 Xuan Zhang
Xuan Zhang Hongjuan Lu
Hongjuan Lu Yichen Ji
Yichen Ji- Department of Neurology, Xuanwu Hospital, Capital Medical University, Beijing, China
Background: Seizures have been identified in most patients with CSNK2B-related Poirer-Bienvenu Neurodevelopment syndrome (POBINDS). Detailed descriptions of seizure phenotypes, various genotypes, and long-term follow-up visits are required for clinicians to provide reasonable clinical management for such patients.
Case summary: We report two new Chinese patients with varying sizes of 6p21.33 deletions encompassing the CSNK2B gene who presented with intellectual disability and seizures. Furthermore, we conducted a literature review of previously reported patients with 6p21.33 deletions or CSNK2B variants. We summarized and analyzed the clinical characteristics of these patients with seizures. The occurrence of a biphasic pattern of epilepsy and pharmacoresistant epilepsy in patients with CSNK2B variants is severely underestimated. One of our patients underwent a long follow-up period and presented with comprehensive disease progression.
Conclusion: Our data suggest that the CSNK2B variant or 6p21.33 deletion should be considered in patients with intellectual disability and epilepsy, especially those characterized by biphasic patterns and digital anomalies.
Introduction
In 2017, Poirier et al. (1) first identified de novo mutations of the CSNK2B gene in two patients with intellectual disability with or without seizures who were later diagnosed with Poirier-Bienvenu neurodevelopmental syndrome (POBINDS) (1). CK 2 beta (CSNK2B), located in chromosome 6p21.33, encodes the β subunit of the CK2 protein complex (2). CK2 comprises two catalytic subunits (α and/or α’) and two regulatory subunits (β) and is ubiquitously expressed in various cells, especially in the brain. The β subunit of CK2 plays roles in recognition and anchoring, ensuring and promoting the phosphorylation reaction (2). CK2 is a serine-threonine protein kinase that phosphorylates hundreds of physiological substrates in human tissue and participates in all important cellular processes, including cell proliferation, differentiation, apoptosis, synaptic transmission, DNA replication, and repair (2, 3).
Poirier-Bienvenu neurodevelopmental syndrome is characterized by developmental delay, intellectual disability, epilepsy, and facial abnormalities. More than 70 cases have been reported, most of which are in the infancy or early stages of disease (4, 5). Genotype-phenotype correlations and comprehensive descriptions of the course of the disease are required. Here, we present two patients harboring varying-sized deletions in 6p21.33, where the CSNK2B gene is located. Further, we offer a comprehensive literature review on detailed seizure descriptions in patients with CSNK2B gene variants, aiming to assist in achieving early diagnosis and intervention.
Cases presentation
Patient 1 was a 24-year-old Chinese male, the second child of healthy, non-consanguineous parents, with a healthy older sister. After an uneventful pregnancy and delivery, the mother gave birth at 28. The patient’s weight, height, and head circumference were normal at birth. The developmental milestone of this patient was within the normal range before the age of three. The patient’s family history was unremarkable. The first seizure occurred at the age of three years, triggered by fever; after that, he experienced febrile seizures approximately two to three times a year. The patient developed afebrile seizures at 16 years of age. The seizures were characterized by sudden episodes of blinking and twitching of the unilateral corners of the mouth, followed by ipsilateral limb twitching that lasted approximately 10 min. He presented with seizures in clusters, with a frequency of more than ten episodes per month. At the age of 18, seizures were controlled with valproate (VPA), lamotrigine (LTG), and oxcarbazepine (OXC) treatment. After a seizure-free period of 1 year, the seizures recurred, and the patient was resistant to antiepileptic medications (AED). A biphasic pattern of epilepsy was prominent in this patient and was characterized by an easy response to AEDs, followed by recurrent refractory seizures in adolescence. After the onset of epilepsy, the patients’ cognitive and language skills gradually decreased. Physical evaluation revealed dysmorphic features, including large low-set ears, a wide-base nose, a prominent forehead, a protuberant eyebrow arch, a high anterior hairline, thin hair, a broad left thumb, clinodactyly of the left fifth digit, and bilateral brachydactyly (Figure 1). The progressive slowing of the electroencephalography (EEG) in the background is prominent. The interictal EEG showed multifocal spikes and spike waves sporadically at an early age and presented continuous spikes and waves during sleep with age. Diffused electrodecremental events were observed over the right frontotemporal region at the beginning of the clinical seizures, and then 5–6 Hz theta activity was recruited over the bilateral anterior regions with interruption of motor artifacts (Figure 2). Brain magnetic resonance imaging (MRI) revealed bilateral temporal horn enlargement and abnormal signals in the right central semiovale (Figure 2).
