
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Med., 29 July 2024
Sec. Regulatory Science
Volume 11 - 2024 | https://doi.org/10.3389/fmed.2024.1425038
This article is part of the Research TopicRepurposing for Small Population NeedsView all 8 articles
 Claudia Fuchs1*
Claudia Fuchs1* Peter A. C. ‘t Hoen2
Peter A. C. ‘t Hoen2 Annelieke R. Müller3,4,5
Annelieke R. Müller3,4,5 Friederike Ehrhart6Clara D. M. Van Karnebeek3,4,5
Friederike Ehrhart6Clara D. M. Van Karnebeek3,4,5Rett syndrome (RTT) and Rett-like syndromes [i.e., CDKL5 deficiency disorder (CDD) and FOXG1-syndrome] represent rare yet profoundly impactful neurodevelopmental disorders (NDDs). The severity and complexity of symptoms associated with these disorders, including cognitive impairment, motor dysfunction, seizures and other neurological features significantly affect the quality of life of patients and families. Despite ongoing research efforts to identify potential therapeutic targets and develop novel treatments, current therapeutic options remain limited. Here the potential of drug repurposing (DR) as a promising avenue for addressing the unmet medical needs of individuals with RTT and related disorders is explored. Leveraging existing drugs for new therapeutic purposes, DR presents an attractive strategy, particularly suited for neurological disorders given the complexities of the central nervous system (CNS) and the challenges in blood-brain barrier penetration. The current landscape of DR efforts in these syndromes is thoroughly examined, with partiuclar focus on shared molecular pathways and potential common drug targets across these conditions.
Rett syndrome (RTT, #312750) and Rett-like syndromes, e.g., CDKL5 deficiency disorder (CDD, #300672) and FOXG1-syndrome (or FOXG1-related encephalopathy, #613454) are rare monogenic neurodevelopmental disorders (NDDs). The relative recent recognition of their distinct clinical entities (1, 2) has deepened our understanding of their underlying pathogenic mechanisms and clinical characteristics (Table 1). Although each disorder exhibits unique clinical features, they share common core symptoms and neurological traits (Table 1), suggesting that these disorders share critical molecular etiology.
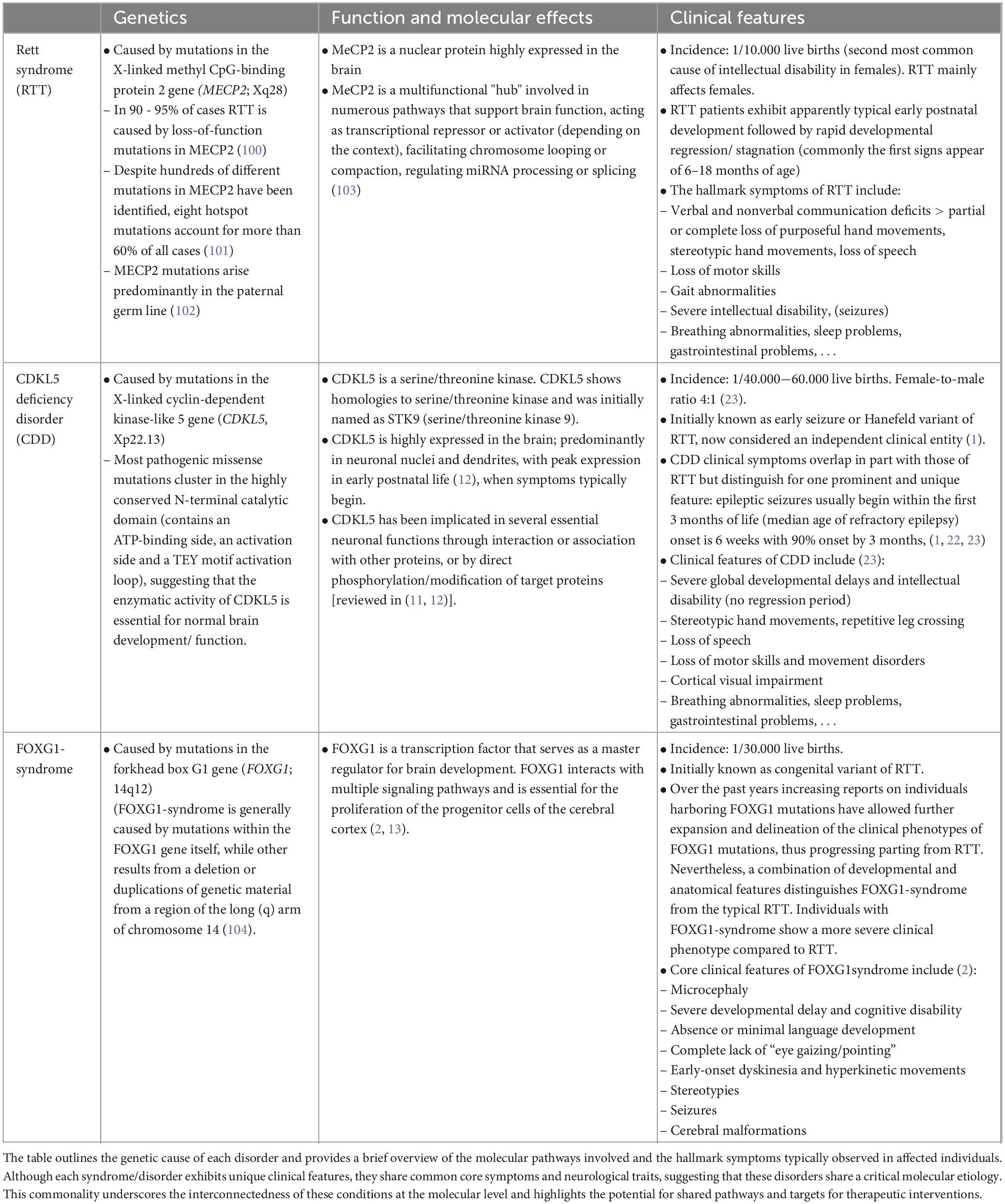
Table 1. Genetic, molecular and symptomatic aspects of Rett syndrome (RTT) and Rett-like syndromes, i.e., CDKL5 deficiency disorder (CDD) and FOXG1-syndrome.
Identifying shared pathways holds significant implications for targeted therapies development and drug repurposing (DR). DR, which involves using existing drugs for new therapeutic purposes, represents a promising approach in the treatment across multiple diseases especially for neurological disorders (3, 4). The complex structure of the central nervous system (CNS), coupled with the challenge of penetrating the blood-brain barrier, poses significant hurdles in the development of new drugs for neuropathological conditions, making DR of particular interest for these disorders. Notable successes of DR in NDDs include e.g., repurposing of fenfluramine in Dravet syndrome (5) or bumetanide (6) and pregnenolone (7) for autism spectrum disorders. These studies validate DR as a valid treatment approach for multiple neuropathological conditions.
We here discuss the current state of art of DR efforts in RTT, CDD and FOXG1-syndrome, with particular emphasis on the shared molecular pathways and the identification of common drug targets across the three conditions. For a more detailed overview on the molecular and circuit mechanisms underlying each syndrome, please refer to (8–10) for RTT, (11, 12) for CDD and (2, 13) for FOXG1-syndrome (2, 13).
