JCI Insight. (2022) 7. doi: 10.1172/jci.insight.160277
36. Wellford, SA, and Moseman, EA. Olfactory immune response to Sars-CoV-2. Cell Mol Immunol. (2024) 21:134–43. doi: 10.1038/s41423-023-01119-5
37. Jiao, L, Yang, Y, Yu, W, Zhao, Y, Long, H, Gao, J, et al. The olfactory route is a potential way for Sars-CoV-2 to invade the central nervous system of rhesus monkeys. Signal Transduct Target Ther. (2021) 6:169. doi: 10.1038/s41392-021-00591-7
38. Zhang, Y, Chen, X, Jia, L, and Zhang, Y. Potential mechanism of Sars-CoV-2-associated central and peripheral nervous system impairment. Acta Neurol Scand. (2022) 146:225–36. doi: 10.1111/ane.13657
39. Meinhardt, J, Radke, J, Dittmayer, C, Franz, J, Thomas, C, Mothes, R, et al. Olfactory transmucosal Sars-CoV-2 invasion as a port of central nervous system entry in individuals with Covid-19. Nat Neurosci. (2021) 24:168–75. doi: 10.1038/s41593-020-00758-5
40. Tanzadehpanah, H, Lotfian, E, Avan, A, Saki, S, Nobari, S, Mahmoodian, R, et al. Role of Sars-Cov-2 and Ace2 in the pathophysiology of peripheral vascular diseases. Biomed Pharmacother. (2023) 166:115321. doi: 10.1016/j.biopha.2023.115321
41. Wu, C-T, Lidsky, PV, Xiao, Y, Cheng, R, Lee, IT, Nakayama, T, et al. Sars-CoV-2 replication in airway epithelia requires motile cilia and microvillar reprogramming. Cell. (2023) 186:112–130.e20. doi: 10.1016/j.cell.2022.11.030
42. Haidar, MA, Shakkour, Z, Reslan, MA, Al-Haj, N, Chamoun, P, Habashy, K, et al. Sars-CoV-2 involvement in central nervous system tissue damage. Neural Regen Res. (2022) 17:1228–39. doi: 10.4103/1673-5374.327323
43. Wan, D, Du, T, Hong, W, Chen, L, Que, H, Lu, S, et al. Neurological complications and infection mechanism of Sars-Cov-2. Signal Transduct Target Ther. (2021) 6:406. doi: 10.1038/s41392-021-00818-7
44. Chen, Y, Yang, W, Chen, F, and Cui, L. COVID-19 and cognitive impairment: neuroinvasive and blood–brain barrier dysfunction. J Neuroinflammation. (2022) 19:222. doi: 10.1186/s12974-022-02579-8
45. Erickson, MA, Rhea, EM, Knopp, RC, and Banks, WA. Interactions of SARS-CoV-2 with the blood–brain barrier. Int J Mol Sci. (2021) 22:2681. doi: 10.3390/ijms22052681
46. Greene, C, Connolly, R, Brennan, D, Laffan, A, Okeeffe, E, Zaporojan, L, et al. Blood–brain barrier disruption and sustained systemic inflammation in individuals with long Covid-associated cognitive impairment. Nat Neurosci. (2024) 27:421–32. doi: 10.1038/s41593-024-01576-9
47. Zhang, L, Zhou, L, Bao, L, Liu, J, Zhu, H, Lv, Q, et al. Sars-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther. (2021) 6:337. doi: 10.1038/s41392-021-00719-9
48. Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell Mol Immunol. (2021) 18:2489–501. doi: 10.1038/s41423-021-00757-x
49. Wang, K, Chen, W, Zhang, Z, Deng, Y, Lian, JQ, Du, P, et al. Cd147-spike protein is a novel route for Sars-CoV-2 infection to host cells. Signal Transduct Target Ther. (2020) 5:283. doi: 10.1038/s41392-020-00426-x
50. Shilts, J, Crozier, TWM, Greenwood, EJD, Lehner, PJ, and Wright, GJ. No evidence for basigin/Cd147 as a direct Sars-CoV-2 spike binding receptor. Sci Rep. (2021) 11:413. doi: 10.1038/s41598-020-80464-1
51. Albornoz, EA, Amarilla, AA, Modhiran, N, Parker, S, Li, XX, Wijesundara, DK, et al. Sars-CoV-2 drives Nlrp3 inflammasome activation in human microglia through spike protein. Mol Psychiatry. (2023) 28:2878–93. doi: 10.1038/s41380-022-01831-0
52. Josephson, SA, and Kamel, H. Neurology and Covid-19. JAMA. (2020) 324:1139–40. doi: 10.1001/jama.2020.14254
53. Lima, M, Siokas, V, Aloizou, AM, Liampas, I, Mentis, AA, Tsouris, Z, et al. Unraveling the possible routes of Sars-Cov-2 invasion into the central nervous system. Curr Treat Options Neurol. (2020) 22:37. doi: 10.1007/s11940-020-00647-z
54. Zhu, Z, Lian, X, Su, X, Wu, W, Marraro, GA, and Zeng, Y. From Sars and Mers to Covid-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir Res. (2020) 21:224. doi: 10.1186/s12931-020-01479-w
55. Vitale-Cross, L, Szalayova, I, Scoggins, A, Palkovits, M, and Mezey, E. Sars-CoV-2 entry sites are present in all structural elements of the human glossopharyngeal and vagal nerves: clinical implications. EBioMedicine. (2022) 78:103981. doi: 10.1016/j.ebiom.2022.103981
56. Zhan, WR, Huang, J, Zeng, PM, Tian, WY, and Luo, ZG. Emerging neurotropic features of Sars-CoV-2. J Mol Cell Biol. (2021) 13:705–11. doi: 10.1093/jmcb/mjab044
57. Merad, M, Blish, CA, Sallusto, F, and Iwasaki, A. The immunology and immunopathology of Covid-19. Science. (2022) 375:1122–7. doi: 10.1126/science.abm8108
58. Mohandas, S, Jagannathan, P, Henrich, TJ, Sherif, ZA, Bime, C, Quinlan, E, et al. Immune mechanisms underlying Covid-19 pathology and post-acute sequelae of Sars-CoV-2 infection (Pasc). eLife. (2023) 12:e86014. doi: 10.7554/eLife.86014
59. Peluso, MJ, Abdel-Mohsen, M, Henrich, TJ, and Roan, NR. Systems analysis of innate and adaptive immunity in Long Covid. Semin Immunol. (2024) 72:101873. doi: 10.1016/j.smim.2024.101873
60. Parotto, M, Gyöngyösi, M, Howe, K, Myatra, SN, Ranzani, O, Shankar-Hari, M, et al. Post-acute sequelae of Covid-19: understanding and addressing the burden of multisystem manifestations. Lancet Respir Med. (2023) 11:739–54. doi: 10.1016/S2213-2600(23)00239-4
61. Pezzini, A, and Padovani, A. Lifting the mask on neurological manifestations of Covid-19. Nat Rev Neurol. (2020) 16:636–44. doi: 10.1038/s41582-020-0398-3
62. Reiss, AB, Greene, C, Dayaramani, C, Rauchman, SH, Stecker, MM, De Leon, J, et al. Long Covid, the brain, nerves, and cognitive function. Neurol Int. (2023) 15:821–41. doi: 10.3390/neurolint15030052
