 Elpida Neofotistou-Themeli1,2
Elpida Neofotistou-Themeli1,2 Panagiota Goutakoli1,2
Panagiota Goutakoli1,2 Theodoros Chanis3,4
Theodoros Chanis3,4 Maria Semitekolou5,6
Maria Semitekolou5,6 Eirini Sevdali1,2
Eirini Sevdali1,2 Prodromos Sidiropoulos1,2*
Prodromos Sidiropoulos1,2*- 1Laboratory of Rheumatology, Autoimmunity and Inflammation, University of Crete, Medical School, Heraklion, Greece
- 2Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology - Hellas (FORTH), Heraklion, Greece
- 3Division of Immunology and Allergy, Department of Medicine, Karolinska Institute, Solna, Sweden
- 4Center for Molecular Medicine, Karolinska Institute, Stockholm, Sweden
- 5Dendritic Cells and Adaptive Immunity Unit, Immunology Department, Pasteur Institute, Paris, France
- 6Developmental Biology and Stem Cells, UMR3738 – National Center for Scientific Research (CNRS), Pasteur Institute, Paris, France
High-throughput technologies in human and animal studies have revealed novel molecular and cellular pathways involved in tissue inflammation of rheumatoid arthritis (RA). Fibroblasts have been in the forefront of research for several decades. Subpopulations with specific phenotypic and functional properties have been characterized both in mouse models and human disease. Data supporting the active involvement of fibroblasts in immune responses and tissue remodeling processes, as well as their central role in promoting clinical relapses and contributing to treatment resistance, have clearly reshaped their role in disease evolution. The lung is an important non-synovial component of RA both from a clinical and an immunopathogenic aspect. Interstitial lung disease (ILD) is a significant contributor to disease burden affecting morbidity and mortality. Although our knowledge of ILD has progressed, significant gaps in both basic and clinical science remain, posing hurdles to efficient diagnosis, prediction of disease course and its effective treatment. The specific role and contribution of fibroblasts to this process has not been clearly defined. The focus of this review is on fibroblasts and their contribution to RA and RA-ILD, presenting data on genetics and immune responses associated with RA-ILD in humans and animal models.
Introduction
Systemic Autoimmune Rheumatic Diseases (SARDs) are a group of immune-mediated disorders characterized by diverse clinical manifestations. SARDs exhibit continuously rising incidence in Western countries and a lifetime prevalence of over 10% in the European Union (1). They contribute substantially to morbidity, mortality and annual health care costs. Common denominators in SARD development include environmental, genetic and immune factors. They orchestrate the initiation, progression and amplification of inflammatory networks and dictate responses to therapy. Although several achievements have been accomplished during recent decades for the diagnosis and treatment of SARDs, there are still significant unmet needs which impede improvements in clinical outcomes, quality of life, and in lowering the cost of treatment.
Rheumatoid arthritis (RA) is a SARD which has a significant societal and financial burden in Europe (2). It is widely recognized that by the time of initial diagnosis of RA, as is the case for many other SARDs, the autoimmune response has already been established and chronic inflammation is mostly irreversible. RA is the most prevalent SARD that primarily affects the joints. In Western Europe, its prevalence is estimated to be 0.63% in females and 0.24% in males, while it increases significantly to up to 2% among individuals over age 60 (3). The treatment goal for RA is remission of the inflammatory process. The management of RA in clinical practice has benefited from recent advances such as early diagnosis and treatment, adoption of the “treat to target” strategy and the introduction of targeted biologic therapies. Nevertheless, a lot of improvement is still needed in disease related outcomes (functional limitation, significant morbidity and increased mortality) (4). Although recent data show that ‘hard’ outcomes like mortality have improved in some cohorts (5, 6), most epidemiological data still indicate a gap compared to the non-RA control population (7, 8). Leading causes of mortality are cardiovascular diseases and lung involvement, primarily interstitial lung disease (ILD) and infections. Clinically significant ILD occurs in approximately 10% of RA patients, while the prevalence of subclinical abnormalities consistent with ILD has been documented as up to 60% (9, 10). Inflammation and fibrosis within the lung parenchyma is the pathophysiological basis of RA-ILD, as it is in many other organs affected in various SARDs. Although clinical, epidemiological and imaging characteristics have been associated with the risk of ILD development and its outcome, they do not represent clinically reliable tools to be used for individual monitoring and clinical decision making.
Fibroblasts are cells of mesenchymal origin present in all tissues. They adopt different phenotypes and functions according to intrinsic characteristics or upon responding to different acute and chronic stimuli (11). High-throughput single-cell RNA sequencing studies in inflammatory arthritis mouse models and in the human RA synovium have revealed several fibroblastic subpopulations with specific phenotypic and functional properties, while spatial diversity has also been identified within the synovium (12–14). The active contribution of fibroblasts to immune and tissue remodeling processes, clinical relapses and treatment resistance has reshaped how we perceive the role of fibroblasts in RA evolution and outcome (15, 16). Functionally distinct subpopulations contribute to lung development, response to injury and repair (17). However, studies focusing on the pathogenesis of RA associated ILD are limited, and no clear pathophysiology concept is accepted.
This review focuses on fibroblasts, presenting recent developments pertinent to their contribution to RA and RA-ILD pathogenesis – from genetic data associated with RA-related lung disease to major discoveries in fibroblast subpopulations – and discussing their contribution to disease evolution, flares and treatment responses in human RA and in experimental models of arthritis.
Overview of RA pathogenesis
The identification of anti-citrullinated protein antibodies (ACPAs) set the stage to advance our understanding of RA pathogenesis. 50–60% of RA patients have ACPA antibodies, which are highly specific for RA, together with rheumatoid factors (RF), which have a lower specificity for RA (18). Most of the knowledge for disease pathogenesis refers to seropositive disease, while data for seronegative disease is much more limited. The current paradigm for RA pathogenesis is that autoimmunity develops over several years before the disease is clinically apparent (Figure 1). In genetically susceptible individuals, early self-reactivity against post-translationally modified proteins develops mostly in mucosal areas, with subsequent targeting of synovial tissue by effector T cells. Amplification of inflammatory responses along with the transition of resident stromal cells into autoaggressive effector cells converts synovitis from acute to chronic destructive joint disease (19).
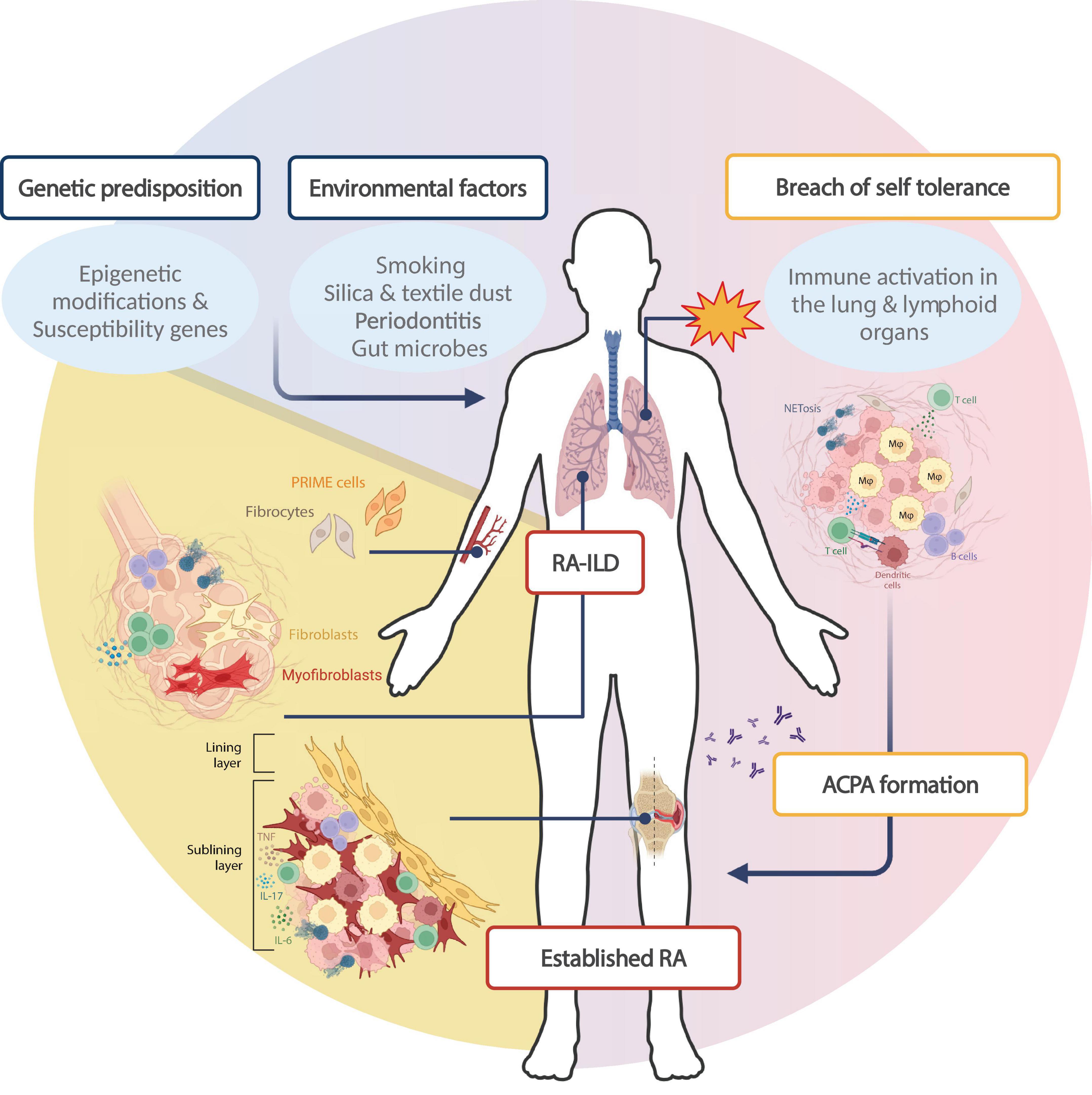
Figure 1. Rheumatoid arthritis pathogenesis: specific contribution of fibroblasts in RA phenotype. The paradigm for RA development stands that environmental parameters trigger primary autoimmune events in genetically predisposed individuals. This interaction leads to loss of self-tolerance in extra-synovial tissues like the lung and lymphoid tissues, through the activation of immune cells of innate and adaptive immunity. Protein citrullination in mucosal areas like the lung combined with RA-associated HLA-DR alleles leads to the production of anti-citrullinated protein antibodies (ACPAs) in 50–60% of patients. Thereafter, autoreactive immune cell homing to the synovial compartment triggers synovitis (early arthritis), characterized by local inflammation and a dramatic expansion of the synovial membrane, characterized by extensive proliferation of fibroblasts and further immune cell recruitment. Established RA is a systemic disease with extra-articular manifestations; approximately 10% of RA patients develop interstitial lung disease (RA-ILD), which has a complex pathogenesis implicating both immune and stromal cells. Within the lung, collagen deposition and subsequent fibrosis is driven by resident macrophages and dendritic cells which favor fibroblast and myofibroblast activation, though the fibroblast subsets involved have not been extensively studied. Of note, circulating pre-inflammatory mesenchymal (PRIME) cells have been proposed to drive disease flares, while circulating fibrocytes could constitute a pathogenic link between joint and lung pathology in RA. Created in BioRender. Neofotistou, E. (2025). https://BioRender.com/l38d140.
