 Sarita Thawanaphong
Sarita Thawanaphong Aswathi Nair
Aswathi Nair Emily Volfson
Emily Volfson Parameswaran Nair1,2
Parameswaran Nair1,2 Manali Mukherjee
Manali Mukherjee- 1Department of Medicine, McMAster University, Hamilton, ON, Canada
- 2Research Institute of St. Joe’s Hamilton, St. Joseph’s Healthcare Hamilton, Hamilton, ON, Canada
- 3Division of Pulmonary and Critical Care Medicine, Department of Medicine, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial Hospital, Thai Red Cross Society, Bangkok, Thailand
The role of interleukin-18 (IL-18) and inflammasomes in chronic inflammatory airway diseases, such as asthma and chronic obstructive pulmonary disease (COPD), has garnered significant attention in recent years. This review aims to provide an overview of the current understanding of IL-18 biology, the associated signaling pathways, and the involvement of inflammasome complexes in airway diseases. We explore the multifaceted role of IL-18 in asthma pathophysiology, including its interactions with other cytokines and contributions to both T2 and non-T2 inflammation. Importantly, emerging evidence highlights IL-18 as a critical player in severe asthma, contributing to chronic airway inflammation, airway hyperresponsiveness (AHR), and mucus impaction. Furthermore, we discuss the emerging evidence of IL-18’s involvement in autoimmunity and highlight potential therapeutic targets within the IL-18 and inflammasome pathways in severe asthma patients with evidence of infections and airway autoimmune responses. By synthesizing recent advancements and ongoing research, this review underscores the importance of IL-18 as a potential novel therapeutic target in the treatment of severe asthma and other related conditions.
Introduction
Asthma is a common respiratory disease that currently affects 338 million individuals worldwide. The disease’s underlying mechanism of airway inflammation and airway hyperresponsiveness, causing airway wall edema, mucus plugging, airway smooth muscle contraction, and/or airway remodeling, leads to clinical symptoms of chronic, episodic cough, phlegm production, chest tightness, wheezing, and shortness of breath (1). Asthma phenotypes are used to characterize patients based on their clinical features, including symptom severity, a history of exacerbation, and lung function in combination with airway inflammation and genetic profiles (2). Majority of asthma patients with mild to moderate disease are controlled by appropriate treatment with high dose Inhaled corticosteroids/Long-acting beta-2 agonist (ICS/LABA) and comorbidities management. Approximately 5–10% suffer from severe uncontrolled asthma and experience symptoms, persistent airway inflammation, and frequent exacerbations despite high-dose ICS treatment (3). Systemic corticosteroids, which have broad anti-inflammatory effects, remain the mainstay of treatment to manage more severe symptoms but are well known for their adverse effects (4).
Airway inflammation and hyperresponsiveness in asthma occur due to both exogenous and endogenous triggers. The key cytokines involved in the Type 2 inflammatory pathways are IL-5 and IL-13 (5). As a steroid-sparing strategy, anti-T2 monoclonal antibody (mAb) therapies were introduced in the past decade, that have shown modest reduction of asthma exacerbations by 50–60% (6). An optimal, personalized treatment is, therefore needed based on the underlying phenotype and associated endotypes (7). Although blood eosinophil counts are widely used in clinical practice, they may not be sufficient to justify treatment decisions. It is crucial to consider the compartmentalization of airway inflammation in asthma (8). The role of airway eosinophils is further corroborated by a recent cohort study showing sputum eosinophil peroxidase (EPX) is superior to blood eosinophils in understanding patients who remain uncontrolled on anti-IL-5 targeted therapy (9). Again, the modest reduction in exacerbations and persistence of symptoms that remain uncontrolled by anti-IL-5 mAbs (10–12) despite depletion of airway eosinophils suggest alternative/residual airway inflammation (13). The measurement of sputum cytokine in this group of patients revealed raised IL-13, IL-18, and/or IL-1β, indicating alternative pathways beyond IL-5 (14). Furthermore, lack of improvement in asthma symptoms on benralizumab, an eosinophil depleting mAb was associated with raised/residual levels of IL-18 in sputum (13). In severe allergic asthma patients, who typically respond to omalizumab (anti-IgE therapy), high baseline serum free IL-18 levels may predict reduced omalizumab efficacy. A 2-year study showed that significantly more patients showed high baseline serum free IL-18 levels among the patients who developed exacerbation in the second year (incomplete responders) than the complete responder group (15). In non-T2 inflammation, IL-18 works in combination with IL-12, stimulating T helper 1 (Th1) cell development, Natural killer (NK) cells, and Natural killer T (NKT) cells (16). Since IL-18 is primarily produced as an inactive precursor, there is a process to proteolyze pro-IL-18 to an active form. This process is involved in the inflammasome complexes cascade (discussed in detail), which plays an important role in our innate immune systems (17).
This review aims to consolidate current knowledge on IL-18 biology and signaling pathways, particularly the involvement of inflammasome complexes in airway disease to provide active IL-18. We also discuss the role of IL-18 in asthma pathophysiology, autoimmunity, interaction with other cytokines, and its clinical perspective, including the potential therapeutic targets.
IL-18 biology
IL-18 was initially identified as an interferon-γ (IFN-γ) inducing factor (IGIF). The IGIF is mainly produced as a 193 amino acids precursor and is cleaved to a mature protein of 157 amino acids (18). IL-18 is classified to be a part of the IL-1 cytokine family due to its shared common beta-pleated sheet structure and amino acid sequence (18–20). Similar to the other cytokines in the IL-1 family, IL-18 exerts active functions by binding to the corresponding receptors on the surface of target cells. The IL-18 receptor belongs to IL-1R family members and The Toll/IL-1 receptor (TIR) domain receptors superfamily (21). The extracellular part is conserved to recognize the cytokine while the intracellular part containing a TIR domain orchestrates the downstream signal transduction. The architectural structure of the IL-18 receptor has a second domain that is unique and contributes to the distinct inter-receptor interaction and binding affinity, not present in the other IL-1Rs (22).
The production of IL-18 occurs in a wide array of cell types, including both hematopoietic and non-hematopoietic cells. The IL-18 precursor is present in several cell types, including circulating monocytes, resident macrophages, dendritic cells, endothelial cells, keratinocytes, osteoblasts, intestinal epithelial cells, and mesenchymal cells (23). The IL-18 mRNA or protein is also found in airway epithelial cells (24, 25). Initially synthesized as an inactive precursor, pro-IL-18 undergoes proteolytic cleavage by the intracellular cysteine protease, caspase (Casp)-1, to yield its biologically active form, IL-18, secreted by the cell. This activation process is intricately linked to the canonical inflammasome pathways, particularly Nucleotide-binding oligomerization domain leucine-rich repeat and pyrin domain-containing protein (NLRP) 3, discussed in detail in subsequent sections (17). Additionally, alternative pathways beyond inflammasome are also identified, such as Fas-mediated signaling via Casp-8 in macrophages and dendritic cells (26).
The IL-18 receptor (IL-18R) is expressed in T cells and NK cells. Non-immune cells like neurons and epithelial cells also express IL-18R that may play a role in their cellular differentiation and survival. The receptor comprises of two subunits, IL-18Rα and IL-18Rβ, forming a high-affinity binding heterodimer upon IL-18 stimulation (23). This complex triggers downstream signaling involving myeloid differentiation primary response 88 (MyD88) that activates nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) through association with the signal adaptor IL-1R-associated kinase (IRAK) 1–4 and tumor necrosis factor (TNF) receptor-associated factor (TRAF) 6 (27, 28). Inhibitors of IL-18 signaling include IL-18-binding protein (IL-18BP) and IL-37. IL-18BP can bind soluble mature IL-18 with a higher affinity and prevent IL-18 binding to IL-18R. Free IL-37 binds to IL-18α with a low affinity and then induces the recruitment of IL-1R8 to form a high-affinity complex. This complex does not recruit MyD88, thus inhibiting IL-18 signaling and inducing an anti-inflammatory effect via signal transducer and activator of transcription (STAT) 3 (23, 29, 30).
IL-18 and immunity
Innate immune response
Innate immunity serves as our first line of defense against a wide array of pathogens, regardless of prior exposure. This system includes physical barriers formed by epithelial cells, phagocytic cells such as neutrophils, macrophages, dendritic cells, and NK cells that target and kill virus-infected and tumor cells, the complement system which enhances pathogen clearance, and the cells that release cytokines that regulate immune responses and inflammation (31).
IL-18 collaborates with IL-12 to stimulate NK cells to produce IL-8, IFN-γ and TNF-α, enhancing their activities against infection and cancer whilst triggering an innate immune response (32). The significance of IL-18 in establishing NK cell activity is evident in IL-18 deficient mice, where susceptibility to infection increases due to impaired NK cell function (33). Additionally, the combined stimulation of IL-18, IL-12, and IL-15 in mice splenic NK cells is associated with the generation of memory-like NK cells, boosting their IFN-γ production when exposed to subsequent repetitive stimuli. This highlights the multifaceted roles of IL-18 in innate immunity (34). Similarly, IL-18, when acting in conjunction with IL-12, activates macrophages, enabling them to produce the crucial cytokine IFN-γ (35). This collaborative action extends to various cell types, including non-polarized T cells, Th1 cells, dendritic cells, and B cells, which can produce IFN-γ in response to the synergistic influence of IL-18 and IL-12 (16).
Airways epithelial cells are the first barrier against inhaled allergens and pathogens. Several extraneous agents, including, fungal agents such as Alternaria extract can cause epithelial damage, cell necrosis, and rapid release of IL-18 (36). This was through autophagy-dependent and Casp-1 and Casp-8-independent pathways (37). Alternaria sensitization is associated with an increased risk of asthma in children (38). A combination of IL-3 and IL-18 can stimulate mast cells and basophils to produce histamine, IL-4, and IL-13 as an innate allergic response process (39). These interactions underscore the paradigm shift from a response to an adaptive Th2 response in asthma orchestrated by IL-18.
Adaptive immune response
The adaptive immune response, also known as acquired immunity, is a specific and delayed response that provides long-lasting protection. The key players include B cells and T cells. The latter lymphocytes can be further divided into two main types: Helper T cells (CD4+) and Cytotoxic T cells (CD8+) (31).
In the adaptive immune system, IL-18 plays a pivotal role in the activation and differentiation of T cells. The combination of IL-18 and IL-12 allows a synergized IFN-γ production from T cells and B cells (19). One of the mechanisms to explain this synergistic effect was demonstrated in Th1 cells, where an increase in IL-18R mRNA expression after IL-12 stimulation was observed (40) along with the reciprocal induction/expression of IL-12R by IL-18 (16).
IL-18 further contributes to the immune response by upregulating the cytotoxic activities of NK and CD8+ T cells. These cells, when activated by IL-18, exhibit enhanced capabilities to eliminate target cells through the release of cytotoxic molecules such as perforin or by inducing apoptosis in Fas-expressing target cells (41, 42). Additionally, IL-18 is implicated in the induction of allergic inflammation, by triggering IgE production from B-cells in a CD4+ T cell-dependent process via IL-4/STAT6 signaling (43). The NKT cells that express high levels of IL-18Rα, when stimulated with IL-18 in combination with IL-2, can generate a Th2 response with IL-4, IL13 production and induction of CD40 ligand expression (44). Intriguingly, IL-18 has the potential to induce plasticity of Th1 to Th2 cells via upregulation of Th2 transcription factor GATA-binding protein 3. After repetitive stimulation with IL-18 and IL-2, Th1 cells differentiated from IL-13+ IFN-γ+ to cells producing primarily IL-13 (45).
The biology of IL-18 and its role in the immune response and asthma pathophysiology are summarized in Figure 1. Given the primary signaling pathway of IL-18 expression is via inflammasome activation, the review will next provide an overview of inflammasome biology, and the different clinically relevant triggers that activate this pathway leading to IL18 abundance in tissue.
