 Ying Liu
Ying Liu Ping Zhu
Ping Zhu Jiajun Tian1,2,3
Jiajun Tian1,2,3- 1Department of Gastroenterology and Hepatology, Tianjin Third Central Hospital, Tianjin, China
- 2Tianjin Key Laboratory of Extracorporeal Life Support for Critical Diseases, Tianjin, China
- 3Artificial Cell Engineering Technology Research Center, Tianjin Institute of Hepatobiliary Disease, Tianjin, China
Congenital hepatic fibrosis (CHF) is considered to be a rare autosomal recessive hereditary fibrocystic liver disease, mainly found in children. However, cases of adult CHF with autosomal dominant polycystic kidney disease (ADPKD) caused by PKD1 gene mutation are extremely rare. We report a 31-year-old female patient admitted for esophageal and gastric variceal bleeding. Physical examination revealed significant splenomegaly, biochemical tests showed a slight increase in liver enzymes, and a decrease in platelet count. Imaging examinations showed significant dilatation of the common bile duct and intrahepatic bile ducts, as well as multiple renal cysts. Liver biopsy revealed enlarged portal areas, bridging fibrosis, and numerous variably shaped small bile ducts. Genetic testing identified two unique mutations in the PKD1 gene, identified as biallelic mutations compound heterozygous mutations composed of a mutation inherited from the father (c.8296 T > C) and one from the mother (c.9653G > C). Based on multiple test results, the patient was diagnosed with the portal hypertension type CHF associated with ADPKD. During her initial hospital stay, the patient underwent endoscopic treatment for gastrointestinal bleeding. To date, the patient has recovered well. Moreover, a significant reduction in varices was observed in a gastroscopy examination 18 months later.
Introduction
In 1961, Kerr (1) first introduced the term “Congenital hepatic fibrosis (CHF)” to describe a unique liver fibrosis condition distinct from cirrhosis. CHF is a rare inherited disorder that typically presents in childhood or adolescence. It is characterized by hepatosplenomegaly and portal hypertension. Interestingly, liver function may remain unaltered or exhibit only minor abnormalities (2). CHF can present as isolated hepatic involvement, but it often co-occurs with Caroli’s syndrome or polycystic kidney disease (PKD) (3). Autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease (ADPKD) are the most prevalent forms of PKD. ARPKD is inherited in an autosomal recessive pattern, usually due to mutations in the PKHD1 gene, while ADPKD is inherited in an autosomal dominant pattern, often caused by mutations in either the PKD1 or PKD2 genes. CHF can be observed in approximately 50% of ARPKD cases (4), whereas it is less common in ADPKD cases. The most prevalent hepatic manifestation of ADPKD is the formation of liver cysts. With advancements in molecular biology techniques, several cases of childhood CHF accompanied by ADPKD, caused by PKD1 gene mutations, have been reported (5–7). However, in the past two decades, there have been relatively fewer reports on adult CHF combined with ADPKD.
Here, we present a rare case of CHF with ADPKD in an adult female, caused by PKD1 variants. This diagnosis was confirmed through medical imaging, liver biopsy, and genetic testing. In addition to presenting the case, we analyze the computed tomography (CT) and magnetic resonance cholangiopancreatography (MRCP) imaging results, discussed the histopathological features, diagnosis, and treatment of CHF, and reviewed relevant literature.
Case presentation
A 31-year-old adult female patient was admitted to our hospital, with a one-day history of massive hematemesis and melena. The patient denied past medical history. The patient’s 61-year-old mother is asymptomatic, while the patient’s father presents with hepatomegaly, splenomegaly, and bilateral multiple renal cysts. Her brother underwent a splenectomy five years ago due to bleeding from esophagogastric varices caused by portal hypertension and also has a history of bilateral multiple renal cysts. Physical examination showed that the patient has pale conjunctiva and an enlarged spleen, with no other abnormalities. Laboratory examination revealed a hemoglobin level of 55 g/L (reference range: 115–150 g/L), and a platelet count of 115 × 109/L (reference range: 125–350 × 109/L). The liver biochemical profile showed elevated alkaline phosphatase of 309 U/L (reference range: 35–100 U/L) and γ glutamyl transpeptidase of 218 U/L (reference range: 7-45 U/L). Levels of white blood cell count, alanine aminotransferase, aspartate aminotransferase, bilirubin, creatinine and prothrombin time were within the normal range. Tests for hepatitis B surface antigen, hepatitis C virus, cytomegalo virus, Epstein–Barr virus antibodies, examination of serum ceruloplasmin, serum iron, transferrin saturation, smooth muscle actin antibodies, liver-kidney microsomal antibodies, anti-nuclear antibodies and immune-globulin (Ig) G and IgM were performed and yielded negative results.
The abdominal ultrasound showed signs of cirrhosis, splenomegaly, and portal vein dilation. An enhanced CT scan showed liver cirrhosis, splenomegaly, polycystic kidneys, a small volume of ascites, and portal hypertension with open collateral circulation (Figures 1A–C). Additionally, MRCP revealed dilation of the common and intrahepatic bile ducts, as well as wall thickening and edema of the gallbladder (Figure 1D). A liver biopsy (Figure 2) showed visible nodules without obvious intralobular inflammation, enlarged portal areas with bridging fibrosis, and a significantly increased number of small biliary ducts of various shapes. Histopathology confirmed CHF.
