 Patricia Guevara-Ramírez1‡†
Patricia Guevara-Ramírez1‡† Viviana A. Ruiz-Pozo1‡†
Viviana A. Ruiz-Pozo1‡† Santiago Cadena-Ullauri1‡
Santiago Cadena-Ullauri1‡ Elius Paz-Cruz1‡
Elius Paz-Cruz1‡ Rafael Tamayo-Trujillo1‡
Rafael Tamayo-Trujillo1‡ Aníbal Gaviria2‡
Aníbal Gaviria2‡ Francisco Cevallos2
Francisco Cevallos2 Ana Karina Zambrano1*‡†
Ana Karina Zambrano1*‡†- 1Centro de Investigación Genética y Genómica, Facultad de Ciencias de la Salud Eugenio Espejo, Universidad UTE, Quito, Ecuador
- 2Hemocentro Nacional, Cruz Roja Ecuatoriana, Quito, Ecuador
Lynch Syndrome (LS) is a hereditary disorder characterized by genetic mutations in DNA mismatch repair genes, affecting approximately 0.35% of the population. LS primarily increases the risk of colorectal cancer (CRC), as well as various other cancer types like endometrial, breast, and gastric cancers. Microsatellite instability, caused by MMR gene mutations, is a key feature of LS, impacting genes such as MLH1, MSH2, MSH6, and PMS2. Pathology tests studying microsatellite instability and immunohistochemical staining are used to diagnose LS. Furthermore, next-generation sequencing (NGS) allows for a thorough investigation of cancer susceptibility genes. This approach is crucial for identifying affected individuals and managing their care effectively. This study evaluated two siblings who harbored a mutation in the MLH1 gene associated with LS. The older brother was diagnosed with CRC at 24, while the younger brother remains asymptomatic at 7 years old. Genetic testing confirmed the presence of the MLH1 mutation in both siblings. Ancestry analysis showed a mix of African, European, and Native American heritage, common among Ecuadorians. Both siblings shared a family history of cancer, suggesting hereditary factors. Treatment involved surgery and chemotherapy for the older brother, emphasizing the importance of genetic testing for siblings with a cancer family history. NGS plays a pivotal role in identifying genetic mutations and guiding treatment decisions, demonstrating its significance in managing LS and other hereditary cancers.
Introduction
Lynch Syndrome (LS) is an autosomal dominant inherited disorder characterized by germline pathogenic variants in DNA mismatch repair (MMR) genes. LS carriers account for 0.35% of the general population (1). Furthermore, the lifetime risk of developing cancer in individuals LS varies depending on the mutated MMR gene, with estimates of 80% for high penetrance genes such as MLH1 and MSH2 (2). Individuals with LS have an elevated risk of various cancers, primarily colorectal cancer (CRC) (up to 80%), as well as other cancers, including endometrial cancer (approximately 60%) (3, 4).
Colorectal cancer is the third most frequent type of cancer worldwide (5). LS represents the predominant hereditary form of CRC. Individuals with LS have an estimated cumulative lifetime risk of developing CRC of up to 52.2% in women and 68.7% in men (6). Approximately 15% of all CRCs exhibit a MMR-deficient phenotype, resulting in microsatellite instability (MSI) and lack of expression of MMR proteins (7, 8). Microsatellite instability is characterized by the accumulation of abnormal lengths of tandemly repeated mono- or dinucleotide sequences, which are caused by mutations in one or both alleles of MMR genes.
The primary MMR genes are MLH1, MSH2, MSH6, or PMS2 and their function involves detecting and repairing DNA mismatches generated during DNA replication (9). The incidence of CRC varies based on the specific MMR gene mutation and the implementation of surveillance colonoscopies. Notably, higher cumulative incidence rates have been reported for individuals with mutations in MLH1 (36–52%) and MSH2 (30–50%) under surveillance, compared to those with mutations in MSH6 (10–17%) and PMS2 (3–11%) (10).
The diagnosis of LS typically relies on a combination of pathology tests including MSI testing and immunohistochemical staining. Furthermore, next-generation sequencing (NGS) has complemented this process by enabling simultaneous analysis of multiple cancer susceptibility genes through multiplex panels. This approach provides a cost-effective alternative compared to single-gene testing (11). Molecular testing, especially for LS, has garnered attention due to its ability to precisely identify affected individuals and its evolving significance in managing prognostic and therapeutic interventions (12, 13).
This study presents a case involving two young siblings from Ecuador, both harboring a mutation in the MLH1 gene associated with LS. The older brother, aged 24 years, has been diagnosed with CRC, while the 7-year-old brother remains asymptomatic. Our study aims to highlight the role of NGS as a fundamental tool in the management and monitoring of cancer patients and their families.
Case presentation
This report presents a case involving two brothers, a 26-year-old man (Individual A) and an 8-year-old boy (Individual B), both of whom have a significant family history of cancer.
Individual A
Individual A reported alterations in his intestinal habits, muscle pain, and spots on the face. Hematological analysis and biochemical parameters revealed hemoglobin in the stool and anemia. A subsequent colonoscopy identified a tumor mass in the cecum, measuring approximately 6 cm. The tumor was partially obstructing the lumen of the cecum, with evidence of ulcers (Figure 1). Furthermore, a gastric biopsy revealed a well-differentiated tubulovillous adenocarcinoma of the colon. Lymph nodes were positive for malignancy, indicating lymphatic spread of the disease. The presumptive diagnosis was CRC. Genomic analyses were performed to confirm the diagnosis and assess the genetic predispositions, including LS.
