 Runzhuo Li
Runzhuo Li Wanyun Tang3†
Wanyun Tang3†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med., 29 July 2024
Sec. Gastroenterology
Volume 11 - 2024 | https://doi.org/10.3389/fmed.2024.1397111
This article is part of the Research TopicDiagnosis and Management of Acute, Chronic, and Autoimmune PancreatitisView all 10 articles
Background: Acute pancreatitis, among the most prevalent gastrointestinal disorders, exhibits a continual rise in its incidence recent years. This study endeavor to explore the correlation between smoking exposure and the severity of acute pancreatitis (AP).
Methods: Five hundred and eight patients diagnosed as acute pancreatitis (AP) were included in our data analysis. Patients were categorized based on their smoking pack-years into four groups: light, moderate, heavy, and non-smokers. Outcomes were classified as two: “mild acute pancreatitis (MAP)” and “moderately severe acute pancreatitis (MSAP) or severe acute pancreatitis (SAP)”. We conducted propensity score matching (PSM) to adjust confounding factors and multivariable logistic regression analysis to determine adjusted odds ratios and 95% confidence intervals. Additionally, a dose-dependent association analysis between smoking exposure and the incidence rate of “MSAP or SAP” was performed.
Results: Smokers exhibited a higher risk of “MSAP or SAP” compared to non-smokers, both before (17.1 vs. 54.9%, p < 0.001) and after (9.4 vs. 24.7%, p < 0.001) PSM. With an area under the ROC curve of 0.708, smoking showed a moderate level of predictive ability. Furthermore, propensity score matching analysis showed that patients who smoked compared to non-smokers had significantly higher risks of “MSAP or SAP” for light smoking (OR 3.76, 95% CI 1.40–10.07, p = 0.008), moderate smoking (OR 4.94, 95% CI 2.23–10.92, p < 0.001), and heavy smoking (OR 8.08, 95% CI 3.39–19.25, p < 0.001).
Conclusion: Smoking is an independent risk factor that can raise the severity of pancreatitis. Moreover, the severity of acute pancreatitis escalates in tandem with the accumulation of pack-years of smoking.
Acute pancreatitis (AP) is an intense inflammatory leading to edema, hemorrhage, and potentially necrosis (1, 2). With an incidence rate estimated at 110–140 cases per 100,000 population, AP has become one of the most prevalent gastrointestinal illnesses requiring hospitalization in the United States (3). Between 2002 and 2013, the number of hospitalizations attributed to AP rose from 9.48 per 1000 cases to 12.19 per 1000 cases (4). According to the 2012 Atlanta guidelines, AP fall under the category as mild (MAP), moderately severe (MSAP), or severe (SAP) in accordance with its severity of onset. The latter two categories, MSAP and SAP, are more severe and often lead to local or systemic complications including pancreatic leakage, pancreatic necrosis, pancreatic abscess, systemic inflammatory response syndrome (SIRS), even multi-organ dysfunction syndrome (MODS) (5). These complications not only prolong hospitalization and increase medical costs but may also result in long-term disabilities and mortality. Approximately 80% of patients present with MAP to MSAP, with one-fifth progressing to severe disease, and a mortality rate of about 20% (6–8). Traditional causes of AP include gallstones, alcohol abuse, and hypertriglyceridemia. Many studies (9–12) has reported smoking also accounting for the incidence of AP. However, within Asian populations, there is limited research on the relationship between smoking and AP, particularly regarding its impact on the severity of the disease.
Currently, smoking is a well-known risk factor for various digestive diseases (13). Nicotine and other harmful substances in tobacco smoke can cause pancreatic damage, inflammation, and impaired pancreatic function. Smoking has been shown to inhibit pancreatic secretion and increase the risk of leakage of pancreatic enzymes into the bloodstream. Prolonged exposure to nicotine increases the content of pancreatic zymogen within cells and induces vacuolization of acinar cells in rats (14). These factors may exacerbate the development of AP and its complications. Therefore, elucidating the link between smoking exposure and the severity of AP is crucial for preventing AP from progressing into an irreversible and lethal disease. The goal in our study is to figure out the link between smoking exposure measured in pack-years and the severity of AP through propensity score matching analysis.
This retrospective investigation utilized anonymized clinical data from electronic medical records of patients admitted for AP at Dandong Central Hospital, China Medical University from October 2017 to October 2023. The Dandong Hospital Ethics Committee approved the study and granted a consent waiver for this retrospective cohort study. In order to protect personal health information, online medical records were limited to the range of anonymized data analysis, which included demographics, message of laboratory findings, imaging analysis results, and various complications.
In this study, we collected variables including demographic characteristics: age, gender, smoking history, drinking history, hypertension, diabetes, hyperlipidemia, cardiovascular disease; and serological indicators: white blood cell count (WBC), red blood cell count (RBC), hemoglobin concentration (HGB), platelet count (PLT), potassium ion (K+), sodium ion (Na+), calcium ion (Ca+), creatinine (Cr), blood glucose (BG), alanine aminotransferase (ALT), lactate dehydrogenase (LDH), total bilirubin (TBIL), albumin (ALB), amylase (AMY), lipopolysaccharide (LPS), D-dimer, C-reactive protein (CRP), prothrombin time (PT), and activated partial thromboplastin time (APTT).
All serological indicators for the patients were obtained from blood samples collected within 24 h of admission.
Both the diagnosis and severity grading of AP referred to the 2012 Atlanta guidelines (5). We included patients in our study who satisfied at least two of the following criteria. (1) Acute, persistent, intense pain in the upper abdomen, with or without radiation to the back; (2) The activity of serum lipase (or amylase) beyond three times the upper limit of normal; (3) Imaging findings match characteristics of AP.
Patients were excluded from the study if meeting any of following criteria. (1) Presence of other pancreatic diseases, such as chronic pancreatitis; (2) Presence of other serious diseases, such as tumors, autoimmune diseases, etc.; (3) Incomplete or unavailable data; (4) Taking medications that may affect the severity of pancreatitis; (5) History of pancreatic surgery; (6) Previous hospitalization for AP.
Before the commencement of the study, four researchers received professional training in data collection, including the diagnosis and severity grading of AP. Three researchers independently collected the variables. If necessary, any differences in variable identification were discussed with or determined by the senior researcher.
Our study's main exposure factor was smoking pack-years, calculated by multiplying the daily pack count by the total number of smoking years. The patients were categorized into smoking and non-smoking groups according to their smoking history. Smokers included in the study were those who had been continuously smoking and were still smoking within 1 month of disease onset. To assess the link between dosage and response of smoking pack-years and AP severity, study population was further split into four groups on the basis of pack-years: nonsmokers (Never smoking), smokers with pack-years ≤ 10 (Light smoking), smokers with pack-years >10 but ≤ 20 (Moderate smoking), and smokers with pack-years >20 (Heavy smoking).
The primary outcome assessed was the severity of AP, graded referring to the 2012 Atlanta guidelines. AP severity categorization was determined on patients' clinical complaints and symptoms, serological indicators, imaging findings, presence of organ failure, and complications, dividing AP into MAP, MSAP, and SAP. Based on previous research (15), we defined the outcome variable as “MAP” and “MSAP or SAP”, categorized as binary variables “yes” or “no”, for our statistical analysis.