The identification of ACPAs has put together the concept that genetic and environmental factors lead to breaching of tolerance to post-translationally modified proteins and to subsequent autoimmunity development. It is known that genetic polymorphisms contribute 30–60% of the overall RA risk (20). Genome-wide association studies (GWAS) data confirmed earlier genetic association studies suggesting that HLA class II alleles, specifically HLA-DRβ1 alleles containing a particular sequence in amino acid residues 71–74 of the β-chain (“shared epitope”, SE), confer the strongest risk (21, 22). Interestingly and further supporting the pathogenetic contribution of HLA-DR genes, it has been shown that proteins encoded by these alleles can bind citrullinated peptides with higher avidity than native peptides, thereby inducing T-cell activation (23, 24). GWAS analysis has revealed more than 100 additional alleles which contribute to the risk of disease development, most of them relevant to innate and adaptive immune response pathways.
Amongst the diverse environmental factors associated with RA development, smoking shows the strongest correlation (25). Smoking is considered to act as an epigenetic modifier and, combined with HLA-DR4 alleles, contributes to ACPA formation. Several human studies in individuals at risk for RA development and early RA patients support that autoimmunity to post-translationally modified proteins develops in the lung parenchyma, resulting in effector CD4 cells and autoreactive B cells (26–28). Data elucidating the mechanisms driving the homing of autoreactive CD4 cells into the synovium, in the absence of any synovium- or joint-specificity of these cells, are limited. Elegant in vitro and in vivo studies have linked the breach in T cell tolerance to cell-endogenous abnormalities present in naive T cells, which drive the differentiation program to favor the generation of effector instead of long-lived memory T cells (19). Impaired DNA repair mechanisms, which compromise telomeric function and mitochondrial fitness, are a major driver of metabolic alterations characterized by decreased glycolysis and favoring catabolic pathways, therefore transforming T cells to tissue-invasive proinflammatory effector cells (29, 30). Among other molecular mechanisms, deficient activity of the nuclease MRE11A in T cells of RA patients has been linked to premature T cell aging, tissue invasiveness and proinflammatory T cell functions (30, 31). Upon synovial localization of inflammatory responses, acute synovitis is considered to transform into a chronic process by maladaptive tissue remodeling mechanisms driven by both immune cells and synovium stromal cells, mostly fibroblasts. Several recent studies based on single-cell RNA sequencing analysis revealed functionally distinct subsets of highly activated synovial fibroblasts, which adopt proinflammatory and tissue-invasive function, and together with tissue-infiltrating myelocytes and lymphocytes contribute to tissue damage. The application of high throughput technologies has provided unprecedented information on the cellular composition of the synovial lesion (12, 14, 32). Thus, several fibroblast, macrophage, T and B cell subpopulations have been identified. Although interesting data concerning the importance of these molecular endotypes for predicting the course of early disease and response to treatment have been shown, mechanistic studies are needed to elucidate their contribution to disease pathogenesis (12, 14, 15, 32).
Synovial fibroblasts in RA pathogenesis
The involvement of fibroblasts in RA development and progression has been extensively studied within the synovium. In the healthy joint, fibroblast-like synoviocytes (FLS) contribute to the formation of the thin synovial membrane enclosing the synovial cavity and are crucial in maintaining cartilage integrity and function by producing extracellular matrix and synovial fluid components (33–35). In the context of RA, these cells become hyperactivated and hyperproliferative, epigenetically imprinted with an aggressive phenotype that alters the joint microenvironment by producing metalloproteases that degrade the extracellular space, invading the synovial cavity and causing cartilage damage and bone erosion directly or via RANKL-mediated osteoclast activation. Moreover, FLS act as a rheostat for immune circuits (36), exacerbating inflammation by facilitating the infiltration of immune cell populations via angiogenesis and redistribution of soluble factors, expression of cell adhesion molecules and secretion of proinflammatory stimuli such as IL-6 and IL-8 (35, 37). Of note, metabolic parameters have been found to contribute to FLS activation, proliferation and aggression in arthritis (38, 39).
Based on research applying high-throughput technologies, knowledge for the phenotypically and functionally distinct subpopulations of fibroblasts in different diseases has been dramatically expanded (Tables 1, 2). Interestingly and besides the pathophysiological correlations, several clinical implications of synovial biology for RA patients are available, concerning disease prognosis, flares and response to treatments. More specifically, early RA patients with a fibroid synovial pathotype are characterized by poor responses to treatment and worse prognosis regardless of the treatment scheme used (32). Analysis of matched peripheral blood and synovial biopsy samples highlighted the importance of synovial tissue evaluation in predicting disease outcome. Indeed, synovial histology correlates with clinical parameters and may aid in early classification of patients (40). In a cohort of established RA patients receiving different biologic agents, synovial composition was able to predict treatment responses (15). More specifically, patients with abundant myeloid cell infiltration responded better to anti-IL6R treatment with tocilizumab than to anti-CD20 treatment with rituximab, while patients with fibroblast-rich synovia displayed multidrug resistance. Elegant studies support the migratory capacity of fibroblasts in humans and mouse models of RA, attributing a role in disease states and its “spread” between different joints. A recent human study identified a B cell-induced circulating population of CD45–CD31–Podoplanin+ pre-inflammatory mesenchymal (PRIME) cells that could predict disease flares (16), while studies in the severe combined immunodeficient (SCID) mice receiving activated human RA synovial fibroblast implants confirmed that FLS can transmigrate, spreading arthritis to unaffected joints (41).
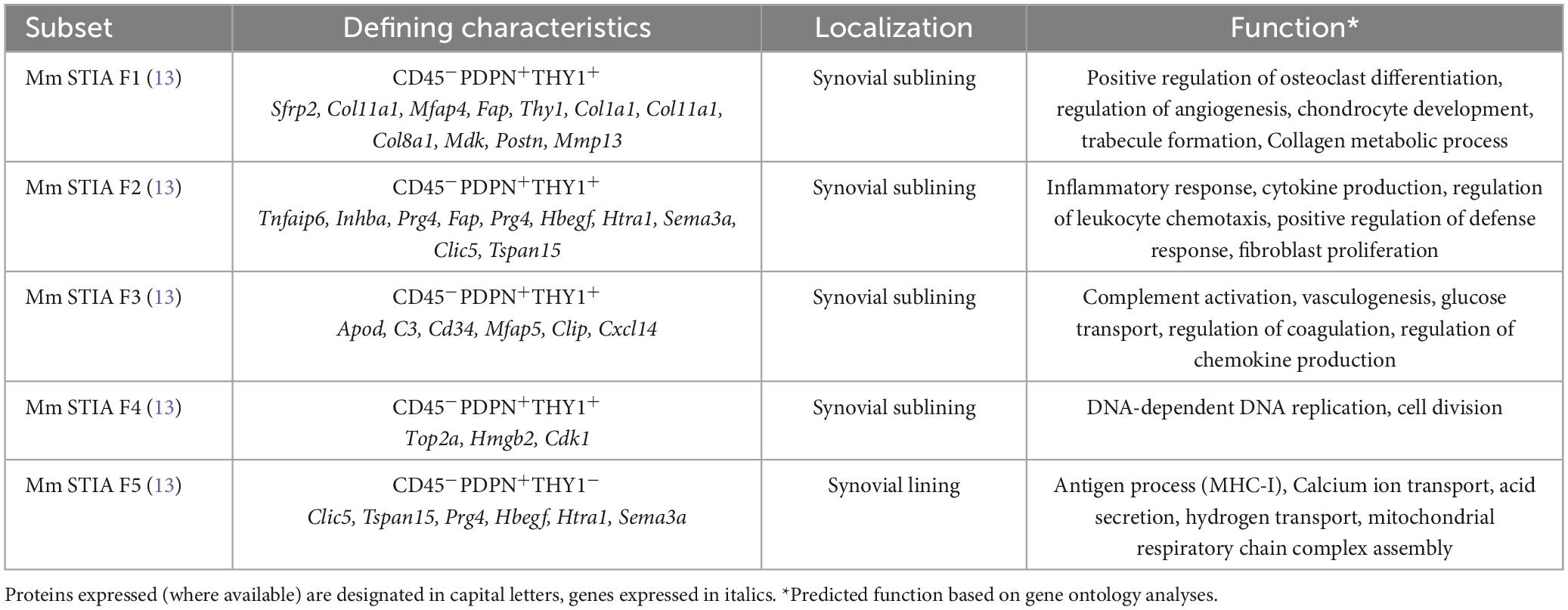
Table 1. Murine synovial fibroblast subsets in experimental arthritis.