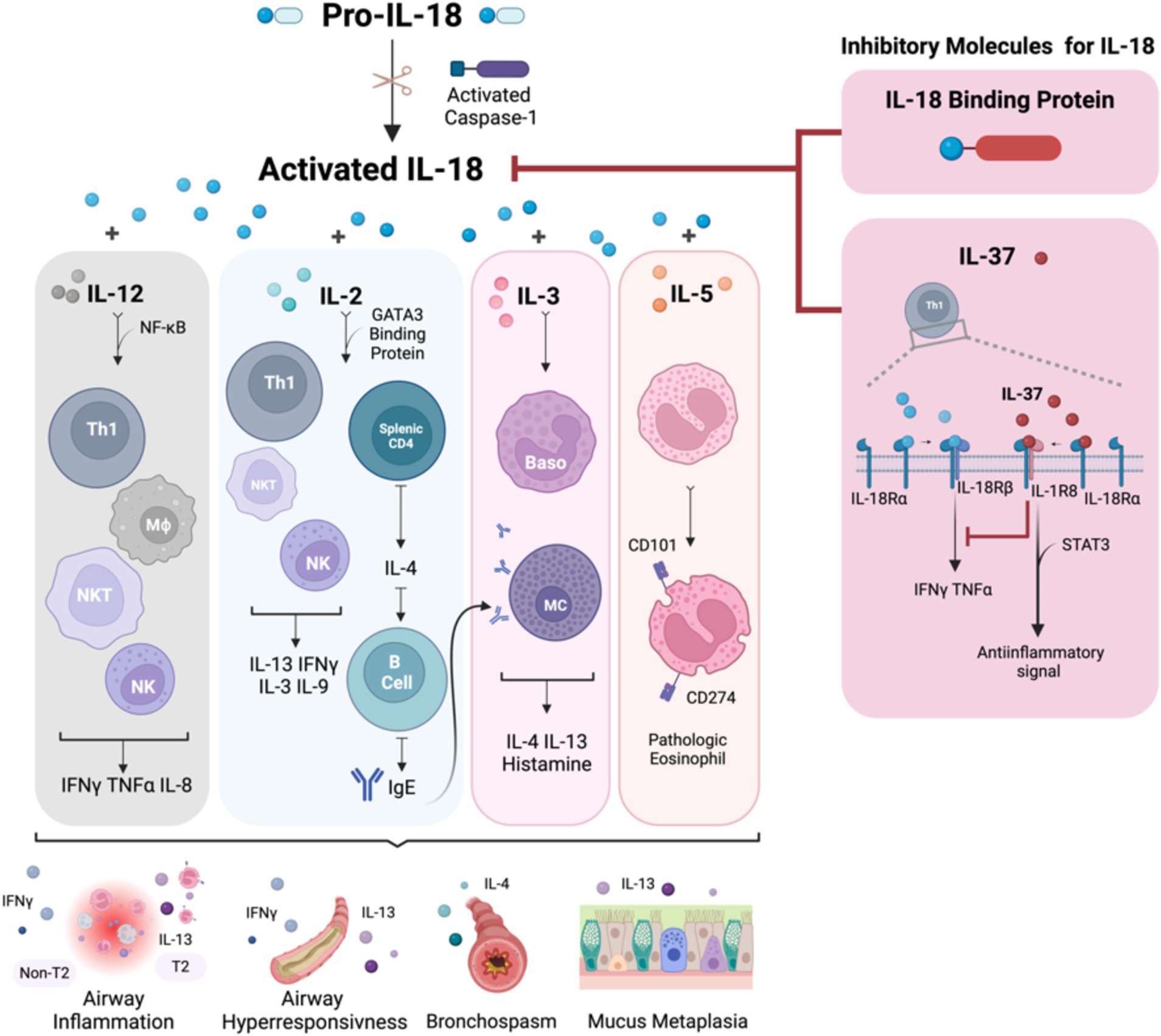
Figure 1. The summary of IL-18 biology and the immune response in association with asthma pathophysiology. IL-18 is activated by caspase-1 and is secreted by both hematopoietic and non-hematopoietic cells. Once activated, IL-18 binds to IL-18R, which is expressed on the surface of T cells, NK cells, neurons, and epithelial cells, triggering downstream inflammatory events. In the Th1 pathway, IL-18, in synergy with IL-12, stimulates Th1 cell development and enhances IFNγ expression. Conversely, in the Th2 pathway, the interaction between IL-18 and IL-2 promotes a Th2 response, which leads to the production of IL-4 and IL-13. This Th2-mediated response increases airway inflammation, hyperresponsiveness, and mucus metaplasia due to IgE production from B cells and heightened IL-13 levels from differentiated cells. Additionally, IL-18 stimulates mast cells and basophils, leading to the release of histamine, IL-4, and IL-13, further promoting a Th2 asthmatic response. IL-18 also induces eosinophils to express CD101 and CD274, transforming IL-5-responsive naive eosinophils into pathogenic eosinophils, contributing to mucus hypersecretion and airway obstruction. IL-18 signaling is regulated by the IL-18 binding protein, which neutralizes IL-18 activity, and inhibitory effects of IL-37, which binds to IL-18α, recruits IL-1R8, and forms a high-affinity complex. This complex inhibits downstream signaling from IL-18 and induces an anti-inflammatory signal via STAT3. Baso, basophil; CD, cluster of differentiation; IFN, interferon; IgE, immunoglobulin E; IL, interleukin; IL-18BP, IL-18 binding protein; MΦ, macrophage; MC, mast cell; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NK, natural killer; NKT, natural killer T-cell; STAT3, signal transducer and activator of transcription 3; Th, T helper cell; TNF, tumor necrosis factor. Created with Biorender.com.
Inflammasome
Inflammasomes are intricate cytosolic complexes that are essential components of the innate immune system. They primarily consist of three components: First, a sensory protein or the pattern recognition receptors (PRRs), including the Nucleotide oligomerization domain (NOD)-like receptors (NLRs); second, an adaptor protein called apoptosis-associated speck-like protein containing a Caspase Activation and Recruitment Domain (CARD) (ASC), and third, an enzymatic effector such as Casp1. The NLR family is characterized by the presence of a central nucleotide-binding and oligomerization (NACHT) domain, which is commonly flanked by C-terminal leucine-rich repeats (LRRs) for ligand sensing and N-terminal effector domain for mediating signal transduction. The NLRs can be further categorized into subfamilies based on the effector domains, including the NLRP with pyrin domain (PYD) and NLRC with CARD domain (46, 47). Several inflammasome moieties have been identified thus far: NLRP1, NLRP3, NLRP6/7/12, NLR family CARD domain-containing protein (NLRC) 4, retinoic acid-inducible gene I (RIG-I), absent in melanoma 2 (AIM-2) and interferon gamma-inducible protein 16 (IFI 16). The specific component and activation mechanisms vary depending on the nature of the individual protein.
As illustrated in Figure 2, when the sensory protein senses the danger signal, including pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPS), and homeostasis-altering molecular processes (HAMPs) (48), the signaling pathway is activated. ASC is recruited and interconnects with pro-Casp1 through the CARD domain, leading to oligomerization. This process triggers autoproteolysis, giving rise to active Casp1. Active Casp1 plays a central role in cleaving pro-IL-1β and pro-IL-18 into their mature forms, facilitating the release of IL-1β, IL-18, high mobility group box 1 (HMGB1) (a known DAMP), and other proteins from the cell. Casp1 also initiates a specific form of highly inflammatory programmed cell death known as pyroptosis, characterized by rapid plasma membrane rupture, leading to the release of intracellular contents. Unlike apoptosis, which is immunologically silent, pyroptosis serves as a defense mechanism against intracellular pathogens. The term “pyroptosis” derives from the Greek words “pyro,” meaning fire or fever, and “ptosis,” meaning falling, emphasizing both its inflammatory nature and the collapse of cellular integrity (49).
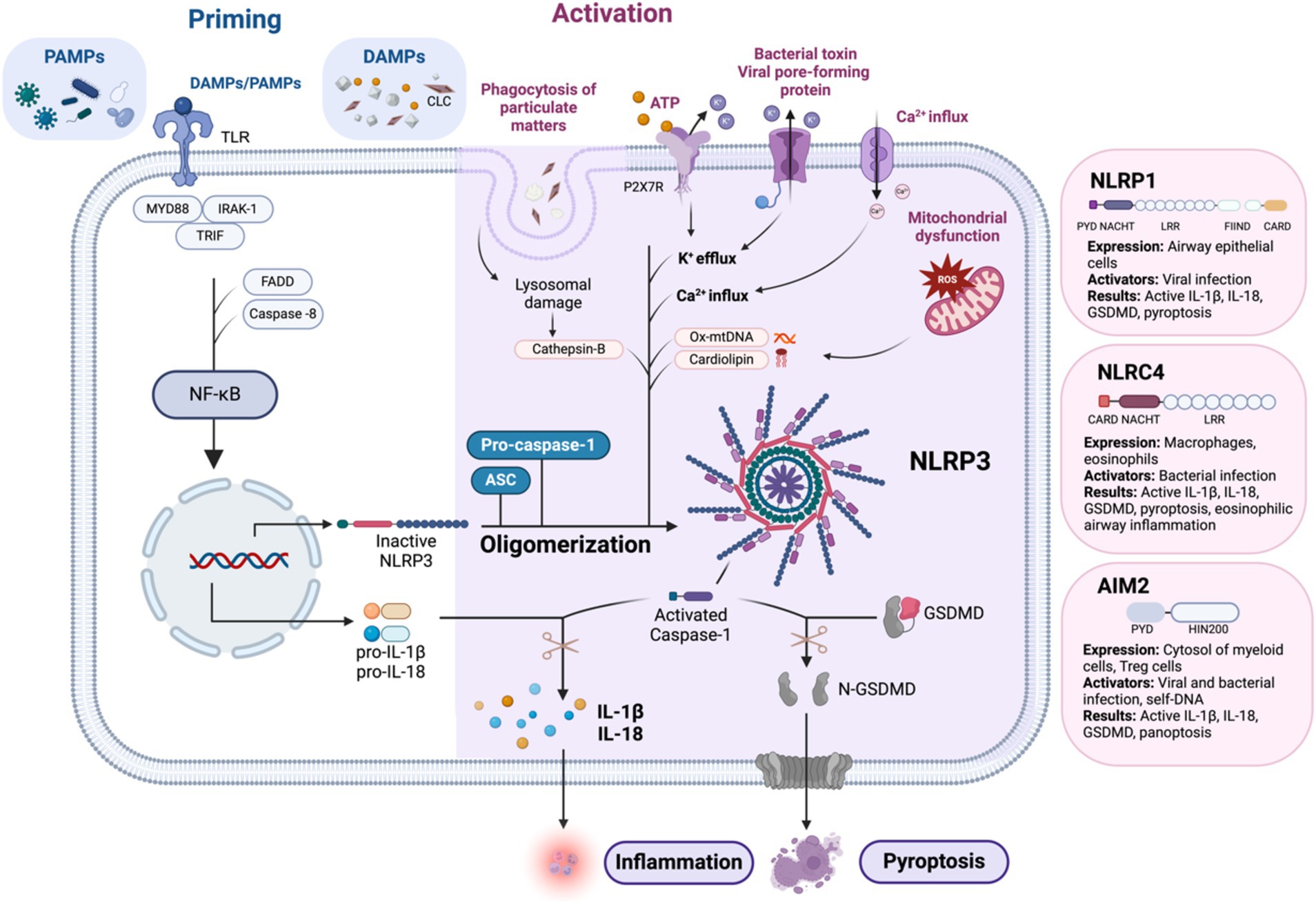
Figure 2. Mechanism of NLRP3 inflammasome activation: the canonical pathway requires two signals: priming and activation. Priming is initiated by the detection of PAMPs and DAMPs, which activate NF-κB by facilitating its translocation into the nucleus via the myddosome complex. This upregulates pro-IL-1β, pro IL-18, and NLRP3 to form the completed NLRP3 inflammasome complex with ASC and pro-Casp-1. Activation of the NLRP3 inflammasome can be triggered by several mechanisms. Phagocytosis of DAMPs can lead to lysosomal damage and the release of cathepsin B. The binding of extracellular ATP to P2X7R, an ATP-gated ion channel, triggers K+ efflux. Additionally, bacterial toxins and viral pore-forming proteins can create cell membrane pores, also leading to K+ efflux. Other mechanisms include Ca2+ influx and mitochondrial dysfunction, which result in the formation of ROS and the secretion of Ox-mtDNA and cardiolipin. Finally, the activation of caspase-1 leads to the conversion of pro-IL-1β and pro-IL-18 into their active forms. Caspase-1 also cleaves GSDMD into N-GSDMD, resulting in pyroptosis. Similar to NLRP3, activation of other airway-relevant inflammasomes, such as NLRP1, NLRC4, and AIM2, also leads to the production of active IL-1β, active IL-18, active GSDMD, and pyroptosis. AIM2, absent in melanoma 2; ASC, apoptosis-associated speck-like protein containing a CARD; ATP, adenosine triphosphate; CARD, caspase recruitment domain; CLC, Charcot-Leyden crystals; DAMPs, damage-associated molecular patterns; DNA, deoxyribonucleic acid; FADD, Fas-associated protein with death domain; FIIND, function-to-find domain; GSDMD, gasdermin D; IL, interleukin; IRAK-1, interleukin-1 receptor-associated kinase 1; LRR, leucine-rich repeat; mtDNA, mitochondrial DNA; MYD88, myeloid differentiation primary response 88; N-GSDMD, N-terminal gasdermin D; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRC4, NOD-like receptor family CARD domain-containing protein 4; NLRP1, NOD-like receptor family pyrin domain-containing protein 1; NLRP3, NOD-like receptor family pyrin domain-containing protein 3; Ox-mtDNA, oxidized mitochondrial DNA; P2X7R, purinergic receptor P2X ligand-gated ion channel 7; PAMPs, pathogen-associated molecular patterns; PYD, pyrin domain; ROS, reactive oxygen species; TLR, toll-like receptor; TRIF, TIR-domain-containing adapter-inducing interferon-β. Created with Biorender.com.