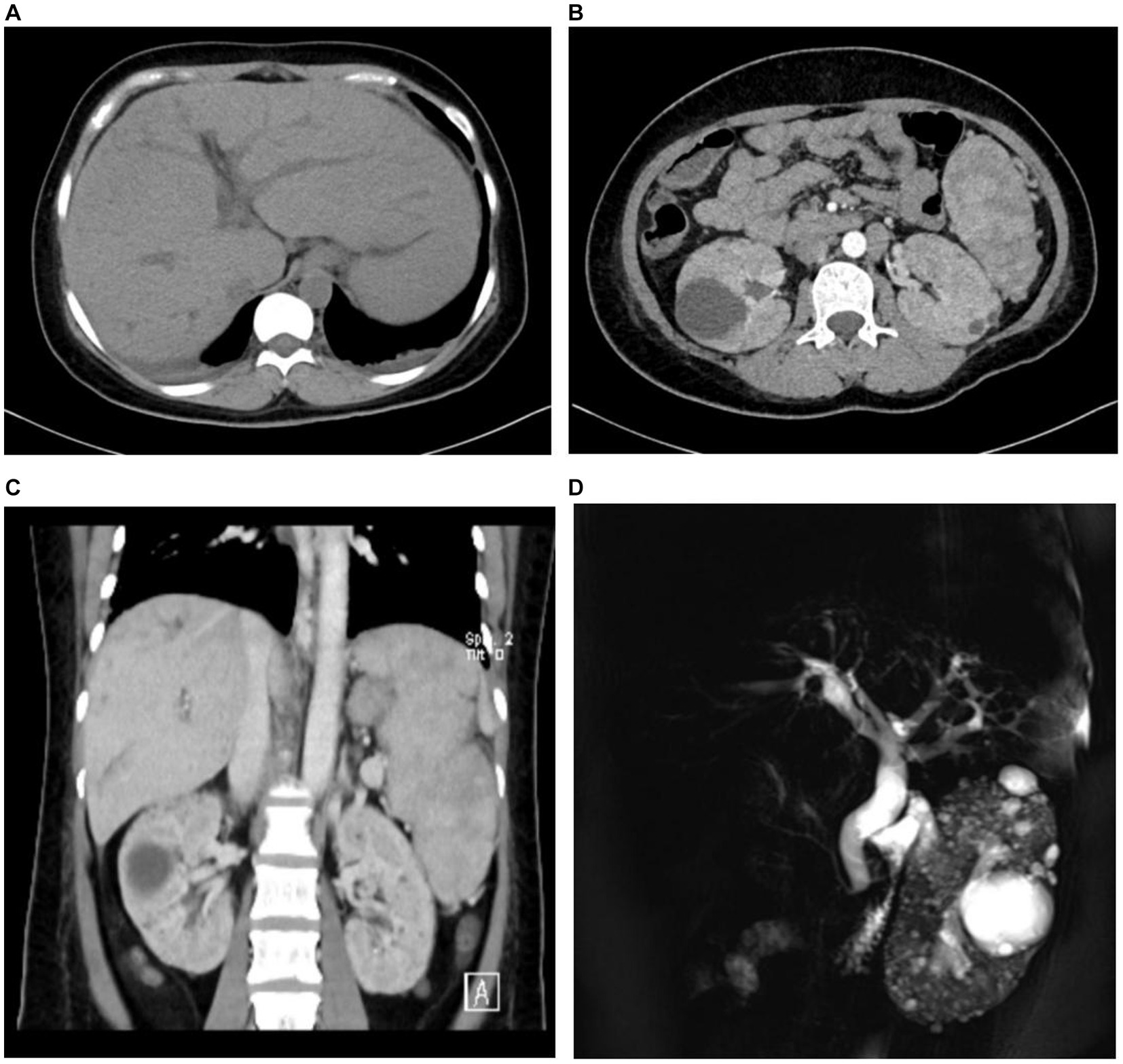
Figure 1. The medical imaging results of the patient. Abdominal contrast-enhanced CT revealed intrahepatic bile ducts dilation (A), multiple renal cysts (B), and splenomegaly (C). Magnetic resonance cholangiopancreatography (MRCP) scan showed the dilation of both extrahepatic and intrahepatic bile ducts, as well as multiple renal cysts (D).