When analyzing patients' baseline characteristics, various types of data necessitate specific methods. Percentages (%) are applicable to categorical variables, with the chi-square test used for group comparisons. Mean ± standard deviation (SD) is suitable for normally distributed continuous measurements, with independent sample t-tests employed for group comparisons. Median and interquartile range are utilized for non-normally distributed data, with the Mann-Whitney U-test used for group comparisons.
We investigate the linkage between smoking and the severity of AP using logistic regression modeling. After possible confounding factors with p-value ≥ 0.05 are excluded using univariable logistic regression, factors with p-value <0.05 are then included in the multivariable regression analysis. Additionally, we computed the area under the ROC curve (AUC) to ascertain discriminative capacity of smoking pack-years for distinguishing between “MAP” and “MSA or SAP”. Then we assessed the relationship between smoking pack-years and AP severity through graphical representations illustrating the association between observed rates and predicted probabilities across various levels of smoking pack-years.
To further mitigate the influence of confounding variables, a propensity score matching (PSM), of which matching ratio was 1:1, was performed by the nearest neighbor algorithm, to make sure the most appropriate balanced distribution of covariates between groups. With a caliper width of 0.25 standard deviations, the matching effect was assessed by standardized mean differences (SMDs). Following PSM, we stratified all covariates and then conducted univariable logistic regression analysis to explore the linkage between smoking pack-years and the severity of AP. Finally, for assessing the strength of the link. we calculated odds ratios (ORs) and 95% confidence intervals (CIs).
SPSS version 27 (IBM Corp., Armonk, NY, USA) and R software version 4.0.3 (R Foundation for Statistical Computing, Vienna, Austria) were our main statistical tools that we used.
Four hundred and twenty-one patients were disqualified in total based on the exclusion criteria. In the end, 508 patients involved into the eventual data analyzing (eFigure). The smoking group had more male patients and a higher proportion of patients with alcohol consumption. Laboratory testing revealed the smoking group had higher levels of LDH, CRP, and WBC count, as well as their median and interquartile range. The two groups had no valid statistical discrepancy in terms of age, hypertension, hyperlipidemia, or cardiovascular disease history. The baseline characteristics of the smoking and non-smoking groups at admission are displayed in Table 1.
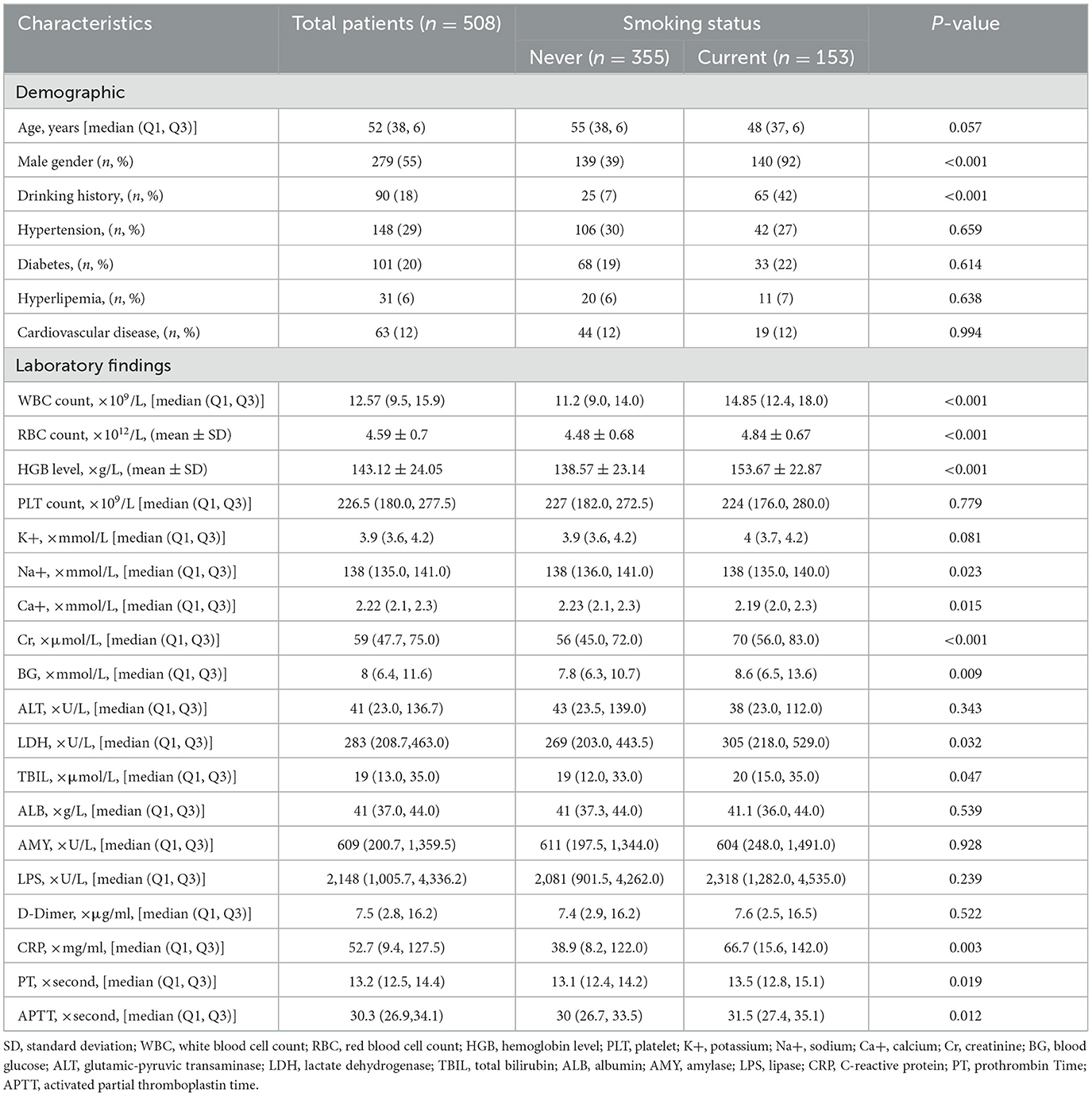
Table 1. Baseline characteristics of the study population by smoking history.
To explore the relationship between the 27 factors and “MSAP or SAP”, we conducted single-factor and multi-factor analyses (Table 2). After adjusting for any confounding variables, we found that seven factors emerged as independent predictors of “MSAP or SAP”: smoking, WBC, HGB level, BG, Ca+ AMY, and PT. Even after adjusting for other variables, these factors remained statistically significant (P < 0.05).