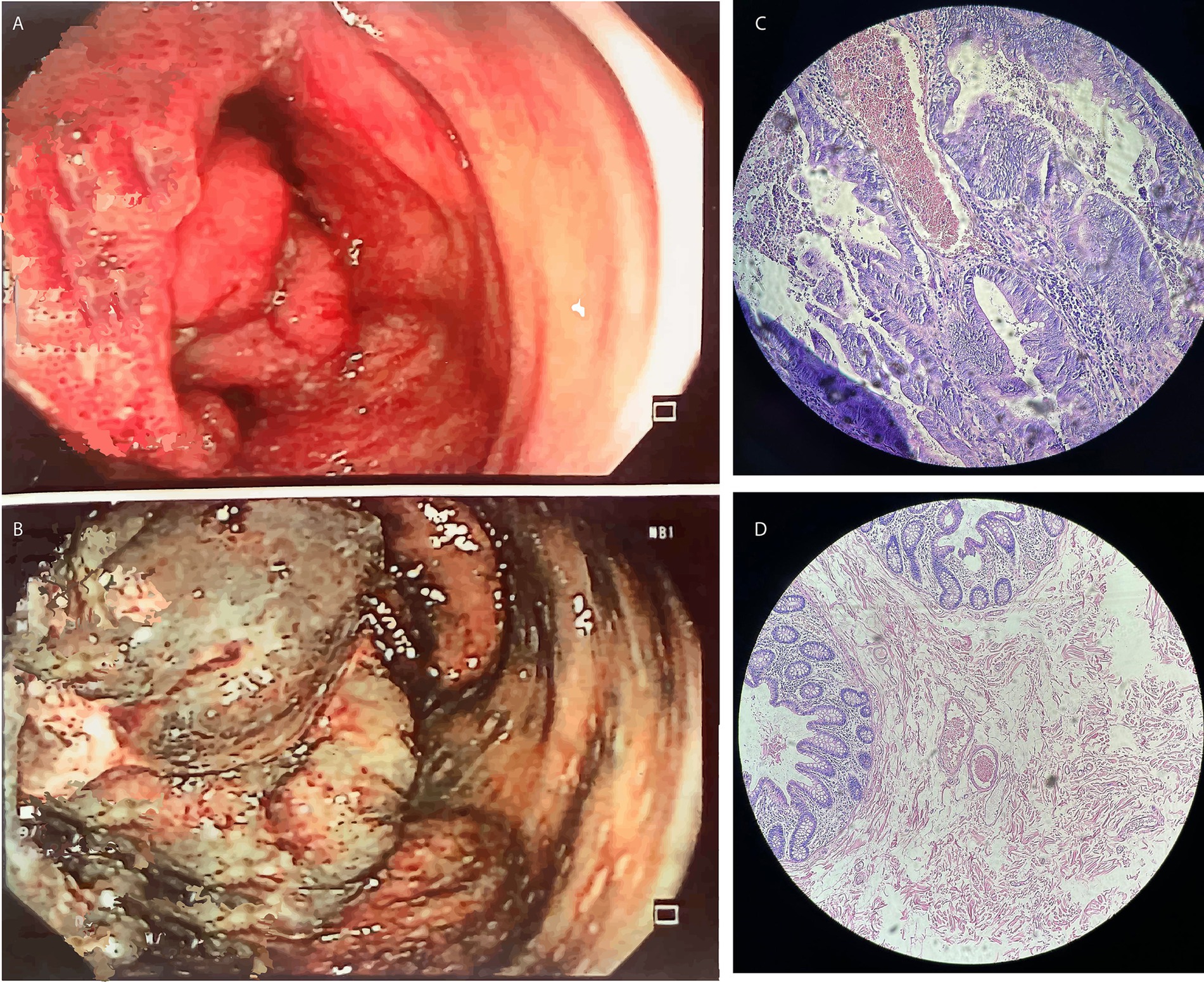
Figure 1. The results of the colonoscopy performed on Individual A indicated (A,B) Tumor mass in the cecum; (C,D) Hematoxylin and eosin-stained slides showing adenocarcinoma of the colon mucosa in Individual A.
Individual B
Individual B did not exhibit any symptoms. However, due to a family history of cancer, the parents authorized genomic testing to investigate potential mutations that could affect his health in the future.
Next generation sequencing (NGS)
The NGS analyses were conducted at the Centro de Investigación de Genética y Genómica (CIGG), Universidad UTE. Individual A and the legal guardians of individual B provided their informed consent before the process.
DNA extraction was performed from peripheral blood samples taken from individuals A and B, using the PureLink™ genomic DNA mini kit (Invitrogen, USA). In the case of Individual A, DNA was extracted from tissue samples in paraffin-embedded blocks with the same kit (PureLink™ genomic DNA mini kit).
The DNA concentrations were obtained using the Qubit™ fluorometer with the 1X dsDNA high-sensitivity (HS) and broad-range (BR) assay kits. NGS was performed on MiSeq platform (Illumina, USA), using the TruSight™ Cancer sequencing panel (Illumina, USA), which includes 255 kb and 94 genes related to different types of cancer. The bioinformatics analyses used were DRAGEN Enrichment v3.9.5, Annotation Engine v3.15, PolyPhen, Sift and Variant Interpreter v2.16.1.300.
Ancestry analysis
For ancestry analysis, a multiplex polymerase chain reaction (PCR) of 46 ancestry-informative INDEL markers (AIMs) was conducted, following the protocol of Zambrano et al. (2019) (14). Fragment detection was performed using Genetic Analyzer 3,500 (Applied Biosystems, USA) equipment. Data Collection v3.3 and Gene Mapper v.5 software were used for data collection and analysis, respectively. The STRUCTURE v.2.3.4 software was used for ancestry study.
Results
In individual A, genomic analyses identified a missense mutation in exon 4/19 (NM_000249.3:c.350C > T) of the MLH1 gene and a Frameshift insertion–deletion (InDel) mutation in exon 8/9 (NM_000314.6c.968dup) of the PTEN gene, these mutations were detected in the paraffin-embedded tissue sample. In addition, the same MLH1 gene mutation was observed in the DNA analysis from a blood sample. The Variant Interpreter Platform (Illumina) classified these mutations as Pathogenic for both the MLH1 and PTEN genes. These analyses suggest a diagnosis consistent with LS, attributed to the variant in the MLH1 gene.
For Individual B, who is asymptomatic and appears healthy, genomic testing revealed the presence of the same MLH1 mutation as individual A (NM_000249.3:c.350C > T) and another mutation in the FH gene (NM_000143.3:c.580G > A). The latter mutation is classified as a variant of uncertain significance (VUS) and it is noteworthy because mutations in the FH gene have been implicated in predisposition to other cancer types (Table 1).