The execution of pyroptosis is mediated by the gasdermin family of proteins, particularly gasdermin D (GSDMD). Upon activation by Casp-1, GSDMD undergoes proteolytic cleavage, liberating its N-terminal domain (50). The N-terminal fragment of GSDMD inserts into the plasma membrane, forming large pores known as “pyroptotic pores.” These pores compromise membrane integrity, leading to osmotic imbalance, cellular swelling, and, ultimately, membrane rupture. Pyroptosis serves as a double-edged sword in host defense, eliminating infected cells to limit pathogen replication while also triggering an inflammatory cascade (51).
Innately, inflammasomes are known for protecting against invading pathogens and initiating adaptive immune responses. However, their dysregulation is implicated in several metabolic disorders (52), autoinflammatory diseases (53, 54), neurodegenerative diseases (55, 56) and of recent, the cytokine storm reported in COVID-19 (57). In the past decade, there has been a growing focus on the role of inflammasomes in chronic airway diseases, particularly asthma and COPD. Mutations of the genes in inflammasome pathway have been associated with eosinophilia in patients with asthma (54). Expression of the NLRP3 inflammasome has been associated with acute exacerbations of COPD (58) and neutrophilic airway inflammation, worsening lung function, and poor asthma control (59). Rhinovirus infection activates RIG-I inflammasome in asthma patients and leads to prolonged viral clearance and unresolved inflammation, demonstrated via both in-vitro and in-vivo investigations (60).
NLRP3 inflammasome
The NLPR3 inflammasome consists of the NLRP3 receptor, the adaptor protein ASC, also known as PYCARD, and Casp-1 as an effector protein. The NLRP3 receptor is a tripartite protein that contains an amino-terminal PYD, a nucleotide-binding NACHT, and a carboxy-terminal LRR domain (61). This inflammasome complex is highly expressed in myeloid cells, including monocytes, neutrophils, macrophages, and dendritic cells, associated with immune responses to various types of infection, including virus [Influenza A (62), SARS-CoV-2 (63)], Bacteria (Listeria monocytogenes) (64), and fungus [Candida albicans (65), Aspergillus fumigatus (66)]. Several PAMPs [viral RNA, muramyl dipeptide from bacteria, bacterial RNA and double-stranded RNA, and Galactosaminoglycan from Aspergillus fumigatus (67)] and DAMPs [adenosine triphosphate (ATP), uric acid crystals and amyloid-b, silica, asbestos, and alum] are identified as activating agents. In context to the current review topic, Charcot-Leyden Crystals (CLCs), that are essentially Galectin-10 sequestered protein crystals resulting from intense eosinophilic airway inflammation, act as DAMPs and trigger inflammasome NLRP3 in macrophages to release IL-1beta (the study did not assess IL-18) (68). Additionally, HAMPs such as potassium (K+) efflux from bacterial toxin, viral pore-forming protein, P2X7R activation by ATP, GSDMD pore formation, calcium (Ca2+) flux, and mitochondrial or lysosomal dysfunction activates NLRP3 (48).
The activation of the canonical NLRP3 inflammasome pathway requires two signals: priming and activation. The priming or transcriptional signal is induced through the NLRs which recognize PAMPs/DAMPs, resulting in the involvement of myddosome complex, which consists of Myeloid differentiation primary response 88 (MyD88), Interleukin-1 receptor-associated kinase 1 (IRAK-1), TIR-domain-containing adaptor-inducing interferon-β (TRIF), Fas-associated protein with death domain (FADD), and Caspase-8. This myddosome complex activates the NF-kB by translocating into the nucleus and further upregulation of NLRP3, pro-IL-1β, and pro-IL-18. Then, the second signal will activate the NLRP3 and assemble it with ASC and pro-Casp-1 to be the NLRP3 inflammasome complex. Many DAMP molecules such as CLCs (68), monosodium urate, silica, asbestos, and amyloid-β, when they are phagocytosed, their physical characteristics can cause lysosomal disruption, releasing their components, including Cathepsin B into the cytoplasm and activate the inflammasome. The PAMPs/DAMPs can trigger the NLRP3 inflammasome complex through reactive oxygen species generation and mitochondrial dysfunction. In addition, there is a noncanonical pathway that responds to intracellular lipopolysaccharides (LPS) of Gram-negative bacteria identified, which is dependent on Casp-4 and 5 (69–71).
NLRP1 inflammasome
NLRP1 was the first described inflammasome-nucleating protein, also called NACHT, LRR, and PYD domains-containing protein 1 (NALP1), that was involved in the caspase activating complex (72). Similar to other NLRs, the NLRP1 consists of the N-terminal PYD, NACHT domain, and LRRs. However, it has a distinguishing structure where these domains are followed by a function-to-find domain (FIIND) and the C-terminal CARD. The FIIND undergoes autoproteolytic cleavage that generates two fragments: N-terminal region and a C-terminal UNC5, PIDD, and ankyrins (UPA)-CARD domain that remain in an inactive state. The activation process called the functional degradation process, occurs when the N-terminal fragment is degraded by the proteasome to release the UPA-CARD fragment that forms an active inflammasome complex. The result of the activation process leads to inflammation similar to the NLRP3 inflammasome with the involvement of active IL-1β, active IL-18, active GSDMD, and pyroptosis. Expression of NLRP1 has been demonstrated mostly in non-myeloid cells, including human airway epithelial cells (73, 74), human keratinocytes of the skin, and in the lining of gastrointestinal tract (75). The first identified trigger factor for NLRP1 was ribotoxic stress from UV radiation, followed by viral proteases and viral dsRNA (48, 76).
NLRC4 inflammasome
The NLRC4 is the first inflammasome sensor identified to activate both Casp-1 and cell death. The structure comprises of NLRC4, ASC, and pro-Casp-1 assembled to form the NLRC4 inflammasome complex. Even though ASC is not deemed necessary, the NLRC4 inflammasome complex without ASC shows inefficient Casp-1 cleavage and diminished IL-1β release. The expression of NLRC4 inflammasome is evident in macrophages, eosinophils, and intestinal epithelial cells. It is known to be triggered by bacterial infection. However, the NLRC4 does not detect bacterial components directly. Activation of NLRC4 inflammasome is associated with eosinophilic airway inflammation due to its expression in human eosinophils. In fact, NLRC4-deficient mice have significantly fewer eosinophils in the bronchoalveolar lavage fluid (BALF) as compared to wild-type mice following induction of allergic airway disease (77).
AIM2 inflammasome
AIM2 terminology expands to “Absent in melanoma 2,” given it was first discovered as a tumor suppressor factor and later found to be involved in the inflammasome pathway. AIM2 is a member of the IFN-inducible HIN-200 family of proteins with an N-terminal PYD and a C-terminal HIN-200 domain. AIM2 exists as an autoinhibited conformation (PYD and the HIN-200 domains) in myeloid cells, keratinocytes, and T regulatory cells. Since its identification as a DNA sensor, AIM2 was found to mediate inflammasome response to bacterial and viral pathogens, including cytomegalovirus (CMV), human papillomavirus (HPV) and L. monocytogenes. The double-stranded DNA must be in a minimal length between 70 and 80 bp for the HIN-200 domain recognition, and the binding occurs in a sequence-independent manner. This binding leads to the structural change, which frees the PYD part to assemble with ASC and pro-Casp-1 to form the AIM2 inflammasome complex (48). Activation of AIM2 can lead to the formation of the AIM2-PANoptosome complex, which is implicated in a hybrid cell death pathway known as PANoptosis, involving the simultaneous activation of pyroptosis, apoptosis, and necroptosis in response to Francisella novicida and HSV1 infections, resulting in the release of cytokines and DAMPs (78). AIM2 inflammasome activation is also evident in COPD lungs and cigarette-exposed mice with an increase of cleaved IL-1β (79). Additionally, AIM2 has an inflammasome-independent role, as it can bind to neutrophils extracellular traps (NETs), leading to DNase-resistant nucleoprotein fibers that can serve as an autoantigen in SLE (80). This is relevant to severe asthma patients who show evidence of airway autoimmune responses (81), associated with smoking (82) and recurrent infections (83), with evidence of NETs.
IL-18 in asthma pathophysiology
Even though IL-18 is not recognized to be a major player in asthma pathobiology, several recent studies have linked IL-18 to diverse immune responses in asthma. As discussed earlier, both clinical and basic science investigations suggest IL-18 to play a significant yet complex role in T2 and non-T2 inflammation, depending on the micro-environmental cues (airways). The studies involving IL-18 with respect to asthma, both in animal models and humans, are summarized in Tables 1, 2.
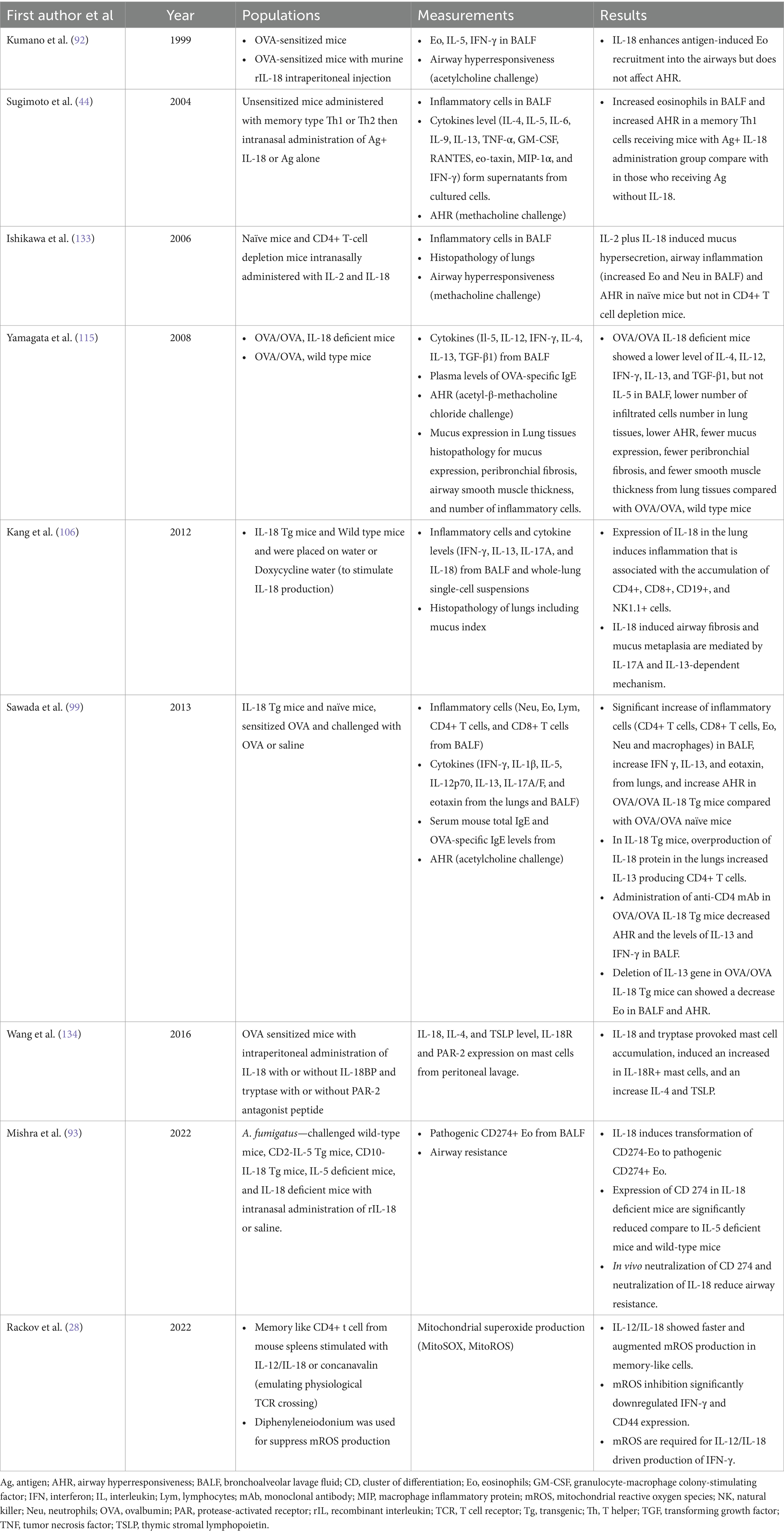
Table 1. Summary of clinical studies about IL-18 and asthma in animal models.

