 Guole Nie
Guole Nie Honglong Zhang
Honglong Zhang Danna Xie
Danna Xie Jun Yan1,2,3,4,5,6
Jun Yan1,2,3,4,5,6 Xun Li
Xun Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 16 January 2024
Sec. Hepatobiliary Diseases
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1320015
This article is part of the Research Topic Reviews in Hepatobiliary Diseases 2023 View all 8 articles
The gut-liver axis refers to the intimate relationship and rigorous interaction between the gut and the liver. The intestinal barrier’s integrity is critical for maintaining liver homeostasis. The liver operates as a second firewall in this interaction, limiting the movement of potentially dangerous compounds from the gut and, as a result, contributing in barrier management. An increasing amount of evidence shows that increased intestinal permeability and subsequent bacterial translocation play a role in liver damage development. The major pathogenic causes in cirrhotic individuals include poor intestinal permeability, nutrition, and intestinal flora dysbiosis. Portal hypertension promotes intestinal permeability and bacterial translocation in advanced liver disease, increasing liver damage. Bacterial dysbiosis is closely related to the development of cirrhosis and its related complications. This article describes the potential mechanisms of dysbiosis in liver cirrhosis and related complications, such as spontaneous bacterial peritonitis, hepatorenal syndrome, portal vein thrombosis, hepatic encephalopathy, and hepatocellular carcinoma, using dysbiosis of the intestinal flora as an entry point.
Liver cirrhosis is a primary cause of morbidity and mortality worldwide. The mortality rate of patients with compensated liver cirrhosis increased five-fold and increased 10-fold in patients with decompensated liver cirrhosis compared with the general population (1). Liver cirrhosis is an advanced chronic liver disease caused by various etiologies, including alcohol abuse, obesity, and hepatitis virus infection, and prevalence of cirrhosis is increasing worldwide (2). Although there are many etiologies of liver cirrhosis, alcoholic liver disease (ALD), nonalcoholic fatty liver disease (NAFLD), and viral hepatitis are the prevalent etiologies of chronic liver disease globally (3, 4). Liver cirrhosis is the end-stage pathological manifestation of chronic liver disease charactered by chronic inflammation, hepatic lobe reconstruction, and the formation of pseudolobule and tubercle (5). The bidirectional interactions of the liver and the intestinal microbiota provide a new perspective for the occurrence and development of liver cirrhosis and its complications (6).
Microbes inhabit specific locations of the body, such as the skin, mouth, respiratory tract, gastrointestinal tract, genitourinary system, etc., and the highest density residing in the gastrointestinal tract (7, 8). The exact time of gut microbial colonization is not known, but intestinal microbial colonization does not occur before birth (9). A variety of internal and external factors will affect the intestinal flora after birth, such as dietary, disease and sleep, etc. (10–13). Metagenomics, metatranscriptomic, and metaproteomic of the human intestinal microbiota show that the intestinal microbiota is an important constituent of the organism and plays a crucial role in human health and disease (14). The portal vein is an important channel for the interaction between the liver and the intestinal microbiota. The portal vein can transport intestinal nutrients, bacteria, and microbial products to the liver (15). Under normal physiological conditions, the intestinal microbiota is essential for liver metabolic functions, maturation and maintenance of immune homeostasis (16), and in turn, the liver regulates the intestinal microbiota and metabolic functions mainly through secreting bile acids (BA) (17).
We review clinical studies on cirrhosis and dysbiosis, and then turn to the topic of cirrhosis-related complications and dysbiosis. Dysbiosis may provide new perspectives on cirrhosis and complications.
Liver cirrhosis affects the intestinal microbiota mainly through two aspects: on the one hand, liver cirrhosis can reduce the synthesis of BA, and resulting in the composition and function of the intestinal microbiota (18, 19). On the other hand, liver cirrhosis can cause portal hypertension, gastrointestinal blood stasis, impair intestinal barrier, and gut dysbiosis. Bacteria and metabolites can enter the portal vein and systemic circulation through translocation of the damaged intestinal barrier, causing an inflammatory state in the body and endotoxemia (20), which have different effects on both cirrhosis and cirrhosis-related complications (21).
Bile acids are the major functional components of bile and are generated by a classical and alternative pathway in the liver (22). BA primarily promote the emulsification of fats and aid the absorption of lipid and fat-soluble vitamins (23, 24). BA and the intestinal microbiota can interact with each other, and BA play an important role in regulating the diversity of intestinal flora and small intestine bacterial overgrowth (25). BA can not only directly affect the integrity of the intestinal barrier via regulating the composition of intestinal flora but promoting the synthesis and antimicrobial peptides secretion of the intestinal epithelial cells (26). Moreover, BA regulate metabolic function, energy consumption, and inflammation through interaction with its receptors, such as farnesoid-X-receptor (FXR) and the vitamin D receptor (VDR) as well as the Takeda G-protein coupled BA receptor (TGR5) (27). The activation of FXR can protect the integrity of the intestinal barrier, reduce bacterial translocation and intestinal inflammation (28, 29).
The intestinal microbiota plays a unique role in BA metabolism and homeostasis in the host (19). The metabolites of the intestinal microbiota with a variety of biological functions, also known as postbiotics, such as short-chain fatty acids, secondary BA, choline metabolites, indole derivatives, vitamins, polyamines, lipids, neurotransmitters, neuroactive compounds, and thalamic-pituitary-adrenal axis hormones play an important role in the body (30). ALD, NAFLD, and viral hepatitis are the leading associated causes of liver cirrhosis worldwide, we have mainly summarized the clinical studies related to dysbiosis in these diseases.
In general, the 16S rRNA gene sequencing analysis showed a significant decrease in the levels of Lachnospiraceae and Ruminococcaceae in patients with liver cirrhosis, while the whole-metagenome shotgun sequencing analysis showed that the level of Faecalibacterium prausnitzii from the Ruminococcaceae and of Coprococcus spp. from the Lachnospiraceae are significantly reduced (31, 32). Dysbiosis of intestinal flora further aggravates liver injury. Studies have shown that the severity of liver injury is closely related to the severity of intestinal flora dysbiosis (33). Changes in the fecal bacterial flora are manifested by changes in the composition of the major Bacteroidetes and Sclerotiniaceae, which produce short-chain fatty acids (SCFA) that are a source of energy for intestinal epithelial cells, and also regulate BA metabolism and induce modulation of the immune function of the intestinal barrier (34). Dysbiosis of the intestinal flora affects changes in intestinal permeability and intestinal metabolites that may be involved in the progression of cirrhosis and its associated complications (35).
Liver cirrhosis is the common pathologic of the advanced stage of ALD and NAFLD (36). Alcohol and its metabolites can affect the tight junction between intestinal epithelial cells, impair intestinal barrier function, induce bacterial translocation and endotoxemia (37, 38). Gut dysbiosis is closely related to the occurrence and development of NAFLD and ALD, but the mechanism is not clear (39, 40). The intestinal microbiota can promote the development of human NAFLD and ALD into end-stage liver disease (41, 42), indicating that gut dysbiosis may be the common changes of these diseases. A higher abundance of Enterobacteriaceae and Halomonadaceae, and lower Lachnospiraceae, Ruminococcaceae, and Clostridialies XIV in alcoholic-related liver cirrhosis than non-alcoholic cirrhotics, whereas non-alcoholic steatohepatitis-related liver cirrhosis (NASH) had a higher level of Porphyromonadaceae, Bacterioidaceae, and lower Veillonellaceae compared to those without NASH etiology (43). The number of Escherichia coli, anaerobes, Lactobacillus, and streptococci in intestinal microflora of patients with NAFLD is higher than that of healthy controls (44). Compared to healthy controls, the proportion of Bacteroidetes was significantly reduced, whereas Proteobacteria and Fusobacteria were highly enriched in the ARLC patients with different etiologies (45). The median abundance of Bacteroidetes was lower and the median abundance of Proteobacteria was higher in the intestines of patients with ALD, and these changes appeared to be associated with higher serum endotoxin levels in some of the samples (46). Studies have shown that alcohol causes a significant increase in Veillonellaceae and a decreasing trend in Bacteroidaceae and Porphyromonadaceae (47). 16S rRNA gene sequencing revealed that Peptostreptococcacae, Proteobacteria, Pasteurellaceae and Bacillales were significantly increased, while Lachnospiraceae, Ruminococcaeae, Clostridiales cluster XIV, Prevotellaceae and Bacteroidaceae significantly decreased in ARLC patients (48). Metagenomic sequencing revealed that Bifidobacterium, Streptococcus and Lactobacillusspecies were significantly increased, while Akkermansia, Coprococcus, Unclassified and Clostridiales significantly decreased in ARLC patients (49).
For NASH patients, there was a significant increase in Clostridium coccoides, Porphyromonadaceae and Actioidaceae and a decrease in Veillonellaceae and Bacteroidetes (43, 50). For NAFLD patients, ClostridiumXI in the Peptostreptococcaceae, the Anaerobacter in the Clostridiaceae, Streptococcus and Lactobacillus were significantly increased, while Lentisphaerae, Ruminococcaceae, Oscillibacter, Flavonifractor and Bacteroidetes decreased (44). Table 1 summarizes the studies about gut dysbiosis in ALD and NAFLD-related liver disease.
