 Natsuko Kameyama
Natsuko Kameyama Aoi Hosaka3
Aoi Hosaka3 Hideki Maeda
Hideki Maeda- 1Department of Regulatory Science, Graduate School of Pharmaceutical Science, Meiji Pharmaceutical University, Tokyo, Japan
- 2CMIC Co., Ltd., Tokyo, Japan
- 3Department of Regulatory Science, Faculty of Pharmacy, Meiji Pharmaceutical University, Tokyo, Japan
Introduction: The system of Risk Management Plan in Japan (J-RMP) is a relatively new system, implemented in 2013; thus, its effect on safety measures is still unclear. One of the purposes of J-RMP is to enhance the postmarketing safety measures to be ensured by publishing J-RMP and sharing information on risk management among healthcare professionals. We hypothesized that this might enable information about postmarketing adverse events to be accumulated rapidly, potentially accelerating the identification of adverse reactions (ARs). Herein, we focused on the speed of adding clinically significant ARs (CSARs) to package inserts (PIs) as an indicator of the rapidity of AR identification, investigated the impact of the J-RMP system on PI revisions.
Methods: We investigated the “Notice of Revision of Precautions” on the website of Pharmaceuticals and Medical Devices Agency (PMDA), targeting PI revisions with the addition of CSARs from April 2003 to March 2023, which corresponds to 10 years before and after J-RMP implementation in April 2013. We created an original database from public information of PMDA and investigated the speed of adding CSARs to PIs.
Results: Comparing the time lapse from drug approvals to PI revisions after J-RMP implementation (149 cases) to that before implementation (318 cases), the median value was 32 months for both. Regarding the time lapse when the additional CSARs were listed and unlisted as safety concerns at the time of approvals, it was 35 months vs. 32 months (14 cases vs. 126 cases, p = 0.7820), with no statistically significant difference. Conversely, there were significant differences within each AR and each drug therapeutic category.
Discussion and conclusions: This study revealed that the rapidity of risk identification as ARs was not affected by J-RMP, and it may be affected by the characteristics of each AR and each drug therapeutic category. It is expected that other J-RMP benefits, such as risk prevention before the occurrence, will be utilized to further develop strategies for the effective utilization of the J-RMP for safety measures in Japan.
1 Introduction
The Risk Management Plan (RMP) in Japan is a document that indicates the risk management of drugs from the development phase to the postmarketing phase. It comprises the following three elements for individual drugs: safety concern, pharmacovigilance activities, and risk minimization activities (1). “Risk Management Plan Guidance,” which was issued in 2012, is applicable to new drugs for which approval applications were submitted on or after April 1, 2013, and requires the creation of RMP in Japan (2). The effect of the J-RMP system (which is a relatively new system) on safety measures is still unclear. Given that it has been more than 10 years since J-RMP was implemented in 2013, we believe it is meaningful to investigate the impact of J-RMP on safety measures and evaluate its effectiveness. One of the purposes of J-RMP is to enhance the postmarketing safety measures to be ensured by publishing J-RMP and sharing information on risk management among healthcare professionals, leading to understand activities as risk management (3). Consequently, it potentially increases spontaneous reports as medical professionals understand the significance of adverse event reporting or recognize it as a risk or insufficient information. Thus, we hypothesized that this might enable information about postmarketing adverse events to be accumulated faster, potentially accelerating the identification of adverse reactions (ARs). However, to the best of our knowledge, no study has been conducted to determine the impact of J-RMP on the rapidity of identifying ARs. In this study, we focused on the speed of adding clinically significant ARs (CSARs) to package inserts (PIs) as an indicator of the rapidity of AR identification and investigated the impact of the J-RMP system on PI revisions.
2 Materials and methods
In this study, we investigated the “Summary of Investigation Results” attached to the “Notice of Revision of Precautions” on the website of Pharmaceuticals and Medical Devices Agency (PMDA) (4). “Notice of Revision of Precautions” is a list of notification based on which manufacturers revise their PIs. We targeted PI revisions with the addition of CSARs from April 2003 to March 2023, which corresponds to the 10 years before and after J-RMP implementation in April 2013. The PI revisions from April 2013 to March 2023 were included as PI revisions after RMP implementation, and PI revisions of drugs with no RMP at the time of approval were excluded from the analysis. The PI revisions from April 2003 to March 2013 were included as PI revisions before RMP implementation. We created an original database and first checked the background characteristics of PI revisions for additional CSARs and therapeutic category of drugs. Next, we compared the speed of adding CSARs to PIs with respect to (1) before and after RMP implementation, and (2) listed and unlisted CSARs as the safety concerns at the time of approval. We also investigated the speed by each CSAR and each therapeutic category of drugs.