Table 2. Summary of clinical studies of IL-18 and asthma.
Airway inflammation
Significantly higher levels of IL-18 were reported in the serum of asthma patients during exacerbations compared to the stable state. These elevated IL-18 levels did not correspond with changes in IFN-γ levels (84). Significantly increased levels of soluble IL-18Rα complex were seen in the serum of atopic asthmatics compared to allergic non-asthmatics and healthy controls. These levels further correlated with increased IgE serum levels, and the authors suggested an antagonistic activity of IL-18Rα levels in a Th2 allergic response (85). The data from lung autopsy obtained from fatal asthma patients had significant expression of IL-18 protein and IL-18R compared to lung tissues from patients with mild asthma and “no asthma” diagnosis. The levels of eosinophils and lymphocytes but not basophils or macrophages were increased in fatal asthma. Increased numbers of activated CD8+ T cells than CD4+ T cells were seen in this population (86). In a separate severe asthma cohort, increased levels of NLRP3 pathway components were documented in sputum macrophages from the neutrophilic asthma endotype (87). Additionally, elevated mRNA levels of NLRP3 were observed in CD14+ monocytes, along with higher levels of IL-1β and IL-18 in cell culture supernatants. These measurements were taken in the non-stimulated state that showed a further significant increase following NLRP3 activation (88).
IL-5 is a well-established differentiation, growth and survival factor for eosinophils, and earlier reports indicate that IL-5 and eotaxin(s) regulate baseline resident eosinophils. However, IL-18, in combination with IL-5, contribute to its pathogenic characteristics (89). A subset of eosinophils expressing CD101 surface marker was identified as lung-specific inflammatory eosinophils (iEOS) in asthmatic mouse models (90). Again, a subset of CD101+/CD274+ double positive iEOS was identified from nasal lavage of asthma patients. The same study also demonstrates that only IL-18 (not IL-13, IL-15, IL-21, and IL-33) can promote the differentiation and transformation of IL-5 responsive naive eosinophils to pathogenic eosinophils (91). In a murine model, intraperitoneal IL-18 injection increased eosinophil recruitment into the airways (92), and intranasal rIL-18 administration also transformed CD274-eosinophils to CD274+ pathogenic eosinophils that were shown to promote mucus hypersecretion and airway obstruction (93). A recent study demonstrated that IL-18 can transdifferentiate innate lymphoid group 2 cell to atypically express ckit ligand and IL-17, particularly relevant in severe asthma patients with recurrent infections (94). Therefore, IL-18 can orchestrate chronic inflammation in severe asthma beyond the canonical T2 pathways.
Airway hyperresponsiveness
Airway hyperresponsiveness (AHR) is one of the key diagnostic features of asthma (95). In patients with asthma, even though airway inflammation is not the sole cause of AHR, eosinophilic airway inflammation can contribute to the variable degrees of AHR through the course of the disease (96). IL-18 has differential effect on eosinophilic inflammation and on AHR as demonstrated in animal models. Kumano and co-workers (93) demonstrated an enhancement of airway eosinophilia, but not AHR in sensitized mice by intraperitoneal administration of recombinant IL-18. This was likely mediated by TNF and not IL-5. Further evidence that the effect of IL-18 is through a Th1 pathway was provided by Sugimoto et al. who administered memory-type Th1 and Th2 cells to non-sensitized mice to avoid the background response of host-derived T-cells. The mice that received Th2 cells developed both airway inflammation and AHR after antigen induction, whereas mice that received Th1 cells exhibited airway inflammation but did not develop AHR. However, the co-administration of IL-18 in the Th1 cell-recipient mice induced both airway inflammation and AHR, highlighting the role of IL-18 in driving AHR, within the context of Th1 cell activity (97). It was demonstrated in a latter study that this process is associated with IL-13 and IFNγ production. Not only can exogenous IL-18 induce AHR, but also the endogenous IL-18 induced by lipopolysaccharide (LPS), can cause the same effect (98). The study in ovalbumin-sensitized and challenged transgenic mice show IL-18 to cause significant increases in AHR and airway inflammatory cells, including CD4+ T cells, CD8+ T cells, eosinophils, neutrophils, and macrophages (99).
Lung function, airway obstruction and mucus impaction
Since asthma is a disease of variable airflow obstruction, patients can have fluctuating lung function over time. However, some patients, usually with long-standing disease, a history of severe exacerbation and/or lack of appropriate treatment, might develop airway remodeling, leading to lung function decline and irreversible airflow obstruction (100, 101). The role of IL-18 or even an association with lung function in asthma is ill-defined. A few studies suggest IL-18 may underlie lower FEV1, but any direct role has not been discerned. For instance, an increase in serum IL-18 levels was documented with a decrease in FEV1 in the patients with asthma-COPD-overlap (102). Data from the Severe Asthma Research Program (SARP) cohort using machine learning validated IL18R1 protein expression in lung tissue and identified downstream NF-κB and activator protein 1 (AP-1) activity. IL-18R1 was negatively correlated to FEV1 in both the SARP and Immune Mechanisms of Severe Asthma (IMSA) cohort (103).
IL-18 may indirectly affect airway obstruction by contributing to mechanisms of mucus plugging. Indeed, mucus impaction underscores airway obstruction (104, 105). A 2012 study by Kang and colleagues found that mucus metaplasia, as well as airway fibrosis and vascular remodeling, can be induced by IL-18 via IL-13 and IL-17A cytokines and not IFN-γ. Further, IL-18 transgenic (Tg) mice that lacked IL-17A and/or IL-13 had a significant decrease in airway fibrosis and mucus metaplasia. Alternately, airway fibrosis and mucus metaplasia increased significantly in IL-18 Tg mice that lacked IFN-γ (106).
Asthma, autoimmunity, and IL-18
Autoimmunity arises when the body’s immune system mistakenly identifies its own cells as foreign, leading to the production of autoantibodies against self-structures. This self-reactivity results in sustained self-immune response and tissue damage (107). The consequence of this phenomenon is demonstrated in a variety of diseases ranging from systemic involvement, such as systemic lupus erythematosus, to organ-specific pathology, such as Crohn’s disease and Hashimoto’s thyroiditis. Even though autoimmunity is theoretically related to Th1/Th17 responses, its possible involvement in the pathogenesis/severity of Th2 diseases such as asthma and chronic rhinosinusitis is being extensively studied (107). Chronic inflammation and subsequent inflammasome signaling may underscore the development and sustenance of airway autoimmune responses described in the airways of patients with complex airways disease (107–109).
In patients with severe asthma with increased airway degranulation evident by the presence of free eosinophil granules, autoantibodies to eosinophil granule proteins such as eosinophil peroxidase (EPX) and anti-nuclear/extranuclear antigens (ANAs) have been reported (81, 109). These sputum autoantibodies were observed in an IL-13-rich micro-environment with increased levels of IL-18 (109). The autoantibodies trigger eosinophil extracellular traps (a similar event to NETs) termed EEtosis (109, 110) which is associated with the release of HMGB1 and crystallization of the periplasmic Galectin-10 to form CLCs (111, 112). The DAMPs activate inflammasomes, leading to the subsequent release of active IL-18 (113), and propagate inflammation and tissue damage beyond the canonical IL-5 and IL-13 pathways. This process leads to self-antigens and activation of self-reactive lymphocytes, resulting in the production of autoantibodies (107), which further triggers and perpetuates EETosis, resulting in a vicious cycle of chronic persistent inflammation in severe asthma (107). Inflammasome signaling and associated IL-18 release may also underlie some of the suboptimal responses documented in prototype severe eosinophilic patients treated with anti-IL-5 (10)/IL-5R biologics (12, 13, 114).
Potential therapeutic targets of IL-18 and Inflammasome in asthma
Recent research has increasingly focused on the role of IL-18 and inflammasomes in a variety of autoimmune and inflammatory diseases, including chronic lung diseases such as asthma and COPD. The IL-18 and inflammasome cascade play crucial roles in the immune response, with IL-18 being intricately involved in both T2 and non-T2 airway inflammation. This has highlighted IL-18 and related proteins as potential therapeutic targets for severe asthma beyond conventional T2 targets.
Animal models have provided encouraging data, indicating that IL-18 suppression can reduce airway inflammation, AHR, and mucus production (93, 115). Therapeutic strategies aimed at inhibiting IL-18 is either by directly blocking its signaling or target its activation/release by modulating the inflammasome pathway. Various molecules have been studied in diverse inflammatory conditions associated with the IL-18 pathways, offering potential avenues for treatment (tabulated in Table 3). For example, the humanized monoclonal antibody GSK 1070806 has been tested in renal transplant delayed graft function (116), Type 2 diabetes patients (117) and is currently in Phase II developmental pipeline for atopic dermatitis indication (NCT05999799). The search for therapeutic targets has now extended to IL-18R blocking agents and IL-18BP activity-enhancing therapies (118). IL-37, which binds to the IL-18Rα, has also shown potential in asthma treatment. Local administration of IL-37 in asthmatic mouse models reduced eosinophils in the airway and improved AHR (119). Another promising therapeutic, APB-R3, a long-acting recombinant human IL-18BP, has shown effectiveness in reducing liver inflammation and splenomegaly in a model of the macrophage activation syndrome and controlled skin inflammation in atopic dermatitis mice model (120). Tadekinig alfa, a recombinant IL-18 binding protein, has completed Phase II trials in adult-onset Still’s disease, showing early signs of efficacy (121).