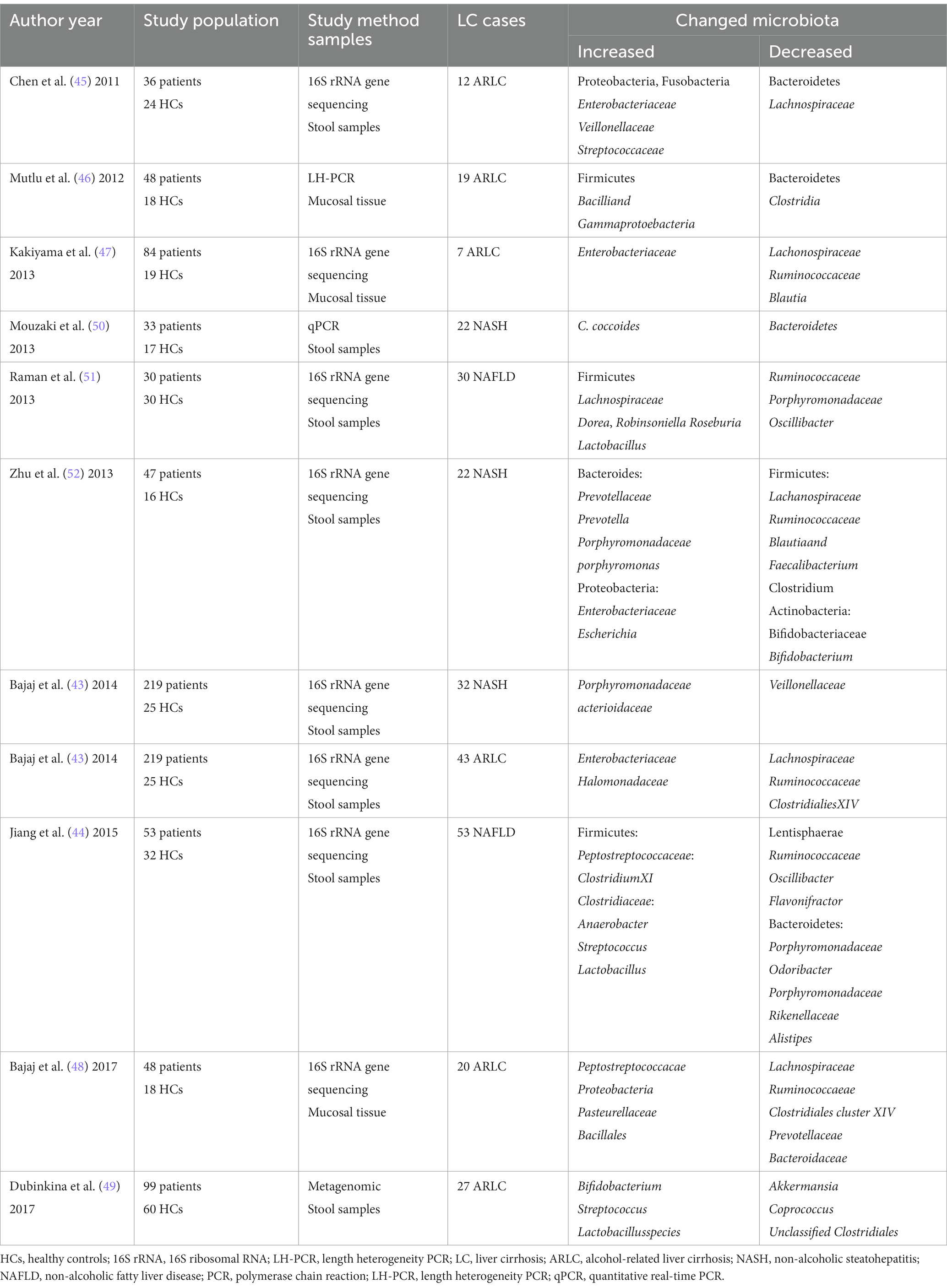
Table 1. The changed gut microbiota in alcohol and NASH-related liver disease.
Infection of hepatitis B and C viruses is the cause of viral hepatitis cirrhosis, which Characterized by chronic inflammation, diffuse liver fibrosis, and pseudolobular formation (53). Recent works have shown that hepatitis virus (mainly hepatitis B virus and hepatitis C virus) related cirrhosis has unique bacterial or fungal microbiota profiles, which include increased numbers of Enterobacteriaceae, Prevotella, Streptococcus, Staphylococcaceae, and Veillonella spp., as well as decreased Firmicutes, Bifidobacteria, Lachnospiraceae, Bacteroidetes, Ruminococcus, and Clostridium (54, 55). Sequencing of the 16S rRNA gene for HBV patients revealed significant increases in Veillonella, Megasphaera, Dialister, Atopobium, and Prevotella, and significant decreases in Neisseria, Haemophilus, and SR1 genera incertae sedis significantly decreased (56). Sequencing of the 16S rRNA gene for HCV revealed a significant increase in Prevotella, Succinivibrio, Catenibacterium and Megasphaera in the Ruminococcaceae and a significant decrease in Enterobacteriaceae, Erysipelotrichaceae and Rikenellaceae (57). But whether the direct-acting antivirals could affect the intestinal microbiota composition in cirrhotic patients is still a matter of controversy (58, 59). Although the gut microbiota varied slightly from study to study, small differences were found after treating patients. Table 2 summarizes the recent studies about gut dysbiosis in viral hepatitis-related disease.
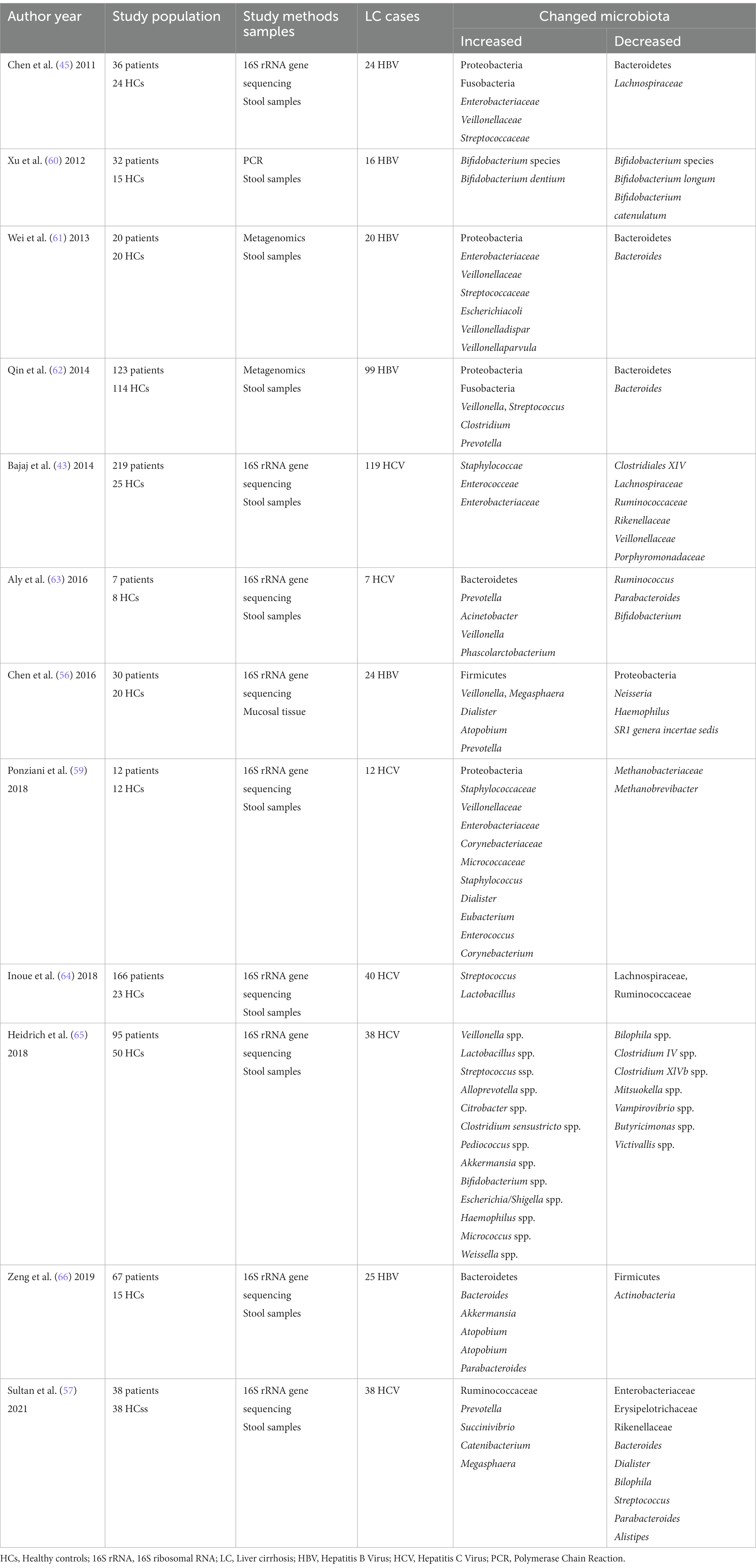
Table 2. The changed gut microbiota in viral hepatitis-related liver disease.
Liver cirrhosis-related complications severely impact the survival rate and mortality of patients with liver cirrhosis, such as portal vein thrombosis (PVT), spontaneous bacterial peritonitis (SBP), hepatic encephalopathy (HE), portal hypertension (PH), hepatorenal syndrome (HRS), hepatopulmonary syndrome (HPS), and hepatocellular carcinoma (HCC). Gut dysbiosis, bacterial translocation, and intestinal barrier injury in liver cirrhosis patients exert crucial roles in liver cirrhosis-related complications (Figure 1).
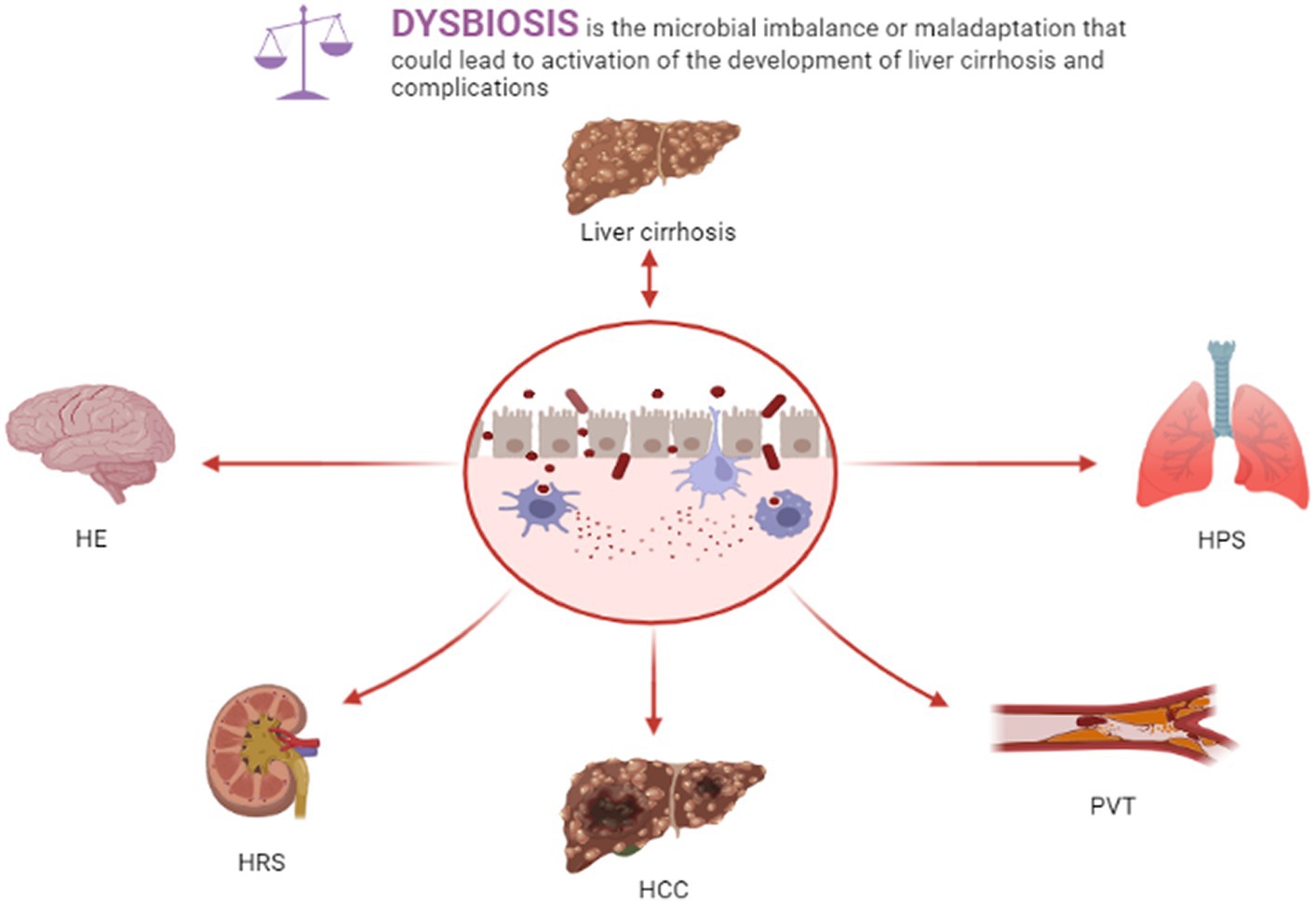
Figure 1. Dysbiosis and liver cirrhosis and complications.