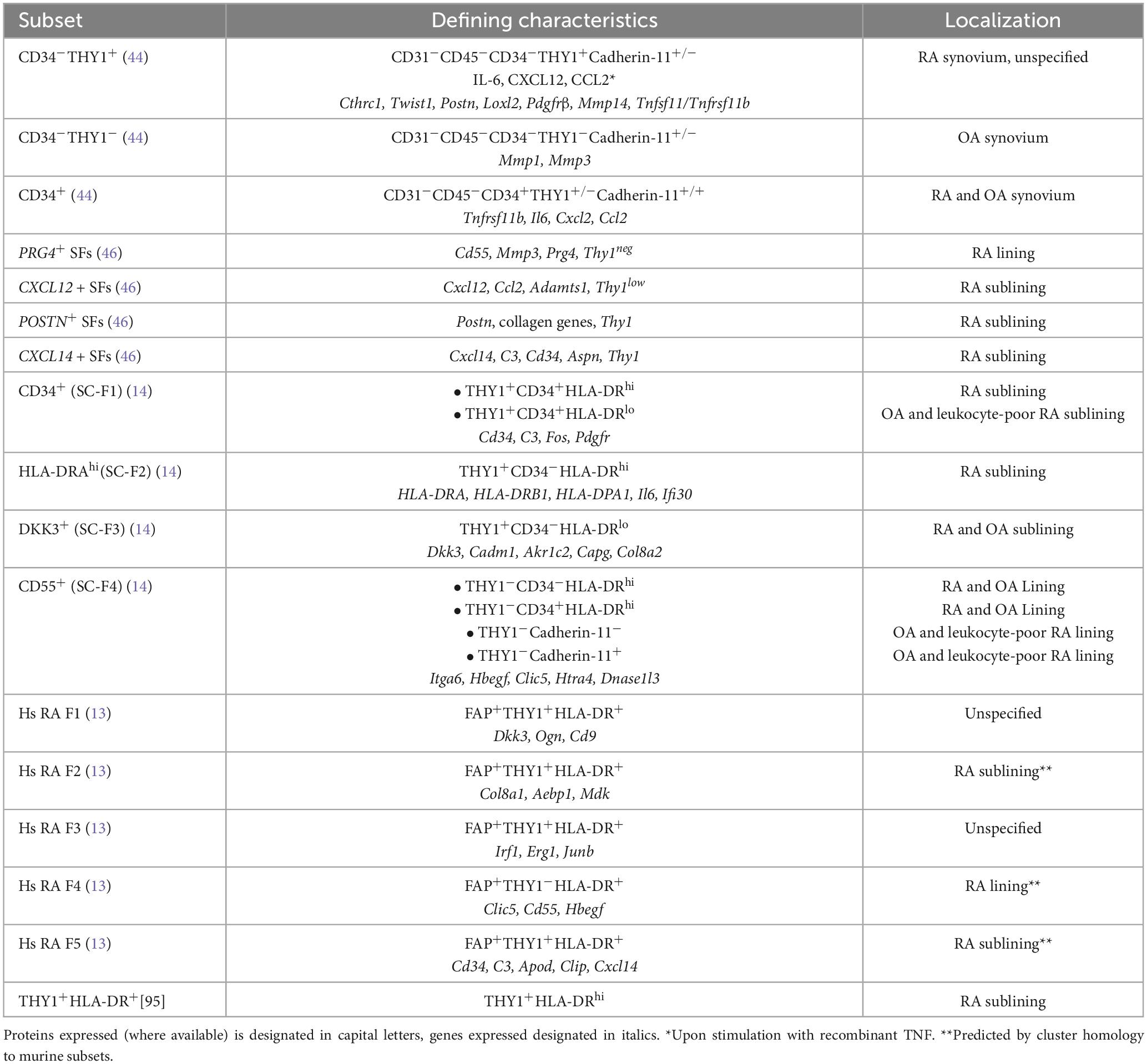
Table 2. Human synovial fibroblast subsets in RA.
Synovial fibroblast heterogeneity
Fibroblasts in the RA synovium exhibit vast heterogeneity and several subsets have been described that differentially contribute to RA-associated pathology in mice and humans (Tables 1, 2). Single-cell transcriptomics in the synovium of serum transfer-induced arthritis (STIA) mice revealed two anatomically distinct fibroblast activation protein-α (FAPα)+ subpopulations, differentiated by the expression of thymus cell antigen 1 (THY1, also known as CD90) (13). FAPα+THY1+ fibroblasts were located in the synovial sublining layer and found to mediate inflammatory responses, whereas FAPα+THY1– fibroblasts of the lining layer exhibited a tissue destructive phenotype. Of note, inducible depletion of both subsets ameliorated experimental arthritis, whereas adoptive transfer of FAPα+THY1+ fibroblasts resulted in more severe, persistent disease. Notably, the inflammatory FAPα+THY1+ subset was enriched in RA patients compared to patients with osteoarthritis (OA). Using single-cell RNA sequencing and synovial tissue organoids, it was further suggested that the positional identity of synovial fibroblasts may be regulated by the endothelium via Notch3 signaling (42).
Three-dimensional spatial transcriptomics in synovial tissue sections from RA patients revealed substantial variation both in architectural organization as well as in cell composition (43). In synovial biopsies from seropositive RA patients, CD55+ lining and HLA-DR+ and CD34+ sublining fibroblasts co-localized with macrophage-rich areas, while THY1+ fibroblasts were in close proximity to plasma cells. Single-cell RNA sequencing in synovial biopsies from RA and OA patients identified seven fibroblast clusters characterized by distinct localization and functions (44). Among those, CD34–THY1+Cadherin-11± perivascular fibroblasts were greatly expanded in RA patients, exhibiting a proliferative, invasive, proinflammatory phenotype evident by the expression of migratory response genes such as CTHRC1, TWIST1, POSTN, LOXL2, PDGFRB and MMP14, transwell matrix invasion assays, and prominent in vitro secretion of IL-6, CXCL12 and CCL2 upon stimulation with recombinant TNF. Additionally, paired single-cell and transposase-accessible chromatin sequencing (ATAC-seq) analysis of synovial biopsies from RA patients, combined with multiplex imaging and spatial transcriptomics, suggested that local TNF, IFN-γ or IL-1β exposure can drive FLS heterogeneity, and also indicated that functional gene expression programs in fibroblasts may be shared across tissues and diseases (45). Combining mass cytometry with transcriptomics showed that THY1(CD90)+HLA-DRhi sublining fibroblasts were expanded in RA synovia, and attributed IL6 expression to this putative key mediator of RA pathogenesis[14]. A deconvolution analysis of published RNAseq and in-house data evaluated the contribution of synovial fibroblast subsets to the synovial pathotypes described for treatment-naïve patients with early RA (46). Interestingly, CXCL12+ and POSTN+ sublining synovial fibroblasts (SFs) constituted the most prominent fibroblast subpopulation in the fibroid, treatment-resistant RA pathotype. Comparing SFs from healthy and transgenic hTNFtg mice overexpressing human TNF, another study identified several homeostatic and disease-associated fibroblast subsets (47). Integrating murine data with publicly available datasets from RA patients, the study revealed conserved networks of gene regulation, while it also suggested a trajectory of transcriptional remodeling culminating in an inflammatory, destructive phenotype. Notably, a spatial transcriptomics study in RA versus spondylarthritis (SpA) patients revealed marked differences in synovial tissue composition, with SpA expressing a more fibrotic profile, enriched in mesenchymal cell signatures suggestive of higher cartilage turnover (48).
The lung as the site of initial autoimmune responses in RA
Though synovial inflammation is a key disease characteristic, the current paradigm for RA development stands that initial autoimmunity against post-translationally modified proteins may develop in tissues outside the joints (see Figure 1). The lung has been accepted as a tissue where in genetically predisposed persons and upon environmental triggers, initial breach of immune tolerance and inflammation may take place. Smoking has – amongst the diverse environmental factors – the strongest association to RA development (25), considered to act as an epigenetic modifier inducing the citrullination of proteins in mucosal areas like the lung, and, combined with HLA-DR4 alleles contributing to ACPA formation. Several studies suggest that pulmonary involvement may be present early in the disease course of RA (49). Specifically, it has been shown that some patients with early RA have airway or parenchymal abnormalities accompanied by decreased lung function (50). Furthermore, pulmonary involvement, including ILD, abnormal high resolution computed tomography (HRCT) findings or abnormal pulmonary function tests (PFT), has been identified in early RA patients with no more than 2 years of disease duration (51). An increased frequency of interstitial abnormalities has been observed in patients with both early and longstanding RA, in contrast to bronchiolar abnormalities which have been shown to be more prominent in longstanding RA (52). Interestingly, ACPA-positive RA patients have a higher prevalence of both parenchymal and airway changes compared to ACPA-negative RA patients. HRCT revealed parenchymal lung abnormalities in ACPA-positive RA patients and further molecular studies on bronchoalveolar lavage (BAL) fluid highlighted the increased levels of ACPAs in lung compartments compared to the sera of ACPA-positive RA patients, suggesting a lung-specific production of ACPAs.
Early human studies supported the concept of the lung as the initial site of environmental-tissue interactions to induce post-translational protein modifications which may act as autoantigens in genetically susceptible individuals. In a case-control study enrolling patients with recent-onset RA, it was shown that previous smoking was dose-dependently associated with the occurrence of ACPAs, and the combination of smoking and the presence of HLA-DR shared epitope (HLA-DR SE) genes increased the risk for ACPA-positive RA (53). Notably, epidemiological studies have proposed that smoking is one of the major environmental factors associated with increased risk of developing seropositive RA even when smoking was discontinued up to 10–19 years before disease onset (54). Apart from smoking exposure (55, 56), strong gene-environment interactions were observed between HLA-SE and silica (57) and HLA-SE and textile dust (58) posing a high risk of ACPA-positive RA.
