 Pradeesh Sivapalan1,2*†
Pradeesh Sivapalan1,2*† Valdemar Rømer1†Tobias Wirenfeldt Klausen1Niklas Dyrby Johansen3
Valdemar Rømer1†Tobias Wirenfeldt Klausen1Niklas Dyrby Johansen3 Manan Pareek3Daniel Modin3Alexander Mathioudakis4,5Jørgen Vestbo1,4,5Josefin Eklöf1Alexander Jordan1
Manan Pareek3Daniel Modin3Alexander Mathioudakis4,5Jørgen Vestbo1,4,5Josefin Eklöf1Alexander Jordan1 John R. Hurst6Tor Biering-Sørensen3,7,8†Jens-Ulrik Jensen1,2†
John R. Hurst6Tor Biering-Sørensen3,7,8†Jens-Ulrik Jensen1,2†- 1Department of Medicine, Respiratory Medicine Section, Copenhagen University Hospital-Herlev and Gentofte, Copenhagen, Denmark
- 2Department of Clinical Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 3Department of Cardiology, Copenhagen University Hospital-Herlev and Gentofte, Copenhagen, Denmark
- 4Division of Infection, Immunity and Respiratory Medicine, University of Manchester, Manchester, United Kingdom
- 5North West Lung Centre, Wythenshawe Hospital, Manchester University NHS Foundation Trust, Manchester, United Kingdom
- 6UCL Respiratory, University College London, London, United Kingdom
- 7Center for Translational Cardiology and Pragmatic Randomized Trials, Department of Biomedical Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Hellerup, Denmark
- 8Steno Diabetes Center Copenhagen, Herlev, Denmark
Rationale: Long-acting muscarinic antagonists (LAMAs) reduce the risk of acute exacerbations of chronic obstructive pulmonary disease (AECOPD), usually taken once daily in the morning. However, the circadian activity of autonomic regulation suggests that the highest need for anticholinergic therapy may be in the late night/early morning. This is supported by evidence that AECOPD most often begins in the morning. Furthermore, the trough spirometry effect of LAMA is lower than the peak effect, which further argues that evening dosing may be more optimal than morning dosing. This trial aims to determine whether evening administration of LAMA reduces hospitalization-requiring AECOPD or death from all causes within 1 year as compared to morning administration of the same LAMA.
Methods: Randomized controlled open-label trial. Persons aged 30 years or older with a once-daily LAMA prescription and a confirmed COPD diagnosis were recruited. Participants were randomized in a 1:1 ratio to either morning or evening LAMA administration. Complete follow-up for the primary outcome, hospitalization-requiring AECOPD, or death from all causes within 1 year was captured from the Danish National Health Register, as were patient-reported outcome assessments at 6 and 12 months.
Results: A total of 10,013 participants were randomized, and the recruitment process started on 9 March 2023. Secondary outcomes include (i) moderate COPD exacerbations; (ii) all-cause hospitalization; (iii) ICU admission; (iv) need for non-invasive ventilation; and (v) all-cause mortality, among others. All outcomes will be evaluated 12 months after recruitment.
Clinical trial registration:ClinicalTrials.gov, NCT05563675.
Introduction
Background and rationale
One of the most serious complications of chronic obstructive pulmonary disease (COPD) is acute exacerbation (AECOPD). On average, each COPD patient experiences 0.5–3.5 acute exacerbations per year, which is an important reason for hospitalization, disease progression, and mortality as well as a decline in health status and lung function (1, 2). Treatment with a long-acting muscarinic antagonist (LAMA) reduces dyspnea and the risk of exacerbations in patients with COPD by binding to muscarinic receptors in bronchial smooth musculature and thus inhibiting cholinergic bronchial constriction. LAMAs are given as inhalation therapy once daily (most often) or twice daily (3). Most COPD patients experience their worst symptoms and experience exacerbations in the early morning hours, before getting out of bed (4). This might be explained by the physiological diurnal changes in the activity of the parasympathetic homeostasis system, which is most active at night to improve digestion and other secretions (5). Correspondingly, the activity of the sympathetic system is physiologically suppressed at night, and stimulation of β-2 receptors is thus also low (and opposite for M-3 receptors). The balance of sympathetic-parasympathetic tone is shifted significantly toward the latter. Most available LAMA treatments are dosed once daily in the morning. Thus, for a COPD patient, being at a trough level of LAMA (which antagonizes the parasympathetic system) at late night/early morning, may carry a hazard for the patient. Studies have found that lung function measured as a forced expiratory volume in 1 s (FEV1) improvement peaks approximately 2 h after LAMA administration and that FEV1 is still significantly improved at 7 h post-treatment but decreases toward the trough level of the LAMA (6). However, as a corollary to the above, when the medicine is probably most needed (02.00 a.m.–07.00 a.m.), the effect is at its lowest level, which may not be desirable, since a low effect of the most important preventive medicine against AECOPD at this time may lead to more exacerbations. Evening administration, on the contrary, would lead to a greater and more certain effect regarding bronchodilation and reduced secretion in the early morning hours, and a maximum effect should be expected during the entire night.