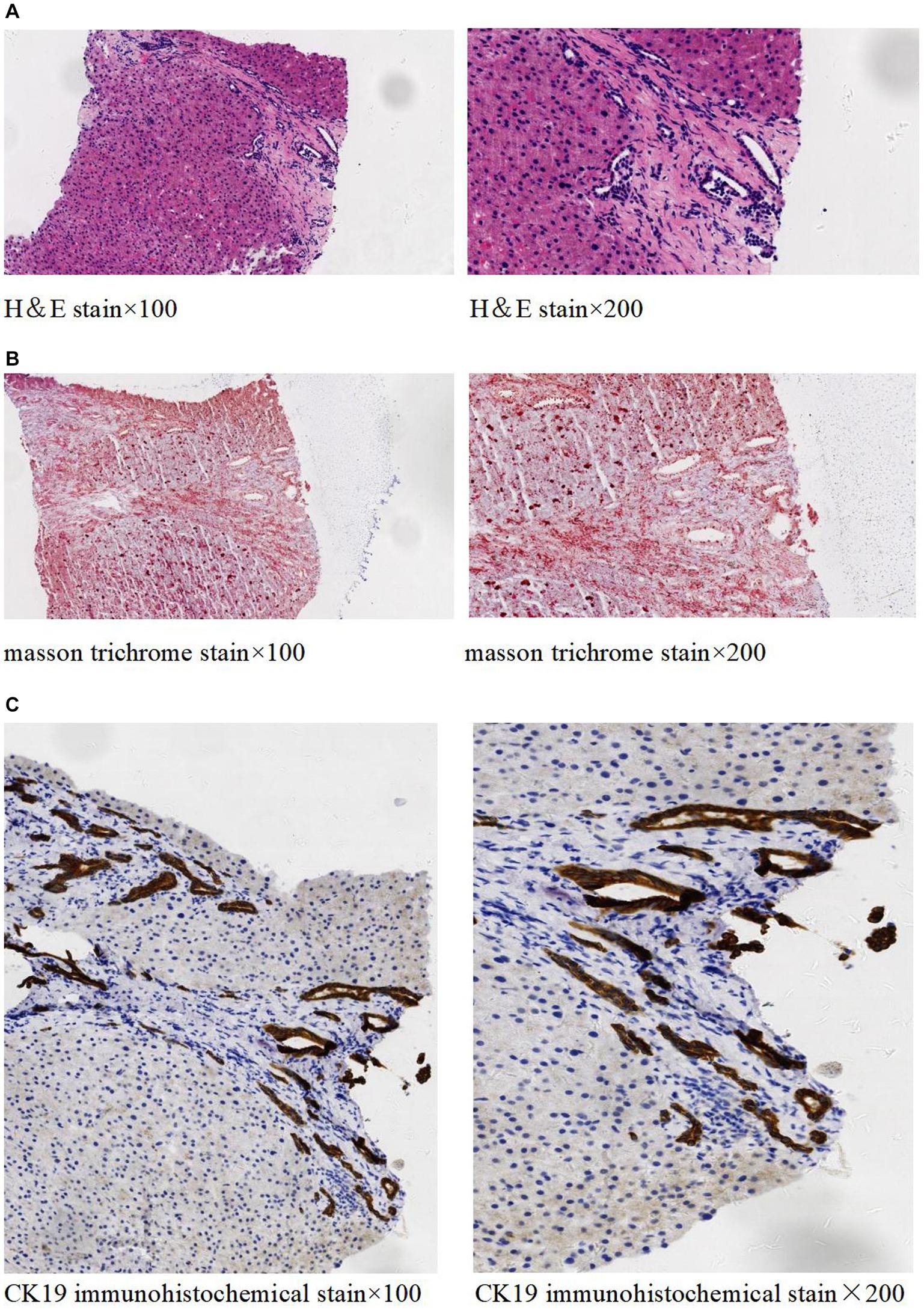
Figure 2. Histopathology of liver tissue. H&E staining (A) showed no significant lobular inflammation, nor signs of cholangitis, but there was portal tract enlargement with bridging fibrosis. Masson trichrome staining (B) demonstrated the enlargement of the portal area along with collagen fiber proliferation. Immunohistochemical staining of CK19 (C) revealed significant small bile duct proliferation in the portal areas.
Genetic analysis revealed two distinct mutations in the PKD1 gene (Figure 3), determined to be biallelic mutations consisting of a paternally inherited mutation (c.8296 T > C) and a maternally inherited mutation (c.9653G > C). The former mutation results in an amino acid change from serine to proline at position 2,766, whereas the latter causes an amino acid change from tryptophan to serine at position 3,218.
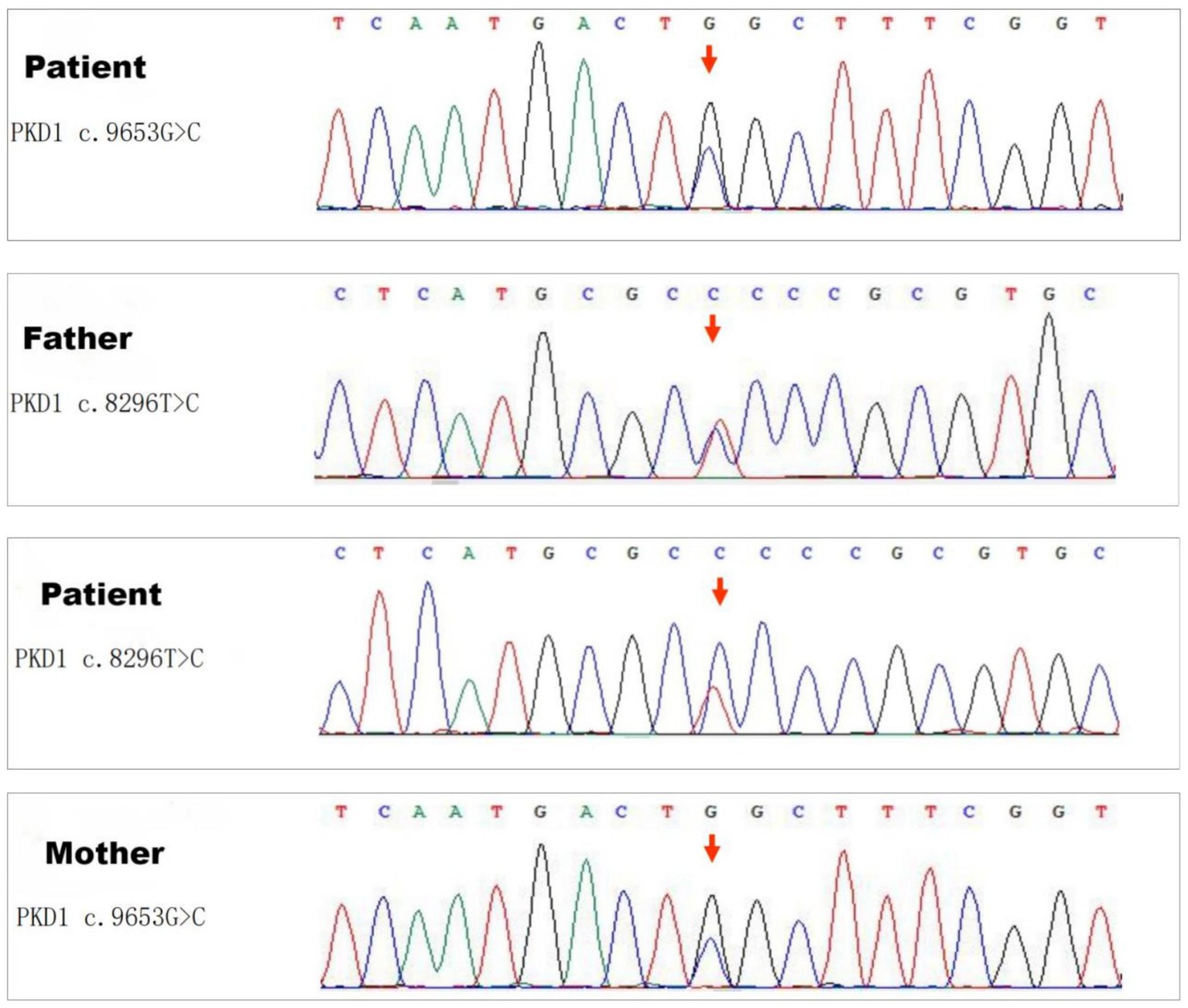
Figure 3. Sequencing results of the PKD1 gene. Genetic analysis revealed two distinct missense mutations in the PKD1 gene: one is a paternally inherited mutation c.8296 T > C (Ser2766Pro), and the other is a maternally inherited variant c.9653G > C (Trp3218Ser).
Gastroscopy revealed severe esophageal and gastric variceal bleeding. Due to her economic status, the patient refused a liver transplant and instead received endoscopic variceal ligation and injection sclerotherapy. After treatment, the patient was discharged in improved condition and survived through the 18-month follow-up period. No variceal re-bleeding was observed during this time. A re-examination gastroscopy after 18 months showed a significant reduction in esophageal and gastric varices.
Discussion and conclusion
Congenital hepatic fibrosis (CHF) is a rare hereditary fibrocystic liver disease with an incidence of about 1/10000 ~ 1/20000 (8). In previous literature reports, CHF has demonstrated a clear familial genetic tendency, and consanguineous marriage can lead to an increase in the prevalence of CHF (9). The pathogenesis of CHF is not fully understood, but the most widely accepted theory attributes it to ductal plate malformation (DPM), which leads to developmental abnormalities in the bile duct system. Proteins encoded by PKD genes (polycystin 1, polycystin 2, and fibrocystin) are known to localize to primary cilia, and the normal development of the portal vein system (10) and renal tubules (11) requires the intact ciliary signaling of PKD proteins. Mutations in PKD genes can cause ciliary dysfunction in biliary epithelial cells, leading to the onset of DPM (12). DPM is the underlying morphological abnormality in all fibrocystic diseases, resulting in an excessive number of embryonic bile ducts and abnormal portal vein branching, characterized by the presence of numerous embryonic-like bile duct structures in the porta hepatis (13). This immature bile duct system triggers inflammation and necrosis, promoting collagen deposition and the formation of fibrous tissue around the portal vein, resulting in the occurrence of CHF and related clinical symptoms such as recurrent cholangitis and complications associated with portal hypertension. The liver function of patients is usually normal or only mildly abnormal due to the absence of significant inflammatory response in hepatocytes.