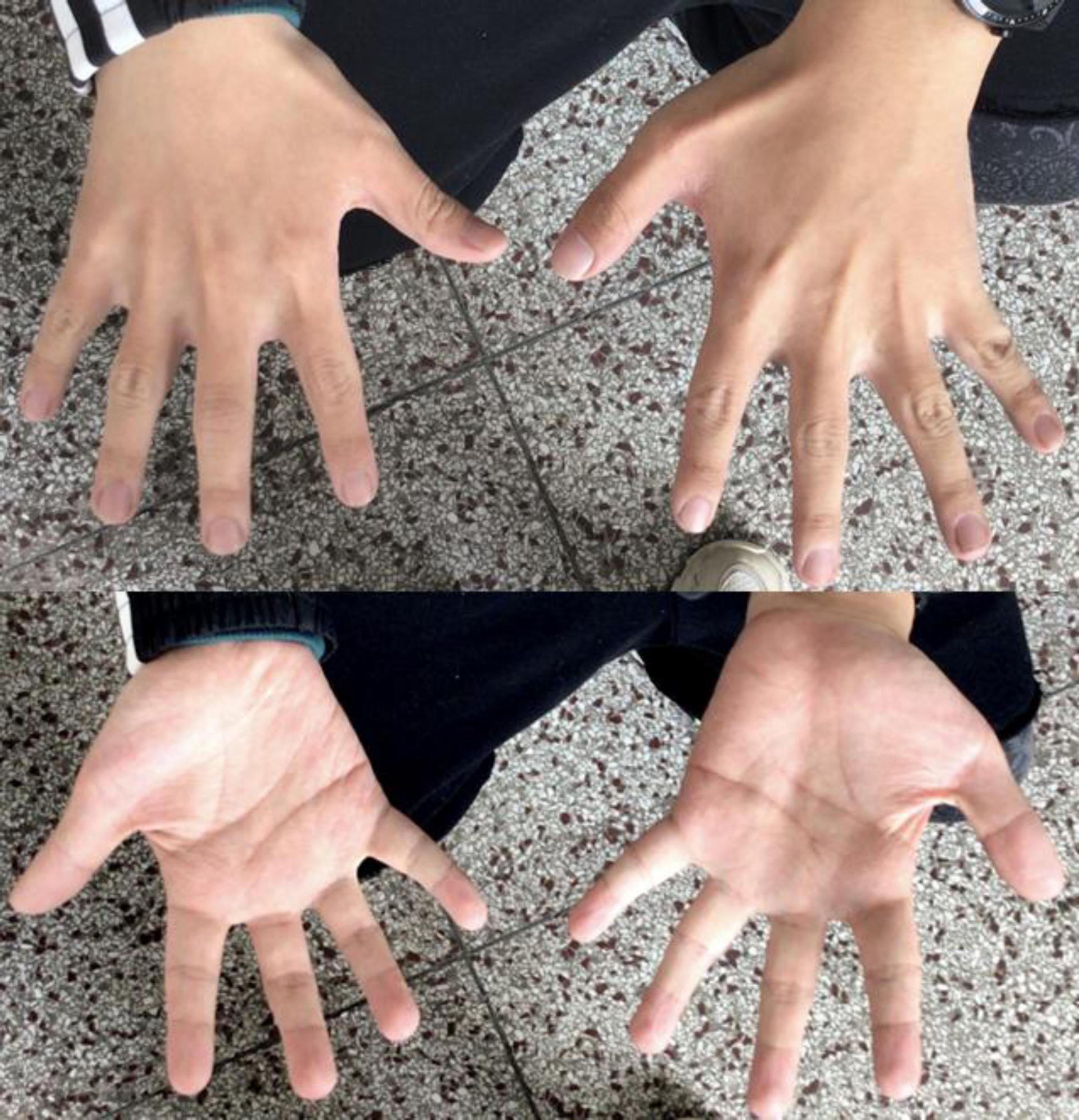
Figure 1. Digital anomalies of patient 1.