63. Burks, SM, Rosas-Hernandez, H, Alejandro Ramirez-Lee, M, Cuevas, E, and Talpos, JC. Can Sars-CoV-2 infect the central nervous system via the olfactory bulb or the blood-brain barrier? Brain Behav Immun. (2021) 95:7–14. doi: 10.1016/j.bbi.2020.12.031
64. Kasuga, Y, Zhu, B, Jang, K-J, and Yoo, J-S. Innate immune sensing of coronavirus and viral evasion strategies. Exp Mol Med. (2021) 53:723–36. doi: 10.1038/s12276-021-00602-1
65. Li, D, and Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. (2021) 6:291. doi: 10.1038/s41392-021-00687-0
66. Thompson, MR, Kaminski, JJ, Kurt-Jones, EA, and Fitzgerald, KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. (2011) 3:920–40. doi: 10.3390/v3060920
67. Yamada, T, and Takaoka, A. Innate immune recognition against Sars-CoV-2. Inflamm Regen. (2023) 43:7. doi: 10.1186/s41232-023-00259-5
68. Fajgenbaum David, C, and June Carl, H. Cytokine Storm. N Engl J Med. (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
69. Rabaan, AA, Al-Ahmed, SH, Muhammad, J, Khan, A, Sule, AA, Tirupathi, R, et al. Role of inflammatory cytokines in Covid-19 patients: A review on molecular mechanisms, immune functions, Immunopathology and Immunomodulatory Drugs to Counter Cytokine Storm. Vaccines (Basel). (2021) 9:9. doi: 10.3390/vaccines9050436
70. Tay, MZ, Poh, CM, Rénia, L, Macary, PA, and Ng, LFP. The trinity of Covid-19: immunity, inflammation and intervention. Nat Rev Immunol. (2020) 20:363–74. doi: 10.1038/s41577-020-0311-8
71. Dey, R, and Bishayi, B. Microglial inflammatory responses to Sars‐CoV‐2 infection: a comprehensive review. Cell Mol Neurobiol. (2023) 44:2. doi: 10.1007/s10571-023-01444-3
72. Vanderheiden, A, and Klein, RS. Neuroinflammation and Covid-19. Curr Opin Neurobiol. (2022) 76:102608. doi: 10.1016/j.conb.2022.102608
73. Queiroz, MAF, Brito, W, Pereira, KAS, Pereira, LMS, Amoras, E, Lima, SS, et al. Severe Covid-19 and long Covid are associated with high expression of Sting, cgas and Ifn-α. Sci Rep. (2024) 14:4974. doi: 10.1038/s41598-024-55696-0
74. Natale, NR, Lukens, JR, and Petri, WA Jr. The nervous system during Covid-19: caught in the crossfire. Immunol Rev. (2022) 311:90–111. doi: 10.1111/imr.13114
75. Visvabharathy, L, Hanson, BA, Orban, ZS, Lim, PH, Palacio, NM, Jimenez, M, et al. Neuro-Pasc is characterized by enhanced Cd4+ and diminished Cd8+ T cell responses to Sars-CoV-2 Nucleocapsid protein. Front Immunol. (2023) 14:1155770. doi: 10.3389/fimmu.2023.1155770
76. Ong, IZ, Kolson, DL, and Schindler, MK. Mechanisms, effects, and Management of Neurological Complications of post-acute sequelae of Covid-19 (Nc-Pasc). Biomedicine. (2023) 11:377. doi: 10.3390/biomedicines11020377
77. Chen, S, Guan, F, Candotti, F, Benlagha, K, Camara, NOS, Herrada, AA, et al. The role of B cells in Covid-19 infection and vaccination. Front Immunol. (2022) 13:988536. doi: 10.3389/fimmu.2022.988536
78. Vahabi, M, Ghazanfari, T, and Sepehrnia, S. Molecular mimicry, hyperactive immune system, and Sars-Cov-2 are three prerequisites of the autoimmune disease triangle following Covid-19 infection. Int Immunopharmacol. (2022) 112:109183. doi: 10.1016/j.intimp.2022.109183
79. Zhao, J, Xia, F, Jiao, X, and Lyu, X. Long Covid and its association with neurodegenerative diseases: pathogenesis, neuroimaging, and treatment. Front Neurol. (2024) 15:1367974. doi: 10.3389/fneur.2024.1367974
80. Walker, KA, Le Page, LM, Terrando, N, Duggan, MR, Heneka, MT, and Bettcher, BM. The role of peripheral inflammatory insults in Alzheimer's disease: a review and research roadmap. Mol Neurodegener. (2023) 18:37. doi: 10.1186/s13024-023-00627-2
81. Amadoro, G, Latina, V, Stigliano, E, and Micera, A. Covid-19 and Alzheimer's disease share common neurological and ophthalmological manifestations: a bidirectional risk in the post-pandemic future. Cells. (2023) 12:2601. doi: 10.3390/cells12222601
82. Maury, A, Lyoubi, A, Peiffer-Smadja, N, De Broucker, T, and Meppiel, E. Neurological manifestations associated with Sars-CoV-2 and other coronaviruses: a narrative review for clinicians. Rev Neurol (Paris). (2021) 177:51–64. doi: 10.1016/j.neurol.2020.10.001
83. Dewanjee, S, Vallamkondu, J, Kalra, RS, Puvvada, N, Kandimalla, R, and Reddy, PH. Emerging Covid-19 neurological manifestations: present outlook and potential neurological challenges in Covid-19 pandemic. Mol Neurobiol. (2021) 58:4694–715. doi: 10.1007/s12035-021-02450-6
84. Ellul, MA, Benjamin, L, Singh, B, Lant, S, Michael, BD, Easton, A, et al. Neurological associations of Covid-19. Lancet Neurol. (2020) 19:767–83. doi: 10.1016/S1474-4422(20)30221-0
85. Islam, MA, Cavestro, C, Alam, SS, Kundu, S, Kamal, MA, and Reza, F. Encephalitis in patients with Covid-19: a systematic evidence-based analysis. Cells. (2022) 11:2575. doi: 10.3390/cells11162575
86. Sharma, R, Nalleballe, K, Shah, V, Haldal, S, Spradley, T, Hasan, L, et al. Spectrum of hemorrhagic encephalitis in Covid-19 patients: a case series and review. Diagnostics (Basel). (2022) 12:924. doi: 10.3390/diagnostics12040924
87. Assi, F, Abdallah, R, Mecheik, A, Rahhal, HH, and Wazne, J. Acute disseminated encephalomyelitis following Covid-19 infection. Cureus. (2023) 15:e33365. doi: 10.7759/cureus.33365
88. Stoian, A, Bajko, Z, Stoian, M, Cioflinc, RA, Niculescu, R, ArbănaȘI, EM, et al. The occurrence of acute disseminated encephalomyelitis in Sars-CoV-2 infection/vaccination: our experience and a systematic review of the literature. Vaccines (Basel). (2023) 11:1225. doi: 10.3390/vaccines11071225
89. El Mawla, Z, El Saddik, G, Zeineddine, M, Hassoun, M, and El Hajj, T. Cerebrovascular disease in patients with Covid-19 infection: a case series from Lebanon. Ann Med Surg (Lond). (2023) 85:3701–8. doi: 10.1097/MS9.0000000000000953