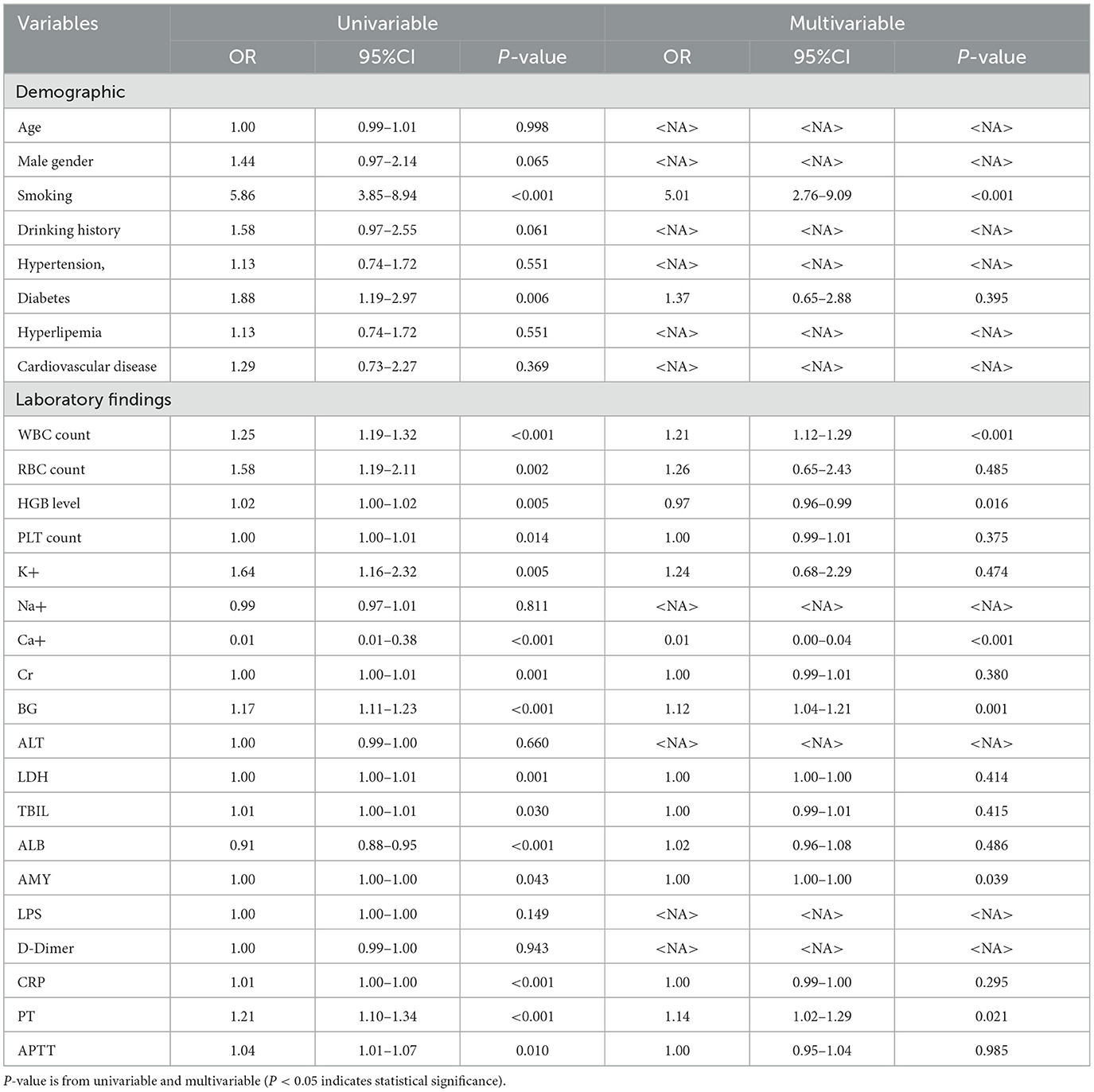
Table 2. Univariable and multivariable analysis for “MSAP or SAP” in AP patients.
Table 3 displays the baseline characteristics of the smoking and non-smoking groups of patients both before and after 1:1 PSM. A satisfactory balance between the smoking and non-smoking groups was attained by PSM, as most variables had standardized mean differences of <0.1.
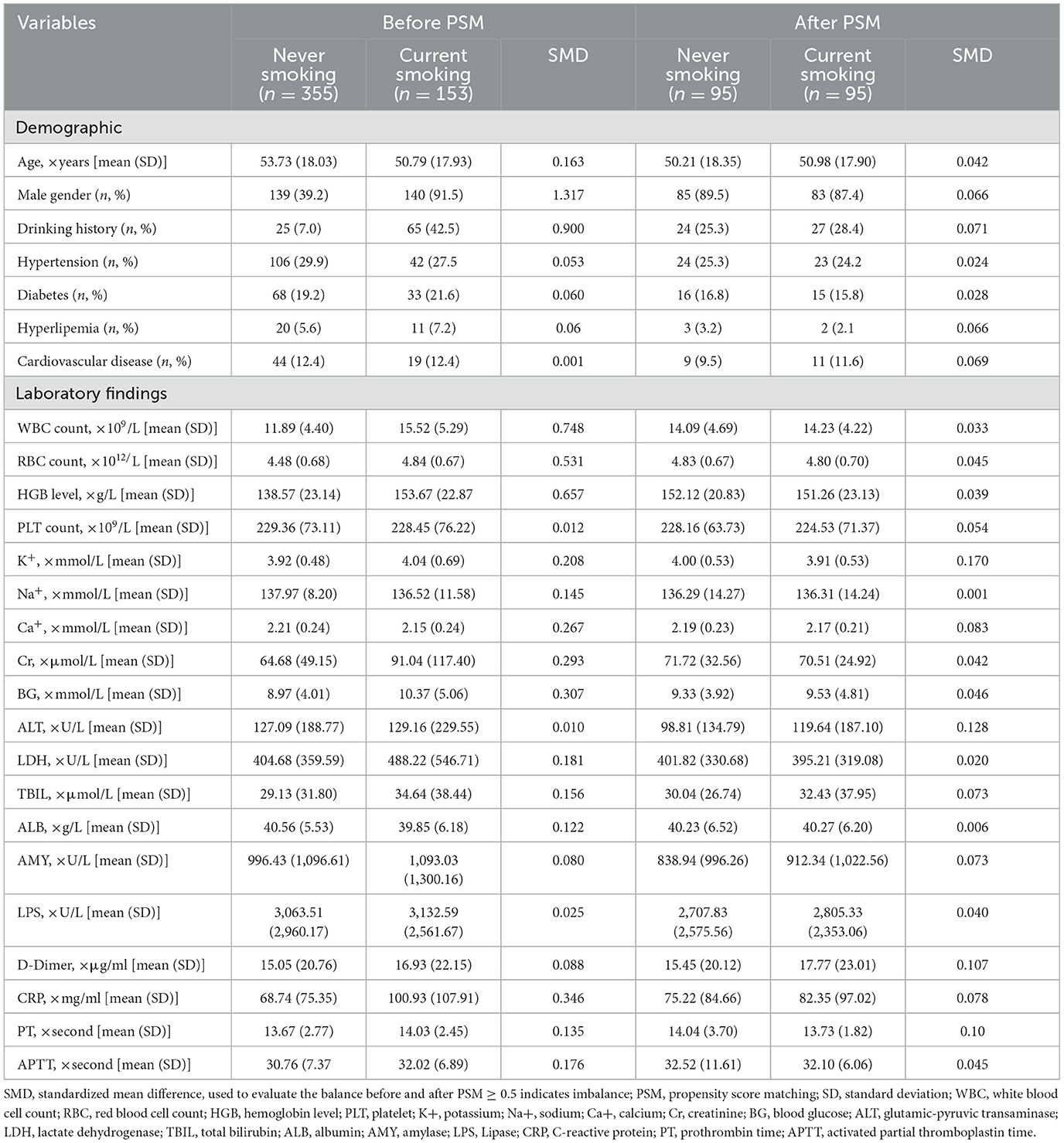
Table 3. Patient baseline characteristics before and after PSM by smoking history.