Objectives
The main objective of this intervention and research protocol is to determine whether evening administration of LAMA, as opposed to morning administration, is superior for the occurrence of hospitalization-requiring AECOPD or death from all causes. The intervention will have the following secondary objectives: (1) moderate COPD exacerbations, (2) all-cause hospitalization, (3) intensive care unit (ICU) admission, (4) need for non-invasive ventilation, (5) all-cause mortality, (6) Short-Acting Beta-Agonist (SABA) use / pick up rate, and exploratory endpoints (1) change in COPD Assessment Test (CAT) score, (2) change in Medical Research Council (MRC) score, and (3) change in the “soundly sleeping” parameter of CAT.
Trial design
The study is a pragmatic, registry-based, open-label, randomized controlled trial combining the utilization of the Danish nationwide health registries and the official Danish electronic letter system (Digital Post/e-Boks) into an innovative, decentralized trial requiring no study visits from participants. The trial will include patients receiving LAMA treatment that will be randomized 1:1 to either LAMA administration in the evening or the morning for 12 months.
Methods: participants, interventions, and outcomes
Study setting
This study will be conducted by the Chronic Obstructive Pulmonary Disease Trial Network (COP:TRIN) at the Section of Respiratory Medicine, Copenhagen University Hospital – Herlev and Gentofte, Copenhagen, Denmark. This group will be responsible for all processes in the study, including recruitment, inclusion, randomization, follow-up, and communication with participants. Registry data will be obtained through the Danish Health Data Authority and accessed through an encrypted remote-access server environment. The study will identify potential participants across Denmark through nationwide health registries, and the electronic letter system (Digital Post/e-Boks) will be used for sending recruitment letters and communicating with patients. The trial recruits and allocates patients electronically, and follow-up consists of electronic questionnaires, while endpoints are assessed using data from national registries. Therefore, there is no list of specific study sites/clinics/hospitals.
Eligibility criteria
Inclusion criteria:
1. Age > 30 years.
2. Current treatment with LAMA once daily (as recorded in the Danish National Prescription Registry and confirmed by the participant via questionnaire).
3. Confirmed COPD diagnosis.
Exclusion criteria:
1. Patients who decline to participate.
Who will obtain informed consent?
We have obtained a waiver from the regional science ethics board for informed consent since the medication and dose will not be changed. The science ethics board argues that the intervention lies beyond the scope of the science ethics legislation in Denmark.
However, only those who provided consent to the written information, and who reported having COPD, are included. We have obtained approval from the Data Protection Agency (P-2022-432) on 1 July 2022.
Additional consent provisions for collection and use of participant data and biological specimens
No biological specimens will be obtained. Regarding informed consent, see previous section.
Interventions
Explanation for the choice of comparators
Patients will be randomized 1:1 to one of the two treatment strategies by computer-generated allocation to either:
• LAMA administration in the morning (control).
• LAMA administration in the evening (intervention).
These treatment strategies have been chosen because morning administration is the most conventional, while evening administration will be investigated to determine if it is superior with respect to the occurrence of hospitalization-requiring AECOPD or death from all causes within 12 months, along with several secondary and exploratory outcomes, as outlined in Table 1.

Table 1. Primary endpoint definition.
Intervention description
Participants randomized to LAMA administration [with or without the combination of inhaled corticosteroids (ICSs) and/or long-acting beta-2-agonists (LABAs)] in the evening will be instructed to take their LAMA-containing inhalation between 8 p.m. and 2 a.m.
Participants randomized to LAMA administration (with or without the combination of ICS and/or LABA) in the morning will be instructed to take their LAMA as usual in the morning between 6 and 12 a.m.
Criteria for discontinuing or modifying allocated interventions
Investigators may pause or interrupt the intervention at any given time in case of medical reasoning, safety risks, or requirements from the authorities.
Furthermore, participating patients can withdraw from the intervention or the entire trial at any given time without providing a reason (Figure 1).