PVT is a common complication observed in liver cirrhosis and occurs in intrahepatic branches of the portal vein, with or without superior mesenteric vein and splenic vein thrombosis (67). The prevalence of PVT is approximately 1%–26% (68). The pathogenesis of liver cirrhosis with PVT is unclear. Slow portal vein blood flow caused by liver cirrhosis is the important factor for PVT (69). The decrease of symbiotic anaerobes and the increase of pathogen abundance after gut dysbiosis, especially the increase of Gram-negative Enterobacteriaceae, are closely related to the occurrence of PVT (70). Bacterial LPS is the glycolipid on the outer membrane of Gram-negative bacteria and is one of the key factors in the hypercoagulable state of liver cirrhosis (71, 72). The translocation of bacteria and pathogen-associated molecular patterns (PAMPs), especially LPS, will cause systemic inflammation, endotoxemia, and platelet activation, at the same time, systemic inflammation and elevated levels of systemic inflammatory factors exacerbate the risk of PVT formation (73, 74). Liver cirrhosis patients usually have higher endotoxemia and systemic inflammation due to gut dysbiosis. Endotoxin can increase thrombosis through the production of tissue factor (TF) (75). With the increase of intestinal permeability and bacterial translocation, the level of LPS in the blood of patients with liver cirrhosis increased significantly. Due to the immune function of the liver, the level of LPS in the portal system was significantly higher than that in the systemic circulation (76, 77).
After entering the circulation system, LPS interacts to toll-like receptors (TLRs) and initiating a series of pathophysiological changes linked to the formation of PVT. On the one hand, LPS binds to TLRs expressed on hepatocytes and immune cells. Activation of these cells will release a huge amount of inflammatory cytokines, chemokines, vasoactive factors, adhesion molecules, and reactive oxygen species (ROS) (78–80), resulting in systemic inflammation, the proliferation of hepatic stellate cells, and the development of liver cirrhosis (81). On the other hand, LPS binds to TLRs receptors expressed on vascular endothelial cells, platelet, and neutrophils, promoting blood hypercoagulability and PVT in the context of liver cirrhosis (70) (Figure 2).
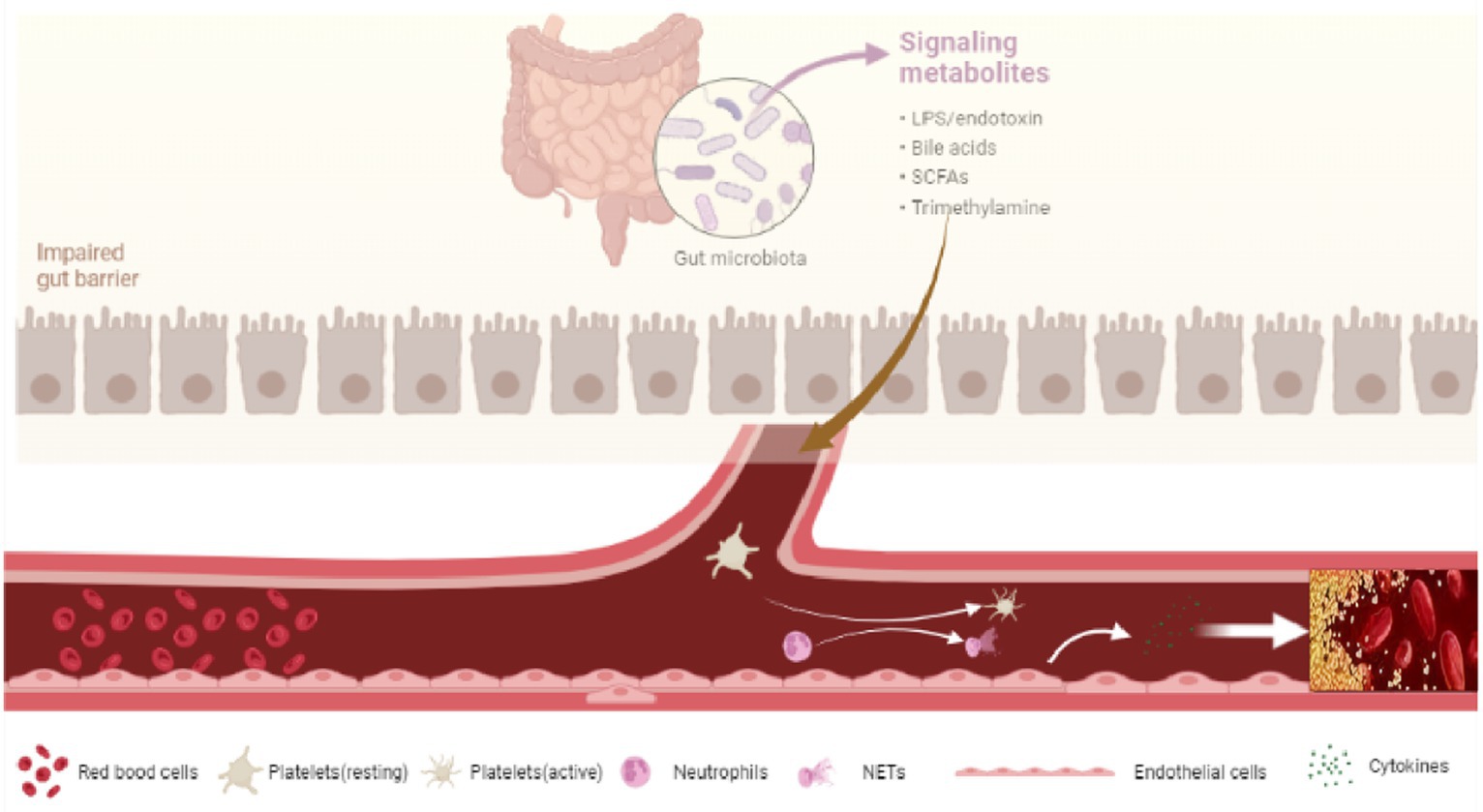
Figure 2. Dysbiosis and PVT.
LPS binds to TLRs receptors on vascular endothelial cells and activates endothelial cells to release von Willebrand factor (vWF) and factor VII (43, 82). Animal experiments showed that LPS could also bind to toll-like receptor 2 (TLR2) on hepatic endothelial cells to promote the synthesis of the vWF precursor and promote platelet integrin-dependent thrombus growth, while the synthesis of vWF in hepatic endothelial cells decreased and the level of plasma vWF decreased significantly in TLR2 knockout mice (83). In vitro studies have shown that LPS can stimulate the formation and secretion of Weibel–Palade bodies in endothelial cells through toll-like receptor 4 (TLR4), and promote the release of VIII and vWf into the blood resulting in a blood hypercoagulable state. TLR receptor blockers can significantly inhibit the release of VIII and vWf, indicating that LPS in the intestinal microbiota increases the level of systemic factor VIII by stimulating the release of endothelial cells (43). The level of Plasminogen activator inhibitor-1 (PAI-1) and tissue plasminogen activator (TPA), key determinants in thrombosis, were shown to be related to the gut microbiota. In vivo and in vitro studies have shown that bacterial lipoprotein TLR2 agonists can increase vascular endothelial cell permeability, increase plasma PAI-1 and decrease TPA (84).
Platelet activation in patients with liver cirrhosis may be associated with increased levels of circulating LPS (85). TLR2 and TLR4 are widely distributed on the surface of platelets. The activation of the TLR2 in platelets causes the activation of platelets, which can cause platelets to release their own stores of α-particles and dense particles to interact with vascular endothelial cells and promote platelet-dependent thrombosis (86, 87). In vitro studies have shown that platelets in patients with liver cirrhosis are more reactive to TLRs agonists than healthy people and have nothing to do with the number of platelets. The reactivity is significantly weakened in the presence of TLR4 blockers, indicating that LPS may activate platelets through TLR4 and may lead to liver cirrhosis-related thrombotic complications (70). Activated platelets release P-selectin to stimulate monocytes and macrophages to release chemokines, promote platelet-monocyte aggregation, and release inflammatory molecules to change the chemotaxis and adhesion of endothelial cells (88).
Activation of neutrophils can induce thrombosis through the neutrophil extracellular bactericidal networks (NETs) (89, 90). In vitro animal models have shown that the elimination of NETs can prevent thrombosis (91). NETs are formed by neutrophils releasing nuclear contents (DNA-histone complex, double-stranded DNA, and neutrophil elastase) into extracellular space (92). Histone-DNA complex can activate coagulation factor XIIa activated by coagulation factor XII, and then activate coagulation factor XI and downstream endogenous coagulation pathway (93). Histone of NETs can promote thrombin production, endothelial cell activation, and thrombus formation through the platelet-dependent mechanism mediated by TLR2 and TLR4 (94).