Due to the nonspecific clinical manifestations of CHF, it is prone to being misdiagnosis or overlooked. The diagnosis of this disease primarily relies on imaging, pathological, and genetic examinations, with liver histopathology being the gold standard. Typical imaging features (14) include the absence of stenosis or occlusion of the portal vein at the porta hepatis, along with decreased, narrowed, or compressed intrahepatic portal vein branches. CT, Magnetic Resonance Imaging (MRI), and other examinations can reveal multiple dilated intrahepatic bile ducts and hepatosplenomegaly. The characteristic pathological findings include enlarged portal areas with bridging fibrosis, proliferating small bile ducts with diverse morphology within fibrous septa, and underdeveloped portal vein branches. In this study, we reported a case of a 31-year-old female patient who was admitted to our hospital with a chief complaint of hematemesis and melena for one day. She had no prior history of gastrointestinal bleeding. The findings of the patient’s examination are consistent with the characteristics of CHF. CHF is usually classified into four types, each with its distinct clinical features: the portal hypertension type, the cholangitis type, the mixed type, and the latent type (15). Given the patient’s clinical symptoms, imaging findings, and the results of the liver histopathological examination-which included portal hypertension and dilated extrahepatic and intrahepatic bile ducts-a diagnosis of portal hypertension type CHF was established.
In the medical literature, it’s well-established that patients with CHF often have concomitant kidney diseases. ARPKD is considered the most common association. Less frequently, there can also be concurrent cases of ADPKD, renal cystic dysplasia, or medullary cystic kidney disease (16). CHF is also linked to a variety of ciliopathies, such as Joubert syndrome (17), Senior-Loken syndrome (18), COACH syndrome (19), Meckel syndrome (20), and Bardet-Biedl syndrome (21). They typically manifest in infancy or early childhood with a spectrum of symptoms including ataxia, developmental delays, and intellectual disabilities, as well as various retinal defects and polydactyly, in addition to CHF. However, in the adult case at hand, the patient did not present with these clinical symptoms but solely with CHF and multiple bilateral renal cysts. Radiological examinations aligned the renal findings with polycystic kidney disease. We advanced our investigation using high-throughput sequencing technology for whole-exome sequencing, which identified two novel missense mutations in the PKD1 gene. It is known that pathogenic variants in PKD1 are responsible for about 85% of ADPKD cases (22). The mutations in PKD1 are diverse, including truncating, missense, splice site, deletions, and nonsense mutations. Although missense mutations sometimes do not alter protein function and may even confer beneficial effects, they most commonly lead to detrimental or lethal consequences.