90. Wang, Z, Yang, Y, Liang, X, Gao, B, Liu, M, Li, W, et al. Covid-19 associated ischemic stroke and hemorrhagic stroke: incidence, potential pathological mechanism, and management. Front Neurol. (2020) 11:571996. doi: 10.3389/fneur.2020.571996
91. Terzioglu, G, and Young-Pearse, TL. Microglial function, Inpp5D/Ship1 signaling, and Nlrp3 inflammasome activation: implications for Alzheimer’s disease. Mol Neurodegener. (2023) 18:89. doi: 10.1186/s13024-023-00674-9
92. Manji, HK, George, U, Mkopi, NP, and Manji, KP. Guillain-Barré syndrome associated with Covid-19 infection. Pan Afr Med J. (2020) 35:118. doi: 10.11604/pamj.supp.2020.35.2.25003
93. Liang, F, and Wang, Y. Covid-19 anosmia: high prevalence, plural Neuropathogenic mechanisms, and scarce Neurotropism of Sars-CoV-2? Viruses. (2021) 13:2225. doi: 10.3390/v13112225
94. Xydakis, MS, Albers, MW, Holbrook, EH, Lyon, DM, Shih, RY, Frasnelli, JA, et al. Post-viral effects of Covid-19 in the olfactory system and their implications. Lancet Neurol. (2021) 20:753–61. doi: 10.1016/S1474-4422(21)00182-4
95. Gold, DM, and Galetta, SL. Neuro-ophthalmologic complications of coronavirus disease 2019 (Covid-19). Neurosci Lett. (2021) 742:135531. doi: 10.1016/j.neulet.2020.135531
96. Kang, K, Lee, SY, and Lee, DC. Neuro-ophthalmologic symptoms after coronavirus disease 2019 vaccination: a retrospective study. BMC Ophthalmol. (2023) 23:11. doi: 10.1186/s12886-022-02747-7
97. Bawor, M, Sairam, S, Rozewicz, R, Viegas, S, Comninos, AN, and Abbara, A. Rhabdomyolysis after Covid-19 infection: a case report and review of the literature. Viruses. (2022) 14:2255. doi: 10.3390/v14102255
98. Daria, S, Katarzyna, S-A, Jerzy, F, and Ivan, K. Muscle pain and muscle weakness in Covid19 patients: Cross-talk with statins – preliminary results. Biomed Pharmacother. (2022) 148:112757. doi: 10.1016/j.biopha.2022.112757
99. Hu, J, Huang, L, Qiu, Z, Liu, Y, Shen, K, Tang, B, et al. Case report: a novel case of Covid-19 triggered tumefactive demyelinating lesions in one multiple sclerosis patient. Front Neurosci. (2023) 17:1287480. doi: 10.3389/fnins.2023.1287480
100. Lotan, I, Nishiyama, S, Manzano, GS, Lydston, M, and Levy, M. Covid-19 and the risk of Cns demyelinating diseases: a systematic review. Front Neurol. (2022) 13:970383. doi: 10.3389/fneur.2022.970383
101. Chasco, EE, Dukes, K, Jones, D, Comellas, AP, Hoffman, RM, and Garg, A. Brain fog and fatigue following Covid-19 infection: an exploratory study of patient experiences of Long Covid. Int J Environ Res Public Health. (2022) 19:15499. doi: 10.3390/ijerph192315499
102. Jiao, T, Huang, Y, Sun, H, and Yang, L. Research progress of post-acute sequelae after Sars-CoV-2 infection. Cell Death Dis. (2024) 15:257. doi: 10.1038/s41419-024-06642-5
103. Duan, T, Du, Y, Xing, C, Wang, HY, and Wang, R-F. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol. (2022) 13:812774. doi: 10.3389/fimmu.2022.812774
104. Nie, L, Cai, SY, Shao, JZ, and Chen, J. Toll-like receptors, associated biological roles, and signaling networks in non-mammals. Front Immunol. (2018) 9:1523. doi: 10.3389/fimmu.2018.01523
105. Kawai, T, and Akira, S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
106. Sameer, AS, and Nissar, S. Toll-like receptors (Tlrs): structure, functions, signaling, and role of their polymorphisms in colorectal Cancer susceptibility. Biomed Res Int. (2021) 2021:1157023. doi: 10.1155/2021/1157023
107. Kim, H-J, Kim, H, Lee, J-H, and Hwangbo, C. Toll-like receptor 4 (Tlr4): new insight immune and aging. Immun Ageing. (2023) 20:67. doi: 10.1186/s12979-023-00383-3
108. Fore, F, Indriputri, C, Mamutse, J, and Nugraha, J. Tlr10 and its unique anti-inflammatory properties and potential use as a target in therapeutics. Immune Netw. (2020) 20:e21. doi: 10.4110/in.2020.20.e21
109. Kawasaki, T, and Kawai, T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
110. Gern, OL, Mulenge, F, Pavlou, A, Ghita, L, Steffen, I, Stangel, M, et al. Toll-like receptors in viral encephalitis. Viruses. (2021) 13:2065. doi: 10.3390/v13102065
111. Mogensen, TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
112. Wang, X, Yuan, T, Yuan, J, Su, Y, Sun, X, Wu, J, et al. Expression of toll-like receptor 2 by dendritic cells is essential for the DnaJ-ΔA146Ply-mediated Th1 immune response against Streptococcus pneumoniae. Infect Immun. (2018) 86:e00651-17. doi: 10.1128/IAI.00651-17
113. Liu, J, Zhang, X, Cheng, Y, and Cao, X. Dendritic cell migration in inflammation and immunity. Cell Mol Immunol. (2021) 18:2461–71. doi: 10.1038/s41423-021-00726-4
114. Wang, X, Guan, F, Miller, H, Byazrova, MG, Cndotti, F, Benlagha, K, et al. The role of dendritic cells in Covid-19 infection. Emerg Microbes Infect. (2023) 12:2195019. doi: 10.1080/22221751.2023.2195019
115. Sanjuan, MA, Dillon, CP, Tait, SWG, Moshiach, S, Dorsey, F, Connell, S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. (2007) 450:1253–7. doi: 10.1038/nature06421
116. Ni, K, Luo, T, Culbert, A, Kaufmann, M, Jiang, X, and Lin, W. Correction to “nanoscale metal–organic framework co-delivers TLR-7 agonists and anti-CD47 antibodies to modulate macrophages and orchestrate Cancer immunotherapy”. J Am Chem Soc. (2022) 144:15907–7. doi: 10.1021/jacs.2c07990
117. Vidyarthi, A, Khan, N, Agnihotri, T, Negi, S, Das, DK, Aqdas, M, et al. Tlr-3 stimulation skews M2 macrophages to M1 through Ifn-αβ signaling and restricts tumor progression. Front Immunol. (2018) 9:1650. doi: 10.3389/fimmu.2018.01650
118. Geng, J, Shi, Y, Zhang, J, Yang, B, Wang, P, Yuan, W, et al. Tlr4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nat Commun. (2021) 12:3519. doi: 10.1038/s41467-021-23683-y
119. Veneziani, I, Alicata, C, Moretta, L, and Maggi, E. Human toll-like receptor 8 (Tlr8) in Nk cells: implication for cancer immunotherapy. Immunol Lett. (2023) 261:13–6. doi: 10.1016/j.imlet.2023.07.003