As shown in Figure 1, pathways across the three diseases can be categorized in three main categories.
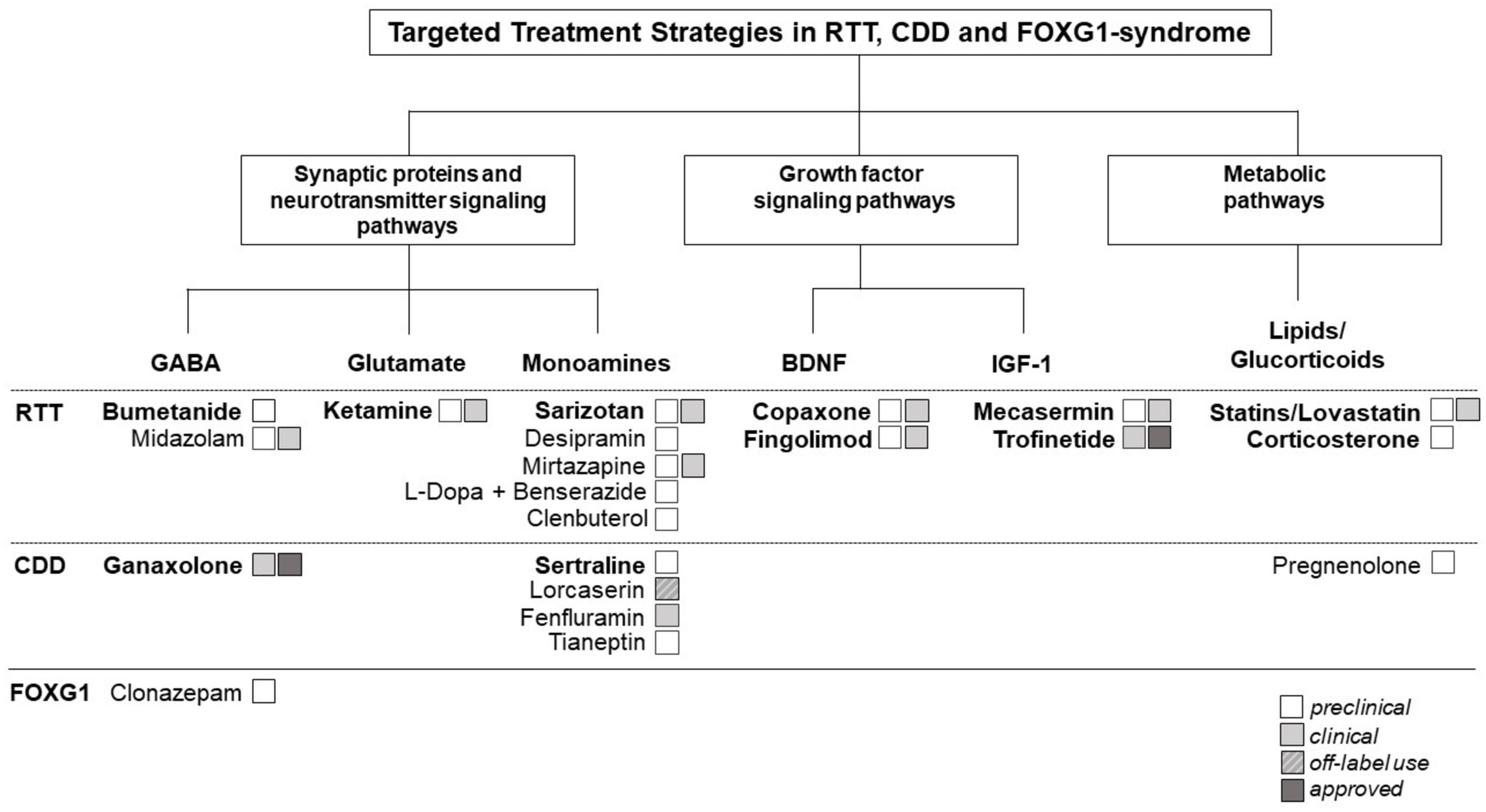
Figure 1. Therapeutic targets and potential repurposing strategies currently being explored in Rett syndrome (RTT) and Rett-like syndromes [i.e., CDKL5 deficiency disorder (CDD)] and FOXG1-syndrome. Shared molecular pathways across these disorders can be categorized in three main categories: (1). Synaptic proteins and neurotransmitter signaling pathways. (2). Growth factor signaling pathways. (3). Metabolic pathways. The figure illustrates the interconnectedness of molecular pathways across these disorders, highlighting common themes and potential targets for therapeutic intervention. Understanding these shared pathways may guide the development of repurposing strategies aimed at addressing the underlying molecular mechanisms of these neurodevelopmental disorders (NDDs). The figure highlights also the current repurposed drugs approved or being investigated for the treatment of RTT, CDD, and/or FOXG-1 syndrome. In bold, repurposing candidates described in detail in the main text of the manuscript.
Synaptic proteins and neurotransmitter signaling pathways are crucial for correcting brain function. Disruptions in these components are linked to alterations in excitatory/inhibitory (E/I) balance, synaptic transmission, and neuronal network activity: common features in many NDDs (14, 15). Neuronal hyperexcitability is a common causative feature contributing to the high prevalence of seizures observed in all three disorders (Table 1). It is intricately linked to the “GABA switch” phenomenon (16): during critical stages of brain development, GABA initially exerts excitatory effects due to high intracellular chloride concentrations. As development progresses, the expression and activity of involved chloride co-transporters shifts, leading to a corresponding change in the polarity of GABAergic signaling from excitatory to inhibitory. Disruptions in this transition, such as delayed maturation or altered expression of chloride co-transporters, can prolong the excitatory phase of GABAergic signaling, resulting in neuronal hyperexcitability. This phenomenon has been extensively studied in NDDs, including RTT (17–20). Multiple studies have investigated the effects of NKCC1 inhibition by the FDA-approved diuretic bumetanide as therapeutic option for multiple NDDs (21) (see section 3.1 - Bumenatide).
Epilepsy is prevalent in RTT and RTT-like disorders (Table 1), particularly in CDD, where seizures commonly manifest within the first 3 months of life (22, 23). Ganaxolone, a synthetic analogue of the neurosteroid allopregnanolone, which is a positive allosteric modulator of the GABAA receptor, has recently been approved for use in CDD, showing promise in managing seizures (see section 3.2 - Ganaxolone).
Dysfunctions in GABAergic signaling pathways contribute to various other RTT-related symptoms, including breathing abnormalities. Respiratory abnormalities in RTT mouse models can be significantly improved by manipulating diverse neurotransmitter systems (24, 25). Benzodiazepine like midazolam, generally used for anesthesia and procedural sedation, alleviate breathing defects in MeCP2-null mice (26) and has also been used for the acute management of prolonged seizures in RTT (27). Interestingly, Chen et al. showed that treatment with clonazepam, a long-acting tranquilizer of the benzodiazepine class, alleviates cognitive defects, limited social interactions, and depression-like behaviors in FOXG1 mutant mice (28).