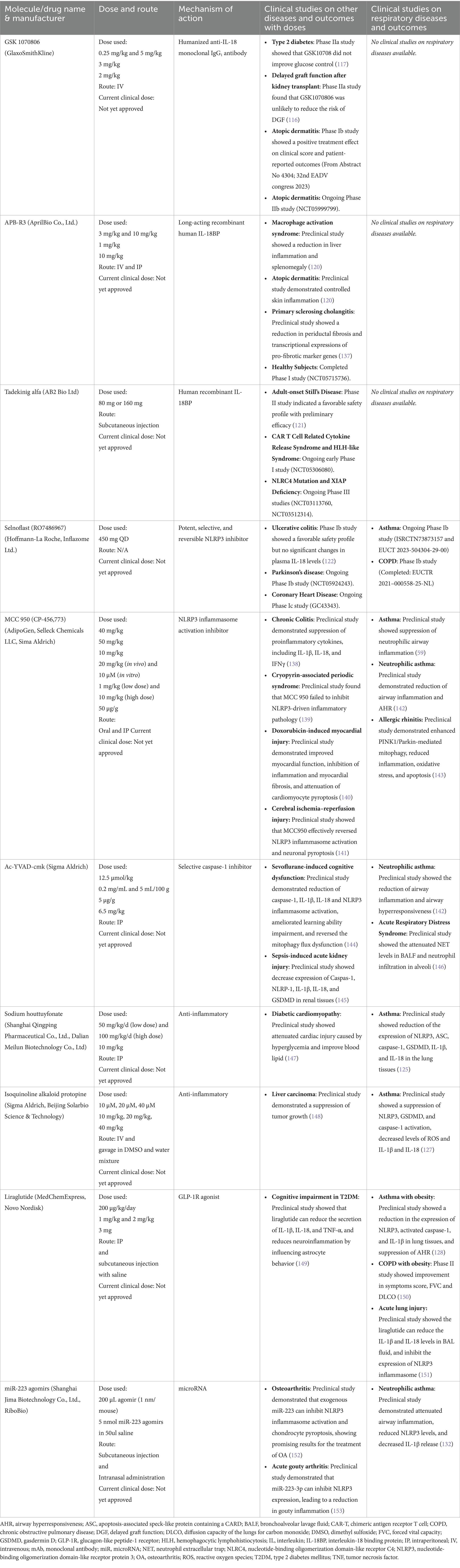
Table 3. Summary of clinical studies targeting IL-18/inflammasome.
Inflammasomes, particularly NLRP3, that cleaves precursor forms of IL-1β and IL-18 into their biologically active forms, have also emerged as therapeutic targets. Numerous agents are under investigation, including Selnoflast, a potent, selective, and reversible NLRP3 blocker, which has completed a Phase Ib study in ulcerative colitis patients (122). Ongoing studies are exploring its potential in diseases such as Parkinson’s (NCT05924243), COPD, and asthma (ISRCTN73873157). Another small-molecule inhibitor, MCC 950 (CP-456,773), a diaryl sulfonylurea-containing compound, that specifically targets NLRP3 inflammasome activation (123) documented significant reductions in NLRP3 and IL-1β production in asthmatic mice (124). Furthermore, sodium houttuyfonate, derived from the Chinese herb Houttuynia cordata, has demonstrated efficacy in reducing the expression of NLRP3, ASC, caspase-1, GSDMD, IL-1β, and IL-18 in the lung tissues of asthmatic mice (125). Similarly, the isoquinoline alkaloid protopine, an anti-inflammatory agent (126), has been shown to reduce airway inflammation in asthmatic rats by inhibiting the TLR4/NF-κB signaling pathways, leading to the suppression of NLRP3, gasdermin D, and caspase-1 activation, along with decreased levels of reactive oxygen species (ROS), IL-1β and IL-18 (127).
Another promising therapeutic approach involves the use of glucagon-like peptide-1 receptor (GLP-1R) agonists, initially approved as anti-diabetic and anti-obesity drugs, have been studied for their potential benefits in obesity-related asthma. Obesity is a major risk factor and disease modifier in asthma (125), and studies in obese asthmatic mouse have revealed that AHR can be NLRP3-dependent. GLP-1R agonists have been shown to suppress peri bronchial inflammation and reduce the expression of NLRP3, activated caspase-1, and IL-1β in lung tissues (128). Additionally, miR-223, a microRNA with evolutionary anti-inflammatory effects, particularly in the lungs, has shown potential as a therapeutic option (129). The microRNA, a non-coding RNA, can function to control the expression of target genes at the post transcriptional level. Overexpression of miR-223 is linked to decreased NLRP3 and NF-κB activity in porcine lungs (130) and bronchial epithelial cells (131). Treatment with miR-223 agomirs in neutrophilic asthmatic mouse models attenuates airway inflammation, reduces NLRP3 levels, and decreases IL-1β release, suggesting miR-223 as a potential therapeutic candidate for severe non-T2 asthma (132).
Conclusion
Ongoing investigations in IL-18 and inflammasomes have revealed their critical roles in the pathophysiology of asthma, particularly in severe forms of the disease. IL-18, traditionally seen as a minor player compared to the key T2 cytokines like IL-5, IL-4, and IL-13, have emerged as a key cytokine involved in both T2 and non-T2 inflammation. Its role in promoting infections, airway inflammation and airway remodeling, as well as its contribution to autoimmune responses within the airways, underscores the complexity of asthma beyond the conventional Th2 paradigm. In this review we have highlighted the multifaceted involvement of IL-18 in asthma, from its interactions with other cytokines to its activation through inflammasome pathways, particularly NLRP3. The evidence linking IL-18 to both airway inflammation, mucus plugging and AHR emphasizes its significance in asthma pathogenesis. Additionally, IL-18’s involvement in autoimmune processes introduces a new dimension to understanding severe asthma, particularly in cases where traditional therapies targeting Th2 inflammation have shown limited effectiveness (107). Furthermore, the current review sheds light on potential therapeutic targets. The identification of novel therapies targeting IL-18 and the associated inflammasome pathways offer promising avenues for treating severe and refractory asthma, addressing the limitations of current biologics. In conclusion, IL-18 is not just an ancillary cytokine in asthma but one of the key components in its pathophysiology, particularly in severe cases. A deeper understanding of its intricate role within the broader network of immune responses can guide the development of more effective treatments, offering new hope for patients with this challenging condition.
Author contributions
ST: Writing – original draft. AN: Writing – original draft. EV: Writing – original draft. PN: Writing – review & editing. MM: Conceptualization, Funding acquisition, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Financial support from the endowed chair funding (AstraZeneca Chair in Respiratory Diseases) available to Dr. Mukherjee.
Acknowledgments
MM holds the AstraZeneca Chair in Respiratory Diseases. PN is supported by the Frederick E. Hargreave Teva Innovation Chair in Airway Diseases. We acknowledge Ms. Yinglan Xie for her help and guidance with creating the figures on Biorender.com.
Conflict of interest
MM reports research grants from Sanofi, Methapharm Specialty Pharamceuticals and Mirimus, consulting fees from AstraZeneca, Sanofi, Respiplus, GSK, Mirimus. PN reports research grants from AstraZeneca, Teva, Sanofi, Foresee, Roche, Genentech and consulting fees from AstraZeneca, Teva, Sanofi, GSK, Methapharm, Arrowhead pharma.
The remaining authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wenzel, S. Severe asthma: from characteristics to phenotypes to endotypes. Clin Exp Allergy. (2012) 42:650–8. doi: 10.1111/j.1365-2222.2011.03929.x
2. Wenzel, SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. (2012) 18:716–25. doi: 10.1038/nm.2678
3. Hekking, PW, Wener, RR, Amelink, M, Zwinderman, AH, Bouvy, ML, and Bel, EH. The prevalence of severe refractory asthma. J Allergy Clin Immunol. (2015) 135:896–902. doi: 10.1016/j.jaci.2014.08.042
4. Fardet, L, Kassar, A, Cabane, J, and Flahault, A. Corticosteroid-induced adverse events in adults: frequency, screening and prevention. Drug Saf. (2007) 30:861–81. doi: 10.2165/00002018-200730100-00005
5. Holgate, ST. Pathogenesis of asthma. Clin Exp Allergy. (2008) 38:872–97. doi: 10.1111/j.1365-2222.2008.02971.x
6. Brusselle, GG, and Koppelman, GH. Biologic therapies for severe asthma. N Engl J Med. (2022) 386:157–71. doi: 10.1056/NEJMra2032506
7. Svenningsen, S, and Nair, P. Asthma endotypes and an overview of targeted therapy for asthma. Front Med. (2017) 4:158. doi: 10.3389/fmed.2017.00158
8. Nair, P. What is an “eosinophilic phenotype” of asthma? J Allergy Clin Immunol. (2013) 132:81–3. doi: 10.1016/j.jaci.2013.05.007
9. Tang, M, Charbit, AR, Johansson, MW, Jarjour, NN, Denlinger, LC, Raymond, WW, et al. Utility of eosinophil peroxidase as a biomarker of eosinophilic inflammation in asthma. J Allergy Clin Immunol. (2024) 154:580–591.e6. doi: 10.1016/j.jaci.2024.03.023
10. Bhalla, A, Zhao, N, Rivas, DD, Ho, T, Perez de Llano, L, Mukherjee, M, et al. Exacerbations of severe asthma while on anti-IL-5 biologics. J Investig Allergol Clin Immunol. (2020) 30:307–16. doi: 10.18176/jiaci.0628
11. McGregor, MC, Krings, JG, Nair, P, and Castro, M. Role of biologics in asthma. Am J Respir Crit Care Med. (2019) 199:433–45. doi: 10.1164/rccm.201810-1944CI
12. Mukherjee, M, Forero, DF, Tran, S, Boulay, ME, Bertrand, M, Bhalla, A, et al. Suboptimal treatment response to anti-IL-5 monoclonal antibodies in severe eosinophilic asthmatics with airway autoimmune phenomena. Eur Respir J. (2020) 56:2000117. doi: 10.1183/13993003.00117-2020
13. Mukherjee, M, Huang, C, Venegas-Garrido, C, Zhang, K, Bhalla, A, Ju, X, et al. Benralizumab normalizes sputum eosinophilia in severe asthma uncontrolled by anti-IL-5 antibodies: a single-blind, placebo-controlled clinical trial. Am J Respir Crit Care Med. (2023) 208:1330–5. doi: 10.1164/rccm.202308-1413LE
14. Hekking, PP, Zhang, K, Garrido, CPV, Lopez-Rodriguez, R, Kjarsgaard, M, Mukherjee, M, et al. Sputum cytokines associated with raised FeNO after anti-IL5 biologic therapy in severe asthma. Allergy. (2024) 79:2244–7. doi: 10.1111/all.16011
15. Morimoto, C, Matsumoto, H, Tajiri, T, Gon, Y, Ito, R, Hashimoto, S, et al. High serum free IL-18 is associated with decreased omalizumab efficacy: findings from a 2-year omalizumab treatment study. J Asthma. (2021) 58:1133–42. doi: 10.1080/02770903.2020.1766061
16. Nakanishi, K. Unique action of interleukin-18 on T cells and other immune cells. Front Immunol. (2018) 9:763. doi: 10.3389/fimmu.2018.00763
17. Schroder, K, and Tschopp, J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
18. Okamura, H, Tsutsi, H, Komatsu, T, Yutsudo, M, Hakura, A, Tanimoto, T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. (1995) 378:88–91. doi: 10.1038/378088a0
19. Nakanishi, K, Yoshimoto, T, Tsutsui, H, and Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. (2001) 19:423–74. doi: 10.1146/annurev.immunol.19.1.423
20. Dinarello, CA, Novick, D, Kim, S, and Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front Immunol. (2013) 4:289. doi: 10.3389/fimmu.2013.00289
21. Zhou, J, Xiao, Y, Ren, Y, Ge, J, and Wang, X. Structural basis of the IL-1 receptor TIR domain-mediated IL-1 signaling. iScience. (2022) 25:104508. doi: 10.1016/j.isci.2022.104508
22. Tsutsumi, N, Kimura, T, Arita, K, Ariyoshi, M, Ohnishi, H, Yamamoto, T, et al. The structural basis for receptor recognition of human interleukin-18. Nat Commun. (2014) 5:5340. doi: 10.1038/ncomms6340
23. Kaplanski, G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol Rev. (2018) 281:138–53. doi: 10.1111/imr.12616
24. Swain, SL. Interleukin 18: tipping the balance towards a T helper cell 1 response. J Exp Med. (2001) 194:F11–4. doi: 10.1084/jem.194.3.F11