The speed of adding the CSAR to the PI was defined as the time from the initial approval for new active ingredients of the drug (s) to the date of issuance of the “Notice of Revision of Precautions.” When comparing such speed of PI revisions, “after RMP implementation” refers to PI revisions from April 2013 to March 2023 for products first approved after April 2013, with RMPs at the time of approval, and “before RMP implementation” refers to PI revisions from April 2003 to March 2013 for products first approved after April 2003, with no RMPs. If “Draft drug risk management plan (5)” was included in the review report at the time of approval, it was determined that the RMP was created at the time of approval. Whether or not the CSARs were listed as the safety concerns was also checked by “Draft drug risk management plan.”
This study was conducted per the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guidelines (6) for cross-sectional studies. All statistical analyses were performed using the analytical tools of JMP Pro 15, with two-sided p-values less than 0.05 being considered statistically significant. The Wilcoxon rank sum test was used to perform comparisons between quantitative data while the Chi-square test was used to perform comparisons between categorical data. CSARs were coded using MedDRA (7) ver. 26.0 and classified by System Organ Class. Therapeutic drug categories were classified according to the Japanese Standard Classification of Products (8).
3 Results
The most common CSARs after RMP implementation were “Infections and infestations,” “Skin and subcutaneous tissue disorders,” and “Hepatobiliary disorders,” classified by System Organ Class with MedDRA (7). As for CSARs before RMP implementation, “Hepatobiliary disorders,” “Nervous system disorders,” and “Skin and subcutaneous tissue disorders” were the most common. Regarding the therapeutic category of the drugs, “Other oncology drugs,” “Antidiabetic drugs,” and “Metabolic drugs not elsewhere classified,” were the most common after RMP implementation, and “Other oncology drugs,” “Metabolic drugs not classified elsewhere” and “Antiviral drugs” were the most common before RMP implementation. Table 1 shows the background characteristics of the PI revisions for the addition of CSARs.
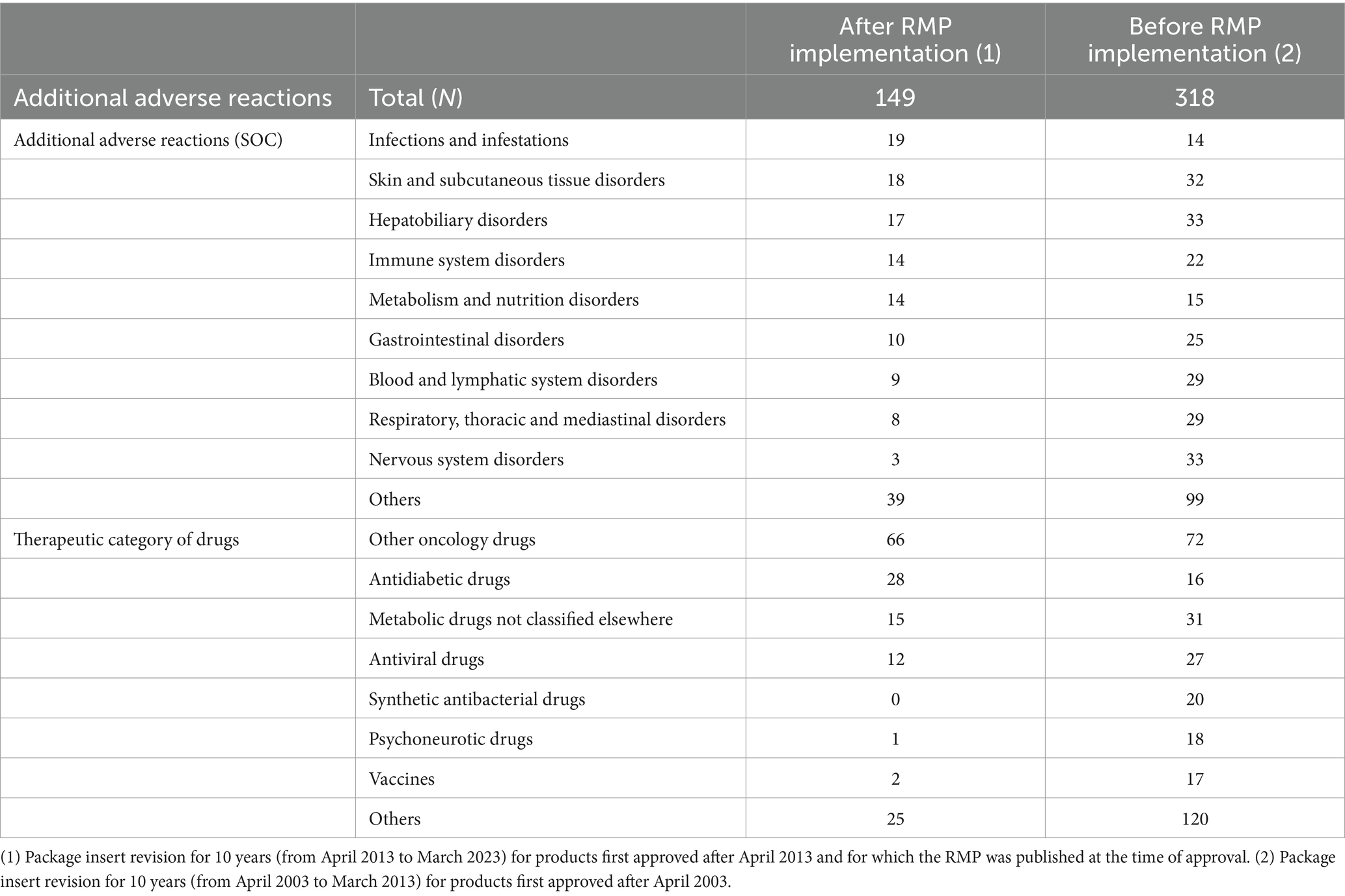
Table 1. Background characteristics of package insert revisions for adding clinically significant adverse reactions.
Comparing the PI revisions after RMP implementation with before implementation, the number of CSARs added to the revised PIs was 149 vs. 318, and the median time from approvals to PI revisions was 32 months for both. Additionally, for 140 of the 149 cases after RMP implementation, excluding nine cases having no information on safety concerns at the time of approvals, we investigated the speed of PI