The average smoking pack-years in the “MSAP or SAP” group were higher than those in the MAP group, both before PSM (3.28 vs. 12.71, p < 0.001) and after PSM (6.64 vs. 14.62, p < 0.001) (Figure 1). The proportion of patients with “MSAP or SAP” in the smoking group was larger than that in the non-smoking group, both before PSM (17.1 vs. 54.9%, p < 0.001) and after PSM (9.4 vs. 24.7%, p < 0.001) (Table 4). The risk of “MSAP or SAP” was higher in the smoking group than in the non-smoking group (unadjusted OR 5.86, 95% CI 3.85–8.94, p < 0.001). After adjusting for confounding factors, this result remained significant (adjusted OR 5.01, 95% CI 2.76–9.09, p < 0.001). With minimized selection bias by PSM, consistent finding was obtained (adjusted OR after PSM: 4.18, 95% CI 2.18–8.03, p < 0.001) (Table 5). Additionally, results from examining smoking pack-years as an ongoing factor were in coincidence: the unadjusted odds increased by 8% and the adjusted odds increased by 7% for every unit increase in smoking pack-years.
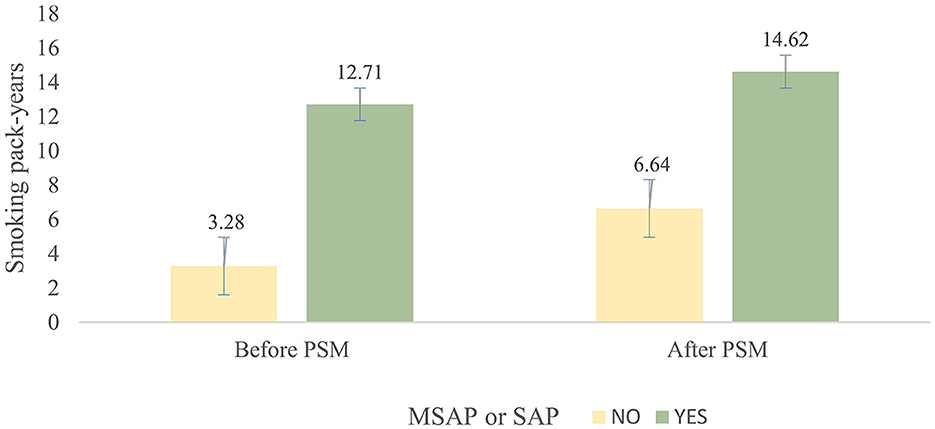
Figure 1. Mean and standard deviation of pack-years between the smoking group and non-smoking group before and after PSM.
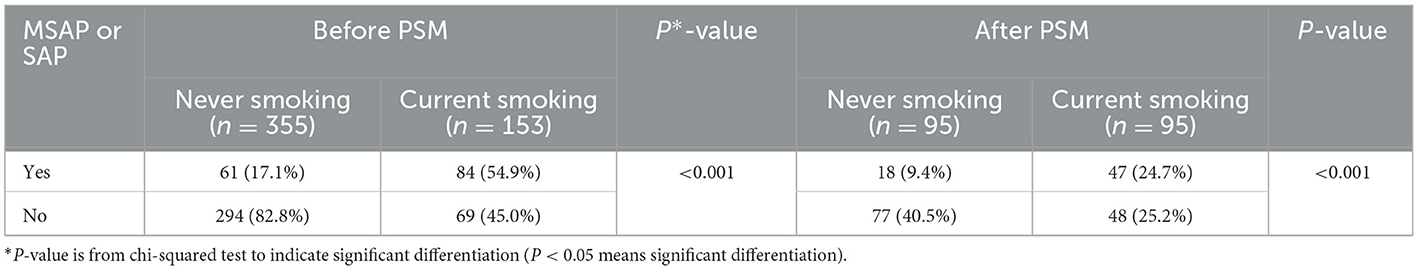
Table 4. Comparison of the incidence of “MSAP or SAP” after PSM based on smoking status.
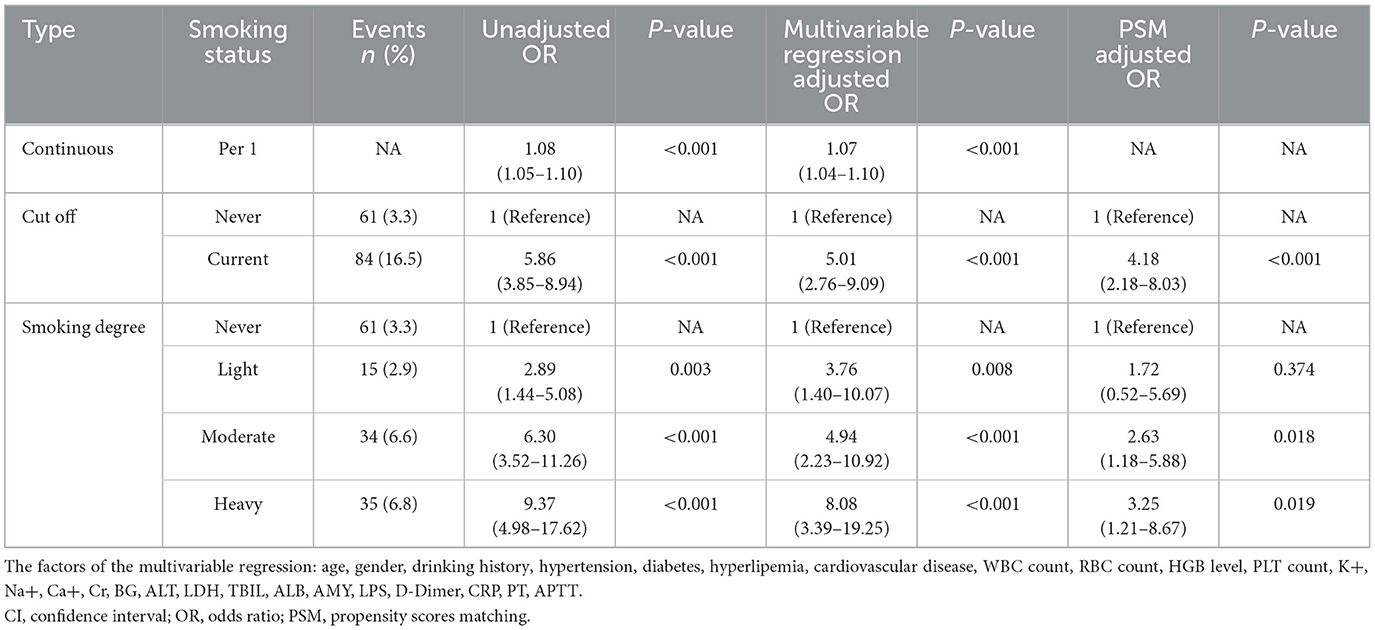
Table 5. Comparison of the unadjusted and risk-adjusted OR by different smoking status.
Compared to non-smoking patients, after adjusting for confounding factors, patients with varying degrees of smoking (light to heavy) exhibited an increased risk of developing “MSAP or SAP” (light: adjusted OR 3.76, 95% CI 1.40–10.07, p = 0.008; moderate: adjusted OR 4.94, 95% CI 2.23–10.92, p < 0.001; heavy: adjusted OR 8.08, 95% CI 3.39–19.25, p < 0.001). This linkage persisted even after PSM (light: adjusted OR after PSM 1.72, 95% CI 0.52–5.69, p = 0.374; moderate: adjusted OR after PSM 2.63, 95% CI 1.18–5.88, p = 0.018; heavy: adjusted OR after PSM 3.25, 95% CI 1.21–8.67, p = 0.019) (Table 5).
Additionally, the AUC of the ROC curve for smoking predicting the severity of AP was 0.708 (Figure 2), showing a risk of “MSAP or SAP” with a moderate predictive value.