In addition to epidemiological studies, analyses of bronchoalveolar lavage (BAL) and histopathology further supported the above concept. More specifically, an enrichment of citrullinated proteins has been found in BAL cells of healthy smokers compared to healthy non-smokers. This was associated with higher expression of the peptidylarginine deiminase (PAD) 2 enzyme, which catalyzes protein citrullination. Increased expression of citrullinating enzymes was also identified in bronchial mucosal biopsies of healthy smokers (59). This study provided evidence that smoking enhances PAD2 expression with consequent generation of citrullinated proteins. Elevated lymphocyte infiltration and germinal center-like structure accumulation/formation, as well as higher immune cell activation markers have been revealed in bronchial biopsies and BAL fluids of untreated patients with early ACPA-positive RA without concomitant lung disease (27). Additionally, RF and ACPAs were found in smokers lacking evidence for RA-related joint involvement. Several studies showed the strong association between smoking, lung disease, and ACPA-positive RA, supporting that lung mucosa may be a site of ACPA generation (60). Finally, neutrophil extracellular trap formation (NETosis) has also been suggested to contribute to early immune responses and lung inflammation in RA. Specifically, ACPA presence was associated with elevated neutrophil cell counts and NET levels in the sputum of persons at risk for RA development, namely first-degree relatives (FDRs) of RA patients, supporting the hypothesis that local airway inflammation and NET formation may drive ACPA production in the lung well before any articular inflammation occurs, and may play a role in the development of RA (61).
Pathophysiology of RA-associated ILD
Pulmonary complications are the most prevalent extra-articular manifestations in RA, clinically apparent in up-to 15% of patients, while up to 60% of patients may have subclinical disease (62), contributing significantly to increased morbidity and mortality (51). The disease is pleomorphic as it can affect distinct compartments of the lungs, including pleura, airways and their vessels, parenchyma and respiratory muscles with varying degrees of inflammation and fibrosis (63, 64). The most common form of RA-associated lung disease is ILD, a group of disorders that affect the pulmonary interstitial spaces leading to reduced lung diffusion capacity (9, 10, 65, 66). ILD develops early during RA progression and has also been associated with more severe joint disease (67), while in some cases it may even precede arthritis (68). The distribution of morphological patterns of RA-ILD as assessed by high-resolution computed tomography (HRCT) differs from the other types of collagen vascular disease (CVD)-ILD (69, 70). The most typical pattern of RA-ILD is that of usual interstitial pneumonia (UIP), which is mainly fibrotic, followed by non-specific interstitial pneumonia (NSIP) that displays both fibrotic and inflammatory manifestations or infiltrates (71). Gender differences also seem to exist among the RA-ILD patterns, with women displaying more frequently NSIP (72), while UIP has positively been associated with male gender, a previous history of smoking, as well as with older age (69, 73–76). RA- ILD patterns are correlated with diverse responses to treatments and prognosis, with retrospective studies showing that UIP is more severe and is associated with worse survival in patients with RA (77). Nevertheless, there are still a few inaccuracies in the diagnosis of RA-ILD, as the association between imaging pattern is not as clearly demonstrated in RA-ILD compared to other types of ILD, highlighting the need of establishing a standard approach to monitor lung structure and function in those patients (78).
A plethora of genetic (MUC5, HLA genes), environmental (smoking), demographic (older age, male sex), clinical (high titers of ACPA and RF) and drug-related risk factors were reported to contribute to the increased risk of developing ILD in the context of RA (79, 80). Nevertheless, the pathophysiological link between these risk factors and the pulmonary changes remains unclear. Recent work shows similar clinical, radiographic and genetic features (MUC5B rs35705950, mutations in genes involved in telomere maintenance) between certain patterns of RA-ILD, in particular UIP, and idiopathic pulmonary fibrosis (IPF), which could indicate shared pathways of pathogenesis and mechanisms of fibrosis (81). Although it is well known that the profibrotic milieu in IPF is established by complex interactions between basaloid cells, invasive fibroblasts and myofibroblasts, profibrotic monocyte-derived macrophages (82)1 and other immune cells (83), it is still not clear whether these findings apply to RA-ILD. There are also histopathological data suggesting that the UIP subtype in RA might be different from the UIP pattern found in IPF (69, 84, 85). It has been proposed that the driving step in RA-ILD pathogenesis is airway and/or alveolar epithelial damage in genetically susceptible individuals, in the context of exposure to environmental factors (smoking, infections and dysbiosis) (86, 87). Collagen deposition and subsequent fibrosis is favored by the production of transforming growth factor (TGF)-β and platelet-derived growth factor (PDGF) by tissue resident macrophages and dendritic cells that promote epithelial- to-mesenchymal (88), and fibroblast-to-myofibroblast transition (89). Local tissue damaged is further promoted by germinal center-like structures – called inducible bronchus-associated lymphoid tissue (iBALT) (90) – which are found in close proximity with alpha-smooth muscle actin (αSMA)+ cells, fibroblasts and collagen. Interestingly, the formation of these structures is not strictly induced by pulmonary inflammation, as patients with IPF do not display iBALTs, suggesting that the pulmonary pathology in RA is much more complicated. Additionally, an upregulation of IL-17RA expression in areas of fibroblastic accumulation and fibrosis was reported in biopsies from RA-ILD patients compared to either IPF or normal lung tissue, suggesting a direct role for IL-17A in lung fibrosis that may be specific to RA-ILD (91). Reduced lung diffusion capacity in a small cohort of RA patients has also been positively correlated with the levels of synovial and circulating fibrocytes (92) – a subset of bone marrow-derived cells expressing both hematopoietic and stromal cell markers which are increased in the circulation and lung biopsies of patients with IPF and ILD (93, 94). These data point out that fibrocytes could be a pathogenic link between joint and lung pathology in RA (92). Until now, studies have shown that anti-fibrotic drugs such as pirfenidone and nintedanib may slow the rate of forced vital capacity in patients with RA-ILD, in particular those with the UIP pattern (95–97). Another study supported the effectiveness of nintedanib in reducing the neutrophil-mediated fibrotic potential of human pulmonary fibroblasts (HPFs), while also reducing the levels of neutrophil extracellular traps (NETs) and soluble C5b-9 in RA-ILD patients (98). Nevertheless, the majority of the trials were underpowered (99–101) or included patients with progressive fibrosing RA-ILD (102) which does not reflect most of the patients in clinical practice. Hence, it is still not clear whether these anti-fibrotic agents could be a first-line treatment option in the management of RA-ILD (103), highlighting the need for further research on their clinical benefits and safety in RA-ILD. Collectively, a better understanding of the role of fibroblast subsets in the pathophysiology of lung involvement in RA would direct future therapeutic strategies targeting both synovial and lung inflammation.
Fibroblasts in lung fibrosis
Within the lung, the unique localization of mesenchymal cells between the epithelium and the stroma facilitates signal transduction both in tissue development as well as in tissue injury (104) (Table 3). Following injury, the lung mesenchyme, itself comprising several different fibroblasts’ subsets, plays a significant part in maintaining a balance between epithelial repair and pathological remodeling due to aberrant activation that may occasionally lead to fibrosis. Of note, lipofibroblasts (LIFs) in murine and human lung tissue have been shown to transdifferentiate to profibrotic myofibroblasts upon nicotine (105) or hyperoxia exposure (106), as well as during bleomycin challenge in fate mapping experiments using reporter mice (107). From a molecular standpoint, lipofibroblast-to-myofibroblasts transition was dependent on TGF-β1 signaling and was attenuated upon forced activation of peroxisome proliferator-activated receptor gamma (PPARγ) in primary human lung fibroblasts. Fibroblasts isolated from IPF patients were characterized by increased invasiveness mediated by hyaluronan synthase 2 (HAS2) and the hyaluronan receptor CD44 (108). This invasiveness was reminiscent of metastatic lung adenocarcinoma fibroblasts, and pathway analysis in fibroblasts from IPF patients suggested this phenotype was mediated by ERBB2 (HER2) signaling (109). These invasive fibroblasts expressed high levels of immune checkpoint ligands Cd274 (PDL1) and Pdcd1lg2 (PDL2), semaphorin 7A (CD108), Itga6 (CD49f) and F3 (CD142), and functional in vitro experiments showed that HER2 inhibition ameliorated lung fibrosis, especially when combined with anti-PDL1 treatment. Single-cell RNA sequencing of mesenchymal cells in lung biopsies from systemic sclerosis-associated interstitial lung disease (SSc-ILD) patients and healthy controls revealed two major and one minor fibroblast subset (SPINT2hi, MFAP5hi and WIF1hi, respectively) in the healthy lung (110). Those subpopulations were also identified in the SSc-ILD patients evaluated, however a distinct ACTA2 expressing and actively proliferative population of myofibroblasts was expanded in patients. The results of this study suggest that within the mesenchymal cell pool, myofibroblasts undergo the most significant phenotypic changes in systemic sclerosis, upregulating collagen-producing and profibrotic genes including but not limited to COL10A1, POSTN and ADAM12.