Figure 2. EEG and Imaging data of patient 1. (A–C) EEG of patient 1 at 20 years old, 23 years old and 24 years old. Background of EEG slows down at 20 years old (A), paroxysmal slow waves lasting for less than 3 s at 23 years old (B), lasting for 20–30 s at 24 years old (C). Progressive slowing of the electroencephalographic (EEG) background is prominent. (D) EEG of patient 1 at 24 years old. The ictal EEG was characterized by right cerebral focus with diffused electrodecremental event, subsequently evolving into bilateral hemispheres. (E) MRI images of patient 1 at 23 years. MRI: revealed abnormal signal in right-sided centrum semiovale.
Next-generation sequencing was performed for the patient and his parents. A size of 0.91 Mbp heterozygous deletion of the short arm of chromosome 6 [(Chr6: 30885220-31795217) × 1] was revealed, deleting the OMIM-listed gene in Figure 3. Neither parent harbored the same deletion as the patient. The deletion regions encompassed 85 genes, and haploinsufficiency of CSNK2B could explain the clinical phenotype of our patient.
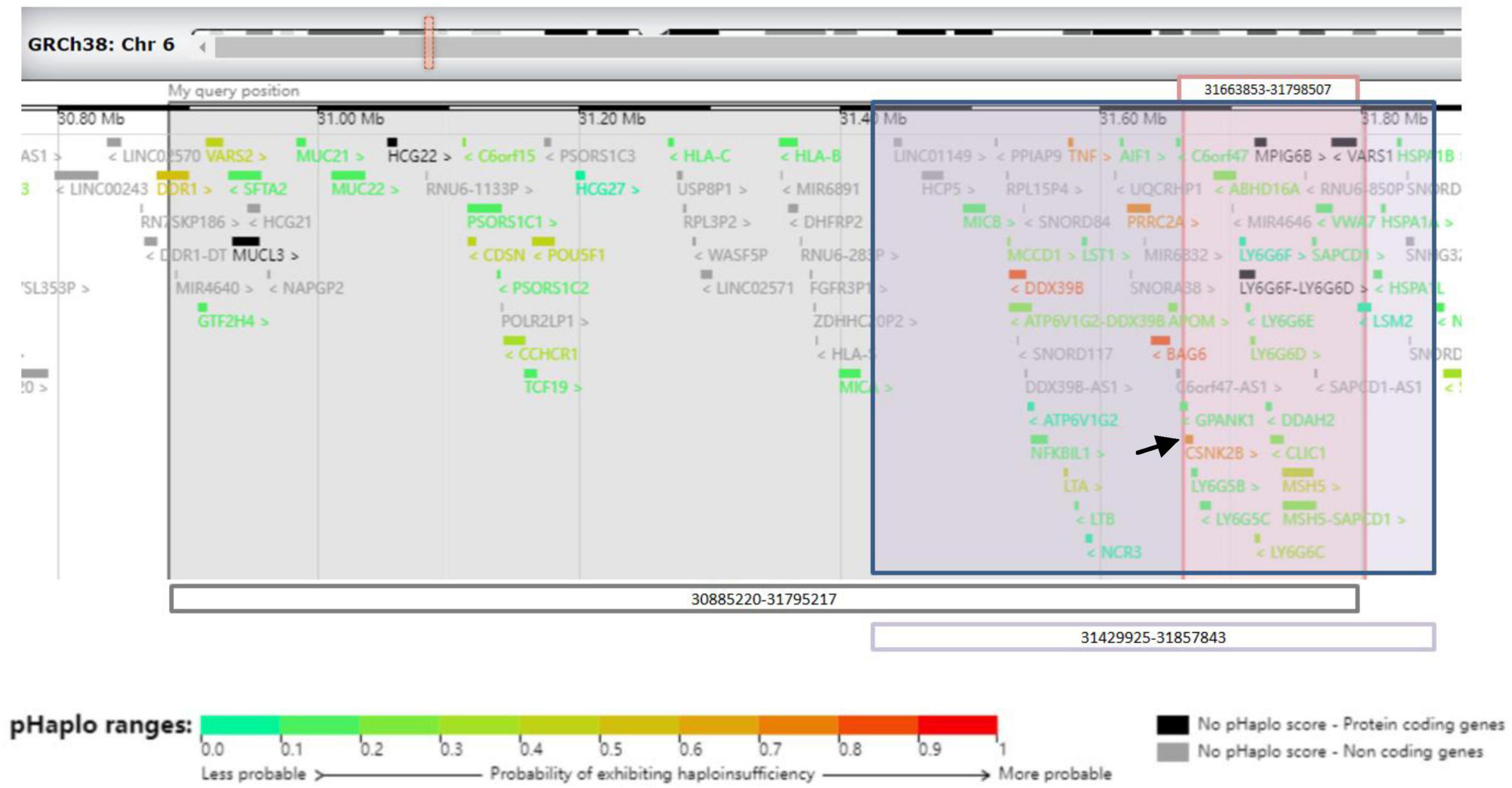
Figure 3. Schematic representation of 6p21.33 deletions in three cases from the UCSC Genome Browser. Gray-shaded area shows the area of 6p21.33 deletion in patient 1. Pink-shaded area shows the area of 6p21.33 deletions in patient 2. Purple-shaded area shows the area of 6p21.33 deletion in patient of Ikuko et al. The black arrow shows CSNK2B gene.