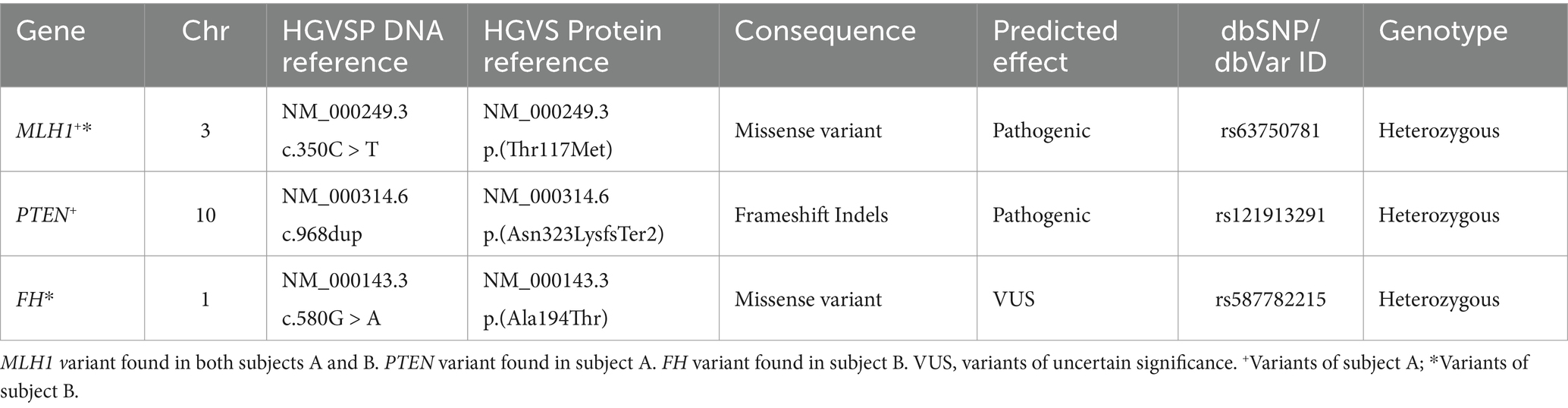
Table 1. Genetic variants identified using TruSight™ Cancer sequencing panel.
Furthermore, the ancestry test revealed that individual A carried 6.6% African, 13.2% European, and 80.2% Native American ancestry proportions, while Individual B showed slightly different proportions with 5.4% African, 17.1% European, and 77.5% Native American ancestry (Supplementary Figure S1).
Family history
Both siblings shared a family history of cancer. Their maternal grandfather was diagnosed with stomach cancer, and their paternal relatives had been diagnosed with pancreatic, liver, esophageal, and breast cancer. The shared genetic mutation in the MLH1 gene between the two siblings suggests hereditary cancer (Figure 2).
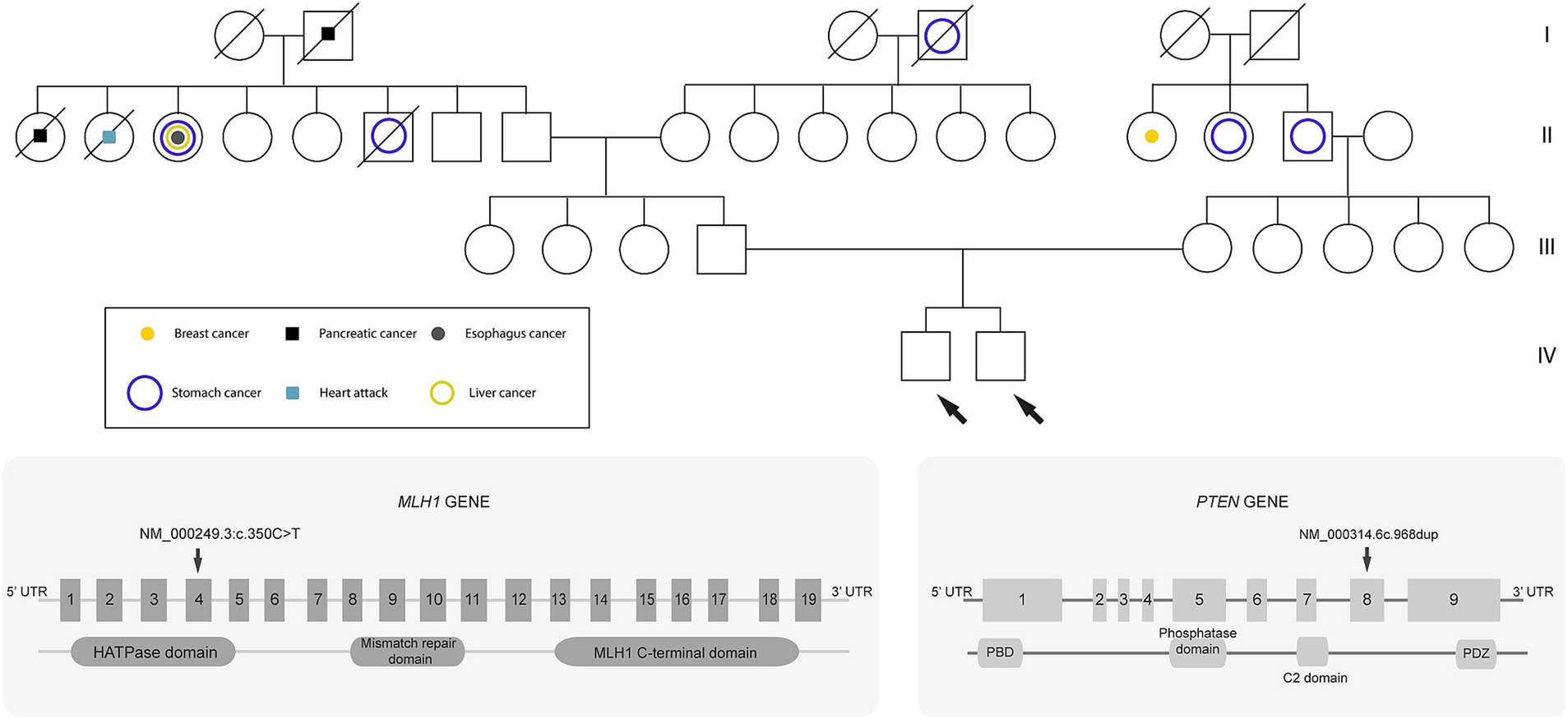
Figure 2. Genealogical tree of the family. Circles represent females, and squares represent males. Diagonal lines through a symbol indicate that the individual is deceased. The white circles or squares represent unaffected (healthy) individuals. Probands are indicated by the arrows.
Treatment
After diagnosis, both patients were informed about the results of the genomic test and the implications of LS. Subsequently, individual A underwent an extended right hemicolectomy and ileotransverse isoperistaltic anastomosis. Individual A was subsequently treated with chemotherapy. On the other hand, Individual B was advised to continue with follow-up medical care. This case highlights the importance of genetic testing for siblings with a family history of cancer (Figure 3).