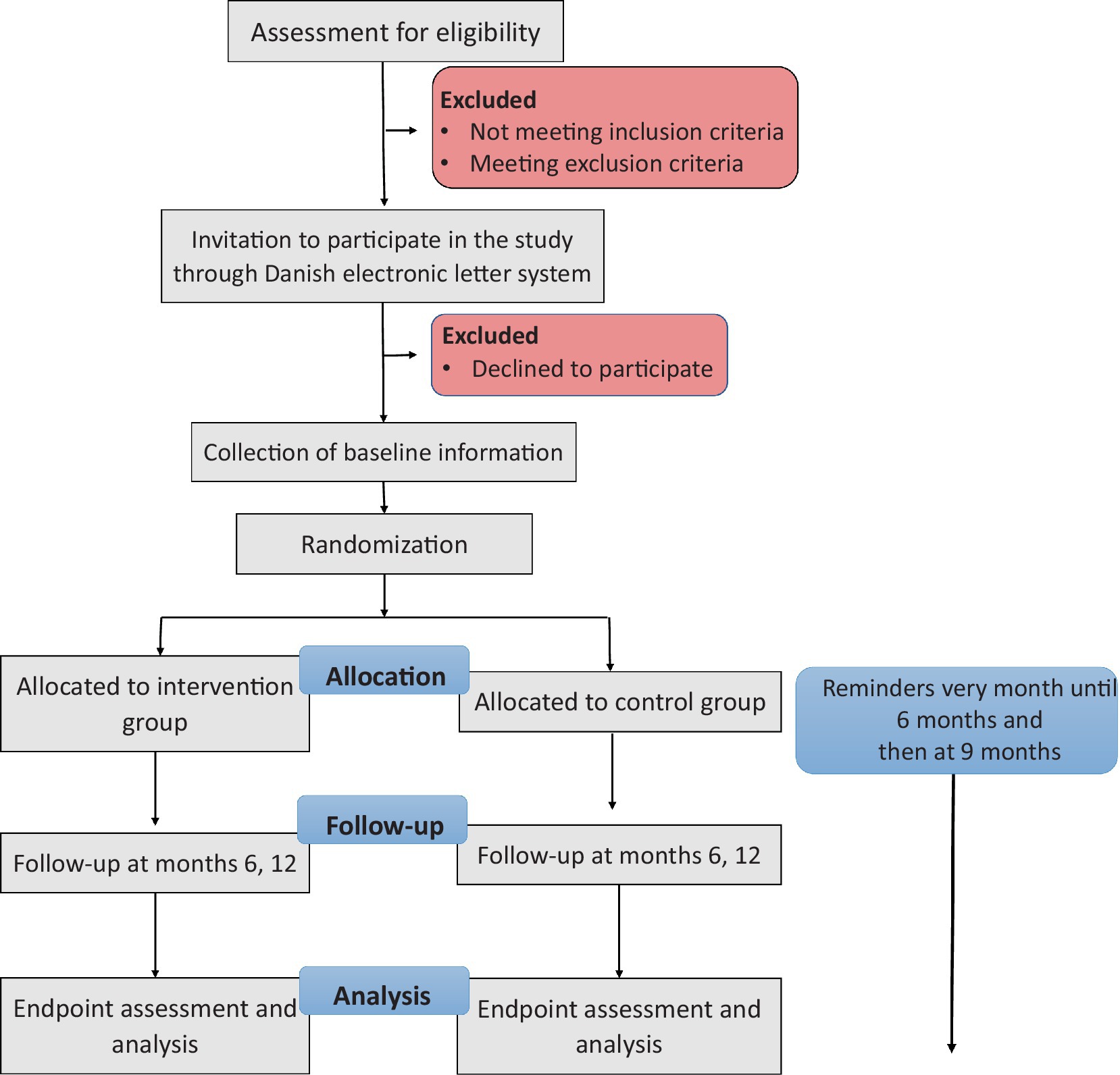
Figure 1. Clinical trial flowchart.
Strategies to improve adherence to interventions
Reminders on LAMA administration time will be sent to participants every month until the first questionnaire (6 months), and then at 9 months.
Relevant concomitant care permitted or prohibited during the trial
N/A: no specific concomitant care or interventions are prohibited during the trial.
Provisions for post-trial care
The trial is covered by the patient compensation scheme if, contrary to expectation, damages occur during the trial and by inclusion.
Outcomes
The primary efficacy endpoint is the occurrence of hospitalization requiring AECOPD or death from all causes within 12 months. Secondary efficacy endpoints included (i) moderate COPD exacerbations; (ii) all-cause hospitalization; (iii) ICU admission; (iv) need for non-invasive ventilation; (v) all-cause mortality; (vi) SABA use/pick-up rate; and exploratory endpoints (i) relation between adherence and AECOPD hospitalization, (ii) change in CAT score, (iii) change in MRC score, and (iv) change in “soundly sleeping” parameter of CAT.
The details on the primary and secondary outcomes of the study can be seen in Tables 1, 2, and exploratory outcomes in Table 3.