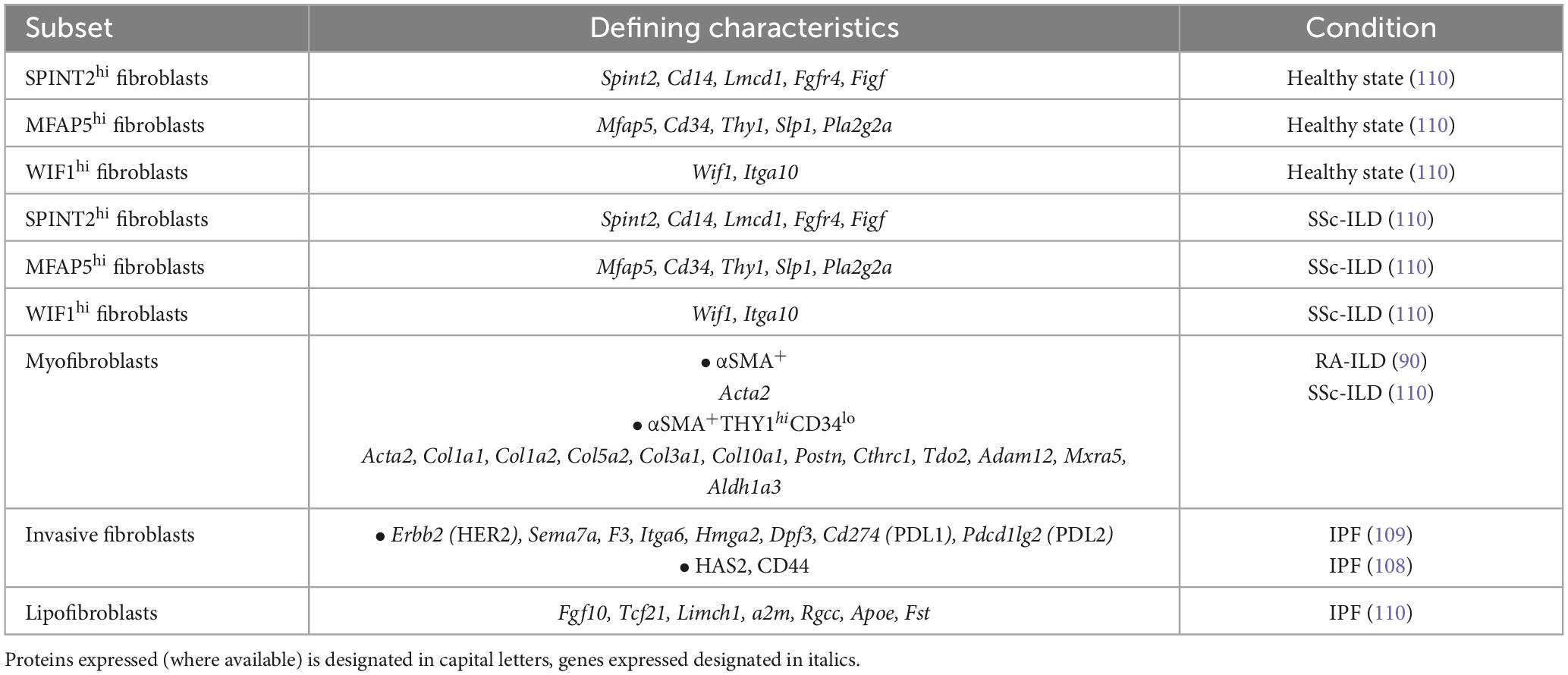
Table 3. Fibroblast subsets in the human lung in health and disease.
This activated myofibroblast state, characterized by expression of αSMA, conferring enhanced migratory and contractile capacity, has been the focus of study in several diseases (36, 37, 110) including pancreatic cancer (111), renal fibrosis (112) and wound healing following reperfusion in myocardial infarction (113). It has been shown that fibroblasts respond to exogenous mechanical loading and cell-generated tension by increasing synthesis of extracellular matrix components, as well as by switching to an αSMA-expressing myofibroblastic phenotype (114, 115). αSMA is required for focal adhesion maturation and for the initial formation of cortical filament bundles in spreading rat lung myofibroblasts (116), thus a potential mechanotransduction circuit has been suggested. According to this, tensile forces applied to cultured RAT-2 lung cells activate MAP kinase p38 through α2β1 integrin, leading to αSMA incorporation in actin filaments which in turn enhances p38 phosphorylation in a feed-forward loop (117). As αSMA expression is regulated by TGFβ canonically via the Smad-dependent signaling pathway or non-canonically by Smad-independent signaling pathways (118, 119), it constitutes a potential target of targeted RA therapies.
Although several animal models of experimental arthritis replicate joint disease of human RA, very few of them were reported to manifest lung pathology, with a pulmonary phenotype characterized by limited fibrosis that resembles more the NSIP than the UIP pattern (120, 121). Indeed, a mixed cellular and fibrotic pulmonary phenotype characterized by infiltrating macrophages, neutrophils and lymphocyte subsets and a lower degree of fibrotic changes has been reported in the SKG mouse model (122) and also in the adjuvant-induced arthritis model in rats (123). Like the iBALT structures of RA patients with ILD, ectopic lymphoid tissue development in the lungs were identified in the K/BxN transgenic mouse model of spontaneous arthritis (124), although they were not sufficient to drive fibrosis at a steady state or upon an inflammatory challenge. So far, several reports have shown that the collagen-induced arthritis model might display a pattern that resembles that of RA-ILD which could be used to study the effect of citrullination and ACPAs on disease initiation and progression (125–127). However, pulmonary lesions spontaneously decrease after the onset of joint pathology, in contrast to humans where exacerbation of the disease occurs in most of the cases as progression of the joint disease proceeds (62, 86, 125, 126). Altogether, further effort is needed for the establishment of reliable animal models of experimental arthritis that could simulate the clinical features and the mechanisms underlying RA-ILD pathogenesis.
Therapeutic targeting of fibroblasts
Although fibroblasts contribute substantially to inflammation and joint destruction, they are not specifically targeted by advanced treatments used in RA. This is in contrast to B- and T-cells which are specifically targeted by anti-CD20 and CTLA4-Ig, respectively. Anti-cytokine treatments like TNF inhibitors and IL-6 receptor blocker may be considered to interfere with fibroblast contribution to RA, since FLS are activated by both cytokines while they are major producers of IL-6 but not TNF. However, an elegant study in mice highlighted the importance of TNF/TNFR signaling in murine arthritis and showcased that synovial fibroblast survival and inflammatory capacity was orchestrated by the p55TNFR–IKK2–Ripk3 axis, suggesting Ikk2 targeting as a potential therapeutic target alongside TNF blockade (128). Moreover, recent elegant in vitro studies have shown that Jak inhibition using upadacitinib in contrast to TNF inhibition with adalimumab, reversed the induction of activated HLA-DR+CD90+ RA synovial fibroblasts by NK cell-derived IFNγ, indicating this disease-associated subpopulation as a therapeutic target of current biologics (129). As previously described, fibroblast depletion in arthritic mice ameliorated disease development, though it remains unclear how this approach could become clinically possible. Interestingly and relevant to RA-ILD pathology, in bleomycin-induced pulmonary fibrosis mouse models, the Jak inhibitor baricitinib attenuated disease severity via inhibition of the TGF-β1/non-Smad and TGF-β1/JAK/STAT signaling pathways, therefore limiting fibroblast activation and epithelial cell injury, respectively (130). Baricitinib administration in collagen-induced arthritis (CIA) on DBA/1 mice also attenuated pulmonary fibrosis by inhibiting the Jak2/Stat3 pathway, which has been found to play a key role in ILD (131).
Conclusion
Considering the rich repository of literature published over the past few decades investigating molecular pathways and cellular networks in RA, it is becoming clear that fibroblasts constitute important players in disease pathogenesis, progression and treatment response. Both within the synovium as well as in the lung, fibroblast subsets mediate events that dramatically alter the tissue microenvironment and affect clinical outcome. Aggressive fibroblast states are associated with severe disease and their abundance has proved to hinder effective responses to treatment. At the same time, studies in experimental animals and cell cultures have offered insights into potential direct or indirect impacts of treatments on fibroblast activation, which have not yet been fully translated to inform disease management. Further, elegant studies are needed to assess the functional importance of fibroblasts implicated in RA, particularly within the lung, where our knowledge of molecular mechanisms mediating disease early stages as well as RA-ILD is very limited (see Figure 1).
Moreover, seeing as several identified fibroblast subsets may overlap to a large extent and mediate similar processes, even across organ systems and diseases, collaborative efforts need to be made in order to fully dissect complex networks of interaction, understand causal relationships and identify precise and informative biomarkers, creating knowledge that can be applied to clinical practice.
Author contributions
EN-T: Conceptualization, Resources, Writing – original draft, Writing – review and editing. PG: Conceptualization, Resources, Writing – original draft, Writing – review and editing. TC: Conceptualization, Resources, Writing – original draft, Writing – review and editing. MS: Conceptualization, Resources, Writing – original draft, Writing – review and editing. ES: Conceptualization, Resources, Writing – original draft, Writing – review and editing. PS: Conceptualization, Resources, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. (2002) 347:911–20. doi: 10.1056/NEJMra020100
2. Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, et al. The global burden of rheumatoid arthritis: Estimates from the Global Burden of Disease 2010 study. Ann Rheum Dis. (2014) 73:1316–22. doi: 10.1136/annrheumdis-2013-204627
3. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
4. Winthrop KL, Mease P, Kerschbaumer A, Voll RE, Breedveld FC, Smolen JS, et al. Unmet need in rheumatology: Reports from the Advances in Targeted Therapies meeting, 2023. Ann Rheum Dis. (2023) 83:409–16. doi: 10.1136/ard-2023-224916
5. Poppelaars PB, van Tuyl LHD, Boers M. Normal mortality of the COBRA early rheumatoid arthritis trial cohort after 23 years of follow-up. Ann Rheum Dis. (2019) 78:586–9. doi: 10.1136/annrheumdis-2018-214618
6. Zhang Y, Lu N, Peloquin C, Dubreuil M, Neogi T, Aviña-Zubieta JA, et al. Improved survival in rheumatoid arthritis: A general population-based cohort study. Ann Rheum Dis. (2017) 76:408–13. doi: 10.1136/annrheumdis-2015-209058
7. Holmqvist M, Ljung L, Askling J. Mortality following new-onset Rheumatoid Arthritis: Has modern Rheumatology had an impact? Ann Rheum Dis. (2018) 77:85–91. doi: 10.1136/annrheumdis-2017-212131
8. Kerola AM, Kazemi A, Rollefstad S, Lillegraven S, Sexton J, Wibetoe G, et al. All-cause and cause-specific mortality in rheumatoid arthritis, psoriatic arthritis and axial spondyloarthritis: A nationwide registry study. Rheumatology. (2022) 61:4656–66. doi: 10.1093/rheumatology/keac210
9. Gabbay E, Tarala R, Will R, Carroll G, Adler B, Cameron D, et al. Interstitial lung disease in recent onset rheumatoid arthritis. Am J Respir Crit Care Med. (1997) 156(2 Pt. 1):528–35. doi: 10.1164/ajrccm.156.2.9609016
10. Bongartz T, Nannini C, Medina-Velasquez YF, Achenbach SJ, Crowson CS, Ryu JH, et al. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: A population-based study. Arthritis Rheum. (2010) 62:1583–91. doi: 10.1002/art.27405
11. Koliaraki V, Prados A, Armaka M, Kollias G. The mesenchymal context in inflammation, immunity and cancer. Nat Immunol. (2020) 21:974–82. doi: 10.1038/s41590-020-0741-2
12. Zhang F, Jonsson AH, Nathan A, Millard N, Curtis M, Xiao Q, et al. Deconstruction of rheumatoid arthritis synovium defines inflammatory subtypes. Nature. (2023) 623:616–24.
13. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. (2019) 570:246–51. doi: 10.1038/s41586-019-1263-7
14. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. (2019) 20: 928–42.
15. Rivellese F, Surace AEA, Goldmann K, Sciacca E, Çubuk C, Giorli G, et al. Rituximab versus tocilizumab in rheumatoid arthritis: Synovial biopsy-based biomarker analysis of the phase 4 R4RA randomized trial. Nat Med. (2022) 28:1256–68.
16. Orange DE, Yao V, Sawicka K, Fak J, Frank MO, Parveen S, et al. RNA identification of PRIME cells predicting rheumatoid arthritis flares. N Engl J Med. (2020) 383:218–28. doi: 10.1056/NEJMoa2004114
17. Ghonim MA, Boyd DF, Flerlage T, Thomas PG. Pulmonary inflammation and fibroblast immunoregulation: From bench to bedside. J Clin Investig. (2023) 133:e170499. doi: 10.1172/JCI170499
18. Myasoedova E, Davis J, Matteson EL, Crowson CS. Is the epidemiology of rheumatoid arthritis changing? Results from a population-based incidence study, 1985–2014. Ann Rheum Dis. (2020) 79:440–4. doi: 10.1136/annrheumdis-2019-216694
19. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol. (2021) 22:10–8. doi: 10.1038/s41590-020-00816-x
20. Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. (2012) 44:1336–40. doi: 10.1038/ng.2462
21. Okada Y, Kim K, Han B, Pillai NE, Ong RTH, Saw WY, et al. Risk for ACPA-positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Hum Mol Genet. (2014) 23:6916–26. doi: 10.1093/hmg/ddu387
22. Viatte S, Plant D, Han B, Fu B, Yarwood A, Thomson W, et al. Association of HLA-DRB1 haplotypes with rheumatoid arthritis severity, mortality, and treatment response. JAMA. (2015) 313:1645. doi: 10.1001/jama.2015.3435
23. Ishigaki K, Lagattuta KA, Luo Y, James EA, Buckner JH, Raychaudhuri S. HLA autoimmune risk alleles restrict the hypervariable region of T cell receptors. Nat Genet. (2022) 54:393–402. doi: 10.1038/s41588-022-01032-z
24. Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: The conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. (2003) 171:538–41. doi: 10.4049/jimmunol.171.2.538
25. Malmström V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: From triggering to targeting. Nat Rev Immunol. (2017) 17:60–75. doi: 10.1038/nri.2016.124
26. Demoruelle MK, Bowers E, Lahey LJ, Sokolove J, Purmalek M, Seto NL, et al. Antibody responses to citrullinated and noncitrullinated antigens in the sputum of subjects with rheumatoid arthritis and subjects at risk for development of rheumatoid arthritis. Arthr Rheumatol. (2018) 70:516–27. doi: 10.1002/art.40401
27. Reynisdottir G, Olsen H, Joshua V, Engström M, Forsslund H, Karimi R, et al. Signs of immune activation and local inflammation are present in the bronchial tissue of patients with untreated early rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1722–7. doi: 10.1136/annrheumdis-2015-208216
28. Reynisdottir G, Karimi R, Joshua V, Olsen H, Hensvold AH, Harju A, et al. Structural changes and antibody enrichment in the lungs are early features of anti–citrullinated protein antibody–positive rheumatoid arthritis. Arthr Rheumatol. (2014) 66:31–9. doi: 10.1002/art.38201
29. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. (2016) 8:331ra38. doi: 10.1126/scitranslmed.aad7151
30. Li Y, Shen Y, Jin K, Wen Z, Cao W, Wu B, et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab. (2019) 30:477.e–92.e. doi: 10.1016/j.cmet.2019.06.016
31. Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, Goodman SB, et al. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity. (2016) 45:903–16. doi: 10.1016/j.immuni.2016.09.013
32. Lewis MJ, Barnes MR, Blighe K, Goldmann K, Rana S, Hackney JA, et al. Molecular portraits of early rheumatoid arthritis identify clinical and treatment response phenotypes. Cell Rep. (2019) 28:2455–70.e5. doi: 10.1016/j.celrep.2019.07.091
33. Ospelt C. Synovial fibroblasts in 2017. RMD Open. (2017) 3:1–10. doi: 10.1136/rmdopen-2017-000471
34. Buckley CD, Ospelt C, Gay S, Midwood KS. Location, location, location: How the tissue microenvironment affects inflammation in RA. Nat Rev Rheumatol. (2021) 17:195–212. doi: 10.1038/s41584-020-00570-2
35. Alivernini S, Firestein GS, McInnes IB. The pathogenesis of rheumatoid arthritis. Immunity. (2022) 55:2255–70. doi: 10.1016/j.immuni.2022.11.009
36. Wong ZY, Nee E, Coles M, Buckley CD. Why does understanding the biology of fibroblasts in immunity really matter? PLoS Biol. (2023) 21:e3001954. doi: 10.1371/journal.pbio.3001954
37. Bhattacharya M, Ramachandran P. Immunology of human fibrosis. Nat Immunol. (2023) 24:1423–33. doi: 10.1038/s41590-023-01551-9
38. Karonitsch T, Kandasamy RK, Kartnig F, Herdy B, Dalwigk K, Niederreiter B, et al. mTOR senses environmental cues to shape the fibroblast-like synoviocyte response to inflammation. Cell Rep. (2018) 23:2157–67. doi: 10.1016/j.celrep.2018.04.044
39. Umar S, Palasiewicz K, Volin MV, Romay B, Rahat R, Tetali C, et al. Metabolic regulation of RA macrophages is distinct from RA fibroblasts and blockade of glycolysis alleviates inflammatory phenotype in both cell types. Cell Mol Life Sci. (2021) 78:7693–707. doi: 10.1007/s00018-021-03978-5
40. Alivernini S, Tolusso B, Gessi M, Gigante MR, Mannocci A, Petricca L, et al. Inclusion of synovial tissue–derived characteristics in a nomogram for the prediction of treatment response in treatment-naive rheumatoid arthritis patients. Arthr Rheumatol. (2021) 73:1601–13. doi: 10.1002/art.41726
41. Lefèvre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. (2009) 15:1414–20. doi: 10.1038/nm.2050
42. Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. (2020) 582:259–64.
43. Vickovic S, Schapiro D, Carlberg K, Lötstedt B, Larsson L, Hildebrandt F, et al. Three-dimensional spatial transcriptomics uncovers cell type localizations in the human rheumatoid arthritis synovium. Commun Biol. (2022) 5:129. doi: 10.1038/s42003-022-03050-3
44. Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun. (2018) 9:789. doi: 10.1038/s41467-018-02892-y
45. Smith MH, Gao VR, Periyakoil PK, Kochen A, DiCarlo EF, Goodman SM, et al. Drivers of heterogeneity in synovial fibroblasts in rheumatoid arthritis. Nat Immunol. (2023) 24:1200–10. doi: 10.1038/s41590-023-01527-9
46. Micheroli R, Elhai M, Edalat S, Frank-Bertoncelj M, Bürki K, Ciurea A, et al. Role of synovial fibroblast subsets across synovial pathotypes in rheumatoid arthritis: A deconvolution analysis. RMD Open. (2022) 8:e001949. doi: 10.1136/rmdopen-2021-001949
47. Armaka M, Konstantopoulos D, Tzaferis C, Lavigne MD, Sakkou M, Liakos A, et al. Single-cell multimodal analysis identifies common regulatory programs in synovial fibroblasts of rheumatoid arthritis patients and modeled TNF-driven arthritis. Genome Med. (2022) 14:78. doi: 10.1186/s13073-022-01081-3
48. Carlberg K, Korotkova M, Larsson L, Catrina AI, Ståhl PL, Malmström V. Exploring inflammatory signatures in arthritic joint biopsies with Spatial Transcriptomics. Sci Rep. (2019) 9:18975. doi: 10.1038/s41598-019-55441-y
49. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
50. Wilsher M, Voight L, Milne D, Teh M, Good N, Kolbe J, et al. Prevalence of airway and parenchymal abnormalities in newly diagnosed rheumatoid arthritis. Respir Med. (2012) 106:1441–6. doi: 10.1016/j.rmed.2012.06.020
51. Habib HM, Eisa AA, Arafat WR, Marie MA. Pulmonary involvement in early rheumatoid arthritis patients. Clin Rheumatol. (2011) 30:217–21. doi: 10.1007/s10067-010-1492-5
52. Mori S, Cho I, Koga Y, Sugimoto M. Comparison of pulmonary abnormalities on high-resolution computed tomography in patients with early versus longstanding rheumatoid arthritis. J Rheumatol. (2008) 35:1513–21.
53. Klareskog L, Stolt P, Lundberg K, Källberg H, Bengtsson C, Grunewald J, et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA–DR (shared epitope)–restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. (2006) 54:38–46. doi: 10.1002/art.21575
54. Stolt P. Quantification of the influence of cigarette smoking on rheumatoid arthritis: Results from a population based case-control study, using incident cases. Ann Rheum Dis. (2003) 62:835–41. doi: 10.1136/ard.62.9.835
55. Karlson EW, Chang SC, Cui J, Chibnik LB, Fraser PA, De Vivo I, et al. Gene–environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis. (2010) 69:54–60. doi: 10.1136/ard.2008.102962
56. Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene–environment interaction between smoking and shared epitope genes in HLA–DR provides a high risk of seropositive rheumatoid arthritis. Arthr Rheum. (2004) 50:3085–92. doi: 10.1002/art.20553
57. Stolt P, Yahya A, Bengtsson C, Källberg H, Rönnelid J, Lundberg I, et al. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheum Dis. (2010) 69:1072–6. doi: 10.1136/ard.2009.114694
58. Too CL, Muhamad NA, Ilar A, Padyukov L, Alfredsson L, Klareskog L, et al. Occupational exposure to textile dust increases the risk of rheumatoid arthritis: Results from a Malaysian population-based case–control study. Ann Rheum Dis. (2016) 75:997–1002. doi: 10.1136/annrheumdis-2015-208278
59. Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJW, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis. (2008) 67:1488–92. doi: 10.1136/ard.2007.075192
60. Fischer A, Solomon JJ, du Bois RM, Deane KD, Olson AL, Fernandez-Perez ER, et al. Lung disease with anti-CCP antibodies but not rheumatoid arthritis or connective tissue disease. Respir Med. (2012) 106:1040–7. doi: 10.1016/j.rmed.2012.03.006
61. Demoruelle MK, Harrall KK, Ho L, Purmalek MM, Seto NL, Rothfuss HM, et al. Anti–citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arthr Rheumatol. (2017) 69:1165–75. doi: 10.1002/art.40066
62. Norton S, Koduri G, Nikiphorou E, Dixey J, Williams P, Young A. A study of baseline prevalence and cumulative incidence of comorbidity and extra-articular manifestations in RA and their impact on outcome. Rheumatology (Oxford). (2013) 52:99–110. doi: 10.1093/rheumatology/kes262
63. Nannini C, Ryu JH, Matteson EL. Lung disease in rheumatoid arthritis. Curr Opin Rheumatol. (2008) 20:340–6. doi: 10.1097/BOR.0b013e3282f798ed
64. Antin-Ozerkis D, Evans J, Rubinowitz A, Homer RJ, Matthay RA. Pulmonary manifestations of rheumatoid arthritis. Clin Chest Med. (2010) 31:451–78. doi: 10.1016/j.ccm.2010.04.003
65. Putman RK, Hatabu H, Araki T, Gudmundsson G, Gao W, Nishino M, et al. Association between interstitial lung abnormalities and all-cause mortality. JAMA. (2016) 315:672–81.
66. Hyldgaard C, Hilberg O, Pedersen AB, Ulrichsen SP, Løkke A, Bendstrup E, et al. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: Comorbidity and mortality. Ann Rheum Dis. (2017) 76:1700–6. doi: 10.1136/annrheumdis-2017-211138
67. Olson AL, Swigris JJ, Sprunger DB, Fischer A, Fernandez-Perez ER, Solomon J, et al. Rheumatoid arthritis–interstitial lung disease–associated mortality. Am J Respir Crit Care Med. (2011) 183:372–8. doi: 10.1164/rccm.201004-0622OC
68. Gizinski AM, Mascolo M, Loucks JL, Kervitsky A, Meehan RT, Brown KK, et al. Rheumatoid arthritis (RA)-specific autoantibodies in patients with interstitial lung disease and absence of clinically apparent articular RA. Clin Rheumatol. (2009) 28:611–3. doi: 10.1007/s10067-009-1128-9
69. Lee HK, Kim DS, Yoo B, Seo JB, Rho JY, Colby TV, et al. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest. (2005) 127:2019–27. doi: 10.1378/chest.127.6.2019
70. Wells AU, Denton CP. Interstitial lung disease in connective tissue disease–mechanisms and management. Nat Rev Rheumatol. (2014) 10:728–39. doi: 10.1038/nrrheum.2014.149
71. Tanaka N, Kim JS, Newell JD, Brown KK, Cool CD, Meehan R, et al. Rheumatoid arthritis-related lung diseases: CT findings. Radiology. (2004) 232:81–91. doi: 10.1148/radiol.2321030174
72. Luqmani R, Hennell S, Estrach C, Basher D, Birrell F, Bosworth A, et al. British society for rheumatology and british health professionals in rheumatology guideline for the management of rheumatoid arthritis (after the first 2 years). Rheumatology (Oxford). (2009) 48:436–9. doi: 10.1093/rheumatology/ken450a
73. Tsuchiya Y, Takayanagi N, Sugiura H, Miyahara Y, Tokunaga D, Kawabata Y, et al. Lung diseases directly associated with rheumatoid arthritis and their relationship to outcome. Eur Respir J. (2011) 37:1411–7. doi: 10.1183/09031936.00019210
74. Kim EJ, Elicker BM, Maldonado F, Webb WR, Ryu JH, Van Uden JH, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. (2010) 35:1322–8. doi: 10.1183/09031936.00092309
75. Gochuico BR, Avila NA, Chow CK, Novero LJ, Wu HP, Ren P, et al. Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch Intern Med. (2008) 168:159–66. doi: 10.1001/archinternmed.2007.59
76. Zamora-Legoff JA, Krause ML, Crowson CS, Ryu JH, Matteson EL. Patterns of interstitial lung disease and mortality in rheumatoid arthritis. Rheumatology (Oxford). (2017) 56:344–50. doi: 10.1093/rheumatology/kex299
77. Solomon JJ, Chung JH, Cosgrove GP, Demoruelle MK, Fernandez-Perez ER, Fischer A, et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. (2016) 47:588–96. doi: 10.1183/13993003.00357-2015
78. Kelly C, Emery P, Dieudé P. Current issues in rheumatoid arthritis-associated interstitial lung disease. Lancet Rheumatol. (2021) 3:e798–807. doi: 10.1016/S2665-9913(21)00250-2
79. Azam AT, Odeyinka O, Alhashimi R, Thoota S, Ashok T, Palyam V, et al. Rheumatoid arthritis and associated lung diseases: A comprehensive review. Cureus. (2022) 14:e22367. doi: 10.7759/cureus.22367
80. Kim Y, Yang HI, Kim KS. Etiology and pathogenesis of rheumatoid arthritis-interstitial lung disease. Int J Mol Sci. (2023) 24:14509. doi: 10.3390/ijms241914509
81. Matson S, Lee J, Eickelberg O. Two sides of the same coin? A review of the similarities and differences between idiopathic pulmonary fibrosis and rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. (2021) 57:2002533. doi: 10.1183/13993003.02533-2020
82. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. (2020) 6:eaba1983. doi: 10.1126/sciadv.aba1983
83. Moss BJ, Ryter SW, Rosas IO. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev Pathol. (2022) 17:515–46. doi: 10.1146/annurev-pathol-042320-030240
84. Song JW, Do KH, Kim MY, Jang SJ, Colby TV, Kim DS. Pathologic and radiologic differences between idiopathic and collagen vascular disease-related usual interstitial pneumonia. Chest. (2009) 136:23–30. doi: 10.1378/chest.08-2572
85. Turesson C, Matteson EL, Colby TV, Vuk-Pavlovic Z, Vassallo R, Weyand CM, et al. Increased CD4+ T cell infiltrates in rheumatoid arthritis-associated interstitial pneumonitis compared with idiopathic interstitial pneumonitis. Arthr Rheum. (2005) 52:73–9. doi: 10.1002/art.20765
86. Lee H, Lee SI, Kim HO. Recent advances in basic and clinical aspects of rheumatoid arthritis-associated interstitial lung diseases. J Rheumatic Dis. (2022) 29:61–70. doi: 10.4078/jrd.2022.29.2.61
87. Wang D, Zhang J, Lau J, Wang S, Taneja V, Matteson EL, et al. Mechanisms of lung disease development in rheumatoid arthritis. Nat Rev Rheumatol. (2019) 15:581–96. doi: 10.1038/s41584-019-0275-x
88. Ba X, Wang H, Huang Y, Yan J, Han L, Lin W, et al. Simiao pill attenuates collagen-induced arthritis and bleomycin-induced pulmonary fibrosis in mice by suppressing the JAK2/STAT3 and TGF-β/Smad2/3 signalling pathway. J Ethnopharmacol. (2023) 309:116274. doi: 10.1016/j.jep.2023.116274
89. Wu C, Lin H, Zhang X. Inhibitory effects of pirfenidone on fibroblast to myofibroblast transition in rheumatoid arthritis-associated interstitial lung disease via the downregulation of activating transcription factor 3 (ATF3). Int Immunopharmacol. (2019) 74:105700. doi: 10.1016/j.intimp.2019.105700
90. Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. (2006) 116:3183–94. doi: 10.1172/JCI28756
91. Zhang J, Wang D, Wang L, Wang S, Roden AC, Zhao H, et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L487–97. doi: 10.1152/ajplung.00301.2018
92. Just SA, Nielsen C, Werlinrud JC, Larsen PV, Hejbøl EK, Tenstad HB, et al. Fibrocytes in early and long-standing rheumatoid arthritis: A 6-month trial with repeated synovial biopsy, imaging and lung function test. RMD Open. (2021) 7:e001494. doi: 10.1136/rmdopen-2020-001494
93. Heukels P, van Hulst JAC, van Nimwegen M, Boorsma CE, Melgert BN, van den Toorn LM, et al. Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir Res. (2018) 19:90. doi: 10.1186/s12931-018-0798-8
94. Fujiwara A, Kobayashi H, Masuya M, Maruyama M, Nakamura S, Ibata H, et al. Correlation between circulating fibrocytes, and activity and progression of interstitial lung diseases. Respirology. (2012) 17:693–8. doi: 10.1111/j.1440-1843.2012.02167.x
95. Yang M, Wu Y, Liu X, Zhao C, Li T, Li T, et al. Efficacy and safety of antifibrotic agents in the treatment of CTD-ILD and RA-ILD: A systematic review and meta-analysis. Respir Med. (2023) 216:107329. doi: 10.1016/j.rmed.2023.107329
96. Juge PA, Hayashi K, McDermott GC, Vanni KMM, Kowalski E, Qian G, et al. Effectiveness and tolerability of antifibrotics in rheumatoid arthritis-associated interstitial lung disease. Semin Arthritis Rheum. (2024) 64:152312. doi: 10.1016/j.semarthrit.2023.152312
97. Jang JH, Ko J, Jung SY, Kim DW, Oh JH, Kim TJ, et al. Antifibrotic agents in rheumatoid arthritis-associated interstitial lung disease: A systematic review and meta-analysis. Life. (2023) 13:2318. doi: 10.3390/life13122318
98. Venetsanopoulou AI, Ntinopoulou M, Papagianni E, Koletsos N, Voulgari PV, Chrysanthopoulou A. Neutrophil extracellular traps as immunofibrotic mediators in RA-ILD; pilot evaluation of the nintedanib therapy. Front Immunol. (2024) 15:1480594. doi: 10.3389/fimmu.2024.1480594
99. Behr J, Prasse A, Kreuter M, Johow J, Rabe KF, Bonella F, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med. (2021) 9:476–86.
100. Solomon JJ, Danoff SK, Woodhead FA, Hurwitz S, Maurer R, Glaspole I, et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: A randomised, double-blind, placebo-controlled, phase 2 study. Lancet Respir Med. (2023) 11:87–96. doi: 10.1016/j.chest.2021.12.293
101. Wang J, Wang X, Qi X, Sun Z, Zhang T, Cui Y, et al. The efficacy and safety of pirfenidone combined with immunosuppressant therapy in connective tissue disease-associated interstitial lung disease: A 24-week prospective controlled cohort study. Front Med (Lausanne). (2022) 9:871861. doi: 10.3389/fmed.2022.871861
102. Matteson EL, Aringer M, Burmester GR, Mueller H, Moros L, Kolb M. Effect of nintedanib in patients with progressive pulmonary fibrosis associated with rheumatoid arthritis: Data from the INBUILD trial. Clin Rheumatol. (2023) 42:2311–9. doi: 10.1007/s10067-023-06623-7
103. Johnson SR, Bernstein EJ, Bolster MB, Chung JH, Danoff SK, George MD, et al. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) guideline for the treatment of interstitial lung disease in people with systemic autoimmune rheumatic diseases. Arthr Rheumatol. (2024) 76:1182–200.
104. El Agha E, Thannickal VJ. The lung mesenchyme in development, regeneration, and fibrosis. J Clin Investig. (2023) 133:e170498. doi: 10.1172/JCI170498
105. Rehan VK, Sakurai R, Wang Y, Santos J, Huynh K, Torday JS. Reversal of nicotine-induced alveolar lipofibroblast-to-myofibroblast transdifferentiation by stimulants of parathyroid hormone-related protein signaling. Lung. (2007) 185:151–9. doi: 10.1007/s00408-007-9007-0
106. Rehan VK, Torday JS. Hyperoxia augments pulmonary lipofibroblast-to-myofibroblast transdifferentiation. Cell Biochem Biophys. (2003) 38:239–50. doi: 10.1385/CBB:38:3:239
107. El Agha E, Moiseenko A, Kheirollahi V, De Langhe S, Crnkovic S, Kwapiszewska G, et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell. (2017) 20:261–73.e3. doi: 10.1016/j.stem.2016.10.004
108. Li Y, Jiang D, Liang J, Meltzer EB, Gray A, Miura R, et al. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med. (2011) 208:1459–71. doi: 10.1084/jem.20102510
109. Liu X, Geng Y, Liang J, Coelho AL, Yao C, Deng N, et al. HER2 drives lung fibrosis by activating a metastatic cancer signature in invasive lung fibroblasts. J Exp Med. (2022) 219:e20220126. doi: 10.1084/jem.20220126
110. Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis. (2019) 78:1379–87. doi: 10.1136/annrheumdis-2018-214865
111. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
112. Meng XM, Wang S, Huang XR, Yang C, Xiao J, Zhang Y, et al. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. (2016) 7:e2495–2495. doi: 10.1038/cddis.2016.402
113. Venugopal H, Hanna A, Humeres C, Frangogiannis NG. Properties and functions of fibroblasts and myofibroblasts in myocardial infarction. Cells. (2022) 11:1386. doi: 10.3390/cells11091386
114. D’Addario M, Arora PD, Ellen RP, McCulloch CAG. Interaction of p38 and Sp1 in a mechanical force-induced, β1 integrin-mediated transcriptional circuit that regulates the actin-binding protein filamin-A. J Biol Chem. (2002) 277:47541–50. doi: 10.1074/jbc.M207681200
115. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. (2001) 410:37–40. doi: 10.1038/35065000
116. Hinz B, Dugina V, Ballestrem C, Wehrle-Haller B, Chaponnier C. α-Smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol Biol Cell. (2003) 14:2508–19. doi: 10.1091/mbc.e02-11-0729
117. Wang J, Fan J, Laschinger C, Arora PD, Kapus A, Seth A, et al. Smooth muscle actin determines mechanical force-induced p38 activation. J Biol Chem. (2005) 280:7273–84. doi: 10.1074/jbc.M410819200
118. Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res. (2009) 19:128–39. doi: 10.1038/cr.2008.328
119. Sanders YY, Cui Z, Le Saux CJ, Horowitz JC, Rangarajan S, Kurundkar A, et al. SMAD-independent down-regulation of caveolin-1 by TGF-β: Effects on proliferation and survival of myofibroblasts. PLoS One. (2015) 10:e0116995. doi: 10.1371/journal.pone.0116995
120. Wu EK, Ambrosini RD, Kottmann RM, Ritchlin CT, Schwarz EM, Rahimi H. Reinterpreting evidence of rheumatoid arthritis-associated interstitial lung disease to understand etiology. Curr Rheumatol Rev. (2019) 15:277–89. doi: 10.2174/1573397115666190116102451
121. Xiong L, Xiong L, Ye H, Ma WL. Animal models of rheumatoid arthritis-associated interstitial lung disease. Immun Inflamm Dis. (2021) 9:37–47. doi: 10.1002/iid3.377
122. Keith RC, Powers JL, Redente EF, Sergew A, Martin RJ, Gizinski A, et al. A novel model of rheumatoid arthritis-associated interstitial lung disease in SKG mice. Exp Lung Res. (2012) 38:55–66. doi: 10.3109/01902148.2011.636139
123. Song LN, Kong XD, Wang HJ, Zhan LB. Establishment of a rat adjuvant arthritis-interstitial lung disease model. Biomed Res Int. (2016) 2016:2970783. doi: 10.1155/2016/2970783
124. Shilling RA, Williams JW, Perera J, Berry E, Wu Q, Cummings OW, et al. Autoreactive T and B cells induce the development of bronchus-associated lymphoid tissue in the lung. Am J Respir Cell Mol Biol. (2013) 48:406–14. doi: 10.1165/rcmb.2012-0065OC
125. Sato T, Satooka H, Ichioka S, Maruo Y, Hirata T. Citrullinated fibrinogen is a target of auto-antibodies in interstitial lung disease in mice with collagen-induced arthritis. Int Immunol. (2020) 32:533–45. doi: 10.1093/intimm/dxaa021
126. Poole JA, Thiele GM, Janike K, Nelson AJ, Duryee MJ, Rentfro K, et al. Combined collagen-induced arthritis and organic dust-induced airway inflammation to model inflammatory lung disease in rheumatoid arthritis. J Bone Miner Res. (2019) 34:1733–43. doi: 10.1002/jbmr.3745
127. Gao B, Lin J, Jiang Z, Yang Z, Yu H, Ding L, et al. Upregulation of chemokine CXCL10 enhances chronic pulmonary inflammation in tree shrew collagen-induced arthritis. Sci Rep. (2018) 8:9993. doi: 10.1038/s41598-018-28404-y
128. Armaka M, Ospelt C, Pasparakis M, Kollias G. The p55TNFR-IKK2-Ripk3 axis orchestrates arthritis by regulating death and inflammatory pathways in synovial fibroblasts. Nat Commun. (2018) 9:618. doi: 10.1038/s41467-018-02935-4
129. Zhao S, Grieshaber-Bouyer R, Rao DA, Kolb P, Chen H, Andreeva I, et al. Effect of JAK inhibition on the induction of proinflammatory HLA-DR+CD90+ rheumatoid arthritis synovial fibroblasts by interferon-γ. Arthr Rheumatol. (2022) 74:441–52. doi: 10.1002/art.41958
130. Gu S, Liang J, Zhang J, Liu Z, Miao Y, Wei Y, et al. Baricitinib attenuates bleomycin-induced pulmonary fibrosis in mice by inhibiting TGF-β1 signaling pathway. Molecules (2023) 28:2195. doi: 10.3390/molecules28052195
Keywords: rheumatoid arthritis, fibroblast, interstitial lung disease (ILD), synovium, lung, ACPA
Citation: Neofotistou-Themeli E, Goutakoli P, Chanis T, Semitekolou M, Sevdali E and Sidiropoulos P (2025) Fibroblasts in rheumatoid arthritis: novel roles in joint inflammation and beyond. Front. Med. 11:1376925. doi: 10.3389/fmed.2024.1376925
Received: 26 January 2024; Accepted: 31 December 2024;
Published: 21 January 2025.
Edited by:
Cong-Qiu Chu, Oregon Health and Science University, United StatesReviewed by:
Apostolos Perelas, VCU Medical Center, United StatesEddie A. James, Benaroya Research Institute, United States
Maria Otaola, University of Buenos Aires, Argentina
Copyright © 2025 Neofotistou-Themeli, Goutakoli, Chanis, Semitekolou, Sevdali and Sidiropoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prodromos Sidiropoulos, c2lkaXJvcHBAdW9jLmdy