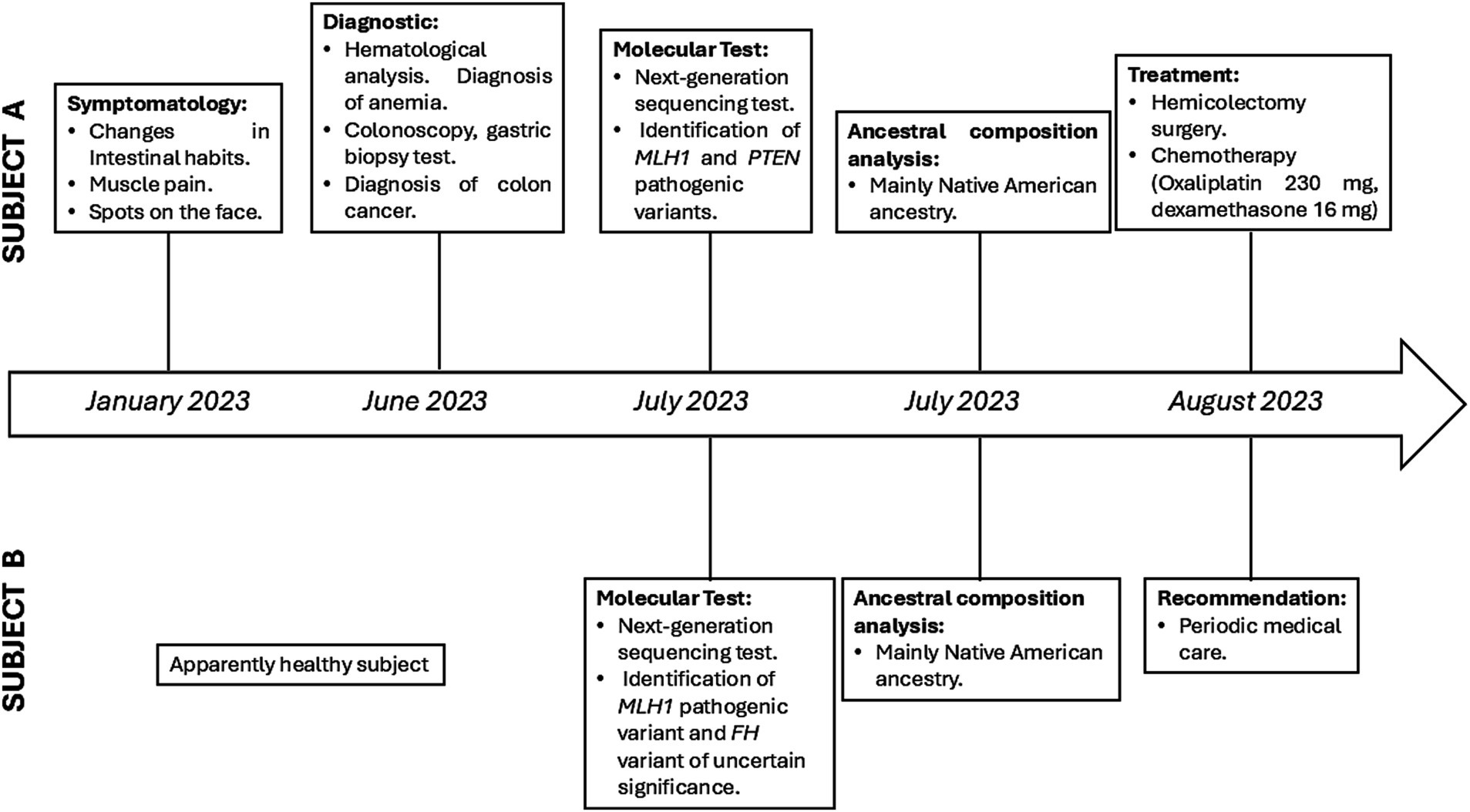
Figure 3. Timeline of subject A and B. The relevant episodes related to the siblings.
Discussion
Lynch Syndrome screening aims to enhance future cancer surveillance for the patient and offer germline variant testing for their at-risk relatives. However, traditional LS screening protocols, such as those based on clinical criteria like Amsterdam I and II or Bethesda guidelines, exhibit limited sensitivity, failing to detect over 40% of LS carriers (15). The advent of NGS has revolutionized clinical laboratory practices, enabling simultaneous sequencing of multiple cancer susceptibility genes with improved efficiency (11). In our investigation, we utilized NGS to analyze individuals with a familial cancer history, successfully identifying various variants, including a pathogenic mutation in the MutL Homolog 1 (MLH1) gene, present in two young siblings.
The MLH1 gene encodes a protein essential for the DNA mismatch repair system forming heterodimer with the mismatch repair endonuclease PMS2, to generate MutL alpha complex. The encoded protein also participates in DNA damage signaling and can heterodimerize with MLH3 protein to form MutL gamma, involved in meiosis (16, 17). Disruption of the MLH1-PMS2 heterodimer compromises MMR, leading to the accumulation of replication errors and increased microsatellite instability. Also, disruption of the heterodimer MLH1-MLH3 could contribute to tumor growth by accumulating errors during meiosis (18–20).
LS is an autosomal dominant disorder caused by germline mutations in MMR genes, primarily MLH1 and MSH2. In families with LS, over 90% of the mutations are found in these two genes (10). Mutations in the MLH1 gene impair the protein’s ability to recognize and repair DNA damage. This leads to the accumulation of genetic errors and hinders apoptosis signaling, which could promote the development of cancer (16, 17). LS is associated with an increased predisposition to different cancer types, primarily colon cancer (occurring in 46–61% of individuals with a pathogenic MLH1 variant) or endometrial cancer (occurring in 34–54% of individuals with a pathogenic MLH1 variant) (2, 21). Colorectal cancer, associated with Lynch syndrome, tends to manifest at an earlier age, with an average age of diagnosis around 40 years (22).
In both subjects, the MLH1 gene variant c.350C > T (p.Thr117Met) was identified, and notably, in Individual A, this variant was present in both blood and tissue samples. According to the InSiGHT classification, the MLH1 variant is classified as class 5 (pathogenic) (23, 24). The c.350C > T variant affects a highly conserved domain in exon 4 of the MLH1 gene. This genetic change arises from cytosine to thymine substitution at position 350 in the nucleotide sequence (25, 26), causing codon 117 undergoes a pathogenic transformation, replacing a polar threonine with a hydrophobic methionine in a highly conserved domain of the MLH1 gene.
Different tests are used for the detection of LS associated with CRC, such as colonoscopy, genetic testing, MSI, immunohistochemistry (IHC) and others. Immunohistochemistry, used for screening patients with suspected LS, can detect approximately 74% of MLH1 germline mutations when using MLH1-specific antibodies (27). This test uses antibodies against four MMR proteins (MLH1, MS2, MSH6, and PMS2) to assess their expression in colorectal cancer tissue (28). For example, the biological interpretation of the MLH1 gene mutation could be shown by IHC that has demonstrated a reduction in DNA repair activity in cells carrying the Thr117Met variant which is associated with increased MSI. Functional analyses reveal reduced protein expression and compromised mismatch repair (MMR) activity linked to the disruption caused by the Thr117Met variant (29, 30). Moreover, this missense mutation likely impairs mismatch repair function by affecting the mutant protein’s ATP binding and hydrolysis, interfering with the formation of protein complex like hPMS2.