Trimethylamine (TMA) lyase from intestinal microorganisms metabolizes phosphatidylcholine, choline and carnitine to TMA, which is further processed to trimethylamine-N-oxide (TMAO) by flavin monooxygenase (FMO) in the liver (95). TMAO not only aggravates cardiovascular events but also is closely related to the formation of thrombosis (96). TMAO activates platelets by increasing the release of Ca2+ stored platelets intracellular, while platelets with hyperreactivity enhance the thrombosis risk (97). The animal experiment showed that a high choline diet can enhance platelet hyperresponsiveness, but this will not occur with the intervention of antibiotics or a high choline diet in germ-free mice (98). Therefore, TMAO acts as a medium to closely connect intestinal microbiota with thrombosis.
Liver cirrhosis is usually accompanying by acquired factors of thrombophilia, such as hyperhomocysteinemia, secondary to vitamin B and folate deficiencies, and antiphospholipid antibody syndrome (67). Moreover, the levels of albumin and protein C were lower in patients with liver cirrhosis compared to healthy controls, and low levels of albumin and protein C were associated with the increased risk of PVT formation (99, 100). Abnormal metabolic state, hemodynamic changes, and PAI-1 in patients with NAFLD may contribute to prethrombotic state and hypercoagulable state (101, 102). In a cross-sectional study, NASH-related cirrhosis was the strongest independent risk factor for the independent diagnosis of PVT in patients undergoing liver transplantation (103).
SBP is a common and serious complication in cirrhotic patients (104). It refers to primary peritonitis that occurs in patients with cirrhosis and ascites without abdominal infection lesions (105). Animal experiments showed that the bacteria strain isolated from intestinal mucosa lymphoid tissue and ascites was similar (106). These studies provide strong evidence for the role of bacterial translocation in SBP. The bacterial translocation and the increase of intestinal permeability is the main mechanism of SBP in the setting of liver cirrhosis, at the same time, the decrease of host immune clearance ability is also closely related to the occurrence of SBP (43).
Gram-negative bacteria, such as Escherichia coli and Klebsiella, and Gram-positive bacteria are common causes of SBP (107, 108). The patients with liver cirrhosis were accompanied by decreased diversity of the gut microbiota, which was characterized by a significant reduction in autochthonous taxa and a significant increase in pathogenic taxa (109). Clinical studies have shown that Streptococcus spp., Klebsiella, Escherichia, and Citrobacter spp. were mainly infectious organisms on routine culture in liver cirrhosis patients with SBP (43).
The gut-liver-immune axis plays a key role in SBP. The changes in intestinal motility, mucosal immunity, and drug usage in patients with liver cirrhosis will lead to significant changes in the composition of gastrointestinal microorganisms and aggravate the translocation of the intestinal microbiota (110, 111). Moreover, interaction between BA and its receptor farnesoid X receptor (FXR) helps to maintain intestinal barrier function and reduce bacterial translocation (112). Animal experiments showed that FXR agonists could reduce intestinal permeability and bacterial translocation through FXR (29, 113). In addition to FXR agonists, selective intestinal decontamination is an important measure for the treatment of SBP. Intestinal decontamination with rifaximin and norfloxacin significantly decreased the incidence of SBP in cirrhotic patients with ascites (114, 115), but rifaximin was more effective than norfloxacin in the secondary prevention of SBP (116). Moreover, non-selective β-blockers (NSBB) can reduce intestinal permeability, markers of bacterial translocation (IL-6/LPS binding protein), and SBP by improving intestinal motility and reducing intestinal bacterial overgrowth (117, 118).
HE is a severe complication of advanced liver cirrhosis and is closely linked to the gut-liver-brain axis (119). The impaired hepatic clearance ability with the progression of liver cirrhosis is reconsider phrasing for clarity, neurotoxic substances, and false neurotransmitters produced by the intestinal microbiota (120). These substances have an important effect on HE. Moreover, the formation of portal shunts further facilitates the entrance of the microbial metabolites into the blood (121).
A series of inflammatory cytokines and endotoxemia caused by gut dysbiosis can impair blood-brain barriers, neuroinflammation, and affect cognition (122). Compared with healthy people, the specific bacteria (Alcaligenes, Porphyromonas, and Enterobacteriaceae) in fecal microbiota were significantly increased in liver cirrhosis patients complicated with HE and were strongly related to the cognition and inflammation of HE (123). A study analyzed the relationship between cognition, Magnetic resonance imaging parameters, and intestinal microflora and found that patients with HE had a significantly lower cognitive ability, systemic inflammation, gut dysbiosis, and hyperammonemia than controls and cirrhotic patients without HE (119). Specific microbial families (autochthonous taxa negatively and Enterobacteriaceae positively) correlated with changes in astrocytes associated with magnetic resonance spectroscopy and hyperammonemia (119). Treatment methods such as fecal microbial transplantation, intestinal decontamination, and diet regulation for the intestinal microbiota can improve the cognitive level of patients with HE (124–126). Therefore, Gut dysbiosis is associated with the development of HE (43).
Emerging evidence also shows that brain cholesterol accumulation contributes to the progression of HE through BA-mediated effects on the FXR (127). Moreover, serotonin and tryptophan metabolism via the gut microbiota is a key factor for the occurrence of central nervous system diseases (128). Recent studies have found that bacteria (Stenotrophomonas pavanii, Methylobacterium extorquens) and metabolites (methanol, threonine) in the blood and feces of patients with liver cirrhosis are positively correlated with HE, while fecal Enterobacteriaceae and TMA were positively correlated with blood proinflammatory cytokines (129). Therefore, bacteria or their metabolites in the blood are correlated with systemic inflammation and HE in patients with liver cirrhosis. Those studies provide new perspectives and treatment strategies for the pathogenesis of HE.
Liver structural disorder and nodular regeneration in liver cirrhosis will cause compression of hepatic sinusoids and blood vessels, and increase intrahepatic resistance, resulting in (PH) (130). PH has an important effect on intestinal permeability, gut microbiota, and bacterial translocation. PH reduces intestinal mucosal blood flow, causes neoangiogenesis, ischemia, and edema of the intestine, destroying intestinal barrier function (131). Therefore, the gut dysbiosis in liver cirrhosis causes the translocation of a large number of bacteria and products, especially LPS, into the blood, leading to activate the immune system and causes systemic inflammation via interacting with TLRs (43, 132), and activation of the liver immune system and systemic inflammation promote the progression of liver cirrhosis and aggravate PH (133). In the animal model, intraperitoneal injection of LPS activates the expression of TLR4 and increases inflammatory mediators, leading to escalating PH (134). The release of systemic inflammatory factors (nitric oxide, NO), can also reduce systemic vascular resistance and induce hyperdynamic circulation, thus affecting PH (135, 136). Therefore, there is an important pathophysiological relationship between PH and the gut-liver axis.
FXR not only plays an important role in intestinal microflora homeostasis, BA metabolism, and intestinal barrier function, but also in anti-fibrosis and reducing PH. In the experimental liver cirrhosis, non-steroidal FXR agonist PX20606 improves PH via reducing intestinal bacterial migration, liver fibrosis, vascular remodeling, and hepatic sinusoid dysfunction (137). Studies in preclinical models of cirrhosis shows that intestinal decontamination can improve the portal vein pressure and hyperdynamic circulation in liver cirrhosis (138). These studies provide insight into molecular mechanisms and novel therapeutic targets in PH.
HRS refers to functional acute renal failure in patients with severe liver disease (139). HRS is a severe complication of advanced liver cirrhosis with a prevalence between 11% and 20% (140). The mechanism of HRS is not clear, but it is mainly related to the following two aspects: on the one hand, the production of ascites increases, and the circulating blood volume decreases after decompensation of liver cirrhosis, resulting in prerenal renal failure (141); on the other hand, the bacterial dysbiosis and the translocation of bacteria and related products after decompensation of liver cirrhosis lead to the endotoxemia, which is closely related to HRS (142, 143). Animal model studies have shown that the increased expression of TLR4 in the kidney tissue of cirrhotic rats increases the susceptibility to LPS, then activates the NF-κB pathway, increases the expression of proinflammatory cytokine tumor necrosis factor-α (TNF-α), and renal tubular injury (144). Selective gut decontamination can improve the systemic hemodynamics and renal function of patients with liver cirrhosis, indicating that the gut microbiota plays an important role in HRS (144, 145). Moreover, the albumin infusion improves renal function in cirrhotic patients and sepsis via affecting endotoxemia, hemodynamics, and oxidative stress (146, 147). Moreover, circadian hemodynamic in cirrhosis is related to renal function (148). As mentioned above, gut dysbiosis has a close relationship with circadian rhythm, it is not clear whether the diurnal rhythm of the microbiota can affect renal function by regulating hemodynamics.
HPS is a pulmonary complication in liver cirrhosis patients and charactered by pulmonary microvascular dilatation and hypoxemia (149). The pathogenesis of HPS is unknown. The bacterial translocation, intestinal endotoxemia, and pulmonary microvascular dilatation may closely relate to the pathogenesis of HPS (150–152). Studies on animal models have shown that bacterial translocation, intestinal endotoxemia, and related inflammatory factors are closely related to the occurrence of HPS (153). Moreover, bacterial translocation can increase the incidence and severity of HPS in cirrhotic rats, and prophylactic norfloxacin usage can reduce the incidence and severity of HPS (154). However, antibiotic norfloxacin usage in clinical patients does not improve the gas exchange of HPS (155). Therefore, there need to be further studies on the effect of antibiotics in patients with HPS and the role of the specific intestinal microbiota in the pathogenesis of HPS.
HCC is the most common type of liver cancer as well as the common cause of death in patients with advanced cirrhosis (156, 157). Moreover, different etiologies may affect the composition of the microbiota in HCC patients (158). As aforementioned, systemic inflammation caused by the translocation of bacteria, LPS, bacterial DNA, and peptidoglycans by activating the TLRs is crucial for the development of HCC (42, 159). LPS activates TLR4 signal to promote the production of interleukin-6 (IL-6) and TNF-α, mediating the differentiation of hepatic progenitor cells into myofibroblasts, and promoting the proliferation and malignant transformation of hepatic progenitor cells (160). Therefore, LPS and TLR4 are closely related to the development of cirrhotic patients to HCC (161). Compared to NAFLD cirrhotic patients without HCC, the NAFLD cirrhotic patients with HCC have a higher abundance of Bacteroidetes at the phylum level, Bacteroidaceae, Streptococcaceae, Enterococcaceae, and Gemellaceae at the family level, and Phascolarctobacterium, Enterococcus, Streptococcus, Gemella, and Bilophila at the genus level (162). In another study, intestinal microbiota constitution in cirrhotic patients with HCC is distinguished from those without HCC. Compared patients without HCC, Haemophilus, Eggerthella, Bifidobacterium, Butyricimonas, Christensella, Odoribacter, an unknown genus phylum Tenericutes, and an unknown genus, phylum Firmicutes, family Erysipelotrichaceae were all elevated in cirrhotic patients with HCC, while Fusobacterium, Prevotella, Streptococcus, S24-7 (Phylum Bacteroidetes) and an unknown genus were all decreased. Thus, gut dysbiosis is a crucial factor in cirrhotic patients with HCC (163).