revisions. Comparing when the additional CSARs were listed and unlisted as safety concerns at the time of approvals, the number of CSARs was 14 vs. 126, and the median time from approvals to PI revisions was 35 months vs. 32 months (p = 0.7820), and these variables did not differ significantly from each other. A comparison of the speed of adding CSAR to the PI is shown in Table 2.

Table 2. Comparisons of the time lapse from drug approval to the addition of adverse reactions in package insert revisions.
Conversely, median interval from approvals to PI revisions for each CSAR was significantly shorter for “Metabolism and nutrition disorders” (14 cases, 10 months, p < 0.0001), and longer for “Blood and lymphatic system disorders” (9 cases, 55 months, p = 0.0413) and “Eye disorders” (4 cases, 79.5 months, p = 0.0234; Table 3).
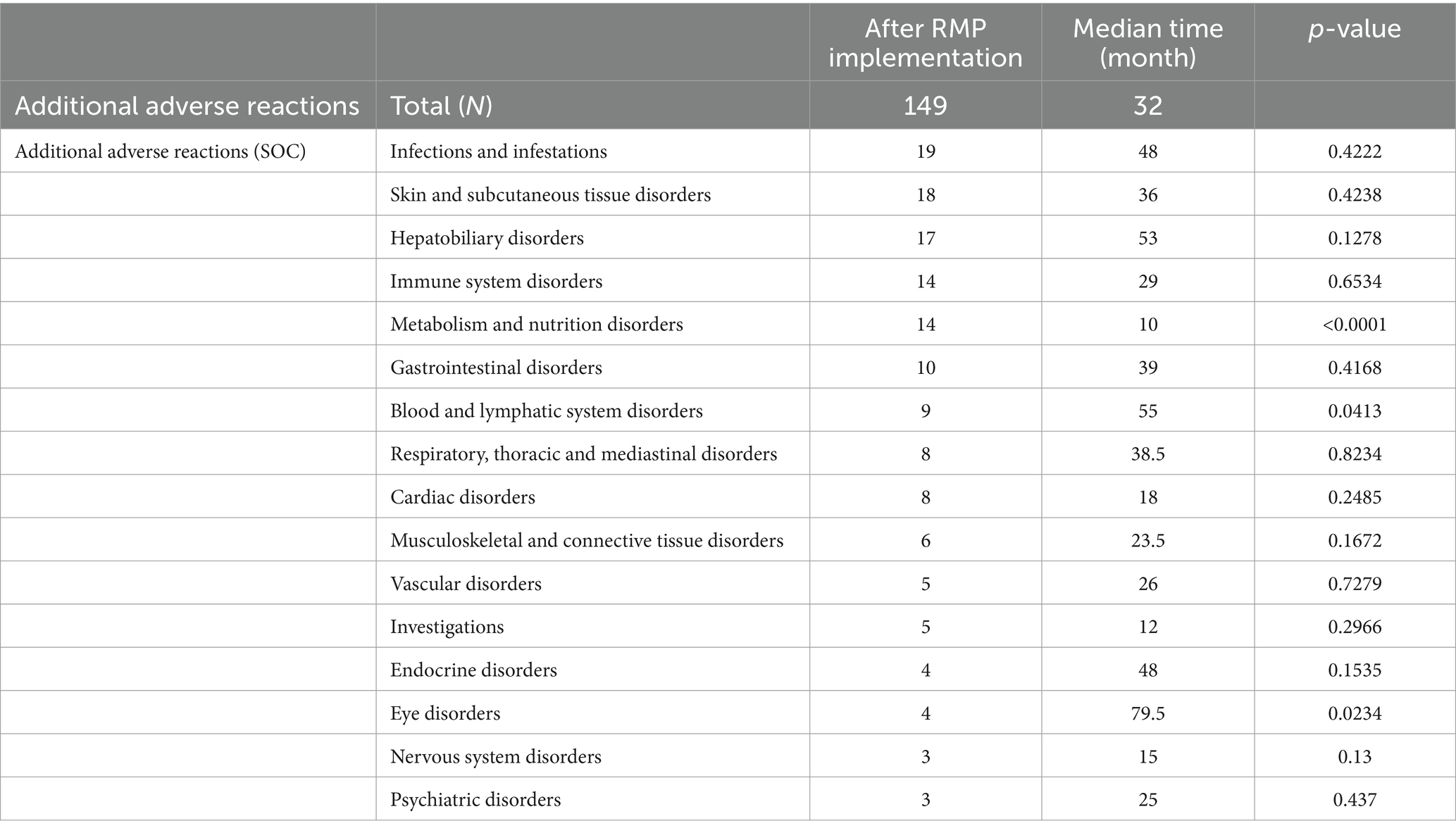
Table 3. Comparisons of the time lapse from drug approval to the addition of adverse reactions in package insert revisions (by each adverse reaction).
The median interval from approval to PI revision by each drug category was significantly shorter for “Antidiabetic drugs” (28 cases, 17 months, p = 0.0106) and “Antiviral drugs” (12 cases, 15 months, p = 0.0088), and significantly longer for “Metabolic drugs not classified elsewhere” (15 cases, 59 months, p = 0.0143) and “Drugs for digestive ulcers” (8 cases, 58 months, p = 0.0087; Table 4).