Considering that individual A has adenocarcinoma of the colon and carries a mutation in the MLH1 gene, MSI analysis of the tumor samples is recommended. This test is essential not only for confirming the diagnosis of LS but also for its prognosis, as MSI has been established as a key biomarker in CRCs. In the general population, the rate of MSI varies between 12 and 17% but has been reported to be as high as 27 to 35% in patients younger than 30 years (31). In the case of 26-year-old A, an evaluation of MSI would not only contribute to a better understanding of his diagnosis but is also essential to guide appropriate follow-up and treatment for both him and his brother.
Determination of MSI has important prognostic value, as tumors with high instability are often associated with a better clinical prognosis. In addition, MSI has been established as a predictive biomarker in the context of immunotherapy, suggesting that immune checkpoint inhibitors may be more effective in these cases. This is because tumors with a higher mutational burden, characteristic of elevated MSI, generate a greater number of neoantigens (32). These neoantigens can stimulate an immune response, making MMR-deficient tumors particularly responsive to immune checkpoint inhibitors, such as anti-PD-1/PD-L1 therapies (33–35). In our case, the MLH1 c.350C > T variant, associated with increased microsatellite instability, could potentially create a pro-inflammatory tumor microenvironment. Understanding the interplay between mismatch repair gene mutations and immune response could lead to personalized immunotherapeutic strategies for Lynch Syndrome patients (14, 15). In this context, performing MSI analysis is not only crucial for the diagnosis and treatment of individual A, but also for the preventive management and follow-up of individual B, given the possible heritable involvement of mutations in the MLH1 gene (36).
This study marks the first report of this variant in Ecuadorian patients. Nevertheless, the variant has also been previously identified in unrelated families from Uruguay and Argentina, suggesting that the c.350C > T variant may be present within the broader Latin American gene pool (37). Notably, the reported frequency of the alternative allele T is 0,000 in the Latin American population (38).
Furthermore, this variant was identified in other populations. For instance, Yanus et al. (39) studied Russian individuals with either colorectal or endometrial cancer, who were referred for LS testing, revealing the MLH1 c.350C > T variant. The results are consistent with the UMD-MMR database, which includes 55 entries for the mutation in French laboratories until 2015 (40). Moreover, another study focused on analyzing the MLH1 gene in Polish and Baltic State families, revealing the mutation in one family with colon cancer (19). Another study reported the mutation in a hereditary nonpolyposis colorectal cancer patient in Slovakia (26). These studies collectively highlight the recurrent nature of the mutation across diverse geographical regions. Despite its global prevalence, the mutation has not been previously reported in Ecuador (14).
In the tumor sample from individual A, a PTEN c.968dup variant was identified. This variant results in a frameshift and a premature stop codon, probably producing absent or truncated PTEN protein. The truncation caused by c.968dup disrupts critical domains such as C2, C-tail, and PDZ, which play pivotal roles in protein localization, stability, and interaction with other proteins (41, 42).
PTEN is a tumor suppressor that regulates the PI3K/Akt signaling pathway, which is crucial for cell growth and survival. In colorectal cancer, loss of PTEN function leads to increased activity in this pathway, promoting tumorigenesis (43, 44). Other research suggests that loss of PTEN in CRC is closely associated with increased genomic instability and worse clinical outcomes, such as more advanced disease stages and the development of liver metastasis (45, 46). In this case study, the pathogenic variants in the PTEN and MLH1 genes identified in individual A could trigger an additive effect from mutations in different genes, leading to a more aggressive disease phenotype.
However, this relationship is not consistently observed across all studies, suggesting that the role of PTEN in tumor progression may be influenced by other genetic or epigenetic factors (45–48). Moreover, in an extensive study involving 1,093 cases of CRC, PTEN mutations were detected in 43 tumors. Among these mutations, 47.3% were identified as nonsense mutations, 38.2% as frameshift mutations, 9.1% as missense mutations, and 5.5% as splice site mutations. Notably, the c.968dup variant was among the frameshift mutations closely related to CRC (49). This variability highlights the molecular complexity of CRC, particularly in cases linked to LS, and underscores the importance of further investigation to fully understand the impact of PTEN on CRC development and progression.
On the other hand, individual B carries a FH c.580G > A mutation, which has been implicated in hereditary cancer predisposition (50). The variant frequency has not been reported among the Latin American population. The c.580G > A variant, also denoted as p.Ala194Thr, is situated within the coding sequence of exon 5 in the FH gene. This genetic alteration arises from the substitution of alanine for threonine at codon 194. Notably, alanine is characterized as neutral and non-polar, whereas threonine possesses a neutral and polar nature. Consequently, advanced protein modeling techniques, coupled with the scrutiny of their physical properties, have revealed that this missense variant disrupts the functionality of FH protein (51). Despite this, the variant has been categorized as a variant of uncertain significance, primarily due to findings from RNA analysis suggesting that its impact on mRNA splicing does not significantly alter splicing patterns. Furthermore, associations have been made between this variant and other types of cancer such as pheochromocytoma, paraganglioma, and renal cell cancer. However, its low population frequency (0.00007), mainly present in individuals of European descent, makes it uninformative for assessing its pathogenicity (51, 52).
Furthermore, the PTEN protein has a physical or functional interaction with MLH1, while the FH protein does not show any interaction with PTEN or MLH1 (Supplementary Figure S2). However, the coexistence of these variants in individuals could have synergistic effects, potentially increasing their cancer risk. Even though the assay employed in this investigation covered some genes implicated in mismatch repair, it would be interesting to utilize a multigene panel encompassing an extensive spectrum of genes or SNPs associated with LS.
The genetic analysis of the ancestral components of the two brothers harboring the MLH1 c.350C > T variant, along with additional variants (PTEN c.968dup for individual A and FH c.580G > A for individual B), has provided a further understanding of the possible relationship between genetic ancestry and specific mutations associated with hereditary cancer predisposition.
Moreover, the ancestral composition of the individuals revealed a mosaic of African, European, and Native American components, as previously reported for the Ecuadorian (14). However, both siblings have high levels of Native American ancestry, which is notable given a lack of data on population specific genetic risks in this group. Although ancestry data does not directly elucidate cancer predisposition in this case, the Native American heritage may offer insights into underrepresented genetic variants relevant to Lynch Syndrome and cancer. Further studies on larger cohorts are needed to explore the relationship between ancestry and cancer risk in Latin American (14).