These specific missense mutations have not been previously recorded in any public genetic variant databases (GnomAD, the 1,000 Genomes Project, or ExAC) or in specialized variant reference databases (ClinVar or Mastermind). However, at codon Trp3218, two missense alterations (Trp3218Arg and Trp3218Gly) and one nonsense mutation (Trp3218Ter) have been reported multiple times (23, 24). To our knowledge, missense mutations at codon Ser2766 have not been reported. Predictive in silico tools such as PolyPhen-2, M-CAP, MutationTaster, and SIFT indicate that these variants could be deleterious. The patient’s mother exhibited no clinical signs of kidney or liver disease, raising the possibility of incomplete penetrance, which could explain the transmission of the mutation without the manifestation of the disease. In contrast, the patient’s father showed clinical signs of hepatomegaly, splenomegaly, and multiple bilateral renal cysts, suggesting that the variant in Exon 23 c.8296 T > c is likely pathogenic. Regrettably, the patient’s brother declined genetic testing for personal reasons. Nevertheless, the familial pattern of the disease suggests an autosomal dominant mode of inheritance. After a thorough diagnostic evaluation, the patient was diagnosed with CHF alongside ADPKD.
Typically, CHF and portal hypertension are not commonly seen together with ADPKD (16). Instead, polycystic liver disease (PLD) represents the most frequent extrarenal manifestation of ADPKD (25, 26), typically not impairing liver function. By the age of 60, approximately 77% of ADPKD patients will have developed liver cysts. PLD is characterized by numerous fluid-filled cysts derived from the bile ducts. When a significant increase in liver volume occurs, it can lead to subjective discomfort due to mass effect (27). Misra A and colleagues described an elderly ADPKD patient with extensive liver and kidney cysts, where a liver biopsy showed that numerous liver cysts were compressing the portal and smaller veins, resulting in portal hypertension (28). In our unique case, the extrarenal manifestation of ADPKD manifested as CHF with accompanying portal hypertension, a relatively rare presentation.
In Table 1, we report the hepatic and renal characteristics of CHF patients with ADPKD as described in nine publications (5–7, 29–34) from 1985 to 2022. Table 1 includes 14 patients from 13 families, comprising 5 males and 9 females. The age of disease onset ranged from 2 to 33 years. Except for one patient (34), all had a definitive family history of ADPKD. Hepatic presentations were categorized as follows: three cases of latent type CHF without portal hypertension or cholangitis, nine cases of CHF with portal hypertension, and two cases of mixed type CHF featuring both portal hypertension and cholangitis. Regarding renal manifestations, one case of ADPKD progressed to end-stage renal disease (31), one patient experienced a decline in renal function during pregnancy necessitating peritoneal dialysis (33), while the remaining 12 patients maintained essentially normal renal function. Given the date range of the articles, including the 1980s and 1990s, genetic testing might not have been widely accessible. Therefore, during that time, six patients did not undergo genetic testing. Additionally, one 8-year-old child opted out of genetic testing for reasons not disclosed. In total, seven patients did not receive genetic testing for ADPKD. The other seven were genetically tested. The findings showed that five patients with CHF and ADPKD had mutations exclusively in the PKD1 gene (6, 7), including two missense mutations, two deletions, and one mutation of unspecified nature within the PKD1 gene. One individual (34) possessed a mutation in the ATP7B gene along with two mutations in the PKD1 gene. Lastly, one case (30) had genetic testing that, likely due to technological limitations of the time, only identified mutations associated with ADPKD onset without specifying whether they were in PKD1, PKD2, or another gene.