The intestinal microbiota plays a significant role in human health and disease. Gut dysbiosis is associated with the onset and progression of liver cirrhotic and its complications. The correlation between specific intestinal microbiota and pathogenesis of liver cirrhosis related complications needs further study. The gut microbiota can be used as a potential diagnosis biomarker and treatment target for liver cirrhosis and its complications. It has been reported that bacteriophage therapy (164), microRNA therapy (165), and carbon nanoparticles (166) that based on targeting the intestinal microbiota in liver cirrhosis. It is important to assess the role of the gut microbiota in the pathogenesis of liver cirrhosis and its complications. With the rise of personalized medicine and nanomedical technology, treatment options that targeting specific intestinal microbiota composition may be the most promising treatment for liver cirrhosis and complications in the future.
GN: Conceptualization. HZ: Conceptualization, Data curation, Writing – original draft. DX: Conceptualization, Data curation, Writing – original draft. JY: Investigation, Writing – review & editing. XL: Project administration, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fleming, KM, Aithal, GP, Card, TR, and West, J. All-cause mortality in people with cirrhosis compared with the general population: a population-based cohort study. Liver Int. (2012) 32:79–84. doi: 10.1111/j.1478-3231.2011.02517.x
2. Ginès, P, Krag, A, Abraldes, JG, Solà, E, Fabrellas, N, and Kamath, PS. Liver cirrhosis. Lancet. (2021) 398:1359–76. doi: 10.1016/S0140-6736(21)01374-X
3. Pimpin, L, Cortez-Pinto, H, Negro, F, Corbould, E, Lazarus, JV, Webber, L, et al. Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies. J Hepatol. (2018) 69:718–35. doi: 10.1016/j.jhep.2018.05.011
4. Xiao, J, Wang, F, Wong, NK, He, J, Zhang, R, Sun, R, et al. Global liver disease burdens and research trends: analysis from a Chinese perspective. J Hepatol. (2019) 71:212–21. doi: 10.1016/j.jhep.2019.03.004
5. Parola, M, and Pinzani, M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
6. Albillos, A, de Gottardi, A, and Rescigno, M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol. (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
7. Sender, R, Fuchs, S, and Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
9. Kennedy, KM, Gerlach, MJ, Adam, T, Heimesaat, MM, Rossi, L, Surette, MG, et al. Fetal meconium does not have a detectable microbiota before birth. Nat Microbiol. (2021) 6:865–73. doi: 10.1038/s41564-021-00904-0
10. Schoeler, M, and Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev Endocr Metab Disord. (2019) 20:461–72. doi: 10.1007/s11154-019-09512-0
11. Dinsmoor, AM, Aguilar-Lopez, M, Khan, NA, and Donovan, SM. A systematic review of dietary influences on fecal microbiota composition and function among healthy humans 1–20 years of age. Adv Nutr. (2021) 12:1734–50. doi: 10.1093/advances/nmab047
12. Han, Q, Wang, J, Li, W, Chen, ZJ, and Du, Y. Androgen-induced gut dysbiosis disrupts glucolipid metabolism and endocrinal functions in polycystic ovary syndrome. Microbiome. (2021) 9:101. doi: 10.1186/s40168-021-01046-5
13. Wang, Z, Chen, WH, Li, SX, He, ZM, Zhu, WL, Ji, YB, et al. Gut microbiota modulates the inflammatory response and cognitive impairment induced by sleep deprivation. Mol Psychiatry. (2021) 26:6277–92. doi: 10.1038/s41380-021-01113-1
14. Li, J, Jia, H, Cai, X, Zhong, H, Feng, Q, Sunagawa, S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. (2014) 32:834–41. doi: 10.1038/nbt.2942
15. Zhou, R, Fan, X, and Schnabl, B. Role of the intestinal microbiome in liver fibrosis development and new treatment strategies. Transl Res. (2019) 209:22–38. doi: 10.1016/j.trsl.2019.02.005
16. Mazagova, M, Wang, L, Anfora, AT, Wissmueller, M, Lesley, SA, Miyamoto, Y, et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. (2015) 29:1043–55. doi: 10.1096/fj.14-259515
17. Hamoud, AR, Weaver, L, Stec, DE, and Hinds, TD Jr. Bilirubin in the liver-gut signaling axis. Trends Endocrinol Metab. (2018) 29:140–50. doi: 10.1016/j.tem.2018.01.002
18. Inagaki, T, Moschetta, A, Lee, YK, Peng, L, Zhao, G, Downes, M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. (2006) 103:3920–5. doi: 10.1073/pnas.0509592103
19. Long, SL, Gahan, C, and Joyce, SA. Interactions between gut bacteria and bile in health and disease. Mol Aspects Med. (2017) 56:54–65. doi: 10.1016/j.mam.2017.06.002
20. Munteanu, D, Negru, A, Radulescu, M, Mihailescu, R, Arama, SS, and Arama, V. Evaluation of bacterial translocation in patients with chronic HCV infection. Rom J Intern Med. (2014) 52:91–6.
21. Acharya, C, Sahingur, SE, and Bajaj, JS. Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight. (2017) 2:e94416. doi: 10.1172/jci.insight.94416
22. Wahlström, A, Sayin, SI, Marschall, HU, and Bäckhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. (2016) 24:41–50. doi: 10.1016/j.cmet.2016.05.005
23. Di Ciaula, A, Garruti, G, Lunardi Baccetto, R, Molina-Molina, E, Bonfrate, L, Wang, DQ, et al. Bile acid physiology. Ann Hepatol. (2017) 16:S4–S14. doi: 10.5604/01.3001.0010.5493
24. Macierzanka, A, Torcello-Gómez, A, Jungnickel, C, and Maldonado-Valderrama, J. Bile salts in digestion and transport of lipids. Adv Colloid Interf Sci. (2019) 274:102045. doi: 10.1016/j.cis.2019.102045
25. Bauer, TM, Schwacha, H, Steinbrückner, B, Brinkmann, FE, Ditzen, AK, Aponte, JJ, et al. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am J Gastroenterol. (2002) 97:2364–70. doi: 10.1111/j.1572-0241.2002.05791.x
26. Swann, JR, Want, EJ, Geier, FM, Spagou, K, Wilson, ID, Sidaway, JE, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci U S A. (2011) 108:4523–30. doi: 10.1073/pnas.1006734107
27. Li, T, and Chiang, JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. (2014) 66:948–83. doi: 10.1124/pr.113.008201
28. Verbeke, L, Farre, R, Verbinnen, B, Covens, K, Vanuytsel, T, Verhaegen, J, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol. (2015) 185:409–19. doi: 10.1016/j.ajpath.2014.10.009
29. Úbeda, M, Lario, M, Muñoz, L, Borrero, MJ, Rodríguez-Serrano, M, Sánchez-Díaz, AM, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol. (2016) 64:1049–57. doi: 10.1016/j.jhep.2015.12.010
30. Usami, M, Miyoshi, M, and Yamashita, H. Gut microbiota and host metabolism in liver cirrhosis. World J Gastroenterol. (2015) 21:11597–608. doi: 10.3748/wjg.v21.i41.11597
31. Milosevic, I, Vujovic, A, Barac, A, Djelic, M, Korac, M, Radovanovic Spurnic, A, et al. Gut-liver axis, gut microbiota, and its modulation in the management of liver diseases: a review of the literature. Int J Mol Sci. (2019) 20:395. doi: 10.3390/ijms20020395
32. Fang, J, Yu, CH, Li, XJ, Yao, JM, Fang, ZY, Yoon, SH, et al. Gut dysbiosis in nonalcoholic fatty liver disease: pathogenesis, diagnosis, and therapeutic implications. Front Cell Infect Microbiol. (2022) 12:997018. doi: 10.3389/fcimb.2022.997018
33. Bajaj, JS, Heuman, DM, Hylemon, PB, Sanyal, AJ, White, MB, Monteith, P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. (2014) 60:940–7. doi: 10.1016/j.jhep.2013.12.019
34. Xu, R, Tan, C, He, Y, Wu, Q, Wang, H, and Yin, J. Dysbiosis of gut microbiota and short-chain fatty acids in encephalitis: a Chinese pilot study. Front Immunol. (2020) 11:1994. doi: 10.3389/fimmu.2020.01994
35. Vallianou, N, Christodoulatos, GS, Karampela, I, Tsilingiris, D, Magkos, F, Stratigou, T, et al. Understanding the role of the gut microbiome and microbial metabolites in non-alcoholic fatty liver disease: current evidence and perspectives. Biomol Ther. (2021) 12:56. doi: 10.3390/biom12010056
36. Brunt, EM, Wong, VW, Nobili, V, Day, CP, Sookoian, S, Maher, JJ, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. (2015) 1:15080. doi: 10.1038/nrdp.2015.80
37. Keshavarzian, A, Farhadi, A, Forsyth, CB, Rangan, J, Jakate, S, Shaikh, M, et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol. (2009) 50:538–47. doi: 10.1016/j.jhep.2008.10.028
38. Dunagan, M, Chaudhry, K, Samak, G, and Rao, RK. Acetaldehyde disrupts tight junctions in Caco-2 cell monolayers by a protein phosphatase 2A-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. (2012) 303:G1356–64. doi: 10.1152/ajpgi.00526.2011
39. Boursier, J, Mueller, O, Barret, M, Machado, M, Fizanne, L, Araujo-Perez, F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. (2016) 63:764–75. doi: 10.1002/hep.28356
40. Safari, Z, and Gérard, P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell Mol Life Sci. (2019) 76:1541–58. doi: 10.1007/s00018-019-03011-w
41. Quigley, EM, Stanton, C, and Murphy, EF. The gut microbiota and the liver. Pathophysiological and clinical implications. J Hepatol. (2013) 58:1020–7. doi: 10.1016/j.jhep.2012.11.023