In addition, changes in excitatory glutamatergic pathways and monoamine neurotransmitters, such as serotonin, dopamine and noradrenaline play a significant role in RTT and RTT-related disorders (26, 27) and several drugs targeting these changes have been tested. Of particular interest is ketamine, a NMDAR antagonist used as dissociative anesthetic and currently repurposed for the treatment of RTT (see section 3.3 - Ketamine).
Among the different monoamine neurotransmitters implicated especially serotonin has gained great attention in RTT and CDD. Different groups have shown a complex dysregulation of the serotoninergic system in mice and individuals with RTT and CDD [RTT: (29–31), CDD: (32, 33)]. Serotoninergic dysregulation in the brainstem contributes to breathing abnormalities and sarizotan, a 5-HT1A and D2-Like Receptor agonist, has been tested on respiratory dysfunction in RTT (34) (see section 3.4 - Sarizotan). A preclinical repurposing study showed that treatment with sertraline, a selective serotonin reuptake inhibitor (SSRI) and commonly used antidepressant improves behavioral abnormalities in CDD mice (33) (see section 3.5 - Sertraline). Many other antidepressants/drugs targeting the serotonergic pathway, including desipramine (35, 36) and tianeptine (37) have shown promising results in RTT and CDD models [reviewed in (35, 36)]. Interestingly, the antidepressant mirtazapine (acts as a potent antagonist of 5-HT2A and 5-HT3 receptors) was shown to ameliorate motor and social behaviors in RTT mice and patients (38, 39).
Different other drugs targeting monoamine neurotransmitter defects in RTT mice were tested. Szczesna and colleagues used the combination of L-Dopa with the dopa-decarboxylase inhibitor (Ddci) benserazide, a combination frequently used in Parkinsonism and related disorders, to stimulate dopaminergic deficiency in RTT mice (37). Mello and colleagues showed that treatment with the adrenergic receptor agonist clenbuterol, primarily used as a bronchodilator to treat conditions like asthma and chronic obstructive pulmonary disease (COPD), increased survival and improved behavioral deficits in RTT mice (40).
Dysregulations in growth factor signaling pathways has been widely implicated in the pathogenesis of RTT and RTT-like disorders. The role of brain-derived neurotrophic factor (BDNF) in promoting neuronal and synaptic development and function [reviewed in (38, 39)], as well as its implication in RTT [reviewed in (41)] and CDD (42) have been widely described. The involvement of BDNF dysregulation in the pathogenesis of FOXG1-syndrome remains to be elucidated (43). Several drugs targeting BDNF dysregulation have been repurposed in addition to different SSRIs and tianeptine, known to also enhance BDNF expression: BDNF boosters such as glatiramer acetate (copaxone) and fingolimod were recently investigated in RTT (see section 3.6 - BDNF-boosters).
Similarly, insulin-like growth factor 1 (IGF-1) is essential for neuronal maturation, survival, and plasticity [reviewed in (44)]. Alterations in IGF signaling pathways have been investigated in both mice and humans with RTT (45) and advanced the development of potential therapeutic strategies targeting IGF signaling in RTT. Interestingly, several studies in RTT mice have shown, that recombinant human IGF-1 (rhIGF-1) rescues RTT-related phenotypes in mice (46, 47). These experimental findings have led to a phase I and II clinical trial with rhIGF-1 (mecaserim) and two phase II trials with the IGF1 peptide analogue trofinetide in RTT patients (see section 3.7 - IGF-1).
Several studies have shown that perturbations in metabolic pathways, influence neurological and non-neurological features in RTT [reviewed in (48–50)]. Alterations in the blood metabolite levels associated with RTT, support the concept that RTT and RTT-related disorders are systemic diseases affecting neurodevelopment.
Abnormalities in lipid metabolism and alterations in lipid profiles, including changes in cholesterol levels have been found in mice and individuals with RTT [reviewed in (51)]. Some studies have suggested that statins, a class of medications primarily used to lower cholesterol levels in the blood may have neuroprotective effects and could potentially offer benefits in RTT and RTT-like syndromes (see section 3.8 - Statins/Lovastin). Disruptions in metabolic pathways are very complex in RTT and RTT-like disorders and offer a broad array of potential therapeutic targets beyond statins. Several studies have investigated abnormalities in brain glucose metabolism in individuals with RTT (52), and investigated the possibility to pharmacologically intervene using for example glucocorticoid (see section 3.9 - Glucorticoids/Corticosterone).
In this section, we will explore specific examples of repurposed drugs used in RTT and RTT-related disorders, drawing from our experience and understanding of common pathways (Figure 1). Rather than conducting a comprehensive review, we will highlight notable instances of DR within this context.
The FDA-approved diuretic bumetanide, acting as an NKCC1 chloride importer antagonist, has garnered great interest for its potential in multiple NDDs (21, 53, 54). Lozovaya et al. have recently demonstrated that in a RTT mouse model the GABA shift is abolished at birth and that this alteration persistent in juvenile offspring can be alleviated by maternal administration of bumetanide (55), suggesting that repurposing of bumetanide might be promising in RTT. However, its use across multiple neurological disorders faces several limitations: bumetanide has low brain penetration and is associated with several collateral issues due to excessive diuresis caused by NKCC2 inhibition in the kidney (21). Ongoing drug discovery efforts seek to develop novel NKCC1 inhibitors and modulators with fewer adverse effects (21) and evaluate their potential across multiple NDDs, including RTT and RTT-related disorders.
Ganaxolone (3-hydroxy-3β-methyl-5δ-pregnane-20-one) belongs to a novel group of neuroactive steroids called epalons that act as positive allosteric modulators of GABAA receptors to enhance GABAergic inhibition (56). FDA approved since 1983, ganaxolone has been investigated in various indications, including epilepsy and psychiatric disorders (57–61). A recent phase III trial (NCT03572933) has shown that ganaxolone reduces the frequency of CDD-associated seizures and is generally well tolerated (62, 63). Ganaxolone has been approved (US: March 2022, Europe: July 2023) for the treatment of seizures in CDD patients aged 2 years and older.
Channel-blocking NMDAR antagonists such as ketamine have shown significant promise in addressing the complex E/I imbalance associated with RTT and RTT-like disorders. Several preclinical studies have shown that ketamine improves cognitive function and behavior in animal models of RTT (64). Ketamine, a dissociative anesthetic initially used for induction and maintenance of anesthesia and repurposed for depression and related disorders (65) has shown promising results in RTT patients. A recently completed phase II clinical trials (NCT03633058) to assess the safety, tolerability and efficacy of low-dose oral ketamine in RTT patients has shown improvements across a broad range of symptoms (unpublished data). Ketamine has currently (in February 2023) been granted Orphan Drug Designation by the FDA and a phase III clinical trial is planned.
Although sarizotan, a phase III 5-HT1A receptor agonist used for the treatment of dyskinesias in Parkinson’s disease (PD) (66, 67) has shown beneficial effects on respiratory dysfunction in different RTT models (34), a phase III clinical trial conducted in 2020 (NCT02790034) has not confirmed these results in a clinical setting. Low and high dose daily oral administration of sarizotan was ineffective in meeting its primary goal of reducing the percentage of apneas in children and adults with RTT-related breathing abnormalities. A new chemical entity acting as selective 5-HT1A agonist (NLX-101), which has shown antidepressant and cognitive enhancing properties, is currently under development (68, 69).