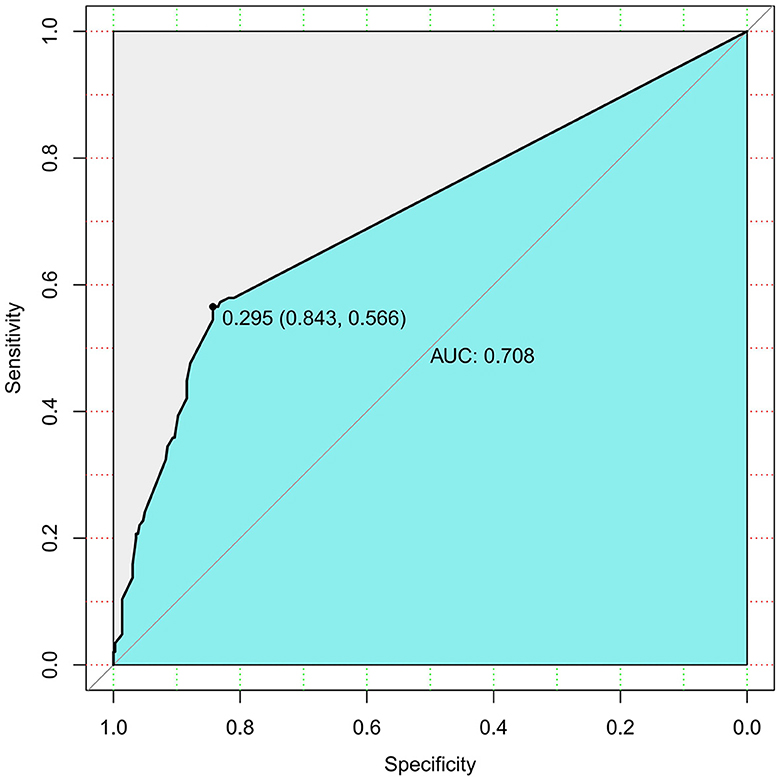
Figure 2. Smoking history prediction model for “MASP or SAP” rate in AP (AUC).
In the AP patients studied, both before PSM (17.1% in non-smokers, 37.5% in light smokers, 56.6% in moderate smokers, and 66% in heavy smokers) and after PSM (18.9% in non-smokers, 31% in light smokers, 56.8% in moderate smokers, and 55.5% in heavy smokers), the incidence of “MSAP or SAP” increased with increasing smoking pack-years (Figure 3). Before and after PSM, there was a dose-response linkage between the risk of “MSAP or SAP” and smoking pack-years (before PSM: Figure 4; after PSM: Figure 5). Non-smokers had a lower risk of “MSAP or SAP” compared to smokers (Figures 4A, 5A). According to the level of smoking pack-years, the predicted and observed rates of MSAP or SAP increased with higher smoking pack-years, and the predicted and observed rates of MSAP or SAP were consistent (Figures 4B, 5B), indicating a dose-response relationship. Additionally, possible interactions between smoking and other factors were assessed in this study (Figure 6). There were no discernible interactions between smoking and the other factors (p > 0.05). Colinearity analysis shows that no collinearity exists between variables (eTable). All in all, these findings suggest that smoking is an independent risk factor for exacerbating AP.

Figure 3. Rate of “MSAP or SAP” in each group by smoking status (never, light, moderate, and heavy smoking group).
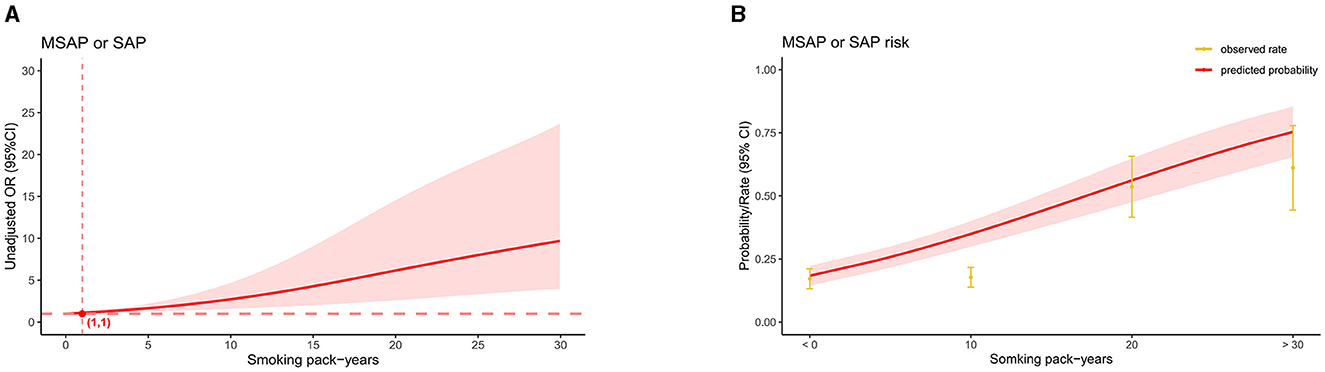
Figure 4. Linkage between smoking pack-years and “MASP or SAP” in patients with AP before PSM. (A) Adjusted odds ratios (ORs) and 95% confidence intervals (CIs) are shown for every five smoking pack-years interval. (B) Predicted probabilities and the observed rate of “MASP or SAP”.
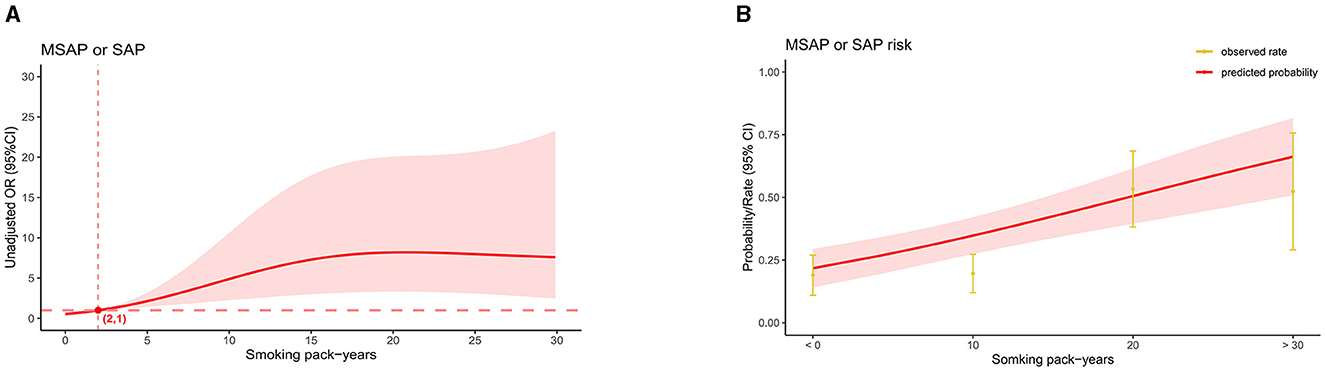
Figure 5. Linkage between smoking pack-years and “MASP or SAP” in patients with AP after PSM. (A) Adjusted odds ratios (ORs) and 95% confidence intervals (CIs) are shown for every five smoking pack-years interval. (B) Predicted probabilities and the observed rate of “MASP or SAP”.