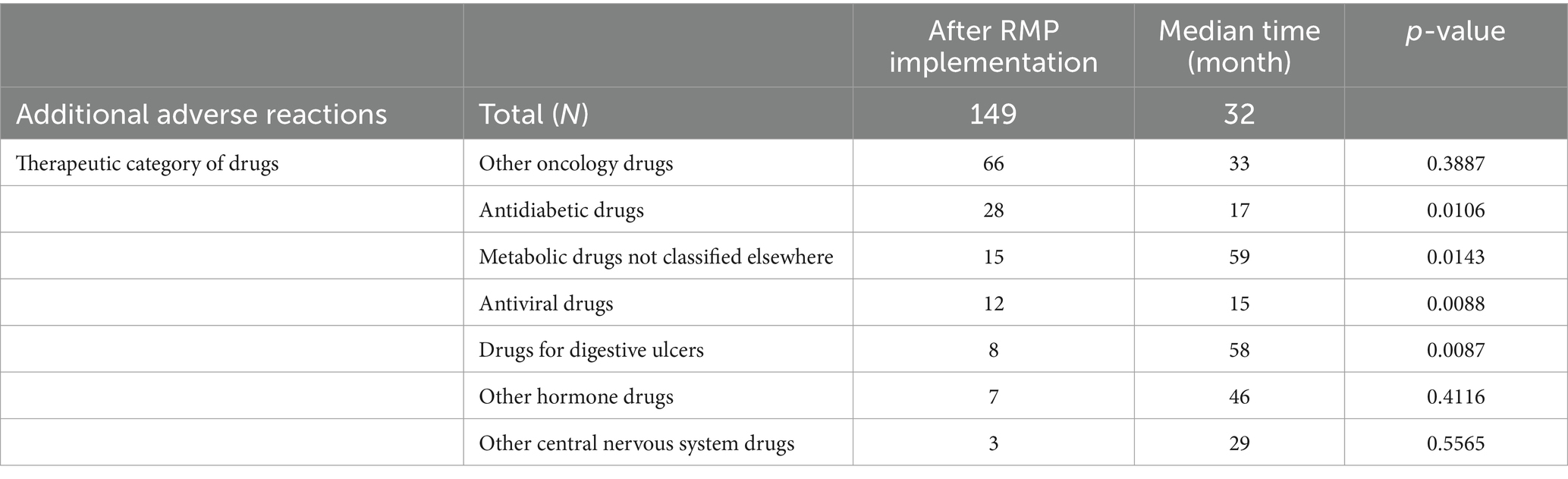
Table 4. Comparisons of the time lapse from drug approval to the addition of adverse reactions in package insert revisions (by each therapeutic category of drugs).
4 Discussion
The speed of PI revisions is instrumental in the prompt identification of risks as ARs to improve awareness and patient safety. In this study, we investigated the impact of the J-RMP system on the revisions of PIs, focusing on PI revision speed. This is because we expected that if risks were appropriately managed using J-RMPs, they could be identified as ARs more rapidly, and PI revisions could be faster. We assumed that J-RMP potentially increase spontaneous reports as healthcare professionals understand the importance of adverse event reporting or recognize it as a risk or insufficient information, leading to fast PI revision speed. However, the results revealed that the implementation of the J-RMP system or description as safety concerns at the time of approvals did not affect the PI revision speed regarding the addition of CSARs. As a side note, in the study examining the relationship between the revision of the information in the CSARs section in PI and the description in J-RMP at the time of drug approval, the median time from drug approval to PI revisions was 29.5 months (9), which was nearly the same as that in our study (32 months). One of the reasons for J-RMP not affecting the rapidity of risk identification as ARs in our study is potentially because healthcare professionals take precautions for reducing the risk, making ARs less likely to occur, and slowing down PI revision speed. Concerning the hypotheses of this study, we focused on the possible publication of the J-RMP that might increase the speed of collecting ARs and PI revision speed; however, risk prevention measures can slow down the PI revision speed, thereby affecting the results. Future studies on the impact of risk minimization measures on PI revision speed will be of interest.
Although the J-RMP did not affect the rapidity of risk identification for ARs, it is known to have other advantages. A study by Saito et al. revealed that there is a strong relationship between ARs listed as safety concerns at the time of approval and those being added to the PIs as CSARs postapproval, indicating that safety concerns could potentially induce severe ARs. This suggests that safety concerns in J-RMPs constitute important drug information, and it is expected that medical professionals will contribute to the prevention of severe ARs in patients by utilizing J-RMPs in addition to PIs (10). Furthermore, “Risk minimization activities” of the J-RMP are also important elements for healthcare professionals because they describe measures to minimize the patient’s risk (11). One of the purposes of the J-RMP is to prevent risks before they occur; however, the low usage rate of the J-RMP in clinical settings has been an issue (12). However, in Japan, the medical fee regulations were recently revised in 2024 to include a provision that medical fee points will be increased if sufficient safety instructions are provided using RMP materials at the time of dispensing (13). According to precedents, regulatory renovation had an obvious effect on Pharmacovigilance Planning (PVP). For example, the publication of the revised Good Post-marketing Study Practice in 2017 (14, 15) and the procedure for developing Postmarketing Surveillance plans in 2018 (16) had a clear impact on PVP shown in J-RMP; the proportion of drugs with efficacy issues decreased, safety issues with additional activity also decreased, and database studies increased in contrast (17). Therefore, it is expected that the revision of medical fee regulation will also promote the use of the J-RMP in clinical settings to mitigate risks. Moreover, J-RMP consolidates risk management into one document to ensure that risk assessments are performed (3), which purpose is different from the RMP in the EU (EU-RMP) (18–21) or risk evaluation and mitigation strategies (REMS) in the US (22), as the EU-RMP lists only safety concerns that require particular attention and REMS are mandatory for only some products. One advantage of the J-RMP is that it allows both regulatory authorities and pharmaceutical companies to conduct risk assessments easily and reliably with one document. However, it has been more than a decade since the implementation of the J-RMP, and there are some preparations for which the J-RMP has been terminated at re-examination. Therefore, a future challenge will be how to implement risk management after J-RMP termination (23).