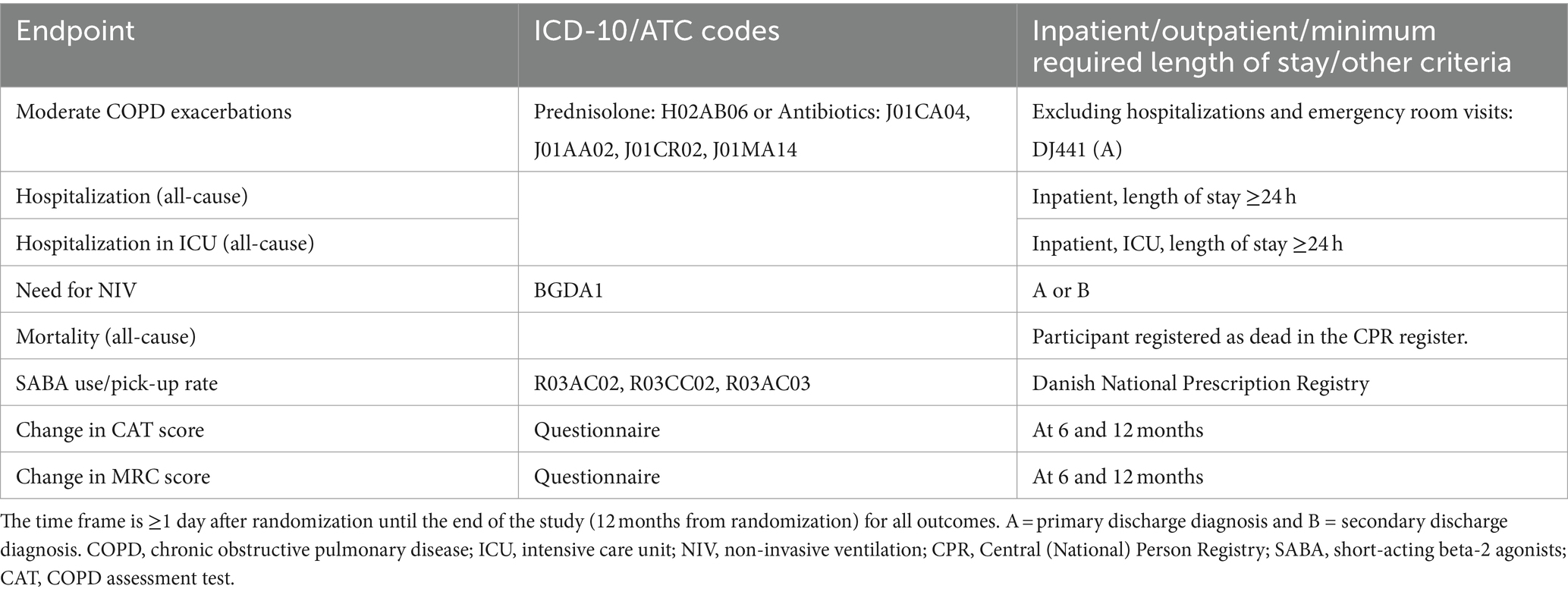
Table 2. Secondary and exploratory endpoint definition.
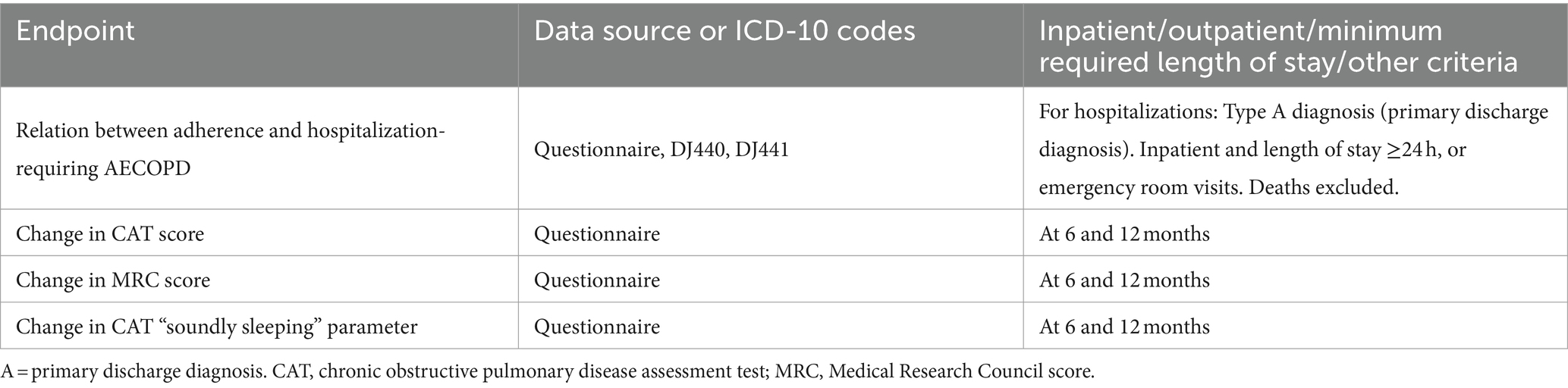
Table 3. Exploratory endpoint definition.