Sertraline, a widely used antidepressant to treat panic, generalized and social anxiety and obsessive compulsive disorders has been tested in a preclinical setting for CDD (70). The study has shown that treatment with sertraline improved several behavioral abnormalities in CDD mice (70). As sertraline is currently not approved for the use in children, it is widely described “off-label” for certain mental health conditions (71, 72). Different studies have evaluated the long-term impact of treatment with sertraline on cognitive, emotional and physical development in pediatric subjects (73–75) and the drug seems quite well tolerated. Interestingly, the sertraline study also included preliminary results on the off-label use of lorcaserin in CDD patients (33). Locaserin, like fenfluramine, acts as a selective 5-HT2C receptor agonist and was initially developed and approved as appetite suppressor. Although the use of both medications was discontinued in the treatment of severe obesity, due to safety and toxicity concerns, lorcaserin and fenfluramine are currently repurposed for the treatment of seizures associated with Dravet syndrome and Lennox-Gastaut syndrome [reviewed in (5, 76)]. A phase II open label trial to investigate the effects of repurposing of fenfluramine to control seizures in CDD patients is currently ongoing (NCT03861871) and preliminary results were published (77).
Glatimarer acetate, sold under the name copaxone is an immunomodulator medication to treat multiple sclerosis (MS). As glatiramer acid stimulates the secretion of BDNF in the brain and the levels of BDNF expression seem to be directly correlated with the severity of RTT related symptoms (78), a small phase II clinical trial (NCT02153723) was run in 2014. Although the trial showed improvements in gait analyses, respiratory dysfunction, electroencephalographic findings, and quality of life, there were severe safety concerns (79, 80).
Like copaxone, fingolimod is an immunomodulating medication used for the treatment of MS. Interestingly this sphingosine-1-phosphate analog, has been shown to upregulate BDNF mRNA levels and increase BDNF protein release (81) and to increase cognitive impairment in Huntington’s disease and Alzheimer’s diseases (82, 83). A recent phase II clinical trial to assess safety and efficacy of fingolimod in children with RTT (NCT02061137) has shown that treatment with fingolimod was safe but had no significative effects on RTT-associated symptoms (84).
Mecasermin is FDA-approved for the long-term treatment of growth failure in children with severe primary IGF-1 deficiency (85). A phase I and II clinical (NCT01777542) study assessed safety and efficacy of mecasermin treatment in RTT patients. Both studies (86, 87) reported that a significant proportion of RTT patients exhibited limited response to mecasermin treatment, suggesting that further studies are needed.
Trofinetide, a synthetic analogue of IGF-1 originally developed as potential treatment for stroke (88) has been successfully repurposed for RTT. A recent phase III clinical trial (NCT04181723) has shown that trofinetide improves behavioral, communication, and physical RTT symptoms (89, 90) and trofinetide has gained FDA approval in 2023. Trofinetide is the first FDA-approved drug for the treatment of RTT.
Statins, commonly used to lower cholesterol, have widely been explored in NDDs due to their ability to reduce neuroinflammation and enhance synaptic function and neuronal survival [reviewed in (91)]. Several studies have suggested that cholesterol metabolism is perturbed in mice and individuals with RTT [reviewed in (58)]. Interestingly, initial research showed promising effects of statin treatment (fluvastatin and lovastatin) in RTT mice (92), but subsequent studies in a mouse model with a different genetic background yielded in conflicting results (93), illustrating the complexity of genetic and pharmacological interactions in NDDs like RTT. Therefore, personalized DR approaches may be necessary to optimize therapeutic outcomes in individuals with RTT. In 2015, a phase II open label dose escalating study of lovastatin in a small group of RTT patients (NCT02563860) has shown some preliminary positive results in improving visual recognition, memory and eye tracking.
Glucocorticoids are steroid hormones well known for their use in the treatment of inflammation, autoimmune diseases, and cancer. Corticosteroids (predominantly prednisolone and hydrocortisone) and adrenocorticotropic hormone (ACTH) are widely used as anti-epileptic drugs in pediatric populations, including RTT and CDD (94, 95). Interestingly, different studies have shown that pharmacological intervention with the glucocorticoid system using low-dose corticosterone has an impact on the symptoms and lifespan in an RTT mice (96, 97). Interesting pre-clinical studies have recently investigated the effects of pregnenolone-methyl-ether, a synthetic neuroactive steroid and derivative of pregnenolone on CDD cell and mouse models (98, 99). However, further research is needed to confirm these findings and determine the safety and efficacy of glucocorticoid therapies in humans with RTT and RTT-like disorders.
RTT and RTT-like syndromes pose significant challenges for therapeutic strategies due to their rarity, complex etiology, and multifaceted symptomatology (Table 1). Traditional drug discovery pipelines often struggle to navigate these complexities. However, given the urgent need for effective treatments in RTT and related disorders, DR emerges as a pragmatic and resource-efficient strategy to expedite clinical translation and enhance treatment availability for affected individuals. In this discussion, we briefly examine the challenges and limitations of DR in RTT and RTT-related disorders and suggest potential solutions to advance the field.
One of the primary hurdles in drug development for RTT and RTT-like disorders undoubtedly lies in their rarity. Traditional drug development necessitates substantial financial investment to design, conduct, and analyze large-scale clinical trials involving significant numbers of participants.
Nevertheless, owing the rarity of RTT and related disorders, recruiting an adequate number of eligible participants for such trials can be extremely challenging and costly.
The heterogeneity and the diverse array of symptoms manifested by these disorders pose additional challenges. While the underlying genetic and molecular mechanisms overlaps to some extent, individual variability demands tailored therapeutic approaches. Grouping patients by shared clinical symptoms instead of genetic diagnosis, and developing DR strategies targeting symptoms that can likely be treated by DR candidates, such as proposed in the recently EU-funded SIMPATHIC project,1 may increase the efficiency of DR.
Repurposing existing drugs with pleiotropic effects or multitargeted mechanisms of action can address multiple aspects of the disorders simultaneously, offering holistic treatment approaches. Furthermore, combinatory repurposing targeting different pathways can be tailored to specific symptoms/domains which may lead to greater therapeutic benefits than any single agent alone. However, it is essential to note that combinatory therapies also pose challenges in terms of optimizing drug combinations, dosing regimens, and potential interactions. Rigorous preclinical and clinical studies are imperative to evaluate their safety and efficacy in RTT and RTT-like syndromes, considering the unique characteristics and variability of these disorders among affected individuals.
Rigorous preclinical and clinical studies are also crucial for better understanding the complex pathophysiology of these syndromes. To date, the precise molecular mechanisms underlying these complex disorders are still not fully understood; hindering the identification and validation of potential drug targets. This specifically applies to CDD and FOXG1-syndrome: both conditions were identified as distinct clinical entities only recently and it is understandable that research efforts initially focused primarily on “classical” RTT. This discrepancy is reflected also in the very different numbers of repurposing studies highlighted in Figure 1. Continued efforts in pre-clinical (identification of valuable cell and animal models etc.) and clinical research (better understanding of the natural history, clinical manifestations, disease progression, biomarkers etc.) will be essential for advancing our understanding and improving outcomes for individuals affected by these syndromes. In particular, better characterizing the shared symptoms and pathways across these entities, will provide valuable insights into the underlying biology and potentially uncover new common mechanisms and targeted therapies. If the disorders demonstrate convergence in their underlying molecular pathways, this provides an opportunity for designing joint DR strategies across RTT and RTT-like disorders. This could reduce the time needed for the development of DR and increase the number of patients benefiting from the treatments, resulting in more attractive business models.