The MLH1 c.350C > T variant, associated with Lynch syndrome, has been extensively studied and reported in different regions, such as Latin America (37) and Europe (19, 26, 39, 40). Besides, a study compared the racial differences in MLH1 mutations between the Caucasian and East Asian races and found that this variant ranks among the top ten mutations in both ethnic groups (53). The prevalence of this mutation in individuals with predominantly Native American ancestry is of particular interest and has not been previously reported, which could provide insights into population-specific genetic risk factors. However, the PTEN and FH variants have not been previously described in other populations.
Consequently, employing multiple gene sequencing may benefit patients, especially those with personal or familial histories. The identification of individuals with hereditary cancer predispositions, such as Lynch syndrome, holds significant promise for reducing cancer incidence and mortality rates. Nonetheless, accessing comprehensive mutation screening may present challenges in certain scenarios. However, once mutations are identified, screening other family members for the same mutation can be achieved using relatively straightforward methods (15).
In this study, we used NGS panels that included the MMR genes (MLH1, MSH2, MSH6, or PMS2), identifying a pathogenic MLH1 mutation in both siblings. This finding underscores the importance of genomic testing in detecting germline mutations within families. Clinically, it enables a more precise diagnosis and treatment for Individual A, who is symptomatic, while providing necessary follow-up for Individual B, who remains asymptomatic.
A limitation of this study is the budget constraint, because NGS could not be performed on all family members. Despite this constraint, the screening process remains informative and contributes significantly to continuous patient monitoring. Consequently, the adoption of massive sequencing approaches may provide valuable insights, enabling the implementation of effective cancer prevention strategies, personalized screening protocols, and tailored therapeutic interventions to mitigate cancer-related morbidity and mortality linked to Lynch Syndrome (11, 54).
In conclusion, the ancestral components, and associated variants of the two siblings provide the first data of a mainly Native American composition and mutations related to LS and cancer predisposition. Further studies on diverse populations and larger cohorts could improve the understanding of population-specific variants, their implications, and healthcare strategies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Human Research Ethics Committee-University UTE. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
PG-R: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. VR-P: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. SC-U: Methodology, Writing – review & editing. EP-C: Methodology, Writing – review & editing. RT-T: Methodology, Writing – review & editing. AG: Conceptualization, Writing – review & editing. FC: Conceptualization, Writing – review & editing. AZ: Conceptualization, Funding acquisition, Methodology, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The experimentation and publication fee of this article are funded by Universidad UTE.
Acknowledgments
We are grateful to Universidad UTE for supporting the researchers. Furthermore, the authors are grateful to Alex Tulcán Cuadros for his help during the project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2024.1498290/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Ancestral composition of the subjects under analysis (in yellow).
SUPPLEMENTARY FIGURE S2 | Protein – protein physical and functional association (constructed with STRING).
References
1. Umemiya, M, Horikawa, N, Kanai, A, Saeki, A, Ida, K, Makio, S, et al. Endometrial Cancer diagnosed at an early stage during lynch syndrome surveillance: a case report. Case Rep Oncol. (2023) 16:634–9. doi: 10.1159/000531837
2. Peltomäki, P, Nyström, M, Mecklin, JP, and Seppälä, TT. Lynch syndrome genetics and clinical implications. Gastroenterology. (2023) 164:783–99. doi: 10.1053/J.GASTRO.2022.08.058
3. Mukucha, K-E, Manase, MT, Muronda, C, Whittaker, J, and Guzha, BT. Challenges managing women with suspected lynch syndrome in Zimbabwe: a case report. South African J Gynaecol Oncol. (2021) 13:42–4. doi: 10.1080/20742835.2021.1991100
4. Ito, T, Kono, K, Eguchi, H, Okazaki, Y, Yamamoto, G, Tachikawa, T, et al. Prevalence of lynch syndrome among patients with upper urinary tract carcinoma in a Japanese hospital-based population. Jpn J Clin Oncol. (2020) 50:80–8. doi: 10.1093/jjco/hyz140
5. Sung, H, Ferlay, J, Siegel, RL, Laversanne, M, Soerjomataram, I, Jemal, A, et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
6. Abu-Ghazaleh, N, Kaushik, V, Gorelik, A, Jenkins, M, and Macrae, F. Worldwide prevalence of lynch syndrome in patients with colorectal Cancer: systematic review and Meta-analysis. Genet Med. (2022) 24:971–85. doi: 10.1016/J.GIM.2022.01.014
7. Bohaumilitzky, L, von Knebel Doeberitz, M, Kloor, M, and Ahadova, A. Implications of hereditary origin on the immune phenotype of mismatch repair-deficient cancers: systematic literature review. J Clin Med. (2020) 9:1741. doi: 10.3390/JCM9061741
8. Boland, CR, and Goel, A. Microsatellite instability in colorectal Cancer. Gastroenterology. (2010) 138:2073–2087.e3. doi: 10.1053/J.GASTRO.2009.12.064
9. Evrard, C, Tachon, G, Randrian, V, Karayan-Tapon, L, and Tougeron, D. Microsatellite instability: diagnosis, heterogeneity, discordance, and clinical impact in colorectal Cancer. Cancers (Basel). (2019) 11:1567. doi: 10.3390/CANCERS11101567
10. Helderman, NC, van Leerdam, ME, Kloor, M, Ahadova, A, and Nielsen, M. Emerge of colorectal Cancer in lynch syndrome despite colonoscopy surveillance: a challenge of Hide and Seek. Crit Rev Oncol Hematol. (2024) 197:104331. doi: 10.1016/J.CRITREVONC.2024.104331
11. Yurgelun, MB, and Hampel, H. Recent advances in lynch syndrome: diagnosis, treatment, and Cancer prevention. Am Soc Clin Oncol Educ Book. (2018) 38:101–9. doi: 10.1200/EDBK_208341
12. Sobocińska, J, Kolenda, T, Teresiak, A, Badziąg-Leśniak, N, Kopczyńska, M, Guglas, K, et al. Diagnostics of Mutations in MMR/ EPCAM Genes and Their Role in the Treatment and Care of Patients with Lynch Syndrome. Diagnostics. (2020) 10:786. doi: 10.3390/diagnostics10100786