120. Noh, JY, Yoon, SR, Kim, TD, Choi, I, and Jung, H. Toll-like receptors in natural killer cells and their application for immunotherapy. J Immunol Res. (2020) 2020:2045860. doi: 10.1155/2020/2045860
121. Akira, S, Takeda, K, and Kaisho, T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. (2001) 2:675–80. doi: 10.1038/90609
122. Vijay, K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol. (2018) 59:391–412. doi: 10.1016/j.intimp.2018.03.002
123. Kircheis, R, and Planz, O. The role of toll-like receptors (Tlrs) and their related signaling pathways in viral infection and inflammation. Int J Mol Sci. (2023) 24:6701. doi: 10.3390/ijms24076701
124. Ma, M, Jiang, W, and Zhou, R. Damps and damp-sensing receptors in inflammation and diseases. Immunity. (2024) 57:752–71. doi: 10.1016/j.immuni.2024.03.002
125. Carson, MJ, Doose, JM, Melchior, B, Schmid, CD, and Ploix, CC. Cns immune privilege: hiding in plain sight. Immunol Rev. (2006) 213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x
126. Lampron, A, Elali, A, and Rivest, S. Innate immunity in the Cns: redefining the relationship between the Cns and its environment. Neuron. (2013) 78:214–32. doi: 10.1016/j.neuron.2013.04.005
127. Facci, L, Barbierato, M, Marinelli, C, Argentini, C, Skaper, SD, and Giusti, P. Toll-like receptors 2, −3 and −4 prime microglia but not astrocytes across central nervous system regions for Atp-dependent interleukin-1β release. Sci Rep. (2014) 4:6824. doi: 10.1038/srep06824
128. Fiebich, BL, Batista, CRA, Saliba, SW, Yousif, NM, and De Oliveira, ACP. Role of microglia Tlrs in neurodegeneration. Front Cell Neurosci. (2018) 12:329. doi: 10.3389/fncel.2018.00329
129. Hanke, ML, and Kielian, T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond). (2011) 121:367–87. doi: 10.1042/CS20110164
130. Gao, C, Jiang, J, Tan, Y, and Chen, S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct Target Ther. (2023) 8:359. doi: 10.1038/s41392-023-01588-0
131. Muzio, L, Viotti, A, and Martino, G. Microglia in Neuroinflammation and neurodegeneration: from understanding to therapy. Front Neurosci. (2021) 15:742065. doi: 10.3389/fnins.2021.742065
132. Ding, X, Jin, S, Tong, Y, Jiang, X, Chen, Z, Mei, S, et al. Tlr4 signaling induces Tlr3 up-regulation in alveolar macrophages during acute lung injury. Sci Rep. (2017) 7:34278. doi: 10.1038/srep34278
133. Nilsen, N, Nonstad, U, Khan, N, Knetter, CF, Akira, S, Sundan, A, et al. Lipopolysaccharide and double-stranded Rna up-regulate toll-like receptor 2 independently of myeloid differentiation factor 88*. J Biol Chem. (2004) 279:39727–35. doi: 10.1074/jbc.M405027200
134. Hanamsagar, R, Hanke, ML, and Kielian, T. Toll-like receptor (Tlr) and inflammasome actions in the central nervous system. Trends Immunol. (2012) 33:333–42. doi: 10.1016/j.it.2012.03.001
135. Yang, J, Wise, L, and Fukuchi, KI. Tlr4 Cross-talk with Nlrp3 Inflammasome and complement signaling pathways in Alzheimer's disease. Front Immunol. (2020) 11:724. doi: 10.3389/fimmu.2020.00724
136. Kelley, N, Jeltema, D, Duan, Y, and He, Y. The Nlrp3 Inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
137. Yang, Y, Wang, H, Kouadir, M, Song, H, and Shi, F. Recent advances in the mechanisms of Nlrp3 inflammasome activation and its inhibitors. Cell Death Dis. (2019) 10:128. doi: 10.1038/s41419-019-1413-8
138. Xu, Y, Jin, MZ, Yang, ZY, and Jin, WL. Microglia in neurodegenerative diseases. Neural Regen Res. (2021) 16:270–80. doi: 10.4103/1673-5374.290881
139. Zhang, W, Xiao, D, Mao, Q, and Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. (2023) 8:267. doi: 10.1038/s41392-023-01486-5
140. Gustin, A, Kirchmeyer, M, Koncina, E, Felten, P, Losciuto, S, Heurtaux, T, et al. Nlrp3 Inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One. (2015) 10:e0130624. doi: 10.1371/journal.pone.0130624
141. Dai, J, Wang, Y, Wang, H, Gao, Z, Wang, Y, Fang, M, et al. Toll-like receptor signaling in severe acute respiratory syndrome coronavirus 2-induced innate immune responses and the potential application value of toll-like receptor Immunomodulators in patients with coronavirus disease 2019. Front Microbiol. (2022) 13:948770. doi: 10.3389/fmicb.2022.948770
142. Adamu, A, Li, S, Gao, F, and Xue, G. The role of neuroinflammation in neurodegenerative diseases: current understanding and future therapeutic targets. Front Aging Neurosci. (2024) 16:1347987. doi: 10.3389/fnagi.2024.1347987
143. Dabi, YT, Ajagbe, AO, and Degechisa, ST. Toll-like receptors in pathogenesis of neurodegenerative diseases and their therapeutic potential. Immun Inflamm Dis. (2023) 11:e839. doi: 10.1002/iid3.839
144. Hanisch, U-K, Johnson, TV, and Kipnis, J. Toll-like receptors: roles in neuroprotection? Trends Neurosci. (2008) 31:176–82. doi: 10.1016/j.tins.2008.01.005
145. Ifuku, M, Hinkelmann, L, Kuhrt, LD, Efe, IE, Kumbol, V, Buonfiglioli, A, et al. Activation of toll-like receptor 5 in microglia modulates their function and triggers neuronal injury. Acta Neuropathol Commun. (2020) 8:159. doi: 10.1186/s40478-020-01031-3
146. Salminen, A, Suuronen, T, and Kaarniranta, K. Rock, Pak, and toll of synapses in Alzheimer's disease. Biochem Biophys Res Commun. (2008) 371:587–90. doi: 10.1016/j.bbrc.2008.04.148
147. Momtazmanesh, S, Perry, G, and Rezaei, N. Toll-like receptors in Alzheimer's disease. J Neuroimmunol. (2020) 348:577362. doi: 10.1016/j.jneuroim.2020.577362
148. Calvo-Rodriguez, M, García-Rodríguez, C, Villalobos, C, and Núñez, L. Role of toll like receptor 4 in Alzheimer’s disease. Front Immunol. (2020) 11:1588. doi: 10.3389/fimmu.2020.01588
149. Wei, J, Zhang, Y, Li, H, Wang, F, and Yao, S. Toll-like receptor 4: a potential therapeutic target for multiple human diseases. Biomed Pharmacother. (2023) 166:115338. doi: 10.1016/j.biopha.2023.115338