Despite promising DR results in preclinical or early-phase clinical trials for RTT and related disorders in our opinion DR is still underrated and underutilized in this kind of disorders. DR holds immense potential for addressing the unmet medical needs and therapeutic challenges posed by such complex NDDs, and recent advancements screening and computational techniques, offer the unique opportunity to predict drug-disease interactions and prioritize candidate compounds for further investigation. By leveraging existing drugs and repurposing them for new indications, this approach offers a pragmatic and efficient strategy to accelerate the development of treatments for individuals affected by these debilitating conditions.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
CF: Writing−review and editing, Writing−original draft, Supervision, Project administration, Conceptualization. P’tH: Writing−review and editing, Writing−original draft. AM: Writing−review and editing, Writing−original draft. FE: Writing−review and editing, Writing−original draft. CV: Writing−review and editing, Writing−original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of the article. The construction of this manuscript was partially funded by a Horizon 2020 Grant to the European Joint Programme on Rare Diseases (Grant no. 825575), Horizon Europe Grants to the REMEDI4ALL (Grant no. 101057442), and SIMPATHIC (Grant no. 101080249) consortia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The views expressed in the article reflect the views of the authors and are not intended to convey the views of their employers or affiliations.
1. Fehr S, Wilson M, Downs J, Williams S, Murgia A, Sartori S, et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet. (2013) 21:266–73. doi: 10.1038/ejhg.2012.156
2. Wong L, Singh S, Wang H, Hsu C, Hu S, Lee W. FOXG1-related syndrome: From clinical to molecular genetics and pathogenic mechanisms. Int J Mol Sci. (2019) 20:4176. doi: 10.3390/ijms20174176
3. Mao X. Drug repurposing in neurological diseases: Opportunities and challenges: Drug repurposing - hypothesis, molecular aspects and therapeutic applications. London: IntechOpen (2020). doi: 10.5772/intechopen.93093
4. Hua Y, Dai X, Xu Y, Xing G, Liu H, Lu T, et al. Drug repositioning: Progress and challenges in drug discovery for various diseases. Eur J Med Chem. (2022) 234:114239. doi: 10.1016/j.ejmech.2022.114239
5. Jonker A, O’Connor D, Cavaller-Bellaubi M, Fetro C, Gogou M, ’T Hoen PAC, et al. Drug repurposing for rare: Progress and opportunities for the rare disease community. Front Med (Lausanne). (2024) 11:1352803. doi: 10.3389/fmed.2024.1352803
6. Ben-Ari Y. Bumetanide to treat autism spectrum disorders: Are complex administrative regulations fit to treat heterogeneous disorders? Rare Dis Orphan Drugs J. (2024) 3:2. doi: 10.20517/rdodj.2023.22
7. Fung L, Libove R, Phillips J, Haddad F, Hardan A. Brief report: An open-label study of the neurosteroid pregnenolone in adults with autism spectrum disorder. J Autism Dev Disord. (2014) 44:2971–7. doi: 10.1007/s10803-014-2144-4
8. Ip J, Mellios N, Sur M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. (2018) 19:368–82. doi: 10.1038/s41583-018-0006-3
9. Banerjee A, Miller M, Li K, Sur M, Kaufmann W. Towards a better diagnosis and treatment of Rett syndrome: A model synaptic disorder. Brain. (2019) 142:239–48. doi: 10.1093/brain/awy323
10. Ehrhart F, Coort S, Cirillo E, Smeets E, Evelo C, Curfs L. Rett syndrome – biological pathways leading from MECP2 to disorder phenotypes. Orphanet J Rare Dis. (2016) 11:158. doi: 10.1186/s13023-016-0545-5
11. Katayama S, Sueyoshi N, Inazu T, Kameshita I. Cyclin-dependent kinase-like 5 (CDKL5): Possible cellular signalling targets and involvement in CDKL5 deficiency disorder. Neural Plast. (2020) 2020:1–14. doi: 10.1155/2020/6970190
12. Van Bergen N, Massey S, Quigley A, Rollo B, Harris A, Kapsa R, et al. CDKL5 deficiency disorder: Molecular insights and mechanisms of pathogenicity to fast-track therapeutic development. Biochem Soc Trans. (2022) 50:1207–24. doi: 10.1042/BST20220791
13. Hou P, Ailín DÓ, Vogel T, Hanashima C. Transcription and beyond: Delineating FOXG1 function in cortical development and disorders. Front Cell Neurosci. (2020) 14:35. doi: 10.3389/fncel.2020.00035
14. Zoghbi H, Bear M. Synaptic Dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. (2012) 4:a009886–009886. doi: 10.1101/cshperspect.a009886
15. Michetti C, Falace A, Benfenati F, Fassio A. Synaptic genes and neurodevelopmental disorders: From molecular mechanisms to developmental strategies of behavioral testing. Neurobiol Dis. (2022) 173:105856. doi: 10.1016/j.nbd.2022.105856
16. Cherubini E, Di Cristo G, Avoli M. Dysregulation of GABAergic signaling in neurodevelomental disorders: Targeting cation-chloride co-transporters to re-establish a proper E/I balance. Front Cell Neurosci. (2022) 15:813441. doi: 10.3389/fncel.2021.813441
17. El-Khoury R, Panayotis N, Matagne V, Ghata A, Villard L, Roux J-C. GABA and glutamate pathways are spatially and developmentally affected in the brain of mecp2-deficient mice. PLoS One. (2014) 9:e92169. doi: 10.1371/journal.pone.0092169
18. Banerjee A, Rikhye RV, Breton-Provencher V, Tang X, Li C, Li K, et al. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc Natl Acad Sci USA. (2016) 113:E7287–96. doi: 10.1073/pnas.1615330113
19. Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci USA. (2016) 113:751–6. doi: 10.1073/pnas.1524013113
20. Hinz L, Torrella Barrufet J, Heine V. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol Commun. (2019) 7:196. doi: 10.1186/s40478-019-0852-x
21. Savardi A, Borgogno M, De Vivo M, Cancedda L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol Sci. (2021) 42:1009–34. doi: 10.1016/j.tips.2021.09.005
22. Fehr S, Wong K, Chin R, Williams S, de Klerk N, Forbes D, et al. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology. (2016) 87:2206–13. doi: 10.1212/WNL.0000000000003352