Participant timeline
The participant timeline is seen in Table 4.
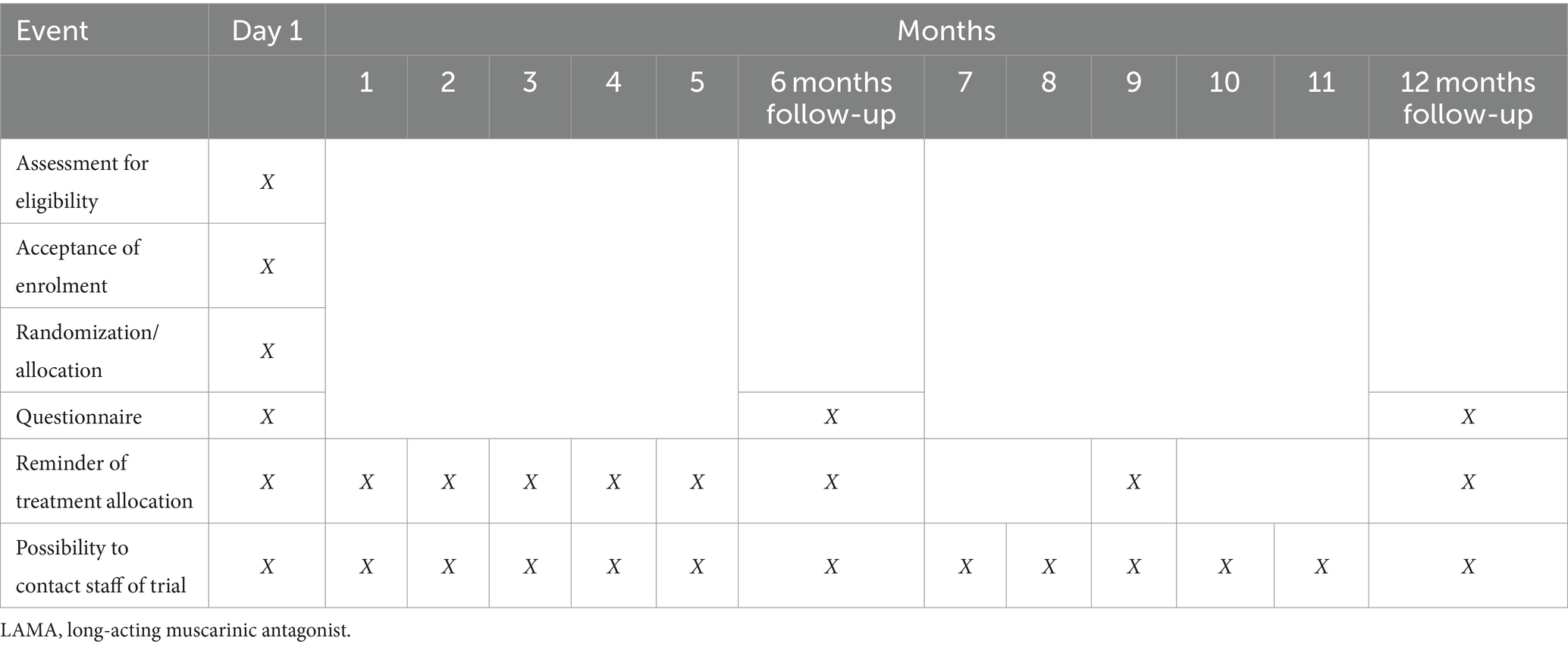
Table 4. Participant timeline.
Every electronic mail to the participants will include a direct hyperlink to a FAQ on https://coptrin.dk/, and an e-mail that they can contact in case of further questions.
Sample size
In Denmark, approximately 400,000 people are assumed to have COPD; however, approximately a quarter of them have not been diagnosed yet. We have data showing that 178,000 citizens have collected LAMA medication within the last 6 months. At the same time, we know that every year, there are approximately 20,000 admissions with AECOPD (severe AECOPD) and 6,000 deaths in patients with COPD. Assuming 50% of the AECOPD admissions are either unique or preceding a death, there are 16,000 annual events corresponding to our primary outcome. Thus, the annual risk of a primary outcome is approx. 16,000/178,000 = 8.9%. We hypothesize that evening LAMA administration will reduce the relative risk of the primary outcome by 25%, corresponding to a 2.2% absolute risk reduction. Assuming 84.4% uniformly distributed adherence to the intervention (= approximately 1 in 6 does not adhere), this corresponds to a relative risk reduction of 21.3% = 1.9% absolute risk reduction (average effect among all) in the ITT population. Alpha and power (1-beta) are set at 0.05 (two-sided) and 0.8, respectively. This leads to a needed sample size of 6,662 (3,331 + 3,331).