42. Tsiaoussis, GI, Assimakopoulos, SF, Tsamandas, AC, Triantos, CK, and Thomopoulos, KC. Intestinal barrier dysfunction in cirrhosis: current concepts in pathophysiology and clinical implications. World J Hepatol. (2015) 7:2058–68. doi: 10.4254/wjh.v7.i17.2058
43. Carnevale, R, Raparelli, V, Nocella, C, Bartimoccia, S, Novo, M, Severino, A, et al. Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implications for hypercoagulability in cirrhosis. J Hepatol. (2017) 67:950–6. doi: 10.1016/j.jhep.2017.07.002
44. Jiang, W, Wu, N, Wang, X, Chi, Y, Zhang, Y, Qiu, X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci Rep. (2015) 5:8096. doi: 10.1038/srep08096
45. Chen, Y, Yang, F, Lu, H, Wang, B, Chen, Y, Lei, D, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. (2011) 54:562–72. doi: 10.1002/hep.24423
46. Mutlu, EA, Gillevet, PM, Rangwala, H, Sikaroodi, M, Naqvi, A, Engen, PA, et al. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G966–78. doi: 10.1152/ajpgi.00380.2011
47. Kakiyama, G, Hylemon, PB, Zhou, H, Pandak, WM, Heuman, DM, Kang, DJ, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol. (2014) 306:G929–37. doi: 10.1152/ajpgi.00315.2013
48. Bajaj, JS, Kakiyama, G, Zhao, D, Takei, H, Fagan, A, Hylemon, P, et al. Continued alcohol misuse in human cirrhosis is associated with an impaired gut-liver axis. Alcohol Clin Exp Res. (2017) 41:1857–65. doi: 10.1111/acer.13498
49. Dubinkina, VB, Tyakht, AV, Odintsova, VY, Yarygin, KS, Kovarsky, BA, Pavlenko, AV, et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome. (2017) 5:141. doi: 10.1186/s40168-017-0359-2
50. Mouzaki, M, Comelli, EM, Arendt, BM, Bonengel, J, Fung, SK, Fischer, SE, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. (2013) 58:120–7. doi: 10.1002/hep.26319
51. Raman, M, Ahmed, I, Gillevet, PM, Probert, CS, Ratcliffe, NM, Smith, S, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2013) 11:868–875.e3. doi: 10.1016/j.cgh.2013.02.015
52. Zhu, L, Baker, SS, Gill, C, Liu, W, Alkhouri, R, Baker, RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. (2013) 57:601–9. doi: 10.1002/hep.26093
53. Tsochatzis, EA, Bosch, J, and Burroughs, AK. Liver cirrhosis. Lancet. (2014) 383:1749–61. doi: 10.1016/S0140-6736(14)60121-5
54. Preveden, T, Scarpellini, E, Milić, N, Luzza, F, and Abenavoli, L. Gut microbiota changes and chronic hepatitis C virus infection. Expert Rev Gastroenterol Hepatol. (2017) 11:813–9. doi: 10.1080/17474124.2017.1343663
55. Wang, Y, Pan, CQ, and Xing, H. Advances in gut microbiota of viral hepatitis cirrhosis. Biomed Res Int. (2019) 2019:1–9. doi: 10.1155/2019/9726786
56. Chen, Y, Ji, F, Guo, J, Shi, D, Fang, D, and Li, L. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci Rep. (2016) 6:34055. doi: 10.1038/srep34055
57. Sultan, S, El-Mowafy, M, Elgaml, A, El-Mesery, M, El Shabrawi, A, Elegezy, M, et al. Alterations of the treatment-naive gut microbiome in newly diagnosed hepatitis C virus infection. ACS Infect Dis. (2021) 7:1059–68. doi: 10.1021/acsinfecdis.0c00432
58. Bajaj, JS, Sterling, RK, Betrapally, NS, Nixon, DE, Fuchs, M, Daita, K, et al. HCV eradication does not impact gut dysbiosis or systemic inflammation in cirrhotic patients. Aliment Pharmacol Ther. (2016) 44:638–43. doi: 10.1111/apt.13732
59. Ponziani, FR, Putignani, L, Paroni Sterbini, F, Petito, V, Picca, A, Del Chierico, F, et al. Influence of hepatitis C virus eradication with direct-acting antivirals on the gut microbiota in patients with cirrhosis. Aliment Pharmacol Ther. (2018) 48:1301–11. doi: 10.1111/apt.15004
60. Xu, M, Wang, B, Fu, Y, Chen, Y, Yang, F, Lu, H, et al. Changes of fecal Bifidobacterium species in adult patients with hepatitis B virus-induced chronic liver disease. Microb Ecol. (2012) 63:304–13. doi: 10.1007/s00248-011-9925-5
61. Wei, X, Yan, X, Zou, D, Yang, Z, Wang, X, Liu, W, et al. Abnormal fecal microbiota community and functions in patients with hepatitis B liver cirrhosis as revealed by a metagenomic approach. BMC Gastroenterol. (2013) 13:175. doi: 10.1186/1471-230X-13-175
62. Qin, N, Yang, F, Li, A, Prifti, E, Chen, Y, Shao, L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. (2014) 513:59–64. doi: 10.1038/nature13568
63. Aly, AM, Adel, A, El-Gendy, AO, Essam, TM, and Aziz, RK. Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog. (2016) 8:42. doi: 10.1186/s13099-016-0124-2
64. Inoue, T, Nakayama, J, Moriya, K, Kawaratani, H, Momoda, R, Ito, K, et al. Gut dysbiosis associated with hepatitis C virus infection. Clin Infect Dis. (2018) 67:869–77. doi: 10.1093/cid/ciy205
65. Heidrich, B, Vital, M, Plumeier, I, Döscher, N, Kahl, S, Kirschner, J, et al. Intestinal microbiota in patients with chronic hepatitis C with and without cirrhosis compared with healthy controls. Liver Int. (2018) 38:50–8. doi: 10.1111/liv.13485
66. Zeng, Y, Chen, S, Fu, Y, Wu, W, Chen, T, Chen, J, et al. Gut microbiota dysbiosis in patients with hepatitis B virus-induced chronic liver disease covering chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. J Viral Hepat. (2020) 27:143–55. doi: 10.1111/jvh.13216
67. Hepatobiliary Disease Study Group, Chinese Society of Gastroenterology, Association CM. Consensus for management of portal vein thrombosis in liver cirrhosis. J Dig Dis. (2020) 22:176–86. doi: 10.1111/1751-2980.12970
68. Loudin, M, and Ahn, J. Portal vein thrombosis in cirrhosis. J Clin Gastroenterol. (2017) 51:579–85. doi: 10.1097/MCG.0000000000000834
69. Stine, JG, Wang, J, Shah, PM, Argo, CK, Intagliata, N, Uflacker, A, et al. Decreased portal vein velocity is predictive of the development of portal vein thrombosis: a matched case-control study. Liver Int. (2018) 38:94–101. doi: 10.1111/liv.13500
70. Hasan, RA, Koh, AY, and Zia, A. The gut microbiome and thromboembolism. Thromb Res. (2020) 189:77–87. doi: 10.1016/j.thromres.2020.03.003
71. Moore, KL, Andreoli, SP, Esmon, NL, Esmon, CT, and Bang, NU. Endotoxin enhances tissue factor and suppresses thrombomodulin expression of human vascular endothelium in vitro. J Clin Invest. (1987) 79:124–30. doi: 10.1172/JCI112772
72. Whitfield, C, and Trent, MS. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem. (2014) 83:99–128. doi: 10.1146/annurev-biochem-060713-035600
73. Saghazadeh, A, and Rezaei, N. Inflammation as a cause of venous thromboembolism. Crit Rev Oncol Hematol. (2016) 99:272–85. doi: 10.1016/j.critrevonc.2016.01.007
74. Branchford, BR, and Carpenter, SL. The role of inflammation in venous thromboembolism. Front Pediatr. (2018) 6:142. doi: 10.3389/fped.2018.00142
75. Holstein, K, Matysiak, A, Witt, L, Sievers, B, Beckmann, L, Haddad, M, et al. LPS-induced expression and release of monocyte tissue factor in patients with haemophilia. Ann Hematol. (2020) 99:1531–42. doi: 10.1007/s00277-020-04075-6
76. Trebicka, J, Krag, A, Gansweid, S, Appenrodt, B, Schiedermaier, P, Sauerbruch, T, et al. Endotoxin and tumor necrosis factor-receptor levels in portal and hepatic vein of patients with alcoholic liver cirrhosis receiving elective transjugular intrahepatic portosystemic shunt. Eur J Gastroenterol Hepatol. (2011) 23:1218–25. doi: 10.1097/MEG.0b013e32834a75dc
77. Balmer, ML, Slack, E, de Gottardi, A, Lawson, MA, Hapfelmeier, S, Miele, L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. (2014) 6:237ra66. doi: 10.1126/scitranslmed.3008618
78. Seki, E, and Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. (2012) 590:447–58. doi: 10.1113/jphysiol.2011.219691
79. Nakamoto, N, and Kanai, T. Role of toll-like receptors in immune activation and tolerance in the liver. Front Immunol. (2014) 5:221. doi: 10.3389/fimmu.2014.00221
80. Custodio-Chablé, SJ, Lezama, RA, and Reyes-Maldonado, E. Platelet activation as a trigger factor for inflammation and atherosclerosis. Cir Cir. (2020) 88:233–43. doi: 10.24875/CIRU.19000725
81. Tsuchida, T, and Friedman, SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
82. Reinhardt, C. The gut microbiota as an influencing factor of arterial thrombosis. Hamostaseologie. (2019) 39:173–9. doi: 10.1055/s-0038-1675357
83. Jäckel, S, Kiouptsi, K, Lillich, M, Hendrikx, T, Khandagale, A, Kollar, B, et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via toll-like receptor-2. Blood. (2017) 130:542–53. doi: 10.1182/blood-2016-11-754416
84. Shin, HS, Xu, F, Bagchi, A, Herrup, E, Prakash, A, Valentine, C, et al. Bacterial lipoprotein TLR2 agonists broadly modulate endothelial function and coagulation pathways in vitro and in vivo. J Immunol. (2011) 186:1119–30. doi: 10.4049/jimmunol.1001647
85. Cognasse, F, Hamzeh, H, Chavarin, P, Acquart, S, Genin, C, and Garraud, O. Evidence of toll-like receptor molecules on human platelets. Immunol Cell Biol. (2005) 83:196–8. doi: 10.1111/j.1440-1711.2005.01314.x