150. Zhou, Y, Chen, Y, Xu, C, Zhang, H, and Lin, C. Tlr4 targeting as a promising therapeutic strategy for Alzheimer disease treatment. Front Neurosci. (2020) 14:602508. doi: 10.3389/fnins.2020.602508
151. Ciesielska, A, Matyjek, M, and Kwiatkowska, K. Tlr4 and Cd14 trafficking and its influence on Lps-induced pro-inflammatory signaling. Cell Mol Life Sci. (2021) 78:1233–61. doi: 10.1007/s00018-020-03656-y
152. Minter, MR, Taylor, JM, and Crack, PJ. The contribution of neuroinflammation to amyloid toxicity in Alzheimer's disease. J Neurochem. (2016) 136:457–74. doi: 10.1111/jnc.13411
153. Zhang, W, Wang, L-Z, Yu, J-T, Chi, Z-F, and Tan, L. Increased expressions of Tlr2 and Tlr4 on peripheral blood mononuclear cells from patients with Alzheimer's disease. J Neurol Sci. (2012) 315:67–71. doi: 10.1016/j.jns.2011.11.032
154. Balistreri, CR, Grimaldi, MP, Chiappelli, M, Licastro, F, Castiglia, L, Listì, F, et al. Association between the polymorphisms of Tlr4 and Cd14 genes and Alzheimer's disease. Curr Pharm Des. (2008) 14:2672–7. doi: 10.2174/138161208786264089
155. Minoretti, P, Gazzaruso, C, Vito, CD, Emanuele, E, Bianchi, M, Coen, E, et al. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer's disease. Neurosci Lett. (2006) 391:147–9. doi: 10.1016/j.neulet.2005.08.047
156. Miron, J, Picard, C, Frappier, J, Dea, D, Théroux, L, and Poirier, J. Tlr4 gene expression and pro-inflammatory cytokines in Alzheimer’s disease and in response to hippocampal Deafferentation in rodents. J Alzheimers Dis. (2018) 63:1547–56. doi: 10.3233/JAD-171160
157. Tang, S-C, Lathia, JD, Selvaraj, PK, Jo, D-G, Mughal, MR, Cheng, A, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid β-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol. (2008) 213:114–21. doi: 10.1016/j.expneurol.2008.05.014
158. Wang, Y, He, H, Li, D, Zhu, W, Duan, K, Le, Y, et al. The role of the Tlr4 signaling pathway in cognitive deficits following surgery in aged rats. Mol Med Rep. (2013) 7:1137–42. doi: 10.3892/mmr.2013.1322
159. Wu, L, Xian, X, Xu, G, Tan, Z, Dong, F, Zhang, M, et al. Toll-like receptor 4: a promising therapeutic target for Alzheimer's disease. Mediat Inflamm. (2022) 2022:7924199. doi: 10.1155/2022/7924199
160. Wang, YL, Tan, MS, Yu, JT, Zhang, W, Hu, N, Wang, HF, et al. Toll-like receptor 9 promoter polymorphism is associated with decreased risk of Alzheimer's disease in Han Chinese. J Neuroinflammation. (2013) 10:101. doi: 10.1186/1742-2094-10-101
161. Cacace, R, Zhou, L, Hendrickx Van De Craen, E, Buist, A, Hoogmartens, J, Sieben, A, et al. Mutated toll-like receptor 9 increases Alzheimer’s disease risk by compromising innate immunity protection. Mol Psychiatry. (2023) 28:5380–9. doi: 10.1038/s41380-023-02166-0
162. Rangasamy, SB, Jana, M, Roy, A, Corbett, GT, Kundu, M, Chandra, S, et al. Selective disruption of Tlr2-MyD88 interaction inhibits inflammation and attenuates Alzheimer's pathology. J Clin Invest. (2018) 128:4297–312. doi: 10.1172/JCI96209
163. Chakrabarty, P, Li, A, Ladd, TB, Strickland, MR, Koller, EJ, Burgess, JD, et al. Tlr5 decoy receptor as a novel anti-amyloid therapeutic for Alzheimer's disease. J Exp Med. (2018) 215:2247–64. doi: 10.1084/jem.20180484
164. Doorn, KJ, Moors, T, Drukarch, B, Van De Berg, W, Lucassen, PJ, and Van Dam, AM. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun. (2014) 2:90. doi: 10.1186/s40478-014-0090-1
165. Xia, Y, Zhang, G, Kou, L, Yin, S, Han, C, Hu, J, et al. Reactive microglia enhance the transmission of exosomal α-synuclein via toll-like receptor 2. Brain. (2021) 144:2024–37. doi: 10.1093/brain/awab122
166. Dzamko, N, Gysbers, A, Perera, G, Bahar, A, Shankar, A, Gao, J, et al. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. (2017) 133:303–19. doi: 10.1007/s00401-016-1648-8
167. Da Silva, DJ, Borges, AF, Souza, PO, Reis De Souza, P, De Barros, R, Cardoso, C, et al. Decreased toll-like receptor 2 and toll-like receptor 7/8-induced cytokines in Parkinson's disease patients. Neuroimmunomodulation. (2016) 23:58–66. doi: 10.1159/000443238
168. Drouin-Ouellet, J, St-Amour, I, Saint-Pierre, M, Lamontagne-Proulx, J, Kriz, J, Barker, RA, et al. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int J Neuropsychopharmacol. (2015) 18:pyu103. doi: 10.1093/ijnp/pyu103
169. Yang, Y, Han, C, Guo, L, and Guan, Q. High expression of the Hmgb1–Tlr4 axis and its downstream signaling factors in patients with Parkinson's disease and the relationship of pathological staging. Brain Behav. (2018) 8:e00948. doi: 10.1002/brb3.948
170. Shin, W-H, Jeon, M-T, Leem, E, Won, S-Y, Jeong, KH, Park, S-J, et al. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: a potential pathogenic mechanism in Parkinson’s disease. Sci Rep. (2015) 5:14764. doi: 10.1038/srep14764
171. Kouli, A, Camacho, M, Allinson, K, and Williams-Gray, CH. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol Commun. (2020) 8:211. doi: 10.1186/s40478-020-01083-5
172. Miri, NS, Saadat, P, Azadmehr, A, Oladnabi, M, and Daraei, A. Toll-like receptor (Tlr)-9 rs352140 polymorphism is an immunopathology protective factor in Parkinson's disease in the northern Iranian population. Iran J Immunol. (2020) 17:313–23. doi: 10.22034/iji.2020.87454.1803
173. Da Rocha Sobrinho, HM, Saar Gomes, R, Da Silva, DJ, Quixabeira, VBL, Joosten, LAB, De Barros, R, et al. Toll-like receptor 10 controls Tlr2-induced cytokine production in monocytes from patients with Parkinson's disease. J Neurosci Res. (2021) 99:2511–24. doi: 10.1002/jnr.24916
174. Miranda-Hernandez, S, and Baxter, AG. Role of toll-like receptors in multiple sclerosis. Am J Clin Exp Immunol. (2013) 2:75–93.