Recruitment
Through the Danish Health Data Authority, COP:TRIN will obtain personal identification numbers for all Danish citizens currently treated with LAMAs (all combinations). Recruitment letters will be sent through the official Danish electronic mailbox system (Digital Post). The letter will contain a link to a REDCap questionnaire for the participants to complete if they are interested. The questionnaire will obtain the participant’s personal identification number, select sociodemographic and lifestyle-related variables, confirm current treatment with LAMAs, and obtain consent to participate.
Assignment of interventions: allocation
Sequence generation
Patients will be randomized 1:1 to one of the two treatment strategies by computer-generated allocation to either LAMA administration in the morning or evening.
Concealment mechanism
The concealment mechanism for investigators will be that the database manager and outcome assessors will be blinded. The allocated group will not be concealed from participants.
Implementation
After providing informed consent, the participant will automatically be provided with a randomization allocation, to either LAMA administration in the evening or the morning, on their screen and sent to their official electronic mailbox, as will the participant be continuously reminded (Table 4).
Assignment of interventions: blinding
Who will be blinded
The trial is open-label. Trial participants will not be blinded. The database manager and the outcome assessors will be blinded. Apart from this, all endpoints are assessed using prespecified registry-based definitions to ensure maximal reduction of the risk of bias.
Procedure for unblinding if needed
N/A, the patient is not blinded to the allocated group.
Data collection and management
Plans for the assessment and collection of outcomes
In addition to what is obtained through the initial questionnaire, all information on baseline characteristics will be collected from Danish nationwide registries. This will be supplemented with data from the initial questionnaire on body mass index, smoking status (including pack-years), FEV1, and asthma. Information on adherence and symptom scores for exploratory outcomes will be obtained through questionnaires. The data from nationwide registries include age, gender, exacerbation history, comorbidities, and relevant medication details. All data used to assess clinical endpoints during the study will be collected from Danish nationwide registries.
Plans to promote participant retention and complete follow-up
The trial will aim to ensure adherence and compliance with the allocated randomized group through electronic reminders (Table 4).
Data management
All data will be saved on REDCap. A statistician will be responsible for the validity of analyses.
Confidentiality
Personal information about potential and enrolled participants will be collected, shared, and maintained according to patient–physician confidentiality and GDPR.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future use
N/A
No biological specimens will be obtained in this trial.
Statistical methods
Statistical methods for primary and secondary outcomes
The primary and all secondary outcomes will be analyzed using the intention-to-treat (ITT) approach, which encompasses the entire cohort of randomized participants in the study. Additionally, per-protocol analyses will be conducted for patients taking inhalation medication 6–7 days per week at the allocated administration time for the full duration of the study, along with modified ITT (mITT) analyses for those who have taken less than 6 days or more than 2 days per week for any amount of time. These analyses will be performed on both the primary and all secondary outcomes, providing a comprehensive evaluation of the study’s results.
The primary outcome (hospitalization-requiring AECOPD or death from all causes within 12) will be presented as a fraction with percentage and relative risk with a 95% confidence interval. The groups will be compared using a two-sided binomial test. The same principles will be applied to secondary binomial outcomes. For the SABA pick-up rate, the incidence rate and ratios will be calculated and presented (including 95% confidence intervals) and compared using the chi-squared test. Changes in CAT and MRC (respectively) will be reported as median differences with IQR and compared within and between groups with a paired two-sided Wilcoxon signed-rank test and an unpaired two-sided Wilcoxon rank-sum test. A subgroup analysis will be performed on the primary outcome with the following subgroups: adherence group (high adherence: ≥4–5 days per week of adherence), age (≥65 years), asthma overlap, coexisting heart failure, body mass index (>30 kg/), frequent exacerbator (>1 severe exacerbation within 12 months prior to baseline), and treatment with systemic beta-2 antagonist at baseline. A p-value of <0.05 will be considered significant.
Interim analyses and stopping guidance
A Data and Safety Monitoring Board (DSMB) will be appointed and act according to a charter agreed by the investigators and approved by the sponsor. Six months after the recruitment of the last patient, an interim analysis, based on the ITT population, will be performed to evaluate the continuation of the trial. As main statistical measures, an O’Brien-Fleming plot of the primary endpoint and mortality (calculating Z-scores) will be performed. The interim analysis will be performed and presented by a sub-investigator not otherwise involved in the data collection or analyses, overlooked by the trial statistician. The data will be presented in a blinded fashion.
At the interim analysis, the event rate and effect size for the primary endpoint, as well as the safety and progress of the study, will be assessed by the DSMB, and follow-up duration may be adjusted accordingly. Safety will be assessed by comparing event rates to the event rate presumption used for the initial sample size determination. The study may stop for futility if it is deemed improbable to achieve a follow-up duration sufficient to detect a clinically important effect size. The study will not stop for superior efficacy at interim analysis. We wish to prioritize increasing the likelihood of having sufficient power to conclude on the secondary endpoints at the final analysis.