86. Blair, P, Rex, S, Vitseva, O, Beaulieu, L, Tanriverdi, K, Chakrabarti, S, et al. Stimulation of toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. (2009) 104:346–54. doi: 10.1161/CIRCRESAHA.108.185785
87. Rex, S, Beaulieu, LM, Perlman, DH, Vitseva, O, Blair, PS, McComb, ME, et al. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein–protein interactions, and alpha-granule release. Thromb Haemost. (2009) 102:97–110. doi: 10.1160/TH08-08-0513
88. Duttaroy, AK. Role of gut microbiota and their metabolites on atherosclerosis, hypertension and human blood platelet function: a review. Nutrients. (2021) 13:13. doi: 10.3390/nu13010144
89. Engelmann, B, and Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
90. Kimball, AS, Obi, AT, Diaz, JA, and Henke, PK. The emerging role of NETs in venous thrombosis and immunothrombosis. Front Immunol. (2016) 7:236. doi: 10.3389/fimmu.2016.00236
91. Brill, A, Fuchs, TA, Savchenko, AS, Thomas, GM, Martinod, K, De Meyer, SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. (2012) 10:136–44. doi: 10.1111/j.1538-7836.2011.04544.x
92. Seo, JD, Gu, JY, Jung, HS, Kim, YJ, and Kim, HK. Contact system activation and neutrophil extracellular trap markers: risk factors for portal vein thrombosis in patients with hepatocellular carcinoma. Clin Appl Thromb Hemost. (2019) 25:107602961882531. doi: 10.1177/1076029618825310
93. Wang, Y, Luo, L, Braun, OÖ, Westman, J, Madhi, R, Herwald, H, et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci Rep. (2018) 8:4020. doi: 10.1038/s41598-018-22156-5
94. Semeraro, F, Ammollo, CT, Morrissey, JH, Dale, GL, Friese, P, Esmon, NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
95. McDonald, D, Ackermann, G, Khailova, L, Baird, C, Heyland, D, Kozar, R, et al. Extreme Dysbiosis of the microbiome in critical illness. mSphere. (2016) 1:e00199. doi: 10.1128/mSphere.00199-16
96. Zhu, Y, Li, Q, and Jiang, H. Gut microbiota in atherosclerosis: focus on trimethylamine N-oxide. APMIS. (2020) 128:353–66. doi: 10.1111/apm.13038
97. Tilg, H. A gut feeling about thrombosis. N Engl J Med. (2016) 374:2494–6. doi: 10.1056/NEJMcibr1604458
98. Zhu, W, Gregory, JC, Org, E, Buffa, JA, Gupta, N, Wang, Z, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cells. (2016) 165:111–24. doi: 10.1016/j.cell.2016.02.011
99. Tripodi, A, Primignani, M, Chantarangkul, V, Dell’Era, A, Clerici, M, de Franchis, R, et al. An imbalance of pro- vs. anti-coagulation factors in plasma from patients with cirrhosis. Gastroenterology. (2009) 137:2105–11. doi: 10.1053/j.gastro.2009.08.045
100. Basili, S, Carnevale, R, Nocella, C, Bartimoccia, S, Raparelli, V, Talerico, G, et al. Serum albumin is inversely associated with portal vein thrombosis in cirrhosis. Hepatol Commun. (2019) 3:504–12. doi: 10.1002/hep4.1317
101. Wang, T, Xu, J, Fu, L, and Li, L. Hypertriglyceridemia is associated with platelet hyperactivation in metabolic syndrome patients. Int J Clin Pract. (2020) 74:e13508. doi: 10.1111/ijcp.13508
102. Ciavarella, A, Gnocchi, D, Custodero, C, Lenato, GM, Fiore, G, Sabbà, C, et al. Translational insight into prothrombotic state and hypercoagulation in nonalcoholic fatty liver disease. Thromb Res. (2021) 198:139–50. doi: 10.1016/j.thromres.2020.12.002
103. Stine, JG, Shah, NL, Argo, CK, Pelletier, SJ, Caldwell, SH, and Northup, PG. Increased risk of portal vein thrombosis in patients with cirrhosis due to nonalcoholic steatohepatitis. Liver Transpl. (2015) 21:1016–21. doi: 10.1002/lt.24134
104. Balan, G, Trifan, A, Botezatu, D, and Anton, C. Spontaneous bacterial peritonitis: a severe complication of liver cirrhosis. Rev Med Chir Soc Med Nat Iasi. (2011) 115:38–44.
105. Song, DS. Spontaneous bacterial peritonitis. Korean J Gastroenterol. (2018) 72:56–63. doi: 10.4166/kjg.2018.72.2.56
106. Llovet, JM, Bartolí, R, March, F, Planas, R, Viñado, B, Cabré, E, et al. Translocated intestinal bacteria cause spontaneous bacterial peritonitis in cirrhotic rats: molecular epidemiologic evidence. J Hepatol. (1998) 28:307–13. doi: 10.1016/0168-8278(88)80018-7
107. Papp, M, Farkas, A, Udvardy, M, and Tornai, I. Bacterial infections in cirrhosis. Orv Hetil. (2007) 148:387–95. doi: 10.1556/oh.2007.27882
108. Kim, JH, Jeon, YD, Jung, IY, Ahn, MY, Ahn, HW, Ahn, JY, et al. Predictive factors of spontaneous bacterial peritonitis caused by Gram-positive bacteria in patients with cirrhosis. Medicine. (2016) 95:e3489. doi: 10.1097/MD.0000000000003489
109. Bajaj, JS, Vargas, HE, Reddy, KR, Lai, JC, O’Leary, JG, Tandon, P, et al. Association between intestinal microbiota collected at hospital admission and outcomes of patients with cirrhosis. Clin Gastroenterol Hepatol. (2019) 17:756–765.e3. doi: 10.1016/j.cgh.2018.07.022
110. Singh, A, Cresci, GA, and Kirby, DF. Proton pump inhibitors: risks and rewards and emerging consequences to the gut microbiome. Nutr Clin Pract. (2018) 33:614–24. doi: 10.1002/ncp.10181
111. Tranah, TH, Edwards, LA, Schnabl, B, and Shawcross, DL. Targeting the gut-liver-immune axis to treat cirrhosis. Gut. (2021) 70:982–94. doi: 10.1136/gutjnl-2020-320786
112. Wang, YD, Chen, WD, Moore, DD, and Huang, W. FXR: a metabolic regulator and cell protector. Cell Res. (2008) 18:1087–95. doi: 10.1038/cr.2008.289
113. Sorribas, M, Jakob, MO, Yilmaz, B, Li, H, Stutz, D, Noser, Y, et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J Hepatol. (2019) 71:1126–40. doi: 10.1016/j.jhep.2019.06.017
114. Hanouneh, MA, Hanouneh, IA, Hashash, JG, Law, R, Esfeh, JM, Lopez, R, et al. The role of rifaximin in the primary prophylaxis of spontaneous bacterial peritonitis in patients with liver cirrhosis. J Clin Gastroenterol. (2012) 46:709–15. doi: 10.1097/MCG.0b013e3182506dbb
115. Moreau, R, Elkrief, L, Bureau, C, Perarnau, JM, Thévenot, T, Saliba, F, et al. Effects of long-term norfloxacin therapy in patients with advanced cirrhosis. Gastroenterology. (2018) 155:1816–1827.e9. doi: 10.1053/j.gastro.2018.08.026
116. Elfert, A, Abo Ali, L, Soliman, S, Ibrahim, S, and Abd-Elsalam, S. Randomized-controlled trial of rifaximin versus norfloxacin for secondary prophylaxis of spontaneous bacterial peritonitis. Eur J Gastroenterol Hepatol. (2016) 28:1450–4. doi: 10.1097/MEG.0000000000000724
117. Madsen, BS, Havelund, T, and Krag, A. Targeting the gut-liver axis in cirrhosis: antibiotics and non-selective β-blockers. Adv Ther. (2013) 30:659–70. doi: 10.1007/s12325-013-0044-1
118. Reiberger, T, Ferlitsch, A, Payer, BA, Mandorfer, M, Heinisch, BB, Hayden, H, et al. Non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J Hepatol. (2013) 58:911–21. doi: 10.1016/j.jhep.2012.12.011
119. Ahluwalia, V, Betrapally, NS, Hylemon, PB, White, MB, Gillevet, PM, Unser, AB, et al. Impaired gut-liver-brain axis in patients with cirrhosis. Sci Rep. (2016) 6:26800. doi: 10.1038/srep26800
120. Wijdicks, EF. Hepatic encephalopathy. N Engl J Med. (2016) 375:1660–70. doi: 10.1056/NEJMra1600561
121. Olde Damink, SW, Deutz, NE, Dejong, CH, Soeters, PB, and Jalan, R. Interorgan ammonia metabolism in liver failure. Neurochem Int. (2002) 41:177–88. doi: 10.1016/S0197-0186(02)00040-2
122. Butterworth, RF. The liver-brain axis in liver failure: neuroinflammation and encephalopathy. Nat Rev Gastroenterol Hepatol. (2013) 10:522–8. doi: 10.1038/nrgastro.2013.99
123. Bajaj, JS, Ridlon, JM, Hylemon, PB, Thacker, LR, Heuman, DM, Smith, S, et al. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G168–75. doi: 10.1152/ajpgi.00190.2011
124. Kaji, K, Takaya, H, Saikawa, S, Furukawa, M, Sato, S, Kawaratani, H, et al. Rifaximin ameliorates hepatic encephalopathy and endotoxemia without affecting the gut microbiome diversity. World J Gastroenterol. (2017) 23:8355–66. doi: 10.3748/wjg.v23.i47.8355
125. Bajaj, JS, Salzman, N, Acharya, C, Takei, H, Kakiyama, G, Fagan, A, et al. Microbial functional change is linked with clinical outcomes after capsular fecal transplant in cirrhosis. JCI Insight. (2019) 4:133410. doi: 10.1172/jci.insight.133410
126. Campion, D, Giovo, I, Ponzo, P, Saracco, GM, Balzola, F, and Alessandria, C. Dietary approach and gut microbiota modulation for chronic hepatic encephalopathy in cirrhosis. World J Hepatol. (2019) 11:489–512. doi: 10.4254/wjh.v11.i6.489
127. Jia, W, Rajani, C, Kaddurah-Daouk, R, and Li, H. Expert insights: the potential role of the gut microbiome-bile acid-brain axis in the development and progression of Alzheimer’s disease and hepatic encephalopathy. Med Res Rev. (2020) 40:1496–507. doi: 10.1002/med.21653
128. O’Mahony, SM, Clarke, G, Borre, YE, Dinan, TG, and Cryan, JF. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res. (2015) 277:32–48. doi: 10.1016/j.bbr.2014.07.027