23. Olson H, Demarest S, Pestana-Knight E, Swanson L, Iqbal S, Lal D, et al. Cyclin-dependent kinase-like 5 deficiency disorder: Clinical review. Pediatr Neurol. (2019) 97:18–25. doi: 10.1016/j.pediatrneurol.2019.02.015
24. Ramirez J, Ward C, Neul J. Breathing challenges in Rett syndrome: Lessons learned from humans and animal models. Respir Physiol Neurobiol. (2013) 189:280–7. doi: 10.1016/j.resp.2013.06.022
25. Katz D, Dutschmann M, Ramirez J, Hilaire G. Breathing disorders in Rett syndrome: Progressive neurochemical dysfunction in the respiratory network after birth. Respir Physiol Neurobiol. (2009) 168:101–8. doi: 10.1016/j.resp.2009.04.017
26. Voituron N, Hilaire G. The benzodiazepine Midazolam mitigates the breathing defects of Mecp2-deficient mice. Respir Physiol Neurobiol. (2011) 177:56–60. doi: 10.1016/j.resp.2011.02.002
27. Germano M. Congress paper AIRETT congress 2017 (2021). Available online at: https://congress.airett.it/wp-content/uploads/2017/01/Germano-MIDAZOLAM-AS-TREATMENT-OF-RECURRENT-SEIZURES-IN-A-CHILD-WITH-RETT-SYNDROME.pdf
28. Chen D, Wang C, Li M, She X, Yuan Y, Chen H, et al. Loss of Foxg1 impairs the development of cortical SST-interneurons leading to abnormal emotional and social behaviors. Cereb Cortex. (2019) 29:3666–82. doi: 10.1093/cercor/bhz114
29. Jellinger K. Rett syndrome – an update. J Neural Transm. (2003) 110:681–701. doi: 10.1007/s00702-003-0822-z
30. Santos M, Summavielle T, Teixeira-Castro A, Silva-Fernandes A, Duarte-Silva S, Marques F, et al. Monoamine deficits in the brain of methylCpG binding protein 2 null mice suggest the involvement of the cerebral cortex in early stages of Rett syndrome. Neuroscience. (2010) 170:453–67. doi: 10.1016/j.neuroscience.2010.07.010
31. Vogelgesang S, Niebert S, Renner U, Möbius W, Hülsmann S, Manzke T, et al. Analysis of the serotonergic system in a mouse model of Rett syndrome reveals unusual upregulation of serotonin receptor 5b. Front Mol Neurosci. (2017) 10:61. doi: 10.3389/fnmol.2017.00061
32. Vigli D, Rusconi L, Valenti D, La Montanara P, Cosentino L, Lacivita E, et al. Rescue of prepulse inhibition deficit and brain mitochondrial dysfunction by pharmacological stimulation of the central serotonin receptor 7 in a mouse model of CDKL5 Deficiency Disorder. Neuropharmacology. (2019) 144:104–14. doi: 10.1016/j.neuropharm.2018.10.018
33. Fuchs C, Gennaccaro L, Ren E, Galvani G, Trazzi S, Medici G, et al. Pharmacotherapy with sertraline rescues brain development and behavior in a mouse model of CDKL5 deficiency disorder. Neuropharmacology. (2020) 167:107746. doi: 10.1016/j.neuropharm.2019.107746
34. Abdala A, Lioy D, Garg S, Knopp S, Paton J, Bissonnette J. Effect of Sarizotan, a 5-HT 1a and D2-like receptor agonist, on respiration in three mouse models of Rett syndrome. Am J Respir Cell Mol Biol. (2014) 50:1031–9. doi: 10.1165/rcmb.2013-0372OC
35. Persico A, Ricciardello A, Cucinotta F. The psychopharmacology of autism spectrum disorder and Rett syndrome. Handb Clin Neurol. (2019) 165:391–414. doi: 10.1016/B978-0-444-64012-3.00024-1
36. Li W. Excitation and inhibition imbalance in Rett syndrome. Front Neurosci. (2022) 16:825063. doi: 10.3389/fnins.2022.825063
37. Szczesna K, de la Caridad O, Petazzi P, Soler M, Roa L, Saez MA, et al. Improvement of the Rett syndrome phenotype in a Mecp2 mouse model upon treatment with levodopa and a Dopa-decarboxylase inhibitor. Neuropsychopharmacology. (2014) 39:2846–56. doi: 10.1038/npp.2014.136
38. Lu B, Nagappan G, Lu Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol. (2014) 220:223–50. doi: 10.1007/978-3-642-45106-5_9
39. Kowiański P, Lietzau G, Czuba E, Waśkow M, Steliga A, Moryś J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol. (2018) 38:579–93. doi: 10.1007/s10571-017-0510-4
40. Mellios N, Woodson J, Garcia R, Crawford B, Sharma J, Sheridan S, et al. β2Adrenergic receptor agonist ameliorates phenotypes and corrects microRNA-mediated IGF1 deficits in a mouse model of Rett syndrome. Proc Natl Acad Sci USA. (2014) 111:9947–52. doi: 10.1073/pnas.1309426111
41. Li W, Pozzo-Miller L. BDNF deregulation in Rett syndrome. Neuropharmacology. (2014) 76:737–46. doi: 10.1016/j.neuropharm.2013.03.024
42. Zhu Z, Li Y, Xu J, Xue H, Feng X, Zhu Y, et al. CDKL5 deficiency in adult glutamatergic neurons alters synaptic activity and causes spontaneous seizures via TrkB signaling. Cell Rep. (2023) 42:113202. doi: 10.1016/j.celrep.2023.113202
43. Ma Z, Zhang R, Rui W, Wang Z, Feng X. Quercetin alleviates chronic unpredictable mild stress-induced depressive-like behaviors by promoting adult hippocampal neurogenesis via FoxG1/CREB/ BDNF signaling pathway. Behav Brain Res. (2021) 406:113245. doi: 10.1016/j.bbr.2021.113245
44. Dyer A, Vahdatpour C, Sanfeliu A, Tropea D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience. (2016) 325:89–99. doi: 10.1016/j.neuroscience.2016.03.056
45. Bray N. Righting Rett syndrome with IGF1. Nat Rev Drug Discov. (2014) 13:653–653. doi: 10.1038/nrd4417
46. Castro J, Garcia R, Kwok S, Banerjee A, Petravicz J, Woodson J, et al. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc Natl Acad Sci USA. (2014) 111:9941–6. doi: 10.1073/pnas.1311685111
47. Yuan Z, Mao S, Shen J, Jiang L, Xu L, Xu J, et al. Insulin-like growth factor-1 downregulates the phosphorylation of FXYD1 and rescues behavioral deficits in a mouse model of Rett syndrome. Front Neurosci. (2020) 14:20. doi: 10.3389/fnins.2020.00020
48. Justice M, Buchovecky C, Kyle S, Djukic A. A role for metabolism in Rett syndrome pathogenesis. Rare Dis. (2013) 1:e27265. doi: 10.4161/rdis.27265
49. Kyle S, Vashi N, Justice M. Rett syndrome: A neurological disorder with metabolic components. Open Biol. (2018) 8:216. doi: 10.1098/rsob.170216
50. Vuu Y, Roberts C, Rastegar M. MeCP2 is an epigenetic factor that links DNA methylation with brain metabolism. Int J Mol Sci. (2023) 24:4218. doi: 10.3390/ijms24044218
51. Segatto M, Tonini C, Pfrieger F, Trezza V, Pallottini V. Loss of mevalonate/cholesterol homeostasis in the brain: A focus on autism spectrum disorder and Rett syndrome. Int J Mol Sci. (2019) 20:3317. doi: 10.3390/ijms20133317