175. Zhou, Y, Fang, L, Peng, L, and Qiu, W. Tlr9 and its signaling pathway in multiple sclerosis. J Neurol Sci. (2017) 373:95–9. doi: 10.1016/j.jns.2016.12.027
176. Hossain, MJ, Morandi, E, Tanasescu, R, Frakich, N, Caldano, M, Onion, D, et al. The soluble form of toll-like receptor 2 is elevated in serum of multiple sclerosis patients: a novel potential disease biomarker. Front Immunol. (2018) 9:457. doi: 10.3389/fimmu.2018.00457
177. Casula, M, Iyer, AM, Spliet, WGM, Anink, JJ, Steentjes, K, Sta, M, et al. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. (2011) 179:233–43. doi: 10.1016/j.neuroscience.2011.02.001
178. Vuono, R, Kouli, A, Legault, EM, Chagnon, L, Allinson, KS, La Spada, A, et al. Association between toll-like receptor 4 (Tlr4) and triggering receptor expressed on myeloid cells 2 (Trem2) genetic variants and clinical progression of Huntington's disease. Mov Disord. (2020) 35:401–8. doi: 10.1002/mds.27911
179. Frederiksen, HR, Haukedal, H, and Freude, K. Cell type specific expression of toll-like receptors in human brains and implications in Alzheimer's disease. Biomed Res Int. (2019) 2019:7420189. doi: 10.1155/2019/7420189
180. Aktar, S, and Amin, S. Sars-CoV-2 mediated dysregulation in cell signaling events drives the severity of Covid-19. Virus Res. (2023) 323:198962. doi: 10.1016/j.virusres.2022.198962
181. Almutairi, MM, Sivandzade, F, Albekairi, TH, Alqahtani, F, and Cucullo, L. Neuroinflammation and its impact on the pathogenesis of Covid-19. Front Med. (2021) 8:745789. doi: 10.3389/fmed.2021.745789
182. Manan, A, Pirzada, RH, Haseeb, M, and Choi, S. Toll-like receptor mediation in Sars-CoV-2: a therapeutic approach. Int J Mol Sci. (2022) 23:10716. doi: 10.3390/ijms231810716
183. Mantovani, S, Oliviero, B, Varchetta, S, Renieri, A, and Mondelli, MU. Tlrs: innate immune sentries against Sars-CoV-2 infection. Int J Mol Sci. (2023) 24:8065. doi: 10.3390/ijms24098065
184. Planès, R, Bert, JB, Tairi, S, Benmohamed, L, and Bahraoui, E. Sars-CoV-2 envelope (E) protein binds and activates Tlr2 pathway: a novel molecular target for Covid-19 interventions. Viruses. (2022) 14:999. doi: 10.3390/v14050999
185. Szabo, MP, Iba, M, Nath, A, Masliah, E, and Kim, C. Does Sars-CoV-2 affect neurodegenerative disorders? Tlr2, a potential receptor for Sars-CoV-2 in the Cns. Exp Mol Med. (2022) 54:447–54. doi: 10.1038/s12276-022-00755-7
186. Frank, MG, Fleshner, M, and Maier, SF. Exploring the immunogenic properties of SARS-CoV-2 structural proteins: PAMP:TLR signaling in the mediation of the neuroinflammatory and neurologic sequelae of COVID-19. Brain Behav Immun. (2023) 111:259–69. doi: 10.1016/j.bbi.2023.04.009
187. Zheng, M, Karki, R, Williams, EP, Yang, D, Fitzpatrick, E, Vogel, P, et al. Tlr2 senses the Sars-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. (2021) 22:829–38. doi: 10.1038/s41590-021-00937-x
188. Nayak, N, Pati, A, Nahak, SK, Sarangi, S, Pradhan, B, Padhi, S, et al. Toll-like receptor-2 (Tlr-2) rs111200466 variant offers protection against Sars-CoV-2 infections and mortality: a worldwide epidemiological correlation analysis. Nucleosides Nucleotides Nucleic Acids. (2023) 42:371–80. doi: 10.1080/15257770.2022.2151015
189. Alhabibi, AM, Hassan, AS, Abd Elbaky, NM, Eid, HA, Khalifa, M, Wahab, MA, et al. Impact of toll-like receptor 2 and 9 gene polymorphisms on Covid-19: susceptibility, severity, and thrombosis. J Inflamm Res. (2023) 16:665–75. doi: 10.2147/JIR.S394927
190. Sahanic, S, Hilbe, R, Dünser, C, Tymoszuk, P, Löffler-Ragg, J, Rieder, D, et al. Sars-CoV-2 activates the Tlr4/MyD88 pathway in human macrophages: a possible correlation with strong pro-inflammatory responses in severe Covid-19. Heliyon. (2023) 9:e21893. doi: 10.1016/j.heliyon.2023.e21893
191. Carreto-Binaghi, LE, Herrera, MT, Guzmán-Beltrán, S, Juárez, E, Sarabia, C, Salgado-Cantú, MG, et al. Reduced Il-8 secretion by nod-like and toll-like receptors in blood cells from Covid-19 patients. Biomedicine. (2023) 11:1078. doi: 10.3390/biomedicines11041078
192. Croci, S, Venneri, MA, Mantovani, S, Fallerini, C, Benetti, E, Picchiotti, N, et al. The polymorphism L412F in Tlr3 inhibits autophagy and is a marker of severe Covid-19 in males. Autophagy. (2022) 18:1662–72. doi: 10.1080/15548627.2021.1995152
193. Lieberum, JN, Kaiser, S, Kalbhenn, J, Bürkle, H, and Schallner, N. Predictive markers related to local and systemic inflammation in severe Covid-19-associated Ards: a prospective single-center analysis. BMC Infect Dis. (2023) 23:19. doi: 10.1186/s12879-023-07980-z
194. Dhangadamajhi, G, and Rout, R. Association of Tlr3 functional variant (rs3775291) with Covid-19 susceptibility and death: a population-scale study. Hum Cell. (2021) 34:1025–7. doi: 10.1007/s13577-021-00510-6
195. Menezes, MCS, Veiga, ADM, Martins De Lima, T, Kunimi Kubo Ariga, S, Vieira Barbeiro, H, De Lucena Moreira, C, et al. Lower peripheral blood toll-like receptor 3 expression is associated with an unfavorable outcome in severe Covid-19 patients. Sci Rep. (2021) 11:15223. doi: 10.1038/s41598-021-94624-4