Conversely, there were significant differences in the PI revision speed by each AR and drug category, suggesting that the speed of AR identification may be influenced by the characteristics of each AR and drug effect. The PI is revised based on AR accumulation in Japan/overseas, revisions of CCDS/overseas labeling, and information on overseas measures (24). Of these, AR accumulation in Japan is recorded in terms of the “number of domestic cases,” the “number of cases in which a causal relationship cannot be ruled out,” and the “number of fatal cases” over the last 3 years. It is possible that the accumulation speed of cases and the ease of causality assessment may influence the PI revision speed (25). In “Metabolism and nutrition disorders,” where the PI revision was faster, 12 out of the 14 CSARs were ketoacidosis and dehydration associated with antidiabetic drugs (SGLT2 inhibitors). Moreover, in “Antidiabetic drugs” with faster PI revision, 25 out of the 28 CSARs were Fournier’s gangrene, ketoacidosis, sepsis, and dehydration associated with SGLT2 inhibitors. The common denominator here is that the patients are many (26) and that causality can be easily assessed based on the drug’s mechanism of action (27, 28), which is likely why the PIs were revised quickly. As for “Antiviral drugs” with faster PI revision, four out of 12 CSARs were associated with drugs for influenza A or B virus infection, and one CSAR was associated with drugs for Herpes zoster infection, and the patients were numerous (29, 30). Regarding the two CSARs of anaphylaxis associated with drugs for SARS-CoV-2 infectious diseases, it may be easier to assess the causal relationship because anaphylaxis occurs immediately after exposure to the causative substances (31). As for CSARs with slower PI revision, seven out of nine CSARs of “Blood and lymphatic system disorders” were associated with molecular-targeted anticancer drugs, five of those were associated with immune checkpoint inhibitors, and three out of four CSARs of “Eye disorder” were uveitis associated with immune checkpoint inhibitors (32). The reason for the slower PI revision could be the relatively new mechanism of action of these drugs, which makes it difficult to assess AR causality with these drugs, and there were not many patients receiving the drugs. However, there were no clear features of other ARs or drug categories. In addition, it is possible that PI revisions regarding similar ARs associated with similar drug classes were coincidentally performed simultaneously. Further investigations using larger samples are necessary.
Nevertheless, our study has some limitations. This study targeted PI revisions with the addition of CSARs, which are clinically important and are frequently revised for analysis, and did not include the revision of other sections such as “Other adverse reactions” as well as “Precautions” or “Warnings,” etc. In addition, of the CSAR section revisions, only the new AR terms was counted, and the revision of frequency, intensity such as severity, or outcomes such as death was not counted, because the different wordings of these elements could have obscured the visual judgment to include them or not; the results may have been affected if these had been included to the analysis. In addition, regarding the comparison between findings before and after J-RMP implementation, there may be differences in the drug safety system or the procedure for PI revisions between the target periods, which may have affected the speed of the PI revision process. To minimize this effect, we also compared the PI revision speed only after J-RMP implementation, between when additional CSARs had been included as safety concerns at the time of drug approvals and when they had not been included. Finally, the COVID-19 pandemic potentially affected the results of this study. The number of PI revisions with the addition of CSARs was 40 in 2019 (prepandemic) and 13 in 2020 (postpandemic). However, considering that the number was around 10 in other years, with not much change, the difference in numbers before and after the pandemic could be a coincidence.
5 Conclusion
In conclusion, the implementation of J-RMP and safety concerns did not affect PI revision and its speed. However, the J-RMP has other benefits, such as the prevention of risks before they occur and the reliability of risk assessment in one document. These benefits are expected to be utilized to further develop strategies for the effective utilization of the J-RMP for safety measures in Japan.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
NK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. AH: Data curation, Investigation, Writing – review & editing. HM: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was partly supported by a grant from Japan Health and Labour Sciences Research Grant (grant number 21KC2006).