25. Cameron, LA, Taha, RA, Tsicopoulos, A, Kurimoto, M, Olivenstein, R, Wallaert, B, et al. Airway epithelium expresses interleukin-18. Eur Respir J. (1999) 14:553–9. doi: 10.1034/j.1399-3003.1999.14c12.x
26. Bossaller, L, Chiang, PI, Schmidt-Lauber, C, Ganesan, S, Kaiser, WJ, Rathinam, VA, et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol. (2012) 189:5508–12. doi: 10.4049/jimmunol.1202121
27. Yasuda, K, Nakanishi, K, and Tsutsui, H. Interleukin-18 in health and disease. Int J Mol Sci. (2019) 20:649. doi: 10.3390/ijms20030649
28. Rackov, G, Tavakoli Zaniani, P, Colomo Del Pino, S, Shokri, R, Monserrat, J, Alvarez-Mon, M, et al. Mitochondrial reactive oxygen is critical for IL-12/IL-18-induced IFN-γ production by CD4(+) T cells and is regulated by Fas/FasL signaling. Cell Death Dis. (2022) 13:531. doi: 10.1038/s41419-022-04907-5
29. Su, Z, and Tao, X. Current understanding of IL-37 in human health and disease. Front Immunol. (2021) 12:696605. doi: 10.3389/fimmu.2021.696605
30. Baggio, C, Bindoli, S, Guidea, I, Doria, A, Oliviero, F, and Sfriso, P. IL-18 in autoinflammatory diseases: focus on adult onset still disease and macrophages activation syndrome. Int J Mol Sci. (2023) 24:11125. doi: 10.3390/ijms241311125
31. Parkin, J, and Cohen, B. An overview of the immune system. Lancet. (2001) 357:1777–89. doi: 10.1016/S0140-6736(00)04904-7
32. Poznanski, SM, Lee, AJ, Nham, T, Lusty, E, Larché, MJ, Lee, DA, et al. Combined stimulation with interleukin-18 and interleukin-12 potently induces interleukin-8 production by natural killer cells. J Innate Immun. (2017) 9:511–25. doi: 10.1159/000477172
33. Freeman, BE, Raué, HP, Hill, AB, and Slifka, MK. Cytokine-mediated activation of NK cells during viral infection. J Virol. (2015) 89:7922–31. doi: 10.1128/JVI.00199-15
34. Cooper, MA, Elliott, JM, Keyel, PA, Yang, L, Carrero, JA, and Yokoyama, WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci USA. (2009) 106:1915–9. doi: 10.1073/pnas.0813192106
35. Munder, M, Mallo, M, Eichmann, K, and Modolell, M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J Exp Med. (1998) 187:2103–8. doi: 10.1084/jem.187.12.2103
36. Murai, H, Qi, H, Choudhury, B, Wild, J, Dharajiya, N, Vaidya, S, et al. Alternaria-induced release of IL-18 from damaged airway epithelial cells: an NF-κB dependent mechanism of Th2 differentiation? PLoS One. (2012) 7:e30280. doi: 10.1371/journal.pone.0030280
37. Murai, H, Okazaki, S, Hayashi, H, Kawakita, A, Hosoki, K, Yasutomi, M, et al. Alternaria extract activates autophagy that induces IL-18 release from airway epithelial cells. Biochem Biophys Res Commun. (2015) 464:969–74. doi: 10.1016/j.bbrc.2015.05.076
38. Halonen, M, Stern, DA, Wright, AL, Taussig, LM, and Martinez, FD. Alternaria as a major allergen for asthma in children raised in a desert environment. Am J Respir Crit Care Med. (1997) 155:1356–61. doi: 10.1164/ajrccm.155.4.9105079
39. Yoshimoto, T, Tsutsui, H, Tominaga, K, Hoshino, K, Okamura, H, Akira, S, et al. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc Natl Acad Sci USA. (1999) 96:13962–6. doi: 10.1073/pnas.96.24.13962
40. Yoshimoto, T, Takeda, K, Tanaka, T, Ohkusu, K, Kashiwamura, S, Okamura, H, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. (1998) 161:3400–7. doi: 10.4049/jimmunol.161.7.3400
41. Kägi, D, Ledermann, B, Bürki, K, Zinkernagel, RM, and Hengartner, H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. (1996) 14:207–32. doi: 10.1146/annurev.immunol.14.1.207
42. Okamoto, I, Kohno, K, Tanimoto, T, Ikegami, H, and Kurimoto, M. Development of CD8+ effector T cells is differentially regulated by IL-18 and IL-12. J Immunol. (1999) 162:3202–11. doi: 10.4049/jimmunol.162.6.3202
43. Yoshimoto, T, Mizutani, H, Tsutsui, H, Noben-Trauth, N, Yamanaka, K, Tanaka, M, et al. IL-18 induction of IgE: dependence on CD4+ T cells, IL-4 and STAT6. Nat Immunol. (2000) 1:132–7. doi: 10.1038/77811
44. Yoshimoto, T, Min, B, Sugimoto, T, Hayashi, N, Ishikawa, Y, Sasaki, Y, et al. Nonredundant roles for CD1d-restricted natural killer T cells and conventional CD4+ T cells in the induction of immunoglobulin E antibodies in response to interleukin 18 treatment of mice. J Exp Med. (2003) 197:997–1005. doi: 10.1084/jem.20021701
45. Nakahira, M, and Nakanishi, K. Requirement of GATA-binding protein 3 for II13 gene expression in IL-18-stimulated Th1 cells. Int Immunol. (2011) 23:761–72. doi: 10.1093/intimm/dxr087
46. Franchi, L, Warner, N, Viani, K, and Nuñez, G. Function of nod-like receptors in microbial recognition and host defense. Immunol Rev. (2009) 227:106–28. doi: 10.1111/j.1600-065X.2008.00734.x
47. Lim, RR, Wieser, ME, Ganga, RR, Barathi, VA, Lakshminarayanan, R, Mohan, RR, et al. NOD-like receptors in the eye: uncovering its role in diabetic retinopathy. Int J Mol Sci. (2020) 21:899. doi: 10.3390/ijms21030899
48. Barnett, KC, Li, S, Liang, K, and Ting, JPY. A 360° view of the inflammasome: mechanisms of activation, cell death, and diseases. Cell. (2023) 186:2288–312. doi: 10.1016/j.cell.2023.04.025
49. Yu, P, Zhang, X, Liu, N, Tang, L, Peng, C, and Chen, X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. (2021) 6:128. doi: 10.1038/s41392-021-00507-5
50. Liu, T, Liu, S, Rui, X, Cao, Y, Hecker, J, Guo, F, et al. Gasdermin B, an asthma-susceptibility gene, promotes MAVS-TBK1 signalling and airway inflammation. Eur Respir J. (2024) 63:2301232. doi: 10.1183/13993003.01232-2023
51. Panek, I, Liczek, M, Gabryelska, A, Rakoczy, I, Kuna, P, and Panek, M. Inflammasome signalling pathway in the regulation of inflammation – its involvement in the development and exacerbation of asthma and chronic obstructive pulmonary disease. Postepy Dermatol Alergol. (2023) 40:487–95. doi: 10.5114/ada.2022.118077
52. Sharma, BR, and Kanneganti, TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol. (2021) 22:550–9. doi: 10.1038/s41590-021-00886-5
53. Moltrasio, C, Romagnuolo, M, and Marzano, AV. NLRP3 inflammasome and NLRP3-related autoinflammatory diseases: from cryopyrin function to targeted therapies. Front Immunol. (2022) 13:1007705. doi: 10.3389/fimmu.2022.1007705
54. Alotaibi, BM, Lopez Rodriguez, R, Garrido, CV, Gonzalez Bravo, L, Khalidi, N, and Nair, P. Autoinflammatory gene mutations associated with eosinophilia and asthma. Allergy Asthma Clin Immunol. (2023) 19:76. doi: 10.1186/s13223-023-00837-9
55. Qin, Y, Qiu, J, Wang, P, Liu, J, Zhao, Y, Jiang, F, et al. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson's disease. Brain Behav Immun. (2021) 91:324–38. doi: 10.1016/j.bbi.2020.10.010
56. Anderson, FL, Biggs, KE, Rankin, BE, and Havrda, MC. NLRP3 inflammasome in neurodegenerative disease. Transl Res. (2023) 252:21–33. doi: 10.1016/j.trsl.2022.08.006
57. van den Berg, DF, and Te Velde, AA. Severe COVID-19: NLRP3 inflammasome dysregulated. Front Immunol. (2020) 11:1580. doi: 10.3389/fimmu.2020.01580
58. Nachmias, N, Langier, S, Brzezinski, RY, Siterman, M, Stark, M, Etkin, S, et al. NLRP3 inflammasome activity is upregulated in an in-vitro model of COPD exacerbation. PLoS One. (2019) 14:e0214622. doi: 10.1371/journal.pone.0214622
59. Kim, RY, Pinkerton, JW, Essilfie, AT, Robertson, AAB, Baines, KJ, Brown, AC, et al. Role for NLRP3 inflammasome-mediated, IL-1beta-dependent responses in severe, steroid-resistant asthma. Am J Respir Crit Care Med. (2017) 196:283–97. doi: 10.1164/rccm.201609-1830OC
60. Radzikowska, U, Eljaszewicz, A, Tan, G, Stocker, N, Heider, A, Westermann, P, et al. Rhinovirus-induced epithelial RIG-I inflammasome suppresses antiviral immunity and promotes inflammation in asthma and COVID-19. Nat Commun. (2023) 14:2329. doi: 10.1038/s41467-023-37470-4
61. Theofani, E, Semitekolou, M, Morianos, I, Samitas, K, and Xanthou, G. Targeting NLRP3 inflammasome activation in severe asthma. J Clin Med. (2019) 8:1615. doi: 10.3390/jcm8101615
62. Pandey, KP, and Zhou, Y. Influenza a virus infection activates NLRP3 inflammasome through trans-Golgi network dispersion. Viruses. (2022) 14:88. doi: 10.3390/v14010088
63. Xu, H, Akinyemi, IA, Chitre, SA, Loeb, JC, Lednicky, JA, McIntosh, MT, et al. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology. (2022) 568:13–22. doi: 10.1016/j.virol.2022.01.003
64. Gao, A, Tang, H, Zhang, Q, Liu, R, Wang, L, Liu, Y, et al. Mst1/2-ALK promotes NLRP3 inflammasome activation and cell apoptosis during Listeria monocytogenes infection. J Microbiol. (2021) 59:681–92. doi: 10.1007/s12275-021-0638-2
65. Lian, H, Fang, X, Li, Q, Liu, S, Wei, Q, Hua, X, et al. NLRP3 Inflammasome-mediated pyroptosis pathway contributes to the pathogenesis of Candida albicans keratitis. Front Med. (2022) 9:845129. doi: 10.3389/fmed.2022.845129
66. Yang, X, Zhao, G, Yan, J, Xu, R, Che, C, Zheng, H, et al. Pannexin 1 channels contribute to IL-1β expression via NLRP3/Caspase-1 inflammasome in Aspergillus Fumigatus keratitis. Curr Eye Res. (2019) 44:716–25. doi: 10.1080/02713683.2019.1584321
67. Briard, B, Fontaine, T, Samir, P, Place, DE, Muszkieta, L, Malireddi, RKS, et al. Galactosaminogalactan activates the inflammasome to provide host protection. Nature. (2020) 588:688–92. doi: 10.1038/s41586-020-2996-z
68. Rodríguez-Alcázar, JF, Ataide, MA, Engels, G, Schmitt-Mabmunyo, C, Garbi, N, Kastenmüller, W, et al. Charcot-Leyden crystals activate the NLRP3 inflammasome and cause IL-1β inflammation in human macrophages. J Immunol. (2019) 202:550–8. doi: 10.4049/jimmunol.1800107
69. Kodi, T, Sankhe, R, Gopinathan, A, Nandakumar, K, and Kishore, A. New insights on NLRP3 inflammasome: mechanisms of activation, inhibition, and epigenetic regulation. J Neuroimmune Pharmacol. (2024) 19:7. doi: 10.1007/s11481-024-10101-5
70. Matikainen, S, Nyman, TA, and Cypryk, W. Function and regulation of noncanonical Caspase-4/5/11 inflammasome. J Immunol. (2020) 204:3063–9. doi: 10.4049/jimmunol.2000373
71. Rathinam, VAK, Zhao, Y, and Shao, F. Innate immunity to intracellular LPS. Nat Immunol. (2019) 20:527–33. doi: 10.1038/s41590-019-0368-3
72. Martinon, F, Burns, K, and Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
73. Robinson, KS, Teo, DET, Tan, KS, Toh, GA, Ong, HH, Lim, CK, et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science. (2020) 370:eaay2002. doi: 10.1126/science.aay2002
74. Planès, R, Pinilla, M, Santoni, K, Hessel, A, Passemar, C, Lay, K, et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol Cell. (2022) 82:2385–400.e9. doi: 10.1016/j.molcel.2022.04.033
75. Barry, K, Murphy, C, and Mansell, A. NLRP1-A CINDERELLA STORY: a perspective of recent advances in NLRP1 and the questions they raise. Commun Biol. (2023) 6:1274. doi: 10.1038/s42003-023-05684-3
76. Bauernfried, S, Scherr, MJ, Pichlmair, A, Duderstadt, KE, and Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science. (2021) 371:eabd0811. doi: 10.1126/science.abd0811
77. Akkaya, I, Oylumlu, E, Ozel, I, Uzel, G, Durmus, L, and Ciraci, C. NLRC4 inflammasome-mediated regulation of eosinophilic functions. Immune Netw. (2021) 21:e42. doi: 10.4110/in.2021.21.e42
78. Pandian, N, and Kanneganti, TD. PANoptosis: a unique innate immune inflammatory cell death modality. J Immunol. (2022) 209:1625–33. doi: 10.4049/jimmunol.2200508
79. Tran, HB, Hamon, R, Jersmann, H, Ween, MP, Asare, P, Haberberger, R, et al. AIM2 nuclear exit and inflammasome activation in chronic obstructive pulmonary disease and response to cigarette smoke. J Inflamm. (2021) 18:19. doi: 10.1186/s12950-021-00286-4
80. Antiochos, B, Trejo-Zambrano, D, Fenaroli, P, Rosenberg, A, Baer, A, Garg, A, et al. The DNA sensors AIM2 and IFI16 are SLE autoantigens that bind neutrophil extracellular traps. eLife. (2022) 11:11. doi: 10.7554/eLife.72103
81. Salter, B, Zhao, N, Son, K, Tan, NS, Dvorkin-Gheva, A, Radford, K, et al. Airway autoantibodies are determinants of asthma severity. Eur Respir J. (2022) 60:2200442. doi: 10.1183/13993003.00442-2022
82. Klein, DK, Silberbrandt, A, Frøssing, L, Hvidtfeldt, M, von Bülow, A, Nair, P, et al. Impact of former smoking exposure on airway eosinophilic activation and autoimmunity in patients with severe asthma. Eur Respir J. (2022) 60:2102446. doi: 10.1183/13993003.02446-2021
83. Son, K, Miyasaki, K, Salter, B, Loukov, D, Chon, J, Zhao, N, et al. Autoantibody-mediated macrophage dysfunction in patients with severe asthma with airway infections. Am J Respir Crit Care Med. (2023) 207:427–37. doi: 10.1164/rccm.202206-1183OC
84. Tanaka, H, Miyazaki, N, Oashi, K, Teramoto, S, Shiratori, M, Hashimoto, M, et al. IL-18 might reflect disease activity in mild and moderate asthma exacerbation. J Allergy Clin Immunol. (2001) 107:331–6. doi: 10.1067/mai.2001.112275
85. Imaoka, H, Takenaka, S, Kawayama, T, Oda, H, Kaku, Y, Matsuoka, M, et al. Increased serum levels of soluble IL-18 receptor complex in patients with allergic asthma. Allergol Int. (2013) 62:513–5. doi: 10.2332/allergolint.13-LE-0548
86. Oda, H, Kawayama, T, Imaoka, H, Sakazaki, Y, Kaku, Y, Okamoto, M, et al. Interleukin-18 expression, CD8(+) T cells, and eosinophils in lungs of nonsmokers with fatal asthma. Ann Allergy Asthma Immunol. (2014) 112:23–8.e1. doi: 10.1016/j.anai.2013.09.004
87. Simpson, JL, Phipps, S, Baines, KJ, Oreo, KM, Gunawardhana, L, and Gibson, PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J. (2014) 43:1067–76. doi: 10.1183/09031936.00105013
88. Theofani, E, Semitekolou, M, Samitas, K, Mais, A, Galani, IE, Triantafyllia, V, et al. TFEB signaling attenuates NLRP3-driven inflammatory responses in severe asthma. Allergy. (2022) 77:2131–46. doi: 10.1111/all.15221
89. Kandikattu, HK, Upparahalli Venkateshaiah, S, and Mishra, A. Synergy of interleukin (IL)-5 and IL-18 in eosinophil mediated pathogenesis of allergic diseases. Cytokine Growth Factor Rev. (2019) 47:83–98. doi: 10.1016/j.cytogfr.2019.05.003
90. Mesnil, C, Raulier, S, Paulissen, G, Xiao, X, Birrell, MA, Pirottin, D, et al. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest. (2016) 126:3279–95. doi: 10.1172/JCI85664
91. Venkateshaiah, SU, Mishra, A, Manohar, M, Verma, AK, Rajavelu, P, Niranjan, R, et al. A critical role for IL-18 in transformation and maturation of naive eosinophils to pathogenic eosinophils. J Allergy Clin Immunol. (2018) 142:301–5. doi: 10.1016/j.jaci.2018.02.011
92. Kumano, K, Nakao, A, Nakajima, H, Hayashi, F, Kurimoto, M, Okamura, H, et al. Interleukin-18 enhances antigen-induced eosinophil recruitment into the mouse airways. Am J Respir Crit Care Med. (1999) 160:873–8. doi: 10.1164/ajrccm.160.3.9805026
93. Mishra, A, Majid, D, Kandikattu, HK, Yadavalli, CS, and Upparahalli, VS. Role of IL-18-transformed CD274-expressing eosinophils in promoting airway obstruction in experimental asthma. Allergy. (2022) 77:1165–79. doi: 10.1111/all.15180
94. Ju, X, Emami Fard, N, Bhalla, A, Xiao, M, Dvorkin-Gheva, A, Radford, K, et al. Sputum c-kit-positive ILC2 in severe asthma express IL-17A: evidence of ILC2 to ILC3 plasticity. Sci Transl Med. (2024)
95. Nair, P, Martin, JG, Cockcroft, DC, Dolovich, M, Lemiere, C, Boulet, LP, et al. Airway hyperresponsiveness in asthma: measurement and clinical relevance. J Allergy Clin Immunol Pract. (2017) 5:649–59.e2. doi: 10.1016/j.jaip.2016.11.030
96. O'Byrne, PM, and Inman, MD. Airway hyperresponsiveness. Chest. (2003) 123:411s–6s. doi: 10.1378/chest.123.3_suppl.411S
97. Sugimoto, T, Ishikawa, Y, Yoshimoto, T, Hayashi, N, Fujimoto, J, and Nakanishi, K. Interleukin 18 acts on memory T helper cells type 1 to induce airway inflammation and hyperresponsiveness in a naive host mouse. J Exp Med. (2004) 199:535–45. doi: 10.1084/jem.20031368
98. Hayashi, N, Yoshimoto, T, Izuhara, K, Matsui, K, Tanaka, T, and Nakanishi, K. T helper 1 cells stimulated with ovalbumin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-gamma and IL-13 production. Proc Natl Acad Sci USA. (2007) 104:14765–70. doi: 10.1073/pnas.0706378104
99. Sawada, M, Kawayama, T, Imaoka, H, Sakazaki, Y, Oda, H, Takenaka, S, et al. IL-18 induces airway hyperresponsiveness and pulmonary inflammation via CD4+ T cell and IL-13. PLoS One. (2013) 8:e54623. doi: 10.1371/journal.pone.0054623
100. O'Byrne, PM, Pedersen, S, Lamm, CJ, Tan, WC, and Busse, WW. Severe exacerbations and decline in lung function in asthma. Am J Respir Crit Care Med. (2009) 179:19–24. doi: 10.1164/rccm.200807-1126OC
101. Kitch, BT, Paltiel, AD, Kuntz, KM, Dockery, DW, Schouten, JP, Weiss, ST, et al. A single measure of FEV1 is associated with risk of asthma attacks in long-term follow-up. Chest. (2004) 126:1875–82. doi: 10.1378/chest.126.6.1875
102. Kubysheva, N, Boldina, M, Eliseeva, T, Soodaeva, S, Klimanov, I, Khaletskaya, A, et al. Relationship of serum levels of IL-17, IL-18, TNF-α, and lung function parameters in patients with COPD, asthma-COPD overlap, and bronchial asthma. Mediat Inflamm. (2020) 2020:1–11. doi: 10.1155/2020/4652898
103. Camiolo, MJ, Zhou, X, Wei, Q, Trejo Bittar, HE, Kaminski, N, Ray, A, et al. Machine learning implicates the IL-18 signaling axis in severe asthma. JCI. Insight. (2021) 6:e149945. doi: 10.1172/jci.insight.149945
104. Rogers, DF. Airway mucus hypersecretion in asthma: an undervalued pathology? Curr Opin Pharmacol. (2004) 4:241–50. doi: 10.1016/j.coph.2004.01.011
105. Morcillo, EJ, and Cortijo, J. Mucus and MUC in asthma. Curr Opin Pulm Med. (2006) 12:1–6. doi: 10.1097/01.mcp.0000198064.27586.37
106. Kang, MJ, Choi, JM, Kim, BH, Lee, CM, Cho, WK, Choe, G, et al. IL-18 induces emphysema and airway and vascular remodeling via IFN-γ, IL-17A, and IL-13. Am J Respir Crit Care Med. (2012) 185:1205–17. doi: 10.1164/rccm.201108-1545OC
107. Mukherjee, M, and Nair, P. Autoimmune responses in severe asthma. Allergy Asthma Immunol Res. (2018) 10:428–47. doi: 10.4168/aair.2018.10.5.428
108. Mukherjee, M, Thomas, SR, Radford, K, Dvorkin-Gheva, A, Davydchenko, S, Kjarsgaard, M, et al. Sputum antineutrophil cytoplasmic antibodies in serum antineutrophil cytoplasmic antibody-negative eosinophilic granulomatosis with polyangiitis. Am J Respir Crit Care Med. (2019) 199:158–70. doi: 10.1164/rccm.201804-0809OC
109. Mukherjee, M, Bulir, DC, Radford, K, Kjarsgaard, M, Huang, CM, Jacobsen, EA, et al. Sputum autoantibodies in patients with severe eosinophilic asthma. J Allergy Clin Immunol. (2018) 141:1269–79. doi: 10.1016/j.jaci.2017.06.033
110. Mukherjee, M, Lacy, P, and Ueki, S. Eosinophil extracellular traps and inflammatory pathologies-untangling the web! Front Immunol. (2018) 9:2763. doi: 10.3389/fimmu.2018.02763
111. Fukuchi, M, Kamide, Y, Ueki, S, Miyabe, Y, Konno, Y, Oka, N, et al. Eosinophil ETosis-mediated release of Galectin-10 in eosinophilic granulomatosis with polyangiitis. Arthritis Rheumatol. (2021) 73:1683–93. doi: 10.1002/art.41727
112. Ueki, S, Tokunaga, T, Melo, RCN, Saito, H, Honda, K, Fukuchi, M, et al. Charcot-Leyden crystal formation is closely associated with eosinophil extracellular trap cell death. Blood. (2018) 132:2183–7. doi: 10.1182/blood-2018-04-842260
113. Aegerter, H, Smole, U, Heyndrickx, I, Verstraete, K, Savvides, SN, Hammad, H, et al. Charcot-Leyden crystals and other protein crystals driving type 2 immunity and allergy. Curr Opin Immunol. (2021) 72:72–8. doi: 10.1016/j.coi.2021.03.013
114. Mukherjee, M, Aleman Paramo, F, Kjarsgaard, M, Salter, B, Nair, G, LaVigne, N, et al. Weight-adjusted intravenous reslizumab in severe asthma with inadequate response to fixed-dose subcutaneous mepolizumab. Am J Respir Crit Care Med. (2018) 197:38–46. doi: 10.1164/rccm.201707-1323OC
115. Yamagata, S, Tomita, K, Sato, R, Niwa, A, Higashino, H, and Tohda, Y. Interleukin-18-deficient mice exhibit diminished chronic inflammation and airway remodelling in ovalbumin-induced asthma model. Clin Exp Immunol. (2008) 154:295–304. doi: 10.1111/j.1365-2249.2008.03772.x
116. Wlodek, E, Kirkpatrick, RB, Andrews, S, Noble, R, Schroyer, R, Scott, J, et al. A pilot study evaluating GSK1070806 inhibition of interleukin-18 in renal transplant delayed graft function. PLoS One. (2021) 16:e0247972. doi: 10.1371/journal.pone.0247972
117. McKie, EA, Reid, JL, Mistry, PC, DeWall, SL, Abberley, L, Ambery, PD, et al. A study to investigate the efficacy and safety of an anti-interleukin-18 monoclonal antibody in the treatment of type 2 diabetes mellitus. PLoS One. (2016) 11:e0150018. doi: 10.1371/journal.pone.0150018
118. Zhang, H, Wang, J, Wang, L, Xie, H, Chen, L, and He, S. Role of IL-18 in atopic asthma is determined by balance of IL-18/IL-18BP/IL-18R. J Cell Mol Med. (2018) 22:354–73. doi: 10.1111/jcmm.13323
119. Zhang, L, Zhang, J, and Gao, P. The potential of interleukin-37 as an effective therapeutic agent in asthma. Respir Res. (2017) 18:192. doi: 10.1186/s12931-017-0675-x
120. Jang, YS, Lee, K, Park, M, Joo Park, J, Choi, GM, Kim, C, et al. Albumin-binding recombinant human IL-18BP ameliorates macrophage activation syndrome and atopic dermatitis via direct IL-18 inactivation. Cytokine. (2023) 172:156413. doi: 10.1016/j.cyto.2023.156413
121. Gabay, C, Fautrel, B, Rech, J, Spertini, F, Feist, E, Kötter, I, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset still’s disease. Ann Rheum Dis. (2018) 77:840–7. doi: 10.1136/annrheumdis-2017-212608
122. Klughammer, B, Piali, L, Nica, A, Nagel, S, Bailey, L, Jochum, C, et al. A randomized, double-blind phase 1b study evaluating the safety, tolerability, pharmacokinetics and pharmacodynamics of the NLRP3 inhibitor selnoflast in patients with moderate to severe active ulcerative colitis. Clin Transl Med. (2023) 13:e1471. doi: 10.1002/ctm2.1471
123. Li, H, Guan, Y, Liang, B, Ding, P, Hou, X, Wei, W, et al. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome. Eur J Pharmacol. (2022) 928:175091. doi: 10.1016/j.ejphar.2022.175091
124. Wang, Y, Zhang, D, Liu, T, Wang, JF, Wu, JX, Zhao, JP, et al. FSTL1 aggravates OVA-induced inflammatory responses by activating the NLRP3/IL-1β signaling pathway in mice and macrophages. Inflamm Res. (2021) 70:777–87. doi: 10.1007/s00011-021-01475-w
125. Li, M, Wang, C, Xu, WT, and Zhong, X. Sodium houttuyfonate plays a protective role in the asthmatic airway by alleviating the NLRP3-related pyroptosis and Th1/Th2 immune imbalance. Mol Immunol. (2023) 160:103–11. doi: 10.1016/j.molimm.2023.06.013
126. Bae, DS, Kim, YH, Pan, CH, Nho, CW, Samdan, J, Yansan, J, et al. Protopine reduces the inflammatory activity of lipopolysaccharide-stimulated murine macrophages. BMB Rep. (2012) 45:108–13. doi: 10.5483/BMBRep.2012.45.2.108
127. Yang, J, Zhang, M, Luo, Y, Xu, F, Gao, F, Sun, Y, et al. Protopine ameliorates OVA-induced asthma through modulatingTLR4/MyD88/NF-κB pathway and NLRP3 inflammasome-mediated pyroptosis. Phytomedicine. (2024) 126:155410. doi: 10.1016/j.phymed.2024.155410
128. Hur, J, Kang, JY, Kim, YK, Lee, SY, and Lee, HY. Glucagon-like peptide 1 receptor (GLP-1R) agonist relieved asthmatic airway inflammation via suppression of NLRP3 inflammasome activation in obese asthma mice model. Pulm Pharmacol Ther. (2021) 67:102003. doi: 10.1016/j.pupt.2021.102003
129. Shi, M, Lu, Q, Zhao, Y, Ding, Z, Yu, S, Li, J, et al. miR-223: a key regulator of pulmonary inflammation. Front Med. (2023) 10:1187557. doi: 10.3389/fmed.2023.1187557
130. Feng, Z, Qi, S, Zhang, Y, Qi, Z, Yan, L, Zhou, J, et al. Ly6G+ neutrophil-derived miR-223 inhibits the NLRP3 inflammasome in mitochondrial DAMP-induced acute lung injury. Cell Death Dis. (2017) 8:e3170. doi: 10.1038/cddis.2017.549
131. Zhou, W, Pal, AS, Hsu, AY, Gurol, T, Zhu, X, Wirbisky-Hershberger, SE, et al. MicroRNA-223 suppresses the canonical NF-κB pathway in basal keratinocytes to dampen neutrophilic inflammation. Cell Rep. (2018) 22:1810–23. doi: 10.1016/j.celrep.2018.01.058
132. Xu, W, Wang, Y, Ma, Y, and Yang, J. MiR-223 plays a protecting role in neutrophilic asthmatic mice through the inhibition of NLRP3 inflammasome. Respir Res. (2020) 21:116. doi: 10.1186/s12931-020-01374-4
133. Ishikawa, Y, Yoshimoto, T, and Nakanishi, K. Contribution of IL-18-induced innate T cell activation to airway inflammation with mucus hypersecretion and airway hyperresponsiveness. Int Immunol. (2006) 18:847–55. doi: 10.1093/intimm/dxl021
134. Wang, J, Zhang, H, Zheng, W, Xie, H, Yan, H, Lin, X, et al. Correlation of IL-18 with tryptase in atopic asthma and induction of mast cell accumulation by IL-18. Mediat Inflamm. (2016) 2016:1–14. doi: 10.1155/2016/4743176
135. Imaoka, H, Gauvreau, GM, Watson, RM, Smith, SG, Dua, B, Baatjes, AJ, et al. Interleukin-18 and interleukin-18 receptor-α expression in allergic asthma. Eur Respir J. (2011) 38:981–3. doi: 10.1183/09031936.00033811
136. Wu, J, Wang, P, Xie, X, Yang, X, Tang, S, Zhao, J, et al. Gasdermin D silencing alleviates airway inflammation and remodeling in an ovalbumin-induced asthmatic mouse model. Cell Death Dis. (2024) 15:400. doi: 10.1038/s41419-024-06777-5
137. Kim, DH, Lee, KJ, Park, J, Chi, S, Han, J, Bang, Y, et al. Disruption of IL-18 signaling via engineered IL-18BP biologics alleviates experimental cholestatic liver disease. Biomed Pharmacother. (2023) 167:115587. doi: 10.1016/j.biopha.2023.115587
138. Perera, AP, Fernando, R, Shinde, T, Gundamaraju, R, Southam, B, Sohal, SS, et al. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. (2018) 8:8618. doi: 10.1038/s41598-018-26775-w
139. Vande Walle, L, Stowe, IB, Šácha, P, Lee, BL, Demon, D, Fossoul, A, et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. (2019) 17:e3000354. doi: 10.1371/journal.pbio.3000354
140. Zhang, L, Jiang, YH, Fan, C, Zhang, Q, Jiang, YH, Li, Y, et al. MCC950 attenuates doxorubicin-induced myocardial injury in vivo and in vitro by inhibiting NLRP3-mediated pyroptosis. Biomed Pharmacother. (2021) 143:112133. doi: 10.1016/j.biopha.2021.112133
141. Wu, X, Wang, B, Zhou, Y, Yang, Z, Jiang, L, Kou, Z, et al. NLRP3 inflammasome inhibitor MCC950 reduces cerebral ischemia/reperfusion induced neuronal ferroptosis. Neurosci Lett. (2023) 795:137032. doi: 10.1016/j.neulet.2022.137032
142. Chen, L, Hou, W, Liu, F, Zhu, R, Lv, A, Quan, W, et al. Blockade of NLRP3/Caspase-1/IL-1β regulated Th17/Treg immune imbalance and attenuated the neutrophilic airway inflammation in an ovalbumin-induced murine model of asthma. J Immunol Res. (2022) 2022:1–11. doi: 10.1155/2022/9444227
143. Ding, H, Lu, X, Wang, H, Chen, W, and Niu, B. NLRP3 inflammasome deficiency alleviates inflammation and oxidative stress by promoting PINK1/Parkin-mediated mitophagy in allergic rhinitis mice and nasal epithelial cells. J Asthma Allergy. (2024) 17:717–31. doi: 10.2147/JAA.S467774
144. Zheng, D, Wang, H, Zhou, Y, Chen, Y, and Chen, G. Ac-YVAD-cmk ameliorated sevoflurane-induced cognitive dysfunction and revised mitophagy impairment. PLoS One. (2023) 18:e0280914. doi: 10.1371/journal.pone.0280914
145. Yang, M, Fang, JT, Zhang, NS, Qin, LJ, Zhuang, YY, Wang, WW, et al. Caspase-1-inhibitor AC-YVAD-CMK inhibits pyroptosis and ameliorates acute kidney injury in a model of sepsis. Biomed Res Int. (2021) 2021:1–9. doi: 10.1155/2021/6636621
146. Li, H, Li, Y, Song, C, Hu, Y, Dai, M, Liu, B, et al. Neutrophil extracellular traps augmented alveolar macrophage pyroptosis via AIM2 inflammasome activation in LPS-induced ALI/ARDS. J Inflamm Res. (2021) 14:4839–58. doi: 10.2147/JIR.S321513
147. Yao, PA, Wei, KZ, Feng, JH, Liu, XN, Xu, X, Cui, HY, et al. Sodium houttuyfonate protects against cardiac injury by regulating cardiac energy metabolism in diabetic rats. Eur J Pharmacol. (2022) 932:175236. doi: 10.1016/j.ejphar.2022.175236
148. Nie, C, Wang, B, Wang, B, Lv, N, Yu, R, and Zhang, E. Protopine triggers apoptosis via the intrinsic pathway and regulation of ROS/PI3K/Akt signalling pathway in liver carcinoma. Cancer Cell Int. (2021) 21:396. doi: 10.1186/s12935-021-02105-5
149. An, JR, Liu, JT, Gao, XM, Wang, QF, Sun, GY, Su, JN, et al. Effects of liraglutide on astrocyte polarization and neuroinflammation in db/db mice: focus on iron overload and oxidative stress. Front Cell Neurosci. (2023) 17:1136070. doi: 10.3389/fncel.2023.1136070
150. Altintas Dogan, AD, Hilberg, O, Hess, S, Jensen, TT, Bladbjerg, EM, and Juhl, CB. Respiratory effects of treatment with a glucagon-like peptide-1 receptor agonist in patients suffering from obesity and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. (2022) 17:405–14. doi: 10.2147/COPD.S350133
151. Zhou, F, Zhang, Y, Chen, J, Hu, X, and Xu, Y. Liraglutide attenuates lipopolysaccharide-induced acute lung injury in mice. Eur J Pharmacol. (2016) 791:735–40. doi: 10.1016/j.ejphar.2016.10.016
152. Liu, W, Liu, A, Li, X, Sun, Z, Sun, Z, Liu, Y, et al. Dual-engineered cartilage-targeting extracellular vesicles derived from mesenchymal stem cells enhance osteoarthritis treatment via miR-223/NLRP3/pyroptosis axis: toward a precision therapy. Bioact Mater. (2023) 30:169–83. doi: 10.1016/j.bioactmat.2023.06.012
Keywords: IL-18, autoimmunity, asthma, inflammasome, eosinophilia
Citation: Thawanaphong S, Nair A, Volfson E, Nair P and Mukherjee M (2024) IL-18 biology in severe asthma. Front. Med. 11:1486780. doi: 10.3389/fmed.2024.1486780
Edited by:
Hsiao-Chi Chuang, Taipei Medical University, TaiwanReviewed by:
Patrizia Pignatti, Scientific Clinical Institute Maugeri (ICS Maugeri), ItalyFara Silvia Yuliani, Gadjah Mada University, Indonesia
Copyright © 2024 Thawanaphong, Nair, Volfson, Nair and Mukherjee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manali Mukherjee, bXVraGVyakBtY21hc3Rlci5jYQ==
†These authors have contributed equally to this work