196. Alseoudy, MM, Elgamal, M, Abdelghany, DA, Borg, AM, El-Mesery, A, Elzeiny, D, et al. Prognostic impact of toll-like receptors gene polymorphism on outcome of Covid-19 pneumonia: a case-control study. Clin Immunol. (2022) 235:108929. doi: 10.1016/j.clim.2022.108929
197. Bagheri-Hosseinabadi, Z, Rezazadeh Zarandi, E, Mirabzadeh, M, Amiri, A, and Abbasifard, M. Mrna expression of toll-like receptors 3, 7, 8, and 9 in the nasopharyngeal epithelial cells of coronavirus disease 2019 patients. BMC Infect Dis. (2022) 22:448. doi: 10.1186/s12879-022-07437-9
198. Sohn, KM, Lee, S-G, Kim, HJ, Cheon, S, Jeong, H, Lee, J, et al. Covid-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial Sepsis. J Korean Med Sci. (2020) 35:e343. doi: 10.3346/jkms.2020.35.e343
199. Pedicillo, MC, De Stefano, IS, Zamparese, R, Barile, R, Meccariello, M, Agostinone, A, et al. The role of toll-like Receptor-4 in macrophage imbalance in lethal Covid-19 lung disease, and its correlation with Galectin-3. Int J Mol Sci. (2023) 24:13259. doi: 10.3390/ijms241713259
200. Carnevale, R, Cammisotto, V, Bartimoccia, S, Nocella, C, Castellani, V, Bufano, M, et al. Toll-like receptor 4-dependent platelet-related thrombosis in Sars-CoV-2 infection. Circ Res. (2023) 132:290–305. doi: 10.1161/CIRCRESAHA.122.321541
201. Flores-Gonzalez, J, Chavez-Galan, L, Falfán-Valencia, R, Roldán, IB, Fricke-Galindo, I, Veronica-Aguilar, A, et al. Variant rs4986790 of toll-like receptor 4 affects the signaling and induces cell dysfunction in patients with severe Covid-19. Int J Infect Dis. (2024) 138:102–9. doi: 10.1016/j.ijid.2023.11.032
202. Zacher, C, Schönfelder, K, Rohn, H, Siffert, W, and Möhlendick, B. The single nucleotide polymorphism rs4986790 (c.896A>G) in the gene Tlr4 as a protective factor in corona virus disease 2019 (Covid-19). Front Immunol. (2024) 15:1355193. doi: 10.3389/fimmu.2024.1355193
203. Taha, SI, Shata, AK, El-Sehsah, EM, Mohamed, MF, Moustafa, NM, and Youssef, MK. Comparison of Covid-19 characteristics in Egyptian patients according to their toll-like Receptor-4 (Asp299Gly) polymorphism. Infez Med. (2022) 30:96–103. doi: 10.53854/liim-3001-11
204. Ali, HN, Niranji, SS, and Al-Jaf, SMA. Association of Toll-like receptor-4 polymorphism with Sars CoV-2 infection in Kurdish population. Hum Gene (Amst). (2022) 34:201115. doi: 10.1016/j.humgen.2022.201115
205. Houssen, ME, Elmaria, MO, Badr, D, El-Mahdy, R, Ghannam, MA, El-Ashwah, S, et al. Serum soluble toll-like receptor 4 and risk for clinical severity in Covid-19 patients. Pneumonia (Nathan). (2024) 16:1. doi: 10.1186/s41479-023-00121-9
206. Alturaiki, W, Alkadi, H, Alamri, S, Awadalla, ME, Alfaez, A, Mubarak, A, et al. Association between the expression of toll-like receptors, cytokines, and homeostatic chemokines in Sars-CoV-2 infection and Covid-19 severity. Heliyon. (2023) 9:e12653. doi: 10.1016/j.heliyon.2022.e12653
207. Fallerini, C, Daga, S, Mantovani, S, Benetti, E, Picchiotti, N, Francisci, D, et al. Association of Toll-like receptor 7 variants with life-threatening Covid-19 disease in males: findings from a nested case-control study. eLife. (2021) 10:e67569. doi: 10.7554/eLife.67569
208. Mantovani, S, Daga, S, Fallerini, C, Baldassarri, M, Benetti, E, Picchiotti, N, et al. Rare variants in toll-like receptor 7 results in functional impairment and downregulation of cytokine-mediated signaling in Covid-19 patients. Genes Immun. (2022) 23:51–6. doi: 10.1038/s41435-021-00157-1
209. Pessoa, NL, Bentes, AA, De Carvalho, AL, De Souza Silva, TB, Alves, PA, De Sousa Reis, EV, et al. Case report: hepatitis in a child infected with Sars-CoV-2 presenting toll-like receptor 7 Gln11Leu single nucleotide polymorphism. Virol J. (2021) 18:180. doi: 10.1186/s12985-021-01656-3
210. El-Hefnawy, SM, Eid, HA, Mostafa, RG, Soliman, SS, Omar, TA, and Azmy, RM. Covid-19 susceptibility, severity, clinical outcome and toll-like receptor (7) mrna expression driven by Tlr7 gene polymorphism (rs3853839) in middle-aged individuals without previous comorbidities. Gene Rep. (2022) 27:101612. doi: 10.1016/j.genrep.2022.101612
211. Zayed, M, Kim, YC, Lee, CS, and Jeong, BH. No association between Sars-CoV-2 infection and the polymorphism of the toll-like receptor 7 (Tlr7) gene in female population. Diagnostics (Basel). (2023) 13:3510. doi: 10.3390/diagnostics13233510
212. Bagci, G, Gundogdu, O, Pektas, AN, Bagci, B, Avci, O, Gursoy, S, et al. The investigation of host genetic variants of toll-like receptor 7 and 8 in Covid-19. Nucleosides Nucleotides Nucleic Acids. (2023) 42:586–602. doi: 10.1080/15257770.2023.2176515
213. Mahallawi, WH, and Suliman, BA. Tlr8 is highly conserved among the Saudi population and its mutations have no effect on the severity of Covid-19 symptoms. Am J Clin Exp Immunol. (2021) 10:71–6.