Methods for additional analyses (e.g., subgroup analyses)
Continuous baseline variables will be summarized using means, 95% confidence intervals, medians, and interquartile ranges according to distribution. Categorical variables will be compared using frequencies and proportions. All statistical tests will be two-sided, and p-values of <0.05 will be considered statistically significant.
Methods in analysis to handle protocol non-adherence and any statistical methods to handle missing data
Secondary analyses will include mITT and per-protocol analyses to provide a comprehensive evaluation of the study outcomes. No data on the primary outcome will be missing as national registries will be used for outcome assessment. Only self-reported outcomes such as MRC and CAT can be expected to be missing. It will be assumed all missing data will be missing at random or completely at random to allow for multiple imputations. No imputed data will be presented in the baseline table.
Plans to give access to the full protocol, participant-level data, and statistical code
The study protocol has been published on www.coptrin.dk.
Participant-level data and statistical code will not be published.
Oversight and monitoring
Composition of the coordinating center and trial steering committee
The coordination center is composed of Pradeesh Sivapalan, Valdemar Rømer, Niklas Dyrby Johansen, Tor Biering-Sørensen, and Jens-Ulrik Stæhr Jensen. The trial steering committee is made up of the COP:TRIN organization and has given input to the design of the study. Information on the study has been published by the coordination center on www.coptrin.dk.
Composition of the data monitoring committee, its role, and reporting structure
The DSMB comprises three independent members who will assess the interim analysis 6 months after the recruitment of the last patient.
Adverse event reporting and harms
N/A as the trial does not alter the type of medication or dosage of medication already being used.
Frequency and plans for auditing trial conduct
There will be an independent data safety monitoring committee, as mentioned in 21a. Otherwise N/A.
Plans for communicating important protocol amendments to relevant parties (e.g., trial participants and ethical committees)
If any changes are made, all relevant parties will be informed promptly by mail and through Trial Setting Committee meetings.
Dissemination plans
Both negative, positive, and inconclusive results will be submitted for publication in peer-reviewed journals and presented at international scientific conferences. Authorship criteria will be based on Recommendations for the Conduct, Reporting, Editing, and Publication of Scholarly Work in Medical Journals by The International Committee of Medical Journal Editors.
Participant-level data and statistical code will not be published.
Discussion
Morning symptoms are common in COPD and have an important impact on patient quality of life (QoL) (7). Nighttime administration may lead to better treatment effects in the morning and thus reduce these symptoms among patients.
In addition to morning symptoms, there is evidence that up to 66% of COPD patients have airway obstruction during their sleep leading to desaturation, poor sleep quality, cardiovascular disease, and more frequent exacerbations (8, 9). A randomized trial with 80 patients showed that tiotropium improved oxygen saturation during sleep compared to placebo (10). The average oxygen saturation during sleep was 3% points higher with nighttime administration and 2% points higher with daytime administration compared to placebo. The difference between the two dosing regimens was not statistically significant; however, only 40 patients were included in this comparison, so it may have been insufficiently powered. Furthermore, this study was not able to examine clinical outcomes such as exacerbations or mortality.
Hospitalization-requiring AECOPD has a significant clinical and socioeconomic impact. A large database review of over 73,000 patients with up to 17-year follow-ups found that fewer than half of patients hospitalized for an exacerbation were still alive after 5 years (11). Patients who survive hospitalization-requiring AECOPD are likely to experience significantly impaired QoL and are at increased risk of further exacerbations. The contribution of non-hospitalized exacerbations to mortality rates (i.e., unreported exacerbations that should have warranted hospital treatment) has not been well-quantified (12). Therefore, COPD treatment to prevent hospitalization-requiring AECOPD or death from all causes is a crucial therapeutic goal for patients with COPD. We will aim to reduce exacerbations and mortality with our simple intervention which can have a great impact on both patients and society, including resource consumption during hospitalizations for this large group of patients.
Ethics statement
The requirement for ethical approval was waived by the Committees on Health Research Ethics for the Capital Region, due to the nature of the study and the absence of alterations in medication or dosage. The studies were conducted in accordance with local legislation and institutional requirements.
Author contributions
PS: Writing – review & editing, Writing – original draft. VR: Writing – review & editing, Writing – original draft. TW: Writing – original draft. ND: Writing – original draft. MP: Writing – original draft. DM: Writing – original draft. AM: Writing – original draft. JV: Writing – original draft. JE: Writing – original draft. AJ: Writing – original draft. JH: Writing – original draft. TB-S: Writing – original draft. J-UJ: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The study was financed by grants from Novo Nordisk Fonden (NO. NNF20OC0060657). The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication. The funders had no role in the study design, data collection and analysis, the decision to publish, or the preparation of the manuscript.