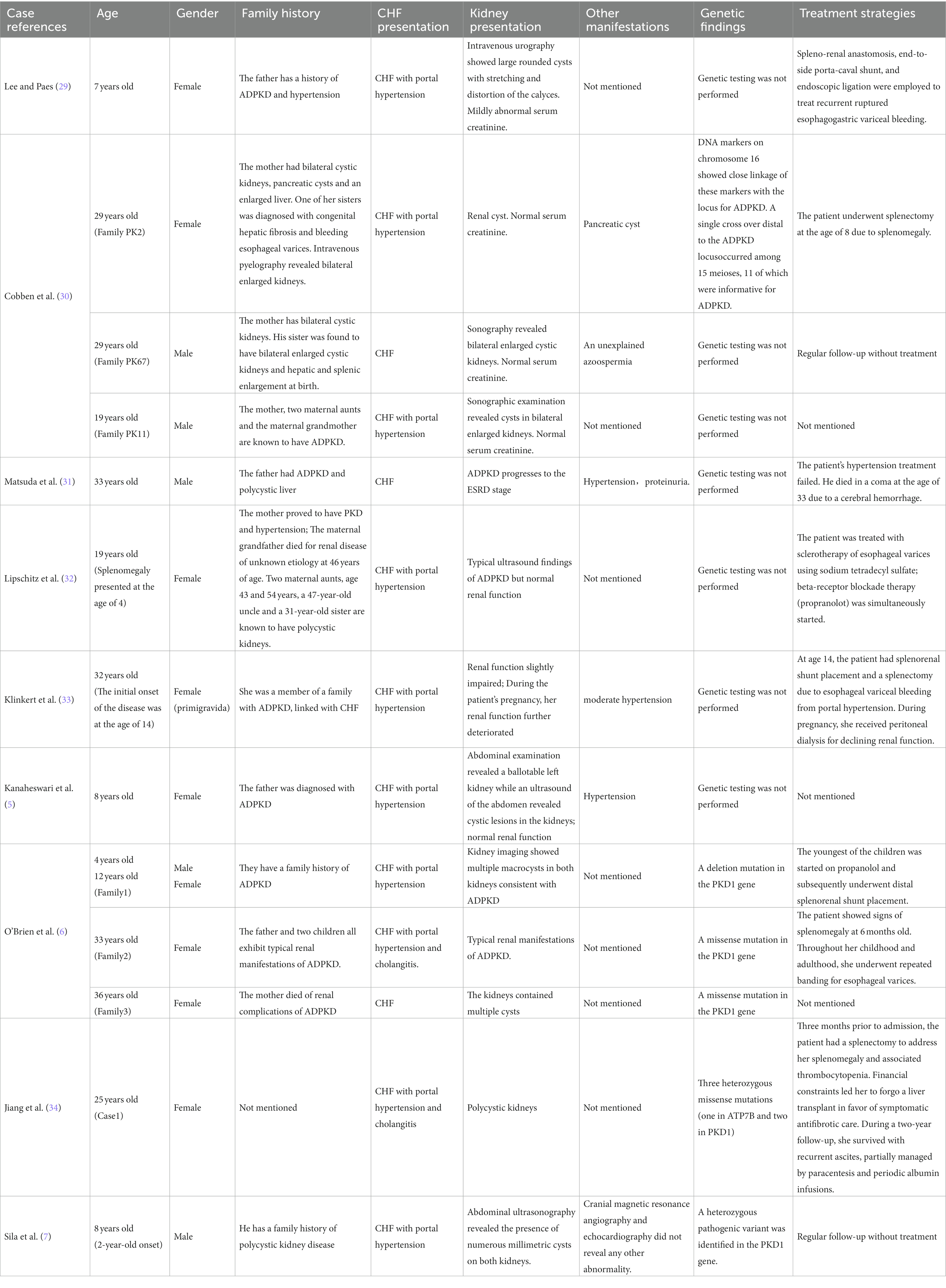
Table 1. Summary of characteristics of congenital liver fibrosis with ADPKD cases.
For patients with CHF, there is currently no specific therapeutic method that can halt or reverse the disease’s progression. Treatment options include supportive care and management of complications related to CHF, such as antifibrotic drug therapy, anti-infection therapy for cholangitis, and interventions to manage portal hypertension (2). The patient in this case was admitted to the hospital due to severe hematemesis and melena. Gastroscopy revealed severe esophageal/gastric variceal hemorrhage as a result of portal hypertension. The patient did not exhibit symptoms of fever or abdominal pain associated with cholangitis. Consequently, symptomatic treatment for this patient could include endoscopic interventions, percutaneous transhepatic portosystemic shunt (TIPS), or surgical procedures to control portal hypertension and arrest the bleeding from the esophageal/gastric varices.
Endoscopic variceal ligation (EVL), often used with gastric variceal obturation (35), is a standard therapy for esophageal and gastric varices caused by portal hypertension, known for its minimal invasiveness and cost-effectiveness. Studies have confirmed its efficacy in controlling bleeding and improving survival (36). Transjugular intrahepatic portosystemic shunt (TIPS) is also a common management strategy for portal hypertension complications (37, 38), providing symptom relief and serving as a bridge to liver transplantation. However, it’s not suited for all due to risks like hepatic encephalopathy. Surgical options remain secondary to transplantation for medication-resistant portal hypertension, with procedures like laparoscopic distal splenorenal shunt offering positive short-term results in children with CHF (39). Liver transplantation emerges as the optimal treatment for patients with CHF in the terminal stage of liver disease (40, 41). In our reported case, the patient with decompensated liver disease faced financial barriers to transplantation. Consequently, the individual pursued EVL in conjunction with tissue adhesives and sclerotherapy, which led to satisfactory short-term results, with no complications observed during an 18-month monitoring period and notable variceal improvement upon subsequent evaluations.
In essence, this report introduces a unique instance of CHF concomitant with ADPKD in an adult female, enhancing the understanding of CHF’s clinical and radiographic presentations linked to ADPKD. This case highlights CHF’s association with ADPKD and underscores the importance of considering CHF in patients with unexplained liver cirrhosis, biliary abnormalities, and polycystic kidneys. Endoscopic management offers an effective alternative for bleeding control in CHF when transplantation is not immediately viable, though transplantation remains the best option for end-stage CHF.
Data availability statement
The datasets presented in this article are not readily available because ethical/privacy restrictions. Requests to access the datasets should be directed to bGl1eWluZ2FnQDEyNi5jb20=.
Ethics statement
The studies involving humans were approved by Tianjin third central hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YL: Writing – original draft. PZ: Writing – review & editing. JT: Data curation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-034A), Tianjin Health Project (no. TJWJ2022XK029), Tianjin Health Project (no. TJWJ2023QN043).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kerr, DN, Harrison, CV, Sherlock, S, and Walker, RM. Congenital hepatic fibrosis. Q J Med. (1961) 30:91–17.