13. Yao, ZG, Lv, BB, Jing, HY, Su, WJ, Li, JM, Fan, H, et al. A practical screening strategy for lynch syndrome and lynch syndrome mimics in colorectal Cancer. J Cancer Res Ther. (2021) 17:790–6. doi: 10.4103/JCRT.JCRT_214_21
14. Zambrano, AK, Gaviria, A, Cobos-Navarrete, S, Gruezo, C, Rodríguez-Pollit, C, Armendáriz-Castillo, I, et al. The three-hybrid genetic composition of an Ecuadorian population using AIMs-InDels compared with autosomes, mitochondrial DNA and Y chromosome data. Sci Rep. (2019) 9:9247. doi: 10.1038/s41598-019-45723-w
15. Pasanen, A, Loukovaara, M, Kaikkonen, E, Olkinuora, A, Pylvänäinen, K, Alhopuro, P, et al. Testing for lynch syndrome in endometrial carcinoma: from universal to age-selective MLH1 methylation analysis. Cancers. (2022) 14:1348. doi: 10.3390/CANCERS14051348
16. Thompson, BA, Goldgar, DE, Paterson, C, Clendenning, M, Walters, R, Arnold, S, et al. A multifactorial likelihood model for MMR gene variant classification incorporating probabilities based on sequence bioinformatics and tumor characteristics: a report from the Colon Cancer family registry. Hum Mutat. (2013) 34:200–9. doi: 10.1002/humu.22213
17. Stelzer, G, Rosen, N, Plaschkes, I, Zimmerman, S, Twik, M, Fishilevich, S, et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. (2016) 54:1.30.1–1.30.33. doi: 10.1002/cpbi.5
18. Frostberg, E, Petersen, AH, Bojesen, A, Rahr, HB, Lindebjerg, J, and Rønlund, K. The prevalence of pathogenic or likely pathogenic germline variants in a Nationwide cohort of Young colorectal Cancer patients using a panel of 18 genes associated with colorectal Cancer. Cancers. (2021) 13:5094. doi: 10.3390/cancers13205094
19. Kurzawski, G, Suchy, J, Kładny, J, Safranow, K, Jakubowska, A, Elsakov, P, et al. Germline MSH2 and MLH1 mutational spectrum in HNPCC families from Poland and the Baltic States. J Med Genet. (2002) 39:65e–665e. doi: 10.1136/jmg.39.10.e65
20. Talbot, A, O’Donovan, E, Berkley, E, Nolan, C, Clarke, R, and Gallagher, D. The contribution of lynch syndrome to early onset malignancy in Ireland. BMC Cancer. (2021) 21:617. doi: 10.1186/s12885-021-08263-z
21. Massachusetts General Hospital Cancer Center. (2023). Lynch syndrome: information for families with a pathogenic variant in the MLH1 Gene Available at: https://www.massgeneral.org/assets/mgh/pdf/cancer-center/genetics/mlh1/mlh1_result_handout_2021.pdf.
22. Momma, T, Gonda, K, Akama, Y, Endo, E, Ujiie, D, Fujita, S, et al. MLH1 germline mutation associated with lynch syndrome in a family followed for more than 45 years. BMC Med Genet. (2019) 20:67. doi: 10.1186/S12881-019-0792-0
23. Pinto, RM, Dragileva, E, Kirby, A, Lloret, A, Lopez, E, St Claire, J, et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet. (2013) 9:e1003930. doi: 10.1371/journal.pgen.1003930
24. Global Variome shared LOVD. (2024). All variants in the MLH1 Gene Available at: https://databases.lovd.nl/shared/variants/MLH1?search_VariantOnGenome/Genetic_origin=SUMMARY#object_id=VariantOnTranscript%2CVariantOnGenome&id=MLH1&order=VariantOnTranscript%2FDNA%2CASC&search_transcriptid=00013676&search_VariantOnGenome/Genetic_origin=S (Accessed on 21 December 2024).
25. Bennett, G, Rudzki, Z, Cole, S, Young, G, and Suthers, G. The impact of molecular diagnosis on familial colorectal. Cancer. (1998) 36:641–4. doi: 10.1515/CCLM.1998.113
26. Zavodna, K, Bujalkova, M, Krivulcik, T, Alemayehu, A, Skorvaga, M, Marra, G, et al. Novel and recurrent germline alterations in the MLH1 and MSH2 genes identified in hereditary nonpolyposis colorectal Cancer patients in Slovakia*. Neoplasma. (2006) 53:269–76. doi: 10.5167/uzh-34290
27. Mojtahed, A, Schrijver, I, Ford, JM, Longacre, TA, and Pai, RK. A two-antibody mismatch repair protein immunohistochemistry screening approach for colorectal carcinomas, skin sebaceous tumors, and gynecologic tract carcinomas. Mod Pathol. (2011) 24:1004–14. doi: 10.1038/modpathol.2011.55
28. Sarode, VR, and Robinson, L. Screening for lynch syndrome by immunohistochemistry of mismatch repair proteins: significance of indeterminate result and correlation with mutational studies. Arch Pathol Lab Med. (2019) 143:1225–33. doi: 10.5858/arpa.2018-0201-OA
29. Jäger, AC, Rasmussen, M, Bisgaard, HC, Singh, KK, Nielsen, FC, and Rasmussen, LJ. HNPCC mutations in the human DNA mismatch repair gene HMLH1 influence assembly of HMutLα and HMLH1–HEXO1 complexes. Oncogene. (2001) 20:3590–5. doi: 10.1038/sj.onc.1204467
30. Trojan, J, Zeuzem, S, Randolph, A, Hemmerle, C, Brieger, A, Raedle, J, et al. Functional analysis of HMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology. (2002) 122:211–9. doi: 10.1053/gast.2002.30296
31. Battaglin, F, Naseem, M, Lenz, HJ, and Salem, ME. Microsatellite instability in colorectal Cancer: overview of its clinical significance and novel perspectives. Clin Adv Hematol Oncol. (2018) 16:735–45.
32. Shaikh, R, Bhattacharya, S, and Prajapati, BG. Microsatellite instability: a potential game-changer in colorectal Cancer diagnosis and treatment. Results Chem. (2024) 7:101461. doi: 10.1016/J.RECHEM.2024.101461
33. Zhao, P, Li, L, Jiang, X, and Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. (2019) 12:1–14. doi: 10.1186/S13045-019-0738-1/TABLES/5
34. Puccini, A, Battaglin, F, Iaia, ML, Lenz, HJ, and Salem, ME. Overcoming resistance to anti-PD1 and anti-PD-L1 treatment in gastrointestinal malignancies. J Immunother Cancer. (2020) 8:e000404. doi: 10.1136/JITC-2019-000404
35. Kim, ST, Klempner, SJ, Park, SH, Park, JO, Park, YS, Lim, HY, et al. Correlating programmed death ligand 1 (PD-L1) expression, mismatch repair deficiency, and outcomes across tumor types: implications for immunotherapy. Oncotarget. (2017) 8:77415–23. doi: 10.18632/ONCOTARGET.20492
36. Abidi, A, Gorris, MAJ, Brennan, E, Jongmans, MCJ, Weijers, DD, Kuiper, RP, et al. Challenges of Neoantigen targeting in lynch syndrome and constitutional mismatch repair deficiency syndrome. Cancers (Basel). (2021) 13:2345. doi: 10.3390/CANCERS13102345
37. Rossi, BM, Palmero, EI, López-Kostner, F, Sarroca, C, Vaccaro, CA, Spirandelli, F, et al. A survey of the Clinicopathological and molecular characteristics of patients with suspected lynch syndrome in Latin America. BMC Cancer. (2017) 17:1–26. doi: 10.1186/s12885-017-3599-4
38. National Library of Medicine Rs63750781 RefSNP Report - DbSNP. (2022). Available at: https://www.ncbi.nlm.nih.gov/snp/rs63750781 (Accessed on 16 December 2023).