Acknowledgments
The authors thank the patients and their families for their participation in the trial; the investigators, trial personnel, all the pulmonary departments, general practitioners, and nurses who have supported this study; and the members of the independent data and safety monitoring committee for their review of the data and COP:TRIN steering committee (https://coptrin.dk/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AECOPD, Acute exacerbations of chronic obstructive pulmonary disease; COPD, Chronic obstructive pulmonary disease; FEV1, Forced expiratory volume in 1 s; LABA, Long-acting beta agonist; LAMA, Long-acting muscarinic antagonist; ICSs, Inhaled corticosteroids.
References
1. Wedzicha, JA, and Seemungal, TA. COPD exacerbations: defining their cause and prevention. Lancet. (2007) 370:786–96. doi: 10.1016/S0140-6736(07)61382-8
2. Seemungal, TAR, Donaldson, GC, Paul, EA, Bestall, JC, Jeffries, DJ, and Wedzicha, JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (1998) 157:1418–22. doi: 10.1164/ajrccm.157.5.9709032
3. Miravitlles, M, and Ribera, A. Understanding the impact of symptoms on the burden of COPD. Respir Res. (2017) 18:67. doi: 10.1186/s12931-017-0682-y
4. van Buul, AR, Kasteleyn, MJ, Chavannes, NH, and Taube, C. Morning symptoms in COPD: a treatable yet often overlooked factor. Expert Rev Respir Med. (2017) 11:311–22. doi: 10.1080/17476348.2017.1305894
5. Fink, AM, Bronas, UG, and Calik, MW. Autonomic regulation during sleep and wakefulness: a review with implications for defining the pathophysiology of neurological disorders. Clin Auton Res. (2018) 28:509–18. doi: 10.1007/s10286-018-0560-9
6. Kerwin, E, Stiler, TM, Korenblat, P, White, A, Eckert, JH, Henley, M, et al. Efficacy and safety of twice-daily Glycopyrrolate versus placebo in patients with COPD: the GEM2 study. Chronic Obstr Pulm Dis. (2016) 3:549–59. doi: 10.15326/jcopdf.3.2.2015.0157
7. Roche, N, Chavannes, NH, and Miravitlles, M. COPD symptoms in the morning: impact, evaluation and management. Respir Res. (2013) 14:112. doi: 10.1186/1465-9921-14-112
8. Shawon, MSR, Perret, JL, Senaratna, CV, Lodge, C, Hamilton, GS, and Dharmage, SC. Current evidence on prevalence and clinical outcomes of co-morbid obstructive sleep apnea and chronic obstructive pulmonary disease: a systematic review. Sleep Med Rev. (2017) 32:58–68. doi: 10.1016/j.smrv.2016.02.007
9. Agusti, A, Hedner, J, Marin, JM, Barbéle, F, Cazzola, M, and Rennard, S. Night-time symptoms: a forgotten dimension of COPD. Eur Respir Rev. (2011) 20:183–94. doi: 10.1183/09059180.00004311
10. McNicholas, WT, Calverley, PMA, Lee, A, Edwards, JC, Williams, A, Rees, J, et al. Long-acting inhaled anticholinergic therapy improves sleeping oxygen saturation in COPD. Eur Res J. (2004) 23:825–31. doi: 10.1183/09031936.04.00085804
11. Suissa, S, Dell’Aniello, S, and Ernst, P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax. (2012) 67:957–63. doi: 10.1136/thoraxjnl-2011-201518
Keywords: long-acting muscarinic antagonists, chronic obstructive pulmonary disease, COPD exacerbation, all-cause mortality, intensive care admission
Citation: Sivapalan P, Rømer V, Wirenfeldt Klausen T, Dyrby Johansen N, Pareek M, Modin D, Mathioudakis A, Vestbo J, Eklöf J, Jordan A, Hurst JR, Biering-Sørensen T and Jensen J-U (2024) AM/PM dosing of LAMA for COPD: a randomized controlled trial protocol using digital recruitment and registries. Front. Med. 11:1430169. doi: 10.3389/fmed.2024.1430169
Edited by:
Chantal Raherison, Centre Hospitalier Universitaire Guadeloupe, FranceReviewed by:
Ourania Papaioannou, General University Hospital of Patras, GreeceMarina Gueçamburu, Centre Hospitalier Universitaire de Bordeaux, France
Copyright © 2024 Sivapalan, Rømer, Wirenfeldt Klausen, Dyrby Johansen, Pareek, Modin, Mathioudakis, Vestbo, Eklöf, Jordan, Hurst, Biering-Sørensen and Jensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pradeesh Sivapalan, cHJhZGVlc2guc2l2YXBhbGFuLjAyQHJlZ2lvbmguZGs=
†These authors have contributed equally to this work