129. Iebba, V, Guerrieri, F, Di Gregorio, V, Levrero, M, Gagliardi, A, Santangelo, F, et al. Combining amplicon sequencing and metabolomics in cirrhotic patients highlights distinctive microbiota features involved in bacterial translocation, systemic inflammation and hepatic encephalopathy. Sci Rep. (2018) 8:8210. doi: 10.1038/s41598-018-26509-y
130. Bosch, J, Groszmann, RJ, and Shah, VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: how changes in paradigm are leading to successful new treatments. J Hepatol. (2015) 62:S121–30. doi: 10.1016/j.jhep.2015.01.003
131. Arab, JP, Martin-Mateos, RM, and Shah, VH. Gut-liver axis, cirrhosis and portal hypertension: the chicken and the egg. Hepatol Int. (2018) 12:24–33. doi: 10.1007/s12072-017-9798-x
132. Trebicka, J, Reiberger, T, and Laleman, W. Gut-liver axis links portal hypertension to acute-on-chronic liver failure. Visc Med. (2018) 34:270–5. doi: 10.1159/000490262
133. Simbrunner, B, Mandorfer, M, Trauner, M, and Reiberger, T. Gut-liver axis signaling in portal hypertension. World J Gastroenterol. (2019) 25:5897–917. doi: 10.3748/wjg.v25.i39.5897
134. Steib, CJ, Hartmann, AC, Hesler, C, Benesic, A, Hennenberg, M, Bilzer, M, et al. Intraperitoneal LPS amplifies portal hypertension in rat liver fibrosis. Lab Investig (2010) 90:1024–1032. doi: 10.1038/labinvest.2010.60
135. Villanueva, C, Albillos, A, Genescà, J, Abraldes, JG, Calleja, JL, Aracil, C, et al. Development of hyperdynamic circulation and response to β-blockers in compensated cirrhosis with portal hypertension. Hepatology. (2016) 63:197–206. doi: 10.1002/hep.28264
136. Reiberger, T, and Mandorfer, M. Beta adrenergic blockade and decompensated cirrhosis. J Hepatol. (2017) 66:849–59. doi: 10.1016/j.jhep.2016.11.001
137. Schwabl, P, Hambruch, E, Seeland, BA, Hayden, H, Wagner, M, Garnys, L, et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol. (2017) 66:724–33. doi: 10.1016/j.jhep.2016.12.005
138. Zhu, Q, Zou, L, Jagavelu, K, Simonetto, DA, Huebert, RC, Jiang, ZD, et al. Intestinal decontamination inhibits TLR4 dependent fibronectin-mediated cross-talk between stellate cells and endothelial cells in liver fibrosis in mice. J Hepatol. (2012) 56:893–9. doi: 10.1016/j.jhep.2011.11.013
139. Francoz, C, Durand, F, Kahn, JA, Genyk, YS, and Nadim, MK. Hepatorenal syndrome. Clin J Am Soc Nephrol. (2019) 14:774–81. doi: 10.2215/CJN.12451018
140. Amin, AA, Alabsawy, EI, Jalan, R, and Davenport, A. Epidemiology, pathophysiology, and management of hepatorenal syndrome. Semin Nephrol. (2019) 39:17–30. doi: 10.1016/j.semnephrol.2018.10.002
141. Adebayo, D, Neong, SF, and Wong, F. Ascites and hepatorenal syndrome. Clin Liver Dis. (2019) 23:659–82. doi: 10.1016/j.cld.2019.06.002
142. Huang, LT, Hung, JF, Chen, CC, Hsieh, CS, Yu, HR, Hsu, CN, et al. Endotoxemia exacerbates kidney injury and increases asymmetric dimethylarginine in young bile duct-ligated rats. Shock. (2012) 37:441–8. doi: 10.1097/SHK.0b013e318244b787
143. Peng, JL, Techasatian, W, Hato, T, and Liangpunsakul, S. Role of endotoxemia in causing renal dysfunction in cirrhosis. J Investig Med. (2020) 68:26–9. doi: 10.1136/jim-2019-001056
144. Shah, N, Dhar, D, El Zahraa, MF, Habtesion, A, Davies, NA, Jover-Cobos, M, et al. Prevention of acute kidney injury in a rodent model of cirrhosis following selective gut decontamination is associated with reduced renal TLR4 expression. J Hepatol. (2012) 56:1047–53. doi: 10.1016/j.jhep.2011.11.024
145. Kalambokis, GN, Mouzaki, A, Rodi, M, Pappas, K, Fotopoulos, A, Xourgia, X, et al. Rifaximin improves systemic hemodynamics and renal function in patients with alcohol-related cirrhosis and ascites. Clin Gastroenterol Hepatol. (2012) 10:815–8. doi: 10.1016/j.cgh.2012.02.025
146. Garcia-Martinez, R, Noiret, L, Sen, S, Mookerjee, R, and Jalan, R. Albumin infusion improves renal blood flow autoregulation in patients with acute decompensation of cirrhosis and acute kidney injury. Liver Int. (2015) 35:335–43. doi: 10.1111/liv.12528
147. Vincent, JL, De Backer, D, and Wiedermann, CJ. Fluid management in sepsis: the potential beneficial effects of albumin. J Crit Care. (2016) 35:161–7. doi: 10.1016/j.jcrc.2016.04.019
148. Liangpunsakul, S, and Agarwal, R. Altered circadian hemodynamic and renal function in cirrhosis. Nephrol Dial Transplant. (2017) 32:333–42. doi: 10.1093/ndt/gfw014
149. Koch, DG, and Fallon, MB. Hepatopulmonary syndrome. Clin Liver Dis. (2014) 18:407–20. doi: 10.1016/j.cld.2014.01.003
150. Zhang, ZJ, and Yang, CQ. Progress in investigating the pathogenesis of hepatopulmonary syndrome. Hepatobiliary Pancreat Dis Int. (2010) 9:355–60.
151. Zhang, J, and Fallon, MB. Hepatopulmonary syndrome: update on pathogenesis and clinical features. Nat Rev Gastroenterol Hepatol. (2012) 9:539–49. doi: 10.1038/nrgastro.2012.123
152. Tumgor, G. Cirrhosis and hepatopulmonary syndrome. World J Gastroenterol. (2014) 20:2586–94. doi: 10.3748/wjg.v20.i10.2586
153. Zhang, HY, Han, DW, Zhao, ZF, Liu, MS, Wu, YJ, Chen, XM, et al. Multiple pathogenic factor-induced complications of cirrhosis in rats: a new model of hepatopulmonary syndrome with intestinal endotoxemia. World J Gastroenterol. (2007) 13:3500–7. doi: 10.3748/wjg.v13.i25.3500
154. Sztrymf, B, Libert, JM, Mougeot, C, Lebrec, D, Mazmanian, M, Humbert, M, et al. Cirrhotic rats with bacterial translocation have higher incidence and severity of hepatopulmonary syndrome. J Gastroenterol Hepatol. (2005) 20:1538–44. doi: 10.1111/j.1440-1746.2005.03914.x
155. Gupta, S, Faughnan, ME, Lilly, L, Hutchison, S, Fowler, R, and Bayoumi, AM. Norfloxacin therapy for hepatopulmonary syndrome: a pilot randomized controlled trial. Clin Gastroenterol Hepatol. (2010) 8:1095–8. doi: 10.1016/j.cgh.2010.08.011
156. Llovet, JM, Zucman-Rossi, J, Pikarsky, E, Sangro, B, Schwartz, M, Sherman, M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. (2016) 2:16018. doi: 10.1038/nrdp.2016.18
157. Marengo, A, Rosso, C, and Bugianesi, E. Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med. (2016) 67:103–17. doi: 10.1146/annurev-med-090514-013832
158. Liu, Q, Li, F, Zhuang, Y, Xu, J, Wang, J, Mao, X, et al. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. (2019) 11:1. doi: 10.1186/s13099-018-0281-6
159. Nicoletti, A, Ponziani, FR, Biolato, M, Valenza, V, Marrone, G, Sganga, G, et al. Intestinal permeability in the pathogenesis of liver damage: from non-alcoholic fatty liver disease to liver transplantation. World J Gastroenterol. (2019) 25:4814–34. doi: 10.3748/wjg.v25.i33.4814
160. Liu, WT, Jing, YY, Gao, L, Li, R, Yang, X, Pan, XR, et al. Lipopolysaccharide induces the differentiation of hepatic progenitor cells into myofibroblasts constitutes the hepatocarcinogenesis-associated microenvironment. Cell Death Differ. (2020) 27:85–101. doi: 10.1038/s41418-019-0340-7
161. Yu, LX, Yan, HX, Liu, Q, Yang, W, Wu, HP, Dong, W, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. (2010) 52:1322–33. doi: 10.1002/hep.23845
162. Ponziani, FR, Bhoori, S, Castelli, C, Putignani, L, Rivoltini, L, Del Chierico, F, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. (2019) 69:107–20. doi: 10.1002/hep.30036
163. Piñero, F, Vazquez, M, Baré, P, Rohr, C, Mendizabal, M, Sciara, M, et al. A different gut microbiome linked to inflammation found in cirrhotic patients with and without hepatocellular carcinoma. Ann Hepatol. (2019) 18:480–7. doi: 10.1016/j.aohep.2018.10.003
164. Duan, Y, Llorente, C, Lang, S, Brandl, K, Chu, H, Jiang, L, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. (2019) 575:505–11. doi: 10.1038/s41586-019-1742-x
165. Tili, E, Michaille, JJ, Piurowski, V, Rigot, B, and Croce, CM. MicroRNAs in intestinal barrier function, inflammatory bowel disease and related cancers-their effects and therapeutic potentials. Curr Opin Pharmacol. (2017) 37:142–50. doi: 10.1016/j.coph.2017.10.010
166. Macnaughtan, J, Ranchal, I, Soeda, J, Sawhney, R, Oben, J, Davies, N, et al. Oral therapy with non-absorbable carbons of controlled porosity (YAQ-001) selectively modulates stool microbiome and its function and this is associated with restoration of immune function and inflammasome activation. J Hepatol. (2015) 62:S240–13. doi: 10.1016/S0168-8278(15)30110-0
Keywords: liver cirrhosis, dysbiosis, complication, portal vein thrombosis, gut-liver axis
Citation: Nie G, Zhang H, Xie D, Yan J and Li X (2024) Liver cirrhosis and complications from the perspective of dysbiosis. Front. Med. 10:1320015. doi: 10.3389/fmed.2023.1320015
Edited by:
Huan Tong, Sichuan University, ChinaReviewed by:
Takahiko Shiba, Tokyo Medical and Dental University, JapanCopyright © 2024 Nie, Zhang, Xie, Yan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xun Li, bHhkcjIxQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.