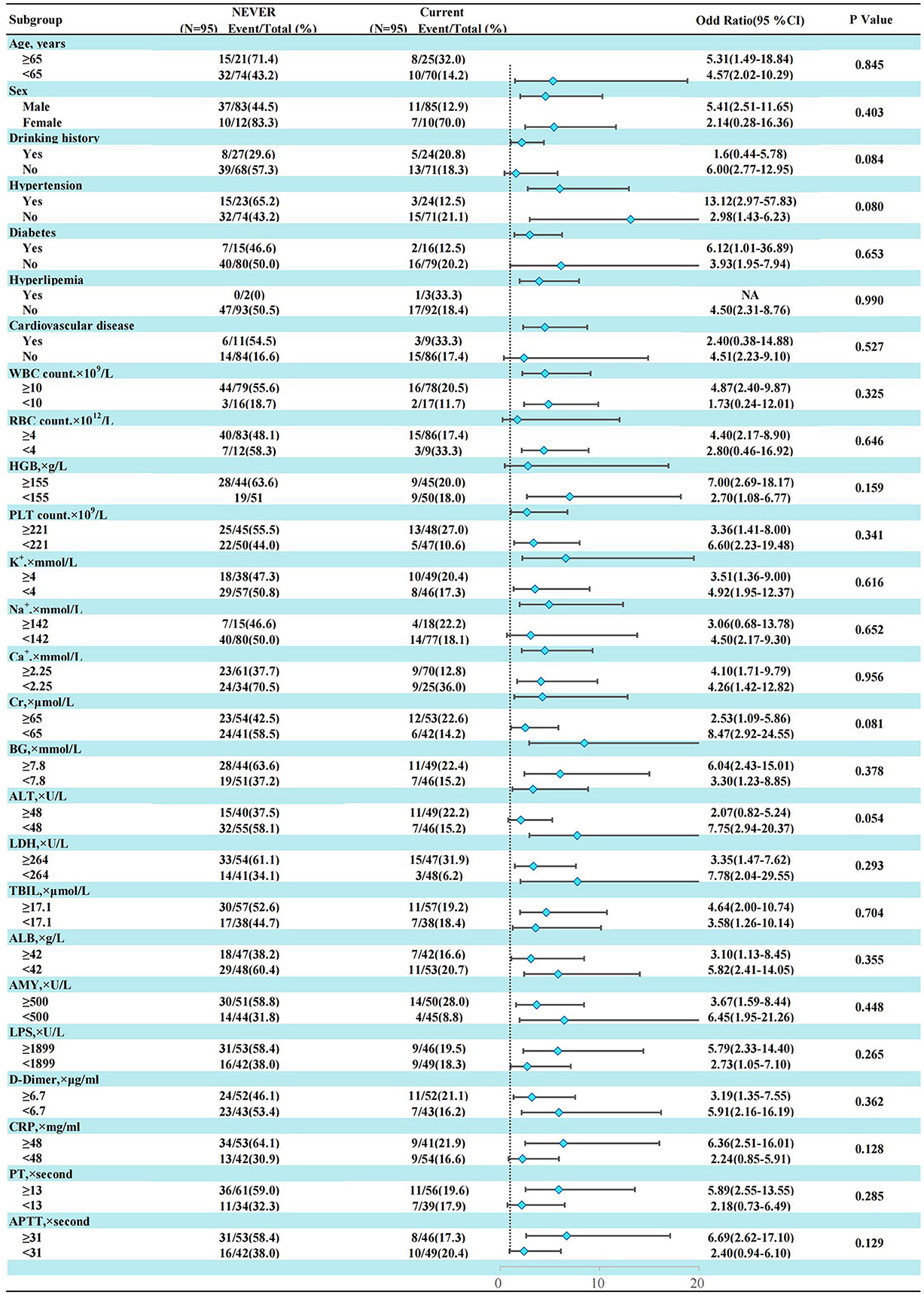
Figure 6. Subgroup analysis of smoking history and “MASP or SAP” in AP patients after propensity score matching.
In our study, we found that smoking is independently associated with the severity of AP. Compared to AP patients who have never smoked, smokers with AP have a more severe condition and a higher incidence of “MSAP or SAP”. Moreover, there is a dose-response linkage between smoking pack-years and the risk of “MSAP or SAP”, with the risk increasing with higher smoking pack-years. Even accounting for possible demographic and clinical variables, the association between smoking and the occurrence of “MSAP or SAP” remained significant. This suggests that smoking itself exacerbates AP. PSM also yielded consistent results, confirming that the linkage between smoking and the severity of pancreatitis cannot be attributable solely to baseline variation within the two groups.
Previous studies have indicated that smoking increases the risk of AP. A cohort study by Tolstrup et al. (10), involving 17,905 patients, observed that 46% of AP cases during the follow-up period could be attributed to smoking. They noted a dose-response relationship between smoking pack-years (15–29 pack-years) and the hazard ratio (HR) for AP (HR 2.2; 95% CI 1.2–3.8). Similarly, a large-scale study by Lee et al. (16) (N = 4,238,822) found that compared to non-smokers, current smokers had an adjusted HR of 1.66 (CI, 1.53–1.8) for acute pancreatitis, higher than that of former smokers (HR 1.34; CI, 1.17–1.54). China's largest prospective cohort study (17) (n = 512,891) also demonstrated an increased risk of AP among current male smokers (HR 1.45; 95% CI 1.28–1.64; P = 0.02). While aforesaid studies revealed that smoking increased the prevalence of AP, few have focused on the linkage between smoking and the severity of AP. Kim et al. (18) conducted a retrospective analysis of 905 AP patients and found that smoking was an independent risk factor for the development of SAP (OR: 7.22; 95% CI: 1.05–49.69; P = 0.04).
Several mechanisms may underlie the observed exacerbation of AP severity by smoking. Firstly, smoking directly damages pancreatic tissue through intricate mechanisms. Overall, chronic smoking exposure promotes pancreatic fibrosis, calcification, and chronic inflammation. This leads to premature activation of pancreatic enzymes and reduced secretion, causing retention of active digestive enzymes within acinar cells, exacerbating pancreatic autodigestion (14, 19, 20). This may increase the risk of complications in AP patients, such as pancreatic leakage, accumulation of necrotic pancreatic material, pseudocysts, and pancreatic abscesses. Additionally, pancreatic necrosis exacerbates systemic inflammation by releasing various inflammatory mediators into the bloodstream, potentially leading to multi-organ failure (21–24). For instance, Garg et al. (25) demonstrated a bidirectional relationship between the severity of pancreatic necrosis and organ failure. The extent of pancreatic necrosis influences the severity of organ failure, while organ failure exacerbates the progression of pancreatic necrosis. In a meta-analysis by Petrov et al. (26) based on 1,478 AP patients, it was found that the impact of organ failure and pancreatic necrosis on mortality was comparable; the presence of either indicates severe disease. Multi-organ dysfunction syndrome, extent of pancreatic necrosis, infection, and sepsis are major determinants of mortality in AP.
Secondly, ingredients of tobacco that can lead to inflammation in a number of different disorders. When acute pancreatitis (AP) occurs, this effect may exacerbate the severity of AP. For instance, a prospective study by Colak et al. (27) involving 98,085 participants found that plasma C-reactive protein levels increased by 4.8% (95% CI 4.4–5.2%) for every 10 pack-years and by 1.6% (95% CI 0.4–2.8%) for each T allele, indicating that both genetically and observably, greater tobacco use is linked to increased systemic inflammation. According to Liu et al. (28), nicotine can cause inflammatory reactions by triggering signaling pathways including STAT3 and NF-κB that are linked to inflammation. This causes increased intracellular inflammation, inflammatory cells to be recruited, inflammatory mediators to be induced, and tissue damage and inflammation to worsen. Nicotine also can activate AChR expressed on immune cell surfaces, thereby reducing the immunological response and blocking macrophages and lymphocytes from functioning (29). This may make inflammation and infection more likely.