52. Villemagne P, Naidu S, Villemagne V, Yaster M, Wagner H, Harris J, et al. Brain glucose metabolism in Rett syndrome. Pediatr Neurol. (2002) 27:117–22. doi: 10.1016/S0887-8994(02)00399-5
53. Ben-Ari Y, Cherubini E. The GABA polarity shift and bumetanide treatment: Making sense requires unbiased and undogmatic analysis. Cells. (2022) 11:396. doi: 10.3390/cells11030396
54. Ben-Ari Y. NKCC1 chloride importer antagonists attenuate many neurological and psychiatric disorders. Trends Neurosci. (2017) 40:536–54. doi: 10.1016/j.tins.2017.07.001
55. Lozovaya N, Nardou R, Tyzio R, Chiesa M, Pons-Bennaceur A, Eftekhari S, et al. Early alterations in a mouse model of Rett syndrome: The GABA developmental shift is abolished at birth. Sci Rep. (2019) 9:9276. doi: 10.1038/s41598-01945635-9
56. Monaghan E, Navalta L, Shum L, Ashbrook D, Lee D. Initial human experience with ganaxolone, a neuroactive steroid with antiepileptic activity. Epilepsia. (1997) 38:1026–31. doi: 10.1111/j.1528-1157.1997.tb01486.x
57. Pieribone V, Tsai J, Soufflet C, Rey E, Shaw K, Giller E, et al. Clinical evaluation of ganaxolone in pediatric and adolescent patients with refractory epilepsy. Epilepsia. (2007) 48:1870–4. doi: 10.1111/j.1528-1167.2007.01182.x
58. Sperling M, Klein P, Tsai J. Randomized, double-blind, placebo-controlled phase 2 study of ganaxolone as add-on therapy in adults with uncontrolled partial-onset seizures. Epilepsia. (2017) 58:558–64. doi: 10.1111/epi.13705
59. Rasmusson A, Marx C, Jain S, Farfel G, Tsai J, Sun X, et al. A randomized controlled trial of ganaxolone in posttraumatic stress disorder. Psychopharmacology. (2017) 234:2245–57. doi: 10.1007/s00213-017-4649-y
60. Kawada T. Effect of ganaxolone in patients with posttraumatic stress disorder. Psychopharmacology. (2018) 235:1619–1619. doi: 10.1007/s00213-018-4892-x
61. Dichtel L, Nyer M, Dording C, Fisher L, Cusin C, Shapero B, et al. Effects of open-label, adjunctive ganaxolone on persistent depression despite adequate antidepressant treatment in postmenopausal women. J Clin Psychiatry. (2020) 81:2887. doi: 10.4088/JCP.19m12887
62. Knight E, Amin S, Bahi-Buisson N, Benke T, Cross J, Demarest S, et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: Results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. Lancet Neurol. (2022) 21:417–27. doi: 10.1016/S1474-4422(22)00077-1
63. Vossler D. Ganaxolone: A new treatment for CDKL5 deficiency disorder. Epilepsy Curr. (2022) 22:348–50. doi: 10.1177/15357597221125238
64. Katz D, Menniti F, Mather RJ. N-Methyl-D-aspartate receptors, ketamine, and Rett syndrome: Something special on the road to treatments? Biol Psychiatry. (2016) 79:710–2. doi: 10.1016/j.biopsych.2016.03.1045
65. Carboni E, Carta A, Carboni E, Novelli A. Repurposing ketamine in depression and related disorders: Can this enigmatic drug achieve success? Front Neurosci. (2021) 15:657714. doi: 10.3389/fnins.2021.657714
66. Olanow C, Damier P, Goetz C, Mueller T, Nutt J, Rascol O, et al. Multicenter, Open-label, trial of sarizotan in parkinson disease patients with levodopainduced dyskinesias (the SPLENDID study). Clin Neuropharmacol. (2004) 27:58–62. doi: 10.1097/00002826-200403000-00003
67. Goetz C, Damier P, Hicking C, Laska E, Müller T, Olanow C, et al. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: A double-blind placebo-controlled trial. Mov Disord. (2007) 22:179–86. doi: 10.1002/mds.21226
68. Levitt E, Hunnicutt B, Knopp S, Williams J, Bissonnette JM. A selective 5-HT 1a receptor agonist improves respiration in a mouse model of Rett syndrome. J Appl Physiol. (2013) 115:1626–33. doi: 10.1152/japplphysiol.00889.2013
69. Sheikh A, Charles I, Bissonnette J, Varney M, Newman-Tancredi A, Paton J. Pre-clinical study of NLX-101 for the treatment of apnoeas in two mouse models of Rett syndrome. (2014). p. 51–60.
70. Hilaire G, Voituron N, Menuet C, Ichiyama R, Subramanian H, Dutschmann M. The role of serotonin in respiratory function and dysfunction. Respir Physiol Neurobiol. (2010) 174:76–88. doi: 10.1016/j.resp.2010.08.017
71. Czaja A, Valuck R. Off-label antidepressant use in children and adolescents compared with young adults: Extent and level of evidence. Pharmacoepidemiol Drug Saf. (2012) 21:997–1004. doi: 10.1002/pds.3312
72. Lai L, Vuong D, Ting A, Dang L, Ngo V, Jo Y, et al. Off-label drug use in children and adolescents with depressive disorder. Value Health. (2018) 21:S146. doi: 10.1016/j.jval.2018.04.1006
73. Cook E, Wagner K, March J, Biederman J, Landau P, Wolkow R, et al. LongTerm sertraline treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. (2001) 40:1175–81. doi: 10.1097/00004583-20011000000011
74. Rynn M, Wagner K, Donnelly C, Ambrosini P, Wohlberg C, Landau P, et al. Long-term sertraline treatment of children and adolescents with major depressive disorder. J Child Adolesc Psychopharmacol. (2006) 16:103–16. doi: 10.1089/cap.2006.16.103
75. Kolitsopoulos F, Ramaker S, Compton S, Broderick S, Orazem J, Bao W, et al. Effects of long-term sertraline use on pediatric growth and development: The sertraline pediatric registry for the evaluation of safety (SPRITES). J Child Adolesc Psychopharmacol. (2023) 33:2–13. doi: 10.1089/cap.2022.0048
76. Bialer M, Perucca E. Lorcaserin for Dravet syndrome: A potential advance over fenfluramine? CNS Drugs. (2022) 36:113–22. doi: 10.1007/s40263-022-00896-3
77. Devinsky O, King L, Schwartz D, Conway E, Price D. Effect of fenfluramine on convulsive seizures in CDKL5 deficiency disorder. Epilepsia. (2021) 62:16923. doi: 10.1111/epi.16923
78. Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The Disease Progression of Mecp2 Mutant Mice Is Affected by the Level of BDNF Expression. Neuron. (2006) 49:341–8. doi: 10.1016/j.neuron.2005.12.027
79. Djukic A, Holtzer R, Shinnar S, Muzumdar H, Rose S, Mowrey W, et al. Pharmacologic treatment of rett syndrome with glatiramer acetate. Pediatr Neurol. (2016) 61:51–7. doi: 10.1016/j.pediatrneurol.2016.05.010
80. Nissenkorn A, Kidon M, Ben-Zeev B. A potential life-threatening reaction to glatiramer acetate in Rett syndrome. Pediatr Neurol. (2017) 68:40–3. doi: 10.1016/j.pediatrneurol.2016.11.006