Conflict of interest
NK is an employee of CMIC Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pharmaceuticals and Medical Devices Agency (2013). Pharmaceuticals and medical devices safety information. Available at: https://www.pmda.go.jp/files/000153064.pdf [Accessed May 28, 2023].
2. Ministry of Health, Labour and Welfare (2012). Risk Management Plan Guidance (PFSB/SD notification no. 0411-1, PFSB/ELD notification no. 0411-2). Pharmaceuticals and Medical Devices Agency. Available at: https://www.pmda.go.jp/files/000153333.pdf [Accessed May 28, 2023].
3. Pharmaceuticals and Medical Devices Agency (n.d.). Risk Management Plan (RMP). Available at: https://www.pmda.go.jp/english/safety/info-services/drugs/rmp/0001.html [Accessed October 03, 2023].
4. Pharmaceuticals and Medical Devices Agency (n.d.). Notice of revision of precautions (Pharmaceuticals). Available at: https://www.pmda.go.jp/safety/info-services/drugs/calling-attention/revision-of-precautions/0001.html [Accessed May 28, 2023].
5. Pharmaceuticals and Medical Devices Agency (n.d.). Information search for prescription drugs. Available at: https://www.pmda.go.jp/PmdaSearch/iyakuSearch/ [Accessed May 28, 2023].
6. Vandenbroucke, JP, von Elm, E, Altman, DG, Gøtzsche, PC, Mulrow, CD, Pocock, SJ, et al. Strengthening the reporting of observational studies in epidemiology (STROBE): explanation and elaboration. Epidemiology. (2007) 18:805–35. doi: 10.1097/EDE.0b013e3181577511
7. MedDRA/J (n.d.). Medical dictionary for regulatory activities. Available at: https://www.jmo.pmrj.jp/english [Accessed May 28, 2023].
8. Ministry of Internal Affairs and Communications (1990). Japanese standard classification of products. Available at: https://www.soumu.go.jp/toukei_toukatsu/index/seido/syouhin/2index.htm [Accessed June 28, 2024].
9. Kakutani, Y, Murayama, T, Kobayashi, E, and Satoh, N. Research on the utilization of risk management plan. Regul Sci Med Prod. (2023) 13:51–61. doi: 10.14982/rsmp.13.51
10. Saito, R, and Miyazaki, S. Analysis of safety specifications in risk management plan at the time of drug approval and addition of clinically significant adverse reactions in the package insert post-approval in Japan. Pharmacol Res Perspect. (2023) 11:e01110. doi: 10.1002/prp2.1110
11. Sato, H, Hirasawa, S, Kadono, S, Haruyama, T, Fujita, K, Kanetaka, Y, et al. Survey of the description of the risk minimization activities in pharmaceutical risk management plans. Jpn J Drug Inform. (2017) 19:32–6. doi: 10.11256/jjdi.19.32
12. Pharmaceuticals and Medical Devices Agency. (2023). Pharmaceuticals and medical devices safety information, no. 401. Available at: https://www.pmda.go.jp/files/000252814.pdf [Accessed 28 May 2023].
13. Ministry of Health, Labour and Welfare (2024). Overview of the FY2024 medical fee revision [dispensing]. Available at: https://www.mhlw.go.jp/content/12400000/001238903.pdf [Accessed June 30, 2024].
14. Ministry of Health, Labour and Welfare (2017). Ministerial ordinance partially revising the ministerial ordinance. Available at: https://www.pmda.go.jp/files/000220720.pdf [Accessed May 28, 2023].
15. Ministry of Health, Labour and Welfare (2017). Partially revising the ministerial ordinance no.116 (PSEHB notification no. 1026–1). Available at: https://www.pmda.go.jp/files/000220721.pdf [Accessed May 28, 2023].
16. Ministry of Health, Labour and Welfare (2019). Procedures for developing postmarking study plan (PSEHB/PED notification no. 0314-4, PSEHB/PSD notification no. 03414-4). Available at: https://www.pmda.go.jp/files/000228612.pdf [Accessed May 28, 2023].
17. Kohama, M, Nonaka, T, Uyama, Y, and Ishiguro, C. Descriptive analysis for the trend of pharmacovigilance planning in risk management plans on new drugs approved during 2016–2019. Ther Innov Regul Sci. (2023) 57:37–47. doi: 10.1007/s43441-022-00437-6
18. European Medicines Agency (n.d.). Risk management plans. Available at: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/pharmacovigilance/risk-management/risk-management-plans [Accessed November 6, 2023].
19. European Medicines Agency (2017). Guideline on good pharmacovigilance practices (GVP) module v-risk management systems (Rev 2). Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-module-v-risk-management-systems-rev-2_en.pdf [Accessed September 5, 2023].
20. Butler, D, Vucic, K, Straus, S, and Cupeli, A. Regulatory experience of handling risk management plans (RMPs) for medicinal products in the EU. Expert Opin Drug Saf. (2021) 20:815–26. doi: 10.1080/14740338.2021.1909569
21. Nakamura, Y, and Maeda, H. (2021). International comparison of risk management plans for drugs approved under the pioneer drug designation system. Ministry of Health, Labour and Welfare (MHLW) Research on rebuilding post-marketing drug safety measures for the next system revision. Available at: https://mhlw-grants.niph.go.jp/system/files/download_pdf/2021/202125035A.pdf [Accessed September 5, 2023].
22. Food and Drug Administration, Guidance. (2011). Medication guides–distribution requirements and inclusion in risk evaluation and mitigation strategies (REMS). Available at: https://www.fda.gov/media/79776/download [Accessed January 08, 2024].
23. Kameyama, N, Hosaka, A, and Maeda, H. Current situation and issues regarding termination of risk management plans in Japan. Front Med. (2024) 11:1387652. doi: 10.3389/fmed.2024.1387652
24. Ministry of Health, Labour and Welfare (2021). Standard workflow for considering safety measures, such as revision of electronic package inserts for pharmaceuticals. Available at: https://www.pmda.go.jp/files/000242993.pdf [Accessed June 30, 2024].
25. Suzuki, Y, Kishi, T, Nakamura, M, and Yamada, H. Evaluation of factors influencing addition of clinically significant adverse reactions section in drug package inserts. Jpn J Drug Inform. (2017) 19:17–23. doi: 10.11256/jjdi.19.17
26. Ministry of Health, Labour and Welfare. (2021). Annual health, labour and welfare report 2021, health and medical services. Available at: https://www.mhlw.go.jp/english/wp/wp-hw14/dl/02e.pdf [Accessed June 30, 2024].
27. Garofalo, C, Borrelli, S, Liberti, ME, Andreucci, M, Conte, G, Minutolo, R, et al. SGLT2 inhibitors: nephroprotective efficacy and side effects. Medicina (Kaunas). (2019) 55:268. doi: 10.3390/medicina55060268
28. Qiu, H, Novikov, A, and Vallon, V. Ketosis and diabetic ketoacidosis in response to SGLT2 inhibitors: basic mechanisms and therapeutic perspectives. Diabetes Metab Res Rev. (2017) 33:e2886. doi: 10.1002/dmrr.2886
29. Sako, A, Gu, Y, Masui, Y, Yoshimura, K, Yanai, H, and Ohmagari, N. Prescription of anti-influenza drugs in Japan, 2014-2020: a retrospective study using open data from the national claims database. PLoS One. (2023) 18:e0291673. doi: 10.1371/journal.pone.0291673
30. Phakey, S, Rogers, SL, Hall, AJ, and Lim, LL. Reduction in herpes zoster antiviral use since the introduction of the live-attenuated zoster vaccine on Australia’s national immunisation program: a population-based study from 1994 to 2019. Infect Dis Ther. (2023) 12:711–26. doi: 10.1007/s40121-022-00749-y
31. Muraro, A, Worm, M, Alviani, C, Cardona, V, DunnGalvin, A, Garvey, LH, et al. Allergy. (2021) 77:357–77. doi: 10.1111/all.15032
Keywords: risk management plan in Japan, risk management plan, package insert revision, adverse reaction, drug safety, pharmacovigilance
Citation: Kameyama N, Hosaka A and Maeda H (2024) Has risk management plan system influenced the speed of package insert revisions in Japan? Front. Med. 11:1465313. doi: 10.3389/fmed.2024.1465313
Edited by:
Simone Grassi, University of Florence, ItalyReviewed by:
Victor M. Rivera, Baylor College of Medicine, United StatesStewart Geary, Eisai, Japan
Copyright © 2024 Kameyama, Hosaka and Maeda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hideki Maeda, bWFlZGFAbXktcGhhcm0uYWMuanA=
†ORCID: Hideki Maeda, https://orcid.org/0000-0002-0117-3961