39. Yanus, GA, Akhapkina, TA, Iyevleva, AG, Kornilov, AV, Suspitsin, EN, Kuligina, ES, et al. The Spectrum of lynch syndrome-associated germ-line mutations in Russia. Eur J Med Genet. (2020) 63:103753. doi: 10.1016/j.ejmg.2019.103753
40. The French National Committee of Informatics and Liberty. (2014). Available at: http://www.umd.be/MLH1/4DACTION/DMD_EX1/4 (Accessed on 24 October 2024).
41. PTEN Regulatory functions in tumor suppression and cell biology - PubMed. (2004). Available at: https://pubmed.ncbi.nlm.nih.gov/15448614/ (Accessed on 22 April 2024).
42. Brito, MB, Goulielmaki, E, and Papakonstanti, EA. Focus on PTEN regulation. Front Oncol. (2015) 5:5. doi: 10.3389/FONC.2015.00166
43. Rascio, F, Spadaccino, F, Rocchetti, MT, Castellano, G, Stallone, G, Netti, GS, et al. The pathogenic role of PI3K/AKT pathway in Cancer onset and drug resistance: an updated review. Cancers (Basel). (2021) 13:3949. doi: 10.3390/CANCERS13163949
44. Salvatore, L, Calegari, MA, Loupakis, F, Fassan, M, Di Stefano, B, Bensi, M, et al. PTEN in colorectal Cancer: shedding light on its role as predictor and target. Cancers (Basel). (2019) 11:1765. doi: 10.3390/CANCERS11111765
45. Guanti, G, Resta, N, Simone, C, Cariola, F, Demma, I, Fiorente, P, et al. Involvement of PTEN mutations in the genetic pathways of colorectal Cancerogenesis. Hum Mol Genet. (2000) 9:283–7. doi: 10.1093/HMG/9.2.283
46. Molinari, F, and Frattini, M. Functions and regulation of the PTEN gene in colorectal Cancer. Front Oncol. (2014) 3:326. doi: 10.3389/FONC.2013.00326
47. Zhou, XP, Loukola, A, Salovaara, R, Nystrom-Lahti, M, Peltomäki, P, De la Chapelle, A, et al. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am J Pathol. (2002) 161:439–47. doi: 10.1016/S0002-9440(10)64200-9
48. List of Variants in Gene PTEN reported as pathogenic for hereditary Cancer-predisposing syndrome - ClinVar miner. (2024). Available at: https://clinvarminer.genetics.utah.edu/variants-by-gene/PTEN/condition/Hereditary%20cancer-predisposing%20syndrome/pathogenic (Accessed on 6 November 2024).
49. Day, FL, Jorissen, RN, Lipton, L, Mouradov, D, Sakthianandeswaren, A, Christie, M, et al. PIK3CA and PTEN gene and Exon mutation-specific Clinicopathologic and molecular associations in colorectal Cancer. Clin Cancer Res. (2013) 19:3285–96. doi: 10.1158/1078-0432.CCR-12-3614
50. NCBI. VCV000142075.34 - ClinVar. (2024). Available at:https://www.ncbi.nlm.nih.gov/clinvar/variation/142075/ (Accessed on 6 November 2024).
51. Jaime Castro-Vega, L, Buffet, A, De Cubas, AA, Cascó, A, lanie Menara, M, Khalifa, E, et al. Germline mutations in FH confer predisposition to malignant Pheochromocytomas and Paragangliomas. Hum Mol Genet. (2014) 23:2440–6. doi: 10.1093/hmg/ddt639
52. Zavoshi, S, Lu, E, Boutros, PC, Zhang, L, Harari, A, Hatchell, KE, et al. Fumarate hydratase variants and their association with Paraganglioma/Pheochromocytoma. Urology. (2023) 176:106–14. doi: 10.1016/J.UROLOGY.2022.11.053
53. Wei, W, Liu, L, Chen, J, Jin, K, Jiang, F, Liu, F, et al. Racial differences in MLH1 and MSH2 mutation: an analysis of yellow race and white race based on the Insight database. J Bioinforma Comput Biol. (2010) 8:111–25. doi: 10.1142/S0219720010005154
Keywords: lynch syndrome, colorectal cancer, genomic, ancestral, Ecuadorian
Citation: Guevara-Ramírez P, Ruiz-Pozo VA, Cadena-Ullauri S, Paz-Cruz E, Tamayo-Trujillo R, Gaviria A, Cevallos F and Zambrano AK (2024) Case report: Exploring Lynch Syndrome through genomic analysis in a mestizo Ecuadorian patient and his brother. Front. Med. 11:1498290. doi: 10.3389/fmed.2024.1498290
Edited by:
Yuanyuan Zheng, Tongji University School of Medicine, ChinaReviewed by:
Guoli Zhu, University of Pennsylvania, United StatesLaixing Zhang, University of California, Los Angeles, United States
Copyright © 2024 Guevara-Ramírez, Ruiz-Pozo, Cadena-Ullauri, Paz-Cruz, Tamayo-Trujillo, Gaviria, Cevallos and Zambrano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Karina Zambrano, YW5hemFtYnJhbm8xN0Bob3RtYWlsLmNvbQ==
‡ORCID: Patricia Guevara-Ramírez, https://orcid.org/0000-0002-4829-3653
Viviana A. Ruiz-Pozo, https://orcid.org/0000-0001-9301-2614
Santiago Cadena-Ullauri, https://orcid.org/0000-0001-8463-6046
Elius Paz-Cruz, https://orcid.org/0000-0003-0062-6030
Rafael Tamayo-Trujillo, https://orcid.org/0000-0001-9059-3281
Aníbal Gaviria, https://orcid.org/0000-0001-5755-4096
Ana Karina Zambrano, https://orcid.org/0000-0003-4102-3965
†These authors have contributed equally to this work