Lastly, Smoking itself can act as a risk factor for various organ diseases (30), such as chronic obstructive pulmonary disease (COPD), atherosclerosis, coronary artery disease, myocardial infarction, heart failure, and stroke. Moreover, both moderately MSAP and SAP can result in varying degrees of organ failure. Prolonged exposure to tobacco may exacerbate this process, leading to an increased risk of susceptible organ infections and failure. For instance, NNK in tobacco suppresses the production of IL-8, which plays a crucial role in acute inflammation by recruiting and activating neutrophils. A reduction in IL-8 may lead to an increased incidence of pulmonary infections (31, 32). Munzel et al. (33) reported that smoking can lead to endothelial dysfunction, increased oxidative stress, and cardiovascular events.
It's worth noting that in our study, the increase in risk and incidence rate of “MSAP or SAP” after PSM exhibits a lower inflection point compared to pre-PSM (1 pack-year before PSM, 2 pack-years after PSM). However, irrespective of pre or post PSM, when smoking pack-years are ≤ 2, the corresponding OR values remain close to 1, consistent with our post-PSM findings regarding the risk of “MSAP or SAP” (smoking pack-years ≤ 10, OR 1.72, 95% CI 0.52–5.69, P = 0.374). This suggests that at lower levels of smoking pack-years, the risk of “MSAP or SAP” occurrence isn't significantly different from that of non-smokers. Smoking needs a certain accumulation time to noticeably exacerbate the severity of AP, with the specific threshold currently unknown. Similar cumulative effects of smoking have been documented in other studies. For instance, a comprehensive study by Tolstrup et al. (10) revealed no significant difference in pancreatitis incidence between smokers consuming 1–14 g/day and never-smokers, yielding a HR of 1.5 (95% CI 0.9–2.5). Pancreatitis incidence attained statistical significance only with increased smoking to 15–24 g/day, showing an HR of 2.5 (95% CI 1.5–3.9). Similarly, Hansen et al. (34), based on a prospective study of 108,438 individuals, reported HRs of 1.1 (95% CI 0.8–1.7, with no statistical significance) and 3.6 (95% CI 1.8–2.5) for smoking pack-years of 0.1–9 and 9.1–24, respectively, for chronic pancreatitis risk. Large-scale prospective studies are imperative to elucidate the underlying mechanisms and determine the threshold for cumulative effects.
Alcohol is considered an independent risk factor for the occurrence of AP (35). According to calculations based on weekly alcohol consumption, drinking ≥5 drinks per day significantly increases the risk of developing AP (36, 37). However, in our study, we did not find a significant correlation between a history of alcohol consumption and the severity of AP (P = 0.061). On one hand, alcohol consumption may be a factor in the onset of pancreatitis rather than exacerbating its severity. On the other hand, this lack of correlation may be related to the drinking patterns of the patients included in our study. We observed a higher proportion of occasional drinkers rather than daily alcohol abusers among our patients. Additionally, in our study, we did not find a significant interaction between a history of alcohol consumption and smoking (P = 0.084).
This study boasts several strengths: we employed propensity score matching and multivariable logistic regression to rigorously control for confounding factors. The dose-response linkage analysis visually depicted the correlation between smoking pack-years and the severity of AP. Nonetheless, certain limitations should be acknowledged: the sample size was relatively small; the single-center retrospective design precludes establishing causality, necessitating further prospective and multicenter studies to validate these findings; our study only included non-smokers and current smokers, excluding former smokers, which limits our ability to determine whether smoking cessation can mitigate the exacerbating effect of smoking on the severity of AP. Additionally, Relative confounding from unmeasured factors may continue even after corrections.
In conclusion, our study provides evidence suggesting that smoking is associated with an increased risk of developing “MSAP or SAP” in patients with AP. We also observed a dose-response relationship between smoking pack-years and the severity of pancreatic involvement, indicating that the impact of smoking on the severity of AP may require a certain amount of time to accumulate before becoming evident. Our findings lay the groundwork for future longitudinal studies. However, whether smoking cessation can mitigate this effect requires further confirmation through new multicenter prospective randomized studies.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Ethics Committee of Dandong Central Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants' legal guardians/next of kin in accordance with the national legislation and institutional requirements.
RL: Supervision, Writing – original draft, Writing – review & editing. WT: Data curation, Writing – original draft, Writing – review & editing. SY: Conceptualization, Supervision, Writing – original draft. XY: Methodology, Writing – original draft. LH: Project administration, Writing – original draft.
The author (s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2024.1397111/full#supplementary-material
1. Uehara S, Honjyo K, Furukawa S, Hirayama A, Sakamoto W. Role of the kallikrein-kinin system in human pancreatitis. Adv Exp Med Biol. (1989) 247B:643–48. doi: 10.1007/978-1-4615-9546-5_106
2. Mayerle J, Sendler M, Hegyi E, Beyer G, Lerch MM, Sahin-Toth M. Genetics, cell biology, and pathophysiology of pancreatitis. Gastroenterology. (2019) 156:1951–68.e1. doi: 10.1053/j.gastro.2018.11.081
3. Sellers ZM, MacIsaac D, Yu H, Dehghan M, Zhang KY, Bensen R, et al. Nationwide trends in acute and chronic pancreatitis among privately insured children and non-elderly adults in the United States, 2007-2014. Gastroenterology. (2018) 155:469–78.e1. doi: 10.1053/j.gastro.2018.04.013
4. Brindise E, Elkhatib I, Kuruvilla A, Silva R. Temporal trends in incidence and outcomes of acute pancreatitis in hospitalized patients in the United States from 2002 to 2013. Pancreas. (2019) 48:169–75. doi: 10.1097/MPA.0000000000001228
5. Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, et al. Classification of acute pancreatitis−2012: revision of the Atlanta classification and definitions by international consensus. Gut. (2013) 62:102–11. doi: 10.1136/gutjnl-2012-302779
6. Forsmark CE, Vege SS, Wilcox CM. Acute pancreatitis. N Engl J Med. (2016) 375:1972–81. doi: 10.1056/NEJMra1505202
7. Krishna SG, Kamboj AK, Hart PA, Hinton A, Conwell DL. The changing epidemiology of acute pancreatitis hospitalizations: a decade of trends and the impact of chronic pancreatitis. Pancreas. (2017) 46:482–8. doi: 10.1097/MPA.0000000000000783