214. Braga, J, Lepra, M, Kish, SJ, Rusjan, PM, Nasser, Z, Verhoeff, N, et al. Neuroinflammation after Covid-19 with persistent depressive and cognitive symptoms. JAMA Psychiatry. (2023) 80:787–95. doi: 10.1001/jamapsychiatry.2023.1321
215. Chen, J, Chen, J, Lei, Z, Zhang, F, Zeng, L-H, Wu, X, et al. Amyloid precursor protein facilitates Sars-CoV-2 virus entry into cells and enhances amyloid-β-associated pathology in app/Ps1 mouse model of Alzheimer’s disease. Transl Psychiatry. (2023) 13:396. doi: 10.1038/s41398-023-02692-z
216. Charnley, M, Islam, S, Bindra, GK, Engwirda, J, Ratcliffe, J, Zhou, J, et al. Neurotoxic amyloidogenic peptides in the proteome of Sars-Cov2: potential implications for neurological symptoms in Covid-19. Nat Commun. (2022) 13:3387. doi: 10.1038/s41467-022-30932-1
217. Chiricosta, L, Gugliandolo, A, and Mazzon, E. Sars-CoV-2 exacerbates Beta-amyloid neurotoxicity, inflammation and oxidative stress in Alzheimer’s disease patients. Int J Mol Sci. (2021) 22:13603. doi: 10.3390/ijms222413603
218. Gonçalves, CA, Bobermin, LD, Sesterheim, P, and Netto, CA. Sars-CoV-2-induced Amyloidgenesis: not one, but three hypotheses for cerebral Covid-19 outcomes. Meta. (2022) 12:1099. doi: 10.3390/metabo12111099
219. Nyström, S, and Hammarström, P. Amyloidogenesis of Sars-CoV-2 spike protein. J Am Chem Soc. (2022) 144:8945–50. doi: 10.1021/jacs.2c03925
220. Zhang, Y, Chen, H, Li, R, Sterling, K, and Song, W. Amyloid β-based therapy for Alzheimer’s disease: challenges, successes and future. Signal Transduct Target Ther. (2023) 8:248. doi: 10.1038/s41392-023-01484-7
221. Mysiris, DS, Vavougios, GD, Karamichali, E, Papoutsopoulou, S, Stavrou, VT, Papayianni, E, et al. Post-Covid-19 parkinsonism and Parkinson's disease pathogenesis: the Exosomal cargo hypothesis. Int J Mol Sci. (2022) 23:9739. doi: 10.3390/ijms23179739
222. Kann, O, Almouhanna, F, and Chausse, B. Interferon γ: a master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci. (2022) 45:913–27. doi: 10.1016/j.tins.2022.10.007
223. Noor Eddin, A, Al-Rimawi, M, Peer-Zada, F, Hundallah, K, and Alhashem, A. Novel Tlr7 hemizygous variant in post-Covid-19 neurological deterioration: a case report with literature review. Front Neurol. (2023) 14:1268035. doi: 10.3389/fneur.2023.1268035
224. Kaushik, D, Bhandari, R, and Kuhad, A. Tlr4 as a therapeutic target for respiratory and neurological complications of Sars-CoV-2. Expert Opin Ther Targets. (2021) 25:491–508. doi: 10.1080/14728222.2021.1918103
225. Zhao, D, Qin, Y, Liu, J, Tang, K, Lu, S, Liu, Z, et al. Orally administered Bzl-srna-20 oligonucleotide targeting Tlr4 effectively ameliorates acute lung injury in mice. Sci China Life Sci. (2023) 66:1589–99. doi: 10.1007/s11427-022-2219-0
226. Klopp-Schulze, L, Shaw, JV, Dong, JQ, Khandelwal, A, Vazquez-Mateo, C, and Goteti, K. Applying modeling and simulations for rational dose selection of novel toll-like receptor 7/8 inhibitor Enpatoran for indications of high medical need. Clin Pharmacol Ther. (2022) 112:297–306. doi: 10.1002/cpt.2606
227. Port, A, Shaw, JV, Klopp-Schulze, L, Bytyqi, A, Vetter, C, Hussey, E, et al. Phase 1 study in healthy participants of the safety, pharmacokinetics, and pharmacodynamics of enpatoran (M5049), a dual antagonist of toll-like receptors 7 and 8. Pharmacol Res Perspect. (2021) 9:e00842. doi: 10.1002/prp2.842
228. Proud, PC, Tsitoura, D, Watson, RJ, Chua, BY, Aram, MJ, Bewley, KR, et al. Prophylactic intranasal administration of a Tlr2/6 agonist reduces upper respiratory tract viral shedding in a Sars-CoV-2 challenge ferret model. EBioMedicine. (2021) 63:103153. doi: 10.1016/j.ebiom.2020.103153
229. Zhou, SH, Zhang, RY, Zhang, HW, Liu, YL, Wen, Y, Wang, J, et al. Rbd conjugate vaccine with a built-in Tlr1/2 agonist is highly immunogenic against Sars-CoV-2 and variants of concern. Chem Commun (Camb). (2022) 58:2120–3. doi: 10.1039/D1CC06520C
230. Evans, SE, Tseng, CK, Scott, BL, Höök, AM, and Dickey, BF. Inducible epithelial resistance against coronavirus pneumonia in mice. Am J Respir Cell Mol Biol. (2020) 63:540–1. doi: 10.1165/rcmb.2020-0247LE
231. Farooq, M, Batool, M, Kim, MS, and Choi, S. Toll-like receptors as a therapeutic target in the era of immunotherapies. Front Cell Develop Biol. (2021) 9:756315. doi: 10.3389/fcell.2021.756315
232. Ono, Y, Maejima, Y, Saito, M, Sakamoto, K, Horita, S, Shimomura, K, et al. Tak-242, a specific inhibitor of toll-like receptor 4 signalling, prevents endotoxemia-induced skeletal muscle wasting in mice. Sci Rep. (2020) 10:694. doi: 10.1038/s41598-020-57714-3
233. Bakkari, MA, Valiveti, CK, Kaushik, RS, and Tummala, H. Toll-like Receptor-4 (Tlr4) agonist-based intranasal Nanovaccine delivery system for inducing systemic and mucosal immunity. Mol Pharm. (2021) 18:2233–41. doi: 10.1021/acs.molpharmaceut.0c01256
234. Abhyankar, MM, Mann, BJ, Sturek, JM, Brovero, S, Moreau, GB, Sengar, A, et al. Development of Covid-19 vaccine using a dual toll-like receptor ligand liposome adjuvant. npj Vaccines. (2021) 6:137. doi: 10.1038/s41541-021-00399-0
235. Abdolmohammadi-Vahid, S, Baradaran, B, Sadeghi, A, Bezemer, GFG, Kiaee, F, Adcock, IM, et al. Effects of toll-like receptor agonists and Sars-CoV-2 antigens on interferon (Ifn) expression by peripheral blood Cd3+ T cells from Covid-19 patients. Exp Mol Pathol. (2024) 137:104897. doi: 10.1016/j.yexmp.2024.104897
236. Liu, ZM, Yang, MH, Yu, K, Lian, ZX, and Deng, SL. Toll-like receptor (Tlrs) agonists and antagonists for Covid-19 treatments. Front Pharmacol. (2022) 13:989664. doi: 10.3389/fphar.2022.989664
237. Walker, DG, Tang, TM, and Lue, L-F. Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer's disease brains. Exp Neurol. (2018) 309:91–106. doi: 10.1016/j.expneurol.2018.07.016
Keywords: toll-like receptors (TLRs), neurodegenerative disorders, post-COVID-19 syndrome, neuroinflammation, SARS-CoV-2
Citation: Satyanarayanan SK, Yip TF, Han Z, Zhu H, Qin D and Lee SMY (2025) Role of toll-like receptors in post-COVID-19 associated neurodegenerative disorders? Front. Med. 12:1458281. doi: 10.3389/fmed.2025.1458281
Edited by:
Marios Kyriazis, National Gerontology Centre, CyprusReviewed by:
Dekai Zhang, Texas A&M University, United StatesCopyright © 2025 Satyanarayanan, Yip, Han, Zhu, Qin and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dajiang Qin, cWluX2RhamlhbmdAZ3pobXUuZWR1LmNu; Suki Man Yan Lee, c3VraS5sZWVAY3JtaC1jYXMub3JnLmhr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.