Patient 2 was a 22-month-old Chinese male with non-consanguineous parents and a healthy elder brother. The patient was born at term after an uneventful pregnancy and delivery. The global development was slightly delayed; he lifted his head at the age of 4 months and sat unassisted at the age of 8 months. The patient cannot walk independently and only speaks “baba” or “mama.” At five months, he experienced episodes of seizures characterized by tonic unilateral extremities lasting for approximately 2–3 s, with a frequency of several times a day. Seizures were controlled with VPA. EEG and MRI were normal. Facial features were not prominent due to her younger age.
Next-generation sequencing was performed for the patient and his parents. A size of 0.135 Mbp de novo heterozygous deletion in the short arm of chromosome 6 [(Chr6: 31663853-31798507) × 1] was observed in patient 2 (Figure 3). Neither parent harbored the same deletion as the patient. The deletion regions encompassed 22 genes, and the haploinsufficiency of CSNK2B could account for the clinical phenotype of our patient.
Approximately 91 cases have been reported in the literature; epilepsy was present in 83/91 of the patients (91%), usually before the age of 2 years. Drug-resistant epilepsy and a biphasic pattern of epilepsy account for 35% of patients with CSNK2B-related epilepsy. We compared the final follow-up age differences between patients with pharmacoresponsive epilepsy and those with pharmacoresistant epilepsy or biphasic epilepsy. The last follow-up age of patients with pharmacoresistant epilepsy or a biphasic pattern of epilepsy was significantly higher than that of patients with pharmacoresponsive epilepsy (P < 0.05). The main clinical, genetic, epilepsy, EEG, and brain MRI features of CSNK2B-related pharmacoresistant epilepsy and the biphasic pattern of epilepsy are summarized in Tables 1, 2.