8. Lankisch PG, Apte M, Banks PA. Acute pancreatitis. Lancet. (2015) 386:85–96. doi: 10.1016/S0140-6736(14)60649-8
9. Sadr-Azodi O, Andren-Sandberg A, Orsini N, Wolk A. Cigarette smoking, smoking cessation and acute pancreatitis: a prospective population-based study. Gut. (2012) 61:262–7. doi: 10.1136/gutjnl-2011-300566
10. Tolstrup JS, Kristiansen L, Becker U, Gronbaek M. Smoking and risk of acute and chronic pancreatitis among women and men: a population-based cohort study. Arch Intern Med. (2009) 169:603–9. doi: 10.1001/archinternmed.2008.601
11. Lindkvist B, Appelros S, Manjer J, Berglund G, Borgstrom A. A prospective cohort study of smoking in acute pancreatitis. Pancreatology. (2008) 8:63–70. doi: 10.1159/000114868
12. Andriulli A, Botteri E, Almasio PL, Vantini I, Uomo G, Maisonneuve P, et al. Smoking as a cofactor for causation of chronic pancreatitis: a meta-analysis. Pancreas. (2010) 39:1205–10. doi: 10.1097/MPA.0b013e3181df27c0
13. Yuan S, Chen J, Ruan X, Sun Y, Zhang K, Wang X, et al. Smoking, alcohol consumption, and 24 gastrointestinal diseases: Mendelian randomization analysis. Elife. (2023) 12:e84051. doi: 10.7554/eLife.84051
14. Thrower E. Pathologic cellular events in smoking-related pancreatitis. Cancers. (2015) 7:723–35. doi: 10.3390/cancers7020723
15. He F, Zhu HM Li BY, Li XC, Yang S, Wang Z, Zhang M. Factors predicting the severity of acute pancreatitis in elderly patients. Aging Clin Exp Res. (2021) 33:183–92. doi: 10.1007/s40520-020-01523-1
16. Lee JM, Han KD, Lee SH, Park JM, Park N, Jeon H, et al. The association between smoking, changes in smoking behavior, and acute pancreatitis: a population-based cohort study in Korea. J Gastroenterol Hepatol. (2023) 38:451–9. doi: 10.1111/jgh.16061
17. Pang Y, Kartsonaki C, Turnbull I, Guo Y, Yang L, Bian Z, et al. Metabolic and lifestyle risk factors for acute pancreatitis in Chinese adults: a prospective cohort study of 05 million people. PLoS Med. (2018) 15:e1002618. doi: 10.1371/journal.pmed.1002618
18. Kim DB, Chung WC, Lee JM, Lee KM, Oh JH, Jeon EJ. Analysis of factors associated with the severity of acute pancreatitis according to etiology. Gastroenterol Res Pract. (2017) 2017:1219464. doi: 10.1155/2017/1219464
19. Chowdhury P, Udupa KB. Effect of nicotine on exocytotic pancreatic secretory response: role of calcium signaling. Tob Induc Dis. (2013) 11:1. doi: 10.1186/1617-9625-11-1
20. van Geenen EJ, Smits MM, Schreuder TC, van der Peet DL, Bloemena E, Mulder CJ. Smoking is related to pancreatic fibrosis in humans. Am J Gastroenterol. (2011) 106:1161–6; quiz 1167. doi: 10.1038/ajg.2011.43
21. Penttila AK, Rouhiainen A, Kylanpaa L, Mustonen H, Puolakkainen P, Rauvala H, et al. Circulating nucleosomes as predictive markers of severe acute pancreatitis. J Intens Care. (2016) 4:14. doi: 10.1186/s40560-016-0135-6
22. Yasuda T, Ueda T, Takeyama Y, Shinzeki M, Sawa H, Nakajima T, et al. Significant increase of serum high-mobility group box chromosomal protein 1 levels in patients with severe acute pancreatitis. Pancreas. (2006) 33:359–63. doi: 10.1097/01.mpa.0000236741.15477.8b
23. Lindstrom O, Tukiainen E, Kylanpaa L, Mentula P, Rouhiainen A, Puolakkainen P, et al. Circulating levels of a soluble form of receptor for advanced glycation end products and high-mobility group box chromosomal protein 1 in patients with acute pancreatitis. Pancreas. (2009) 38:e215–20. doi: 10.1097/MPA.0b013e3181bb59a7
24. Liu T, Huang W, Szatmary P, Abrams ST, Alhamdi Y, Lin Z, et al. Accuracy of circulating histones in predicting persistent organ failure and mortality in patients with acute pancreatitis. Br J Surg. (2017) 104:1215–25. doi: 10.1002/bjs.10538
25. Garg PK, Singh VP. Organ failure due to systemic injury in acute pancreatitis. Gastroenterology. (2019) 156:2008–23. doi: 10.1053/j.gastro.2018.12.041
26. Petrov MS, Shanbhag S, Chakraborty M, Phillips AR, Windsor JA. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology. (2010) 139:813–20. doi: 10.1053/j.gastro.2010.06.010
27. Colak Y, Afzal S, Lange P, Nordestgaard BG. Smoking, systemic inflammation, and airflow limitation: a Mendelian Randomization analysis of 98 085 individuals from the general population. Nicotine Tob Res. (2019) 21:1036–44. doi: 10.1093/ntr/nty077
28. Liu Y, Lu L, Yang H, Wu X, Luo X, Shen J, et al. Dysregulation of immunity by cigarette smoking promotes inflammation and cancer: a review. Environ Pollut. (2023) 339:122730. doi: 10.1016/j.envpol.2023.122730
29. McComb S, Thiriot A, Akache B, Krishnan L, Stark F. Introduction to the immune system. Methods Mol Biol. (2019) 2024:1–24. doi: 10.1007/978-1-4939-9597-4_1
30. Lushniak BD, Samet JM, Pechacek TF, Norman LA, Taylor PA. The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General. Atlanta, GA: Centers for Disease Control and Prevention (2014).
31. Rioux N, Castonguay A. 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone modulation of cytokine release in U937 human macrophages. Cancer Immunol Immunother. (2001) 49:663–70. doi: 10.1007/s002620000157
32. Proulx LI, Gaudreault M, Turmel V, Augusto LA, Castonguay A, Bissonnette EY. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone, a component of tobacco smoke, modulates mediator release from human bronchial and alveolar epithelial cells. Clin Exp Immunol. (2005) 140:46–53. doi: 10.1111/j.1365-2249.2005.02739.x
33. Munzel T, Hahad O, Kuntic M, Keaney JF, Deanfield JE, Daiber A. Effects of tobacco cigarettes, e-cigarettes, and waterpipe smoking on endothelial function and clinical outcomes. Eur Heart J. (2020) 41:4057–70. doi: 10.1093/eurheartj/ehaa460
34. Hansen SEJ, Nordestgaard BG, Langsted A. Smoking as the most important risk factor for chronic pancreatitis in the general population. Eur J Epidemiol. (2023) 38:95–107. doi: 10.1007/s10654-022-00945-7
35. Sahin-Toth M, Hegyi P. Smoking and drinking synergize in pancreatitis: multiple hits on multiple targets. Gastroenterology. (2017) 153:1479–81. doi: 10.1053/j.gastro.2017.10.031
36. Yadav D, Hawes RH, Brand RE, Anderson MA, Money ME, Banks PA, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med. (2009) 169:1035–45. doi: 10.1001/archinternmed.2009.125
Keywords: smoking, acute pancreatitis, propensity score-matching, severity grading, dose–response relationship
Citation: Li R, Tang W, Yan S, Yu X and Hu L (2024) A dose–response correlation between smoking and severity of acute pancreatitis: a propensity score-matched study. Front. Med. 11:1397111. doi: 10.3389/fmed.2024.1397111
Received: 06 March 2024; Accepted: 15 July 2024;
Published: 29 July 2024.
Edited by:
Ravi Kumar Sharma, Chandigarh University, IndiaReviewed by:
Wenjian Mao, Nanjing University, ChinaCopyright © 2024 Li, Tang, Yan, Yu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lian Hu, MjUzMDYwNTY4NUBxcS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.