81. Deogracias R, Yazdani M, Dekkers M, Guy J, Ionescu M, Vogt K, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci USA. (2012) 109:14230–5. doi: 10.1073/pnas.1206093109
82. Miguez A, García-Díaz Barriga G, Brito V, Straccia M, Giralt A, Ginés S, et al. Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75 NTR up-regulation and astrocyte-mediated inflammation. Hum Mol Genet. (2015) 24:4958–70. doi: 10.1093/hmg/ddv218
83. Fukumoto K, Mizoguchi H, Takeuchi H, Horiuchi H, Kawanokuchi J, Jin S, et al. Fingolimod increases brain-derived neurotrophic factor levels and ameliorates amyloid βinduced memory impairment. Behav Brain Res. (2014) 268:88–93. doi: 10.1016/j.bbr.2014.03.046
84. Naegelin Y, Kuhle J, Schädelin S, Datta A, Magon S, Amann M, et al. Fingolimod in children with Rett syndrome: The FINGORETT study. Orphanet J Rare Dis. (2021) 16:19. doi: 10.1186/s13023-020-01655-7
85. Backeljauw P, Chernausek S. The insulin-like growth factors and growth disorders of childhood. Endocrinol Metab Clin North Am. (2012) 41:265–82. doi: 10.1016/j.ecl.2012.04.010
86. Khwaja O, Ho E, Barnes KV, O’Leary H, Pereira L, Finkelstein Y, et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci USA. (2014) 111:4596–601. doi: 10.1073/pnas.1311141111
87. O’Leary H, Kaufmann W, Barnes KV, Rakesh K, Kapur K, Tarquinio D, et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann Clin Transl Neurol. (2018) 5:323–32. doi: 10.1002/acn3.533
88. Bickerdike M, Thomas G, Batchelor D, Sirimanne E, Leong W, Lin H, et al. NNZ-2566: A Gly–Pro–Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke. J Neurol Sci. (2009) 278:85–90. doi: 10.1016/j.jns.2008.12.003
89. Neul J, Percy A, Benke T, Berry-Kravis E, Glaze D, Peters S, et al. Design and outcome measures of LAVENDER, a phase 3 study of trofinetide for Rett syndrome. Contemp Clin Trials. (2022) 114:106704. doi: 10.1016/j.cct.2022.106704
90. Neul J, Percy A, Benke T, Berry-Kravis E, Glaze D, Marsh, et al. Trofinetide for the treatment of Rett syndrome: A randomized phase 3 study. Nat Med. (2023) 29:1468–75. doi: 10.1038/s41591-023-02398-1
91. Fracassi A, Marangoni M, Rosso P, Pallottini V, Fioramonti M, Siteni S, et al. Statins and the brain: More than lipid lowering agents? Curr Neuropharmacol. (2018) 17:59–83. doi: 10.2174/1570159X15666170703101816
92. Buchovecky C, Turley S, Brown H, Kyle S, McDonald J, Liu B, et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat Genet. (2013) 45:1013–20. doi: 10.1038/ng.2714
93. Villani C, Sacchetti G, Bagnati R, Passoni A, Fusco F, Carli M, et al. Lovastatin fails to improve motor performance and survival in methyl-CpG-binding protein2-null mice. Elife. (2016) 5:e22409. doi: 10.7554/eLife.22409
94. Gupta R. Corticosteroids in the management of the paediatric epilepsies. Arch Dis Child. (2005) 90:379–84. doi: 10.1136/adc.2004.051375
95. Hong W, Haviland I, Pestana-Knight E, Weisenberg J, Demarest S, Marsh ED, et al. CDKL5 deficiency disorder-related epilepsy: A review of current and emerging treatment. CNS Drugs. (2022) 36:591–604. doi: 10.1007/s40263-022-00921-5
96. Braun S, Kottwitz D, Nuber U. Pharmacological interference with the glucocorticoid system influences symptoms and lifespan in a mouse model of Rett syndrome. Hum Mol Genet. (2012) 21:1673–80. doi: 10.1093/hmg/ddr602
97. De Filippis B, Ricceri L, Fuso A, Laviola G. Neonatal exposure to low dose corticosterone persistently modulates hippocampal mineralocorticoid receptor expression and improves locomotor/exploratory behaviour in a mouse model of Rett syndrome. Neuropharmacology. (2013) 68:174–83. doi: 10.1016/j.neuropharm.2012.05.048
98. Barbiero I, Zamberletti E, Tramarin M, Gabaglio M, Peroni D, De Rosa R, et al. Pregnenolone-methyl-ether enhances CLIP170 and microtubule functions improving spine maturation and hippocampal deficits related to CDKL5 deficiency. Hum Mol Genet. (2022) 31:2738–50. doi: 10.1093/hmg/ddac067
99. De Rosa R, Valastro S, Cambria C, Barbiero I, Puricelli C, Tramarin M, et al. Loss of CDKL5 causes synaptic GABAergic defects that can be restored with the neuroactive steroid pregnenolone-methyl-ether. Int J Mol Sci. (2022) 24:68. doi: 10.3390/ijms24010068
100. Laurvick C, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, et al. Rett syndrome in Australia: A review of the epidemiology. J Pediatr. (2006) 148:347–52. doi: 10.1016/j.jpeds.2005.10.037
101. Neul J, Fang P, Barrish J, Lane J, Caeg E, Smith E, et al. Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome. Neurology. (2008) 70:1313–21. doi: 10.1212/01.wnl.0000291011.54508.aa
102. Trappe R, Laccone F, Cobilanschi J, Meins M, Huppke P, Hanefeld F, et al. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet. (2001) 68:1093–101. doi: 10.1086/320109
103. Sharifi O, Yasui D. The molecular functions of MeCP2 in Rett syndrome pathology. Front Genet. (2021) 12:624290. doi: 10.3389/fgene.2021.624290
104. Ponzi E, Gentile M, Agolini E, Matera E, Palumbi R, Buonadonna A, et al. 14q12q13.2 microdeletion syndrome: Clinical characterization of a new patient, review of the literature, and further evidence of a candidate region for CNS anomalies. Mol Genet Genomic Med. (2020) 8:1289. doi: 10.1002/mgg3.1289
Keywords: Rett syndrome, CDKL5 deficiency disorder, FOXG1-syndrome, shared molecular pathways, common drug targets, drug repurposing
Citation: Fuchs C, ‘t Hoen PAC, Müller AR, Ehrhart F and Van Karnebeek CDM (2024) Drug repurposing in Rett and Rett-like syndromes: a promising yet underrated opportunity? Front. Med. 11:1425038. doi: 10.3389/fmed.2024.1425038
Received: 29 April 2024; Accepted: 12 July 2024;
Published: 29 July 2024.
Edited by:
Viviana Giannuzzi, Fondazione per la Ricerca Farmacologica Gianni Benzi Onlus, ItalyReviewed by:
Michela Palmieri, San Raffaele Hospital (IRCCS), ItalyCopyright © 2024 Fuchs, ‘t Hoen, Müller, Ehrhart and Van Karnebeek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Fuchs, Y2xhdWRpYS5mdWNoc0BldXJvcmRpcy5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.