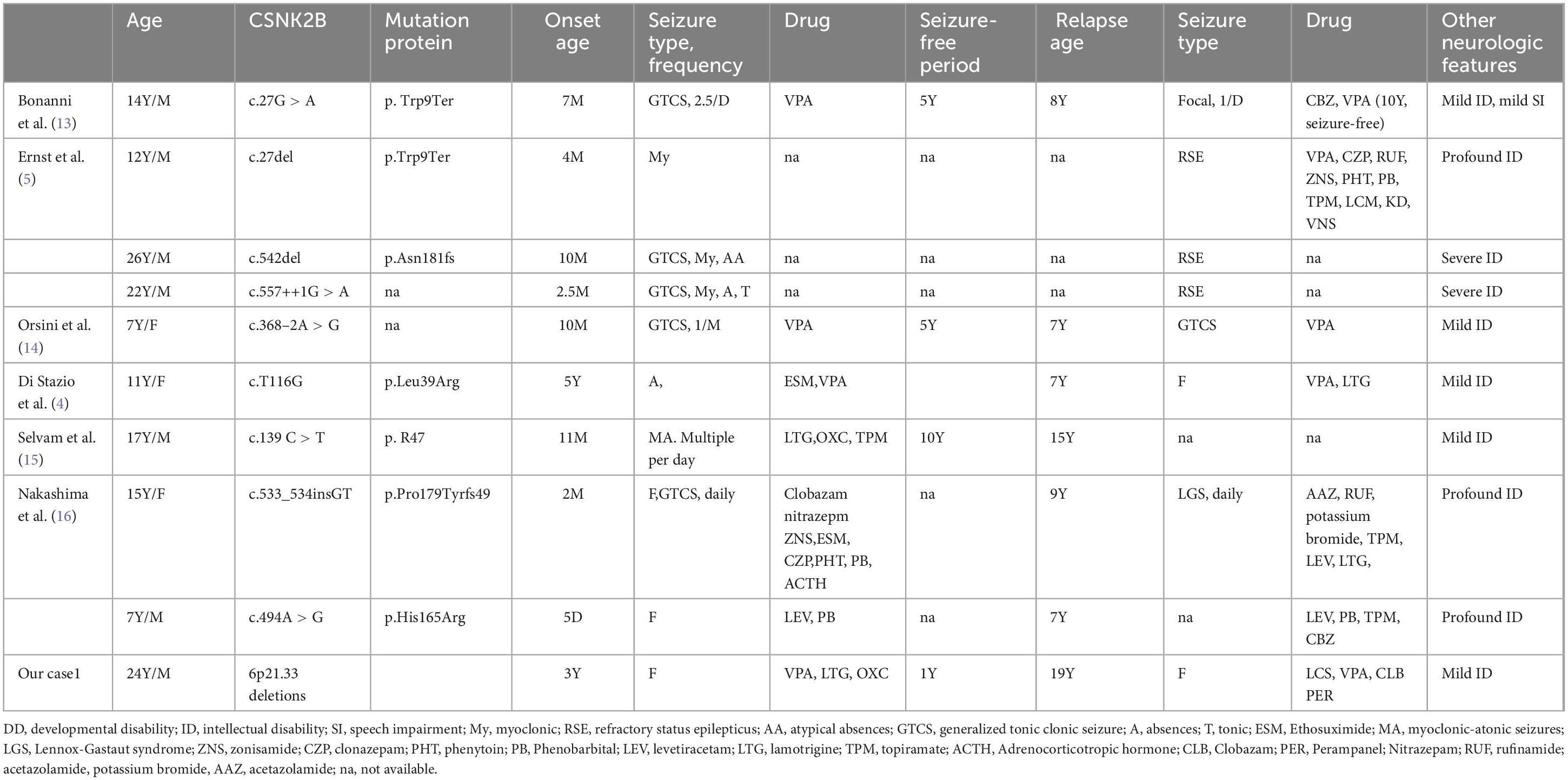
Table 1. Summary of clinical features of patients with CSNK2B related biphasic pattern of epilepsy.

Table 2. Summary of clinical features of patients with CSNK2B related pharmaco-resistant epilepsy.
Literature review
A systematic literature search was performed using PubMed and the Web of Science. MeSH and title/abstract were used for all eligible studies that mainly focused on the CSNK2B mutation or 6p21.33 deletion. We included (1) publications written in English, (2) clinical case reports and series with sufficient clinical data, and (3) diagnoses confirmed by genetic testing. Data from all eligible studies was analyzed and discussed by two reviewers. We additionally reported our two patients.
We carefully read all of the articles to extract the clinical information for the patients and we included all the patients with CSNK2B mutation or 6p21.33 deletion described in the analysis. Approximately 91 cases have been reported in the literature; epilepsy was present in 83/91 of the patients (91%), usually before the age of 2 years. Drug-resistant epilepsy and a biphasic pattern of epilepsy account for 35% of patients with CSNK2B-related epilepsy. The last follow-up age of patients with pharmacoresistant epilepsy or a biphasic pattern of epilepsy was significantly higher than that of patients with pharmacoresponsive epilepsy (P < 0.05). The main clinical, genetic, epilepsy, EEG, and brain MRI features of CSNK2B-related pharmacoresistant epilepsy and the biphasic pattern of epilepsy are summarized in Tables 1, 2.
Discussion
We present two patients with varying sizes of 6p21.33 deletions, including 0.135 Mbp and 0.91 Mbp, respectively, and provide a literature review of all the reported clinical cases on this subject. To date, only one case has been reported in the literature: Ikuko et al. reported a 7-year-old boy with a 0.43 Mbp deletion of 6p21.33 (6). A set of overlapping phenotypes with mild developmental delays, seizures, or febrile convulsions was observed in these three patients. According to the data from DECIPHER,1 the overlapping deleted regions encompassing 22 genes (Figure 3). Among them, CSNK2B showed high predictive scores for haploinsufficiency. CSNK2B gene was first identified by Poirier in patients with POBINDS (1). Thus, the haploinsufficiency of CSNK2B gene accounts for these clinical features. In addition, we observed more clinical phenotypes along with the evolution of the disease in Patient 1, suggesting a potentially progressive disease nature of CSNK2B-related POBINDS.
Despite the variable size of 6p21.33 deletions, all three patients shared an overlapping region (Figure 1) with a size of 0.135 Mb deletion. Terminal 4p deletion leads to Wolf–Hirschhorn syndrome (WHS), a contiguous gene deletion syndrome. The phenotypic severity of WHS is closely related to the size of the 4 p deletion (7). In contrast to WHS, all phenotypes presented by our patients could be explained by haploinsufficiency of the CSNK2B gene, which is located in an overlapping region (mentioned above). We speculated that the size of the 6p21.33 deletion does not correspond with the severity of the clinical phenotype.
Maria et al. reported that the c.94G > A variant of CSNK2B spawned a distinct phenotype characterized by cranial and digital anomalies, termed Intellectual Disability-Craniodigital Syndrome (IDCS), which is distinguishable from POBINDS (8). They suggested that haploinsufficiency of CSNK2B variants is the underlying pathomechanism of POBINDS. In contrast, a dominant-negative effect of CSNK2B variants contributes to the IDCS phenotype (8). In Patient 1, in addition to cranial anomalies and intellectual disability with seizures, digital anomalies were prominent, suggesting that both IDCS and POBINDS belong to a continuous spectrum of CSNK2B-associated phenotypes. The 6p21.33 deletion in this patient indicated that haploinsufficiency of CSNK2B had a predominant effect on the phenotype. The underlying pleiotropic effects of the variants may contribute to the variability of CSNK2B-associated phenotypes.
Up to date, approximately 91 patients with CSNK2B variants have been reported (5, 9–12). Epilepsy is a common symptom (approximately 91%). Epilepsy in patients with POBINDS begins early, usually before the age of 2 years, and generalized tonic or tonic-clonic seizures (GTCS) are the most common seizure type. Approximately 14.3% of patients had febrile convulsions or seizures. Drug-resistant epilepsy accounts for 37% (31/83) of patients with CSNK2B-related epilepsy. Similar to the observation of Ernst et al. (5) that generally seizure diminishes with age, but described 3 patients with worsening seizure frequency aged 7–12 years, we also find a biphasic pattern of epilepsy, which is characterized by an early age onset that is easily responsive to ASMs, followed by recurrent refractory seizures in adolescence, and was documented in 12% (10/83) of patients with epilepsy. We compared the final follow-up age differences between patients with pharmacoresponsive epilepsy and those with pharmacoresistant epilepsy or biphasic epilepsy. The last follow-up age of patients with pharmacoresistant epilepsy or a biphasic pattern of epilepsy was significantly higher than that of patients with pharmacoresponsive epilepsy. Although it has been reported that CSNK2B-related epilepsy outcomes are highly variable, we believe that the occurrence of a biphasic pattern of epilepsy and pharmacoresistant epilepsy in patients with CSNK2B variants is severely underestimated because the diagnosed age and last follow-up time of most patients are usually infants. Approximately 10 patients with a biphasic pattern of epilepsy were reported, and all of them had a relatively long follow-up duration, averaging 14.5 years (Tables 1, 2). The main clinical, genetic, epilepsy, EEG, and other neurological features are summarized in Table 1. Among these patients, we observed that seizure onset started in the first year of life in 80% of patients and within 5 years of age in 100%. During this phase, seizures were responsive to ASMs. After various periods of seizure freedom, ranging from 1 to 10 years, seizures recur in adolescence and tend to be pharmaco-resistant (Tables 1, 2). ID/DD was observed in 90% of the patients with CNSK2B-related epilepsy. Patients with pharmacoresistant epilepsy frequently showed more severe cognitive impairment, especially in situations in which seizures were not controlled for a long time (Tables 1, 2). Clinical management of patients with CNSK2B-related epilepsy requires aggressive treatment of seizures to avoid worsening neurodevelopmental outcomes.
In conclusion, our study identified two patients with rare 6p21.33 deletions encompassing the CSNK2B gene. Drawing from our observations, it seems that CSNK2B haploinsufficiency might play a decisive role in clinical manifestations. Our study further expands the phenotype-genotype spectrum of CNSK2B, thereby facilitating better the clinical management of this condition. Future researchs should focus on developing more effective therapies for CNSK2B-related epilepsy.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the Xuanwu Hospital ethics committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
XZ: Writing – original draft. HL: Writing – review and editing, Investigation. YJ: Writing – review and editing, Visualization, Investigation. WS: Writing – review and editing, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Xuanwu Hospital Science Program for Fostering Young Scholars (grant number: QNPY2021006) and National Natural Science Foundation of China (grant number: 81571267).
Acknowledgments
We sincerely thank the patient for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Poirier K, Hubert L, Viot G, Rio M, Billuart P, Besmond C, et al. CSNK2B splice site mutations in patients cause intellectual disability with or without myoclonic epilepsy. Hum Mutat. (2017) 38:932–41. doi: 10.1002/humu.23270
2. Borgo C, D’Amore C, Sarno S, Salvi M, Ruzzene M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct Target Ther. (2021) 6:183. doi: 10.1038/s41392-021-00567-7
3. Kravic B, Harbauer A, Romanello V, Simeone L, Vögtle F, Kaiser T, et al. In mammalian skeletal muscle, phosphorylation of TOMM22 by protein kinase CSNK2/CK2 controls mitophagy. Autophagy. (2018) 14:311–35. doi: 10.1080/15548627.2017.1403716
4. Di Stazio M, Zanus C, Faletra F, Pesaresi A, Ziccardi I, Morgan A, et al. Haploinsufficiency as a foreground pathomechanism of Poirer-Bienvenu syndrome and novel insights underlying the phenotypic continuum of CSNK2B-associated disorders. Genes (Basel). (2023) 14:250. doi: 10.3390/genes14020250
5. Ernst M, Baugh E, Thomas A, Bier L, Lippa N, Stong N, et al. CSNK2B: A broad spectrum of neurodevelopmental disability and epilepsy severity. Epilepsia. (2021) 62:e103–9. doi: 10.1111/epi.16931
6. Ohashi I, Kuroda Y, Enomoto Y, Murakami H, Masuno M, Kurosawa K. 6p21.33 deletion encompassing CSNK2B is associated with relative macrocephaly, facial dysmorphism, and mild intellectual disability. Clin Dysmorphol. (2021) 30:139–41. doi: 10.1097/MCD.0000000000000372
7. Battaglia A, Carey JC, South ST. Wolf-Hirschhorn syndrome: A review and update. American journal of medical genetics. Part C Semin Med Genet. (2015) 169:216–23. doi: 10.1002/ajmg.c.31449
8. Asif M, Kaygusuz E, Shinawi M, Nickelsen A, Hsieh T, Wagle P, et al. De novo variants of CSNK2B cause a new intellectual disability-craniodigital syndrome by disrupting the canonical Wnt signaling pathway. HGG Adv. (2022) 3:100111. doi: 10.1016/j.xhgg.2022.100111
9. Chen X, Han Y, Li X, Huang S, Yuan H, Qin Y. Case report: Two cases of Poirier-Bienvenu neurodevelopmental syndrome and review of literature. Front Pediatr. (2023) 11:967701. doi: 10.3389/fped.2023.967701
10. Hu C, Liu D, Luo T, Wang Y, Liu Z. Phenotypic spectrum and long-term outcome of children with genetic early-infantile-onset developmental and epileptic encephalopathy. Epileptic Disord. (2022) 24:343–52. doi: 10.1684/epd.2021.1394
11. Ballardin D, Cruz-Gamero J, Bienvenu T, Rebholz H. comparing two neurodevelopmental disorders linked to CK2: Okur-Chung neurodevelopmental syndrome and Poirier-Bienvenu neurodevelopmental syndrome-two sides of the same coin? Front Mol Biosci. (2022) 9:850559. doi: 10.3389/fmolb.2022.850559
12. Li D, Zhou B, Tian X, Chen X, Wang Y, Hao S, et al. Genetic analysis and literature review of a Poirier-Bienvenu neurodevelopmental syndrome family line caused by a de novo frameshift variant in CSNK2B. Mol Genet Genomic Med. (2024) 12:e2327. doi: 10.1002/mgg3.2327
13. Bonanni P, Baggio M, Duma G, Negrin S, Danieli A, Giorda R. Developmental and epilepsy spectrum of Poirier-Bienvenu neurodevelopmental syndrome: Description of a new case study and review of the available literature. Seizure. (2021) 93:133–9. doi: 10.1016/j.seizure.2021.10.019
14. Orsini A, Santangelo A, Bravin F, Bonuccelli A, Peroni D, Battini R, et al. Expanding phenotype of Poirier-Bienvenu syndrome: New evidence from an Italian multicentrical cohort of patients. Genes (Basel). (2022) 13:276. doi: 10.3390/genes13020276
15. Selvam P, Jain A, Cheema A, Atwal H, Forghani I, Atwal P. Poirier-Bienvenu neurodevelopmental syndrome: A report of a patient with a pathogenic variant in CSNK2B with abnormal linear growth. Am J Med Genet A. (2021) 185:539–43. doi: 10.1002/ajmg.a.61960
16. Nakashima M, Tohyama J, Nakagawa E, Watanabe Y, Siew C, Kwong C, et al. Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures. J Hum Genet. (2019) 64:313–22. doi: 10.1038/s10038-018-0559-z
17. Li J, Gao K, Cai S, Liu Y, Wang Y, Huang S, et al. Germline de novo variants in CSNK2B in Chinese patients with epilepsy. Sci Rep. (2019) 9:17909. doi: 10.1038/s41598-019-53484-9
18. Wilke M, Oliveira B, Pereira A, Doriqui M, Kok F, Souza C. Two different presentations of de novo variants of CSNK2B: Two case reports. J Med Case Rep. (2022) 16:4. doi: 10.1186/s13256-021-03184-8
19. Zhang W, Ye F, Chen S, Peng J, Pang N, Yin F. Splicing interruption by intron variants in CSNK2B causes Poirier-Bienvenu neurodevelopmental syndrome: A focus on genotype-phenotype correlations. Front Neurosci. (2022) 16:892768. doi: 10.3389/fnins.2022.892768
Keywords: 6p21.33 deletion, CNSK2B, POBINDS, epilepsy, biphasic patterns, digital anomalies
Citation: Zhang X, Lu H, Ji Y and Sun W (2024) Case report: Novel deletions in the 6p21.33 involving the CSNK2B gene in patients with Poirier-Bienvenu neurodevelopmental syndrome and literature review. Front. Med. 11:1441573. doi: 10.3389/fmed.2024.1441573
Received: 31 May 2024; Accepted: 09 September 2024;
Published: 18 October 2024.
Edited by:
Andrea Venerando, University of Udine, ItalyReviewed by:
Heike Rebholz, INSERM U1266 Institut de Psychiatrie et Neurosciences de Paris, FranceChristian Borgo, University of Padua, Italy
Copyright © 2024 Zhang, Lu, Ji and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Sun, Ym11c3Vubnl3QDE2My5jb20=