2. Zhu, B, du, Z, Wang, Z, Li, Y, Zhang, J, and Zhu, H. Congenital hepatic fibrosis in children and adults: clinical manifestations, management, and outcome-case series and literature review. Gastroenterol Res Pract. (2020) 2020:8284274. doi: 10.1155/2020/8284274
3. Abdul Majeed, N, Font-Montgomery, E, Lukose, L, Bryant, J, Veppumthara, P, Choyke, PL, et al. Prospective evaluation of kidney and liver disease in autosomal recessive polycystic kidney disease-congenital hepatic fibrosis. Mol Genet Metab. (2020) 131:267–76. doi: 10.1016/j.ymgme.2020.08.006
4. Gunay-Aygun, M, Font-Montgomery, E, Lukose, L, Tuchman Gerstein, M, Piwnica-Worms, K, Choyke, P, et al. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. (2013) 144:112–21.e2. doi: 10.1053/j.gastro.2012.09.056
5. Kanaheswari, Y, Hamzaini, AH, and Wong, SW. Congenital hepatic fibrosis in a child with autosomal dominant polycystic kidney disease. Med J Malaysia. (2008) 63:251–3.
6. O'Brien, K, Font-Montgomery, E, Lukose, L, Bryant, J, Piwnica-Worms, K, Edwards, H, et al. Congenital hepatic fibrosis and portal hypertension in autosomal dominant polycystic kidney disease. J Pediatr Gastroenterol Nutr. (2012) 54:83–9. doi: 10.1097/MPG.0b013e318228330c
7. Sila, L, Velmishi, V, Saraci, B, Dervishi, E, Sila, S, Shtiza, D, et al. Congenital hepatic fibrosis as an early sign of presentation of ADPKD. Balkan J Med Genet. (2023) 25:91–5. doi: 10.2478/bjmg-2022-0024
8. Parkash, A, Cheema, HA, Malik, HS, and Fayyaz, Z. Congenital hepatic fibrosis: clinical presentation, laboratory features and management at a tertiary care hospital of Lahore. J Pak Med Assoc. (2016) 66:984–8.
9. Farahmand, F, Soleimani, K, Hashemi, M, Shafieyoun, A, Rezaei, N, and Yousefi, A. Familial congenital hepatic fibrosis: report of a family with three affected children. Acta Med Iran. (2013) 51:655–6.
10. Masyuk, T, Masyuk, A, and LaRusso, N. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Curr Opin Gastroenterol. (2009) 25:265–71. doi: 10.1097/MOG.0b013e328328f4ff
11. Yoder, BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. (2007) 18:1381–8. doi: 10.1681/ASN.2006111215
12. Desmet, VJ. Ludwig symposium on biliary disorders: part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. (1998) 73:80–9. doi: 10.4065/73.1.80
13. Mirza, H, Besse, W, Somlo, S, Weinreb, J, Kenney, B, and Jain, D. An update on ductal plate malformations and fibropolycystic diseases of the liver. Hum Pathol. (2023) 132:102–13. doi: 10.1016/j.humpath.2022.06.022
14. Chen, IY, Whitney-Miller, CL, and Liao, X. Congenital hepatic fibrosis and its mimics: a clinicopathologic study of 19 cases at a single institution. Diagn Pathol. (2021) 16:81. doi: 10.1186/s13000-021-01142-y
15. Desmet, VJ. What is congenital hepatic fifibrosis? Histopathology. (1992) 20:465–78. doi: 10.1111/j.1365-2559.1992.tb01031.x
16. Arnold, HL, and Harrison, SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol. (2005) 100:2569–82. doi: 10.1111/j.1572-0241.2005.00263.x
17. Spahiu, L, Behluli, E, Grajçevci-Uka, V, Liehr, T, and Temaj, G. Joubert syndrome: molecular basis and treatment. J Mother Child. (2023) 26:118–23. doi: 10.34763/jmotherandchild.20222601.d-22-00034, eCollection 2022 Mar 1
18. Kaur, A, Dhir, SK, Goyal, G, Mittal, N, and Goyal, RK. Senior Loken Syndrome. J Clin Diagn Res. (2016) 10:SD03–4. doi: 10.7860/JCDR/2016/21832.8816
19. Gentile, M, Di Carlo, A, Susca, F, Gambotto, A, Caruso, ML, Panella, C, et al. COACH syndrome: report of two brothers with congenital hepatic fibrosis, cerebellar vermis hypoplasia, oligophrenia, ataxia, and mental retardation. Am J Med Genet. (1996) 64:514–20. doi: 10.1002/(SICI)1096-8628(19960823)64:3<514::AID-AJMG13>3.0.CO;2-O
20. Salonen, R, and Paavola, P. Meckel syndrome. J Med Genet. (1998) 35:497–01. doi: 10.1136/jmg.35.6.497
21. Forsythe, E, and Beales, PL. Bardet-Biedl syndrome. Eur J Hum Genet. (2013) 21:8–13. doi: 10.1038/ejhg.2012.115
22. Paul, BM, and Vanden Heuvel, GB. Kidney: polycystic kidney disease. Wiley Interdiscip Rev Dev Biol. (2014) 3:465–87. doi: 10.1002/wdev.152
23. Rossetti, S, Consugar, MB, Chapman, AB, Torres, VE, Guay-Woodford, LM, Grantham, JJ, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. (2007) 18:2143–60. doi: 10.1681/ASN.2006121387
24. Raj, S, Singh, RG, and Das, P. Mutational screening of PKD1 and PKD2 in Indian ADPKD patients identified 95 genetic variants. Mutat Res. (2020) 821:111718. doi: 10.1016/j.mrfmmm.2020.111718
25. Bae, KT, Zhu, F, Chapman, AB, Torres, VE, Grantham, JJ, Guay-Woodford, LM, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the consortium for radiologic imaging studies of polycystic kidney disease cohort. Clin J Am Soc Nephro. (2006) 1:64–9. doi: 10.2215/CJN.00080605. Epub 2005 Oct 26
26. Hogan, MC, Abebe, K, Torres, VE, Chapman, AB, Bae, KT, Tao, C, et al. Liver involvement in early autosomal dominant polycystic kidney disease. Clin Gastroenterol Hepatol. (2015) 13:155–64.e6. doi: 10.1016/j.cgh.2014.07.051. Epub 2014 Aug 9
27. Lemaigre, FP. Molecular mechanisms of biliary development. Prog Mol Biol Trans Sci. (2010) 97:103–26. doi: 10.1016/B978-0-12-385233-5.00004-0
28. Misra, A, Loyalka, P, and Alva, F. Portal hypertension due to extensive hepatic cysts in autosomal dominant polycystic kidney disease. South Med J. (1999) 92:626–7. doi: 10.1097/00007611-199906000-00015
29. Lee, FI, and Paes, AR. Congenital hepatic fibrosis and adult-type autosomal dominant polycystic kidney disease in a child. Postgrad Med J. (1985) 61:641–2. doi: 10.1136/pgmj.61.717.641
30. Cobben, JM, Breuning, MH, Schoots, C, ten Kate, LP, and Zerres, K. Congenital hepatic fibrosis in autosomal-dominant polycystic kidney disease. Kidney Int. (1990) 38:880–5. doi: 10.1038/ki.1990.286
31. Matsuda, O, Ideura, T, Shinoda, T, Shiigai, T, Takeuchi, H, Chen, WC, et al. Polycystic kidney of autosomal dominant inheritance, polycystic liver and congenital hepatic fibrosis in a single kindred. Am J Nephrol. (1990) 10:237–41. doi: 10.1159/000168088
32. Lipschitz, B, Berdon, WE, Defelice, AR, and Levy, J. Association of congenital hepatic fibrosis with autosomal dominant polycystic kidney disease. Report of a family with review of literature. Pediatr Radiol. (1993) 23:131–3. doi: 10.1007/BF02012406
33. Klinkert, J, Koopman, MG, and Wolf, H. Pregnancy in a patient with autosomal-dominant polycystic kidney disease and congenital hepatic fibrosis. Eur J Obstet Gynecol Reprod Biol. (1998) 76:45–7. doi: 10.1016/s0301-2115(97)00153-x
34. Jiang, C, Zhou, Q, Jin, M, Niu, J, and Gao, Y. Congenital hepatic fibrosis with polycystic kidney disease: two case reports. Medicine (Baltimore). (2019) 98:e15600. doi: 10.1097/MD.0000000000015600
35. Wang, S, Xiao, M, Hua, L, Jia, Y, Chen, S, and Zhang, K. Endoscopic therapy for gastro-oesophageal varices of Caroli's syndrome: a case report. J Int Med Res. (2020) 48:300060519877993. doi: 10.1177/0300060519877993
36. Su, J, Zhang, H, Ren, M, Xing, Y, Yin, Y, and Liu, L. Efficacy and safety of ligation combined with sclerotherapy for patients with acute esophageal variceal bleeding in cirrhosis: a meta-analysis. Front Surg. (2021) 8:664454. doi: 10.3389/fsurg.2021.664454. eCollection 2021
37. Verbeeck, S, Mekhali, D, Cassiman, D, Maleux, G, and Witters, P. Long-term outcome of transjugular intrahepatic portosystemic shunt for portal hypertension in autosomal recessive polycystic kidney disease. Dig Liver Dis. (2018) 50:707–12. doi: 10.1016/j.dld.2018.03.009
38. Wang, J, Shu, J, Wu, F, Song, ZT, Gan, HY, Yu, J, et al. A case of congenital hepatic fibrosis diagnosed and treated by transjugular intrahepatic portosystemic shunt. Zhonghua Gan Zang Bing Za Zhi. (2021) 29:373–6. doi: 10.3760/cma.j.cn501113-20190527-00190
39. Zhang, JS, Cheng, W, and Li, L. Laparoscopic distal splenoadrenal shunt for the treatment of portal hypertension in children with congenital hepatic fibrosis: a case report. Medicine (Baltimore). (2017) 96:e5843. doi: 10.1097/MD.0000000000005843
40. Wu, WK, Ziogas, IA, Izzy, M, Pai, AK, Hafberg, ET, Matsuoka, LK, et al. Liver transplantation for congenital hepatic fibrosis. Transpl Int. (2021) 34:1281–92. doi: 10.1111/tri.13884
Keywords: PKD1 gene mutation, congenital hepatic fibrosis, ADPKD, adult female, case report
Citation: Liu Y, Zhu P and Tian J (2024) Case report: Rare genetic liver disease - a case of congenital hepatic fibrosis in adults with autosomal dominant polycystic kidney disease. Front. Med. 11:1344151. doi: 10.3389/fmed.2024.1344151
Edited by:
Joel Edward Lavine, Columbia University, United StatesReviewed by:
Pietro Vajro, University of Salerno, ItalyRavi Kumar Sharma, Chandigarh University, India
Copyright © 2024 Liu, Zhu and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Liu, bGl1eWluZ2FnQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship