 Jing Zhuang1
Jing Zhuang1 Ailima Aierken1
Ailima Aierken1 Dilina Yalikun1
Dilina Yalikun1 Jun Zhang1
Jun Zhang1 Xiaoqin Wang1
Xiaoqin Wang1 Yongfang Ren2
Yongfang Ren2 Xuefei Tian3*
Xuefei Tian3* Hong Jiang1*
Hong Jiang1*- 1Division of Nephrology, Department of Internal Medicine, People’s Hospital of Xinjiang Uygur Autonomous Region, Ürümqi, China
- 2Department of Radiology and Medical Imaging, People’s Hospital of Xinjiang Uygur Autonomous Region, Ürümqi, China
- 3Section of Nephrology, Department of Internal Medicine, Yale University School of Medicine, New Haven, CT, United States
Introduction: Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic disorder. The PKD1 gene is responsible for the majority of ADPKD cases, and the mutations in this gene exhibit high genetic diversity. This study aimed to investigate the association between genotype and phenotype in ADPKD patients with PKD1 gene mutations through pedigree analysis.
Methods: Eight Chinese pedigrees affected by ADPKD were analyzed using whole-exome sequencing (WES) on peripheral blood DNA. The identified variants were validated using Sanger sequencing, and clinical data from the patients and their families were collected and analyzed.
Results: Nine novel mutation sites in PKD1 were discovered across the pedigrees, including c.4247T > G, c.3298_3301delGAGT, c.4798A > G, c.7567G > A, c.11717G > C, c.7703 + 5G > C, c.3296G > A, c.8515_8516insG, and c.5524C > A. These mutations were found to be associated with a range of clinical phenotypes, including chronic kidney disease, hypertension, and polycystic liver. The age of onset and disease progression displayed significant heterogeneity among the pedigrees, with some individuals exhibiting early onset and rapid disease progression, while others remained asymptomatic or had milder disease symptoms. Inheritance patterns supported autosomal dominant inheritance, as affected individuals inherited the mutations from affected parents. However, there were instances of individuals carrying the mutations who remained asymptomatic or exhibited milder disease phenotypes.
Conclusion: This study highlights the importance of comprehensive genotype analysis in understanding the progression and prognosis of ADPKD. The identification of novel mutation sites expands our knowledge of PKD1 gene mutations. These findings contribute to a better understanding of the disease and may have implications for personalized therapeutic strategies.
Introduction
Polycystic kidney disease (PKD) is a genetic disease that can be inherited in two different ways: autosomal dominant inheritance and autosomal recessive inheritance. Autosomal dominant polycystic kidney disease (ADPKD) is the most common single-gene hereditary kidney disease, with an incidence rate of about 1/1000-1/2500 (1, 2). ADPKD is classified into two types, polycystic kidney disease type 1 (OMIM173900) and polycystic kidney disease type 2 (OMIM613095). The discovery and cloning of the PKD1 and PKD2 genes have facilitated the development of molecular diagnostics based on DNA sequencing (3). About 78% of ADPKD cases are caused by PKD1 gene mutations on chromosome 16, and about 15% of cases are caused by PKD2 gene mutations on chromosome 4 (4, 5). The PKD1 and PKD2 genes encode polycystin 1 (PC1) and polycystin 2 (PC2), respectively, which are primarily located in the primary cilium of renal tubular epithelial cells. In addition to mutations in the PKD1 and PKD2 genes, other mutations have been found in some ADPKD patients, including GANAB, DNAJB11, IFT140 and other gene mutations that can cause atypical ADPKD (6–8). Mutations in genes related to autosomal dominant polycystic liver disease (PRKCSH, SEC63, LRP5, ALG8, and SEC61B) may also cause mild kidney phenotypes (9). Clinical manifestations of ADPKD include hypertension, hematuria, proteinuria, and decreased kidney function. Patients with ADPKD may also experience other complications involving multiple organs or tissues, such as liver cysts, pancreatic cysts, spleen cysts, and intracranial aneurysms.
About 50% of ADPKD patients usually progress to end-stage kidney disease (ESKD) by the age of 60 (10, 11). Evidence shows that about 5−10% of patients receiving kidney replacement therapy (KRT) worldwide have ADPKD, and it accounts for about 5% of ESKD patients in China, affecting about 1.25 million people (12). ADPKD is the fourth most common cause of ESKD, and it brings a heavy burden to society and families (3).
Currently, there have been significant advancements in understanding the pathogenesis of ADPKD in the fields of cell biology and molecular mechanisms. Mutations in the main genes PKD1 and PKD2 lead to various abnormalities in downstream cell signal transduction, including intracellular calcium disorders, abnormalities in cAMP and mTOR signaling pathways, and classical and non-classical Wnt pathway signal activation. These abnormalities partially explain the pathogenesis and progression of kidney cysts. In addition to the genetically abnormal PKD genes, several studies have shown that the formation of cysts requires the normal haplotype of secondary acquired somatic cell loss, known as the “second hit” hypothesis. Additionally, a “threshold” model for cyst formation has been proposed, suggesting that cyst formation may be triggered when the level of polycystin protein in renal tubular epithelial cells falls below a critical threshold (13). Despite significant progress in understanding the pathogenesis of ADPKD, several issues remain unresolved. For instance, while PC2 is known to function as a TRP-like calcium channel and interact with PC1 to regulate primary cilia signaling pathways, the exact role of calcium ions in the pathogenesis of ADPKD is still not fully understood. Moreover, the mechanism by which decreased polycystin signaling in cilia leads to the formation of cystic lesions remains unclear, highlighting the need for further basic and clinical research to expand our understanding of ADPKD’s pathogenesis (13, 14).
Early diagnosis of PKD patients, identification of pathogenic mutations in ADPKD families, regular monitoring of clinical indicators, and timely symptomatic treatment can help delay the progression of ADPKD. Genetic diagnosis is increasingly being used to provide information for the clinical management of PKD nephropathy patients. This approach can result in a more accurate diagnosis, targeted disease monitoring, treatment, and family genetic counseling (15). Studies have shown a high correlation between the phenotype and genotype of ADPKD patients, with pathogenic gene mutations being the primary determinant of disease progression (16). Several studies have shown that the type of mutation is closely related to the severity of ADPKD. Patients with PKD2 mutations generally have a milder form of kidney disease compared to those with PKD1 mutations (17). Additionally, frameshift mutations, nonsense mutations, and splice site changes in PKD1 truncated mutations are associated with more severe disease than non-truncated mutations, such as missense mutations and in-frame insertion/deletion (18–20). Furthermore, mutations in non-PKD1 or PKD2 genes can also result in the development of renal cysts. For example, the deletion of the tuberous sclerosis complex 2 (TSC2) gene adjacent to the PKD1 gene can cause a rare and severe form of polycystic kidney disease that often leads to ESKD in childhood (21).
In some cases of ADPKD, mutations in PKD1 or PKD2 genes are not detected, and somatic mosaicism may be present (22). These mutations are not concentrated in any specific location of the gene. Identifying gene mutations associated with ADPKD can aid in diagnosis and predicting disease progression. Ultrasound, the most commonly used diagnostic method, may have low sensitivity, particularly in high-risk individuals under 40 years of age, hindering early or presymptomatic diagnosis (23). With the advancements in precision medicine and increased understanding of cystic diseases, whole-exome sequencing (WES) using next-generation sequencing may become a vital tool in screening for ADPKD and providing accurate diagnoses and effective treatments.
This study involved the use of whole-exome sequencing (WES) to screen eight Chinese patients who had been diagnosed with ADPKD and then verified the results with Sanger sequencing in their family members. We also analyzed the pathogenicity of the mutations to identify the specific pathogenic mutation gene responsible for the disease in each patient and their family. Some of the gene mutations identified in this study have not been previously reported, which has important implications for both clinical practice and further research on ADPKD.
Materials and methods
Patients
From November 2021 to November 2022, we conducted a study on ADPKD patients admitted to the People’s Hospital of Xinjiang Uygur Autonomous Region (Ürümqi, China). The diagnosis of ADPKD was made based on the criteria outlined in Table 1, as previously described (12). A total of eight patients from distinct families were enrolled in the study. Blood samples were obtained from each patient and their immediate family members via peripheral venous collection.
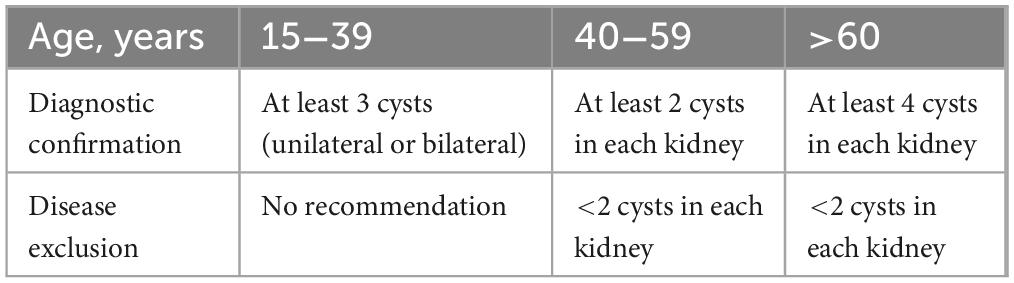
Table 1. Criteria for diagnosis and exclusion of autosomal dominant polycystic kidney disease.
The study was approved by the Medical Ethics Committee of the People’s Hospital of the Autonomous Region and was conducted in compliance with the Declaration of Helsinki. The study involving human participants was reviewed and approved by the Medical Ethics Committee of the People’s Hospital of Xinjiang Uygur Autonomous Region, and written informed consent was obtained from all participants (Approval number: KY2023032201).
Acquisition of clinical data
Abdominal ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI) were conducted on the proband and their family members to gather clinical data based on their medical history and clinical manifestations. Laboratory data, including blood and urine sample analysis, as well as biochemical tests, were also collected.
Target gene enrichment, library construction, and capture sequencing
A peripheral blood sample (5 mL) was drawn from participants, and genomic DNA (gDNA) was extracted using the MagPure Buffy Coat DNA Midi KF Kit as per the manufacturer’s instructions. BGI’s enzyme kit (Segmentase, BGI) was used to fragment the gDNA into fragments of 100−500 bp, which were then subjected to magnetic bead selection to collect fragments of 280−320 bp. After repairing the ends of the fragments, “A” bases were added to the 3′ overhangs, enabling them to pair with a special adapter containing “T” bases. The library with adapters was then amplified using pre-capture ligation-mediated PCR (LM-PCR), purified, and enriched by array hybridization (Roche KAPA HyperExome, Madison, WI, USA) for 16−24 h at 47°C, followed by elution and post-capture amplification. The enriched products were assessed for the level of enrichment using the Agilent 2100 Bioanalyzer and BMG. The qualified products were pooled and quantified based on different library quantities, and the single-stranded library products were circularized to generate DNA nanoballs (DNBs) before being sequenced using PE100 + 100 sequencing on the MGISEQ-2000 platform.
Analysis of sequencing results
Upon receipt of the primary sequencing data, we conducted bioinformatics processing and data analysis to identify potential variants in the family. To perform further analysis, we used previously established filtering criteria to generate “clean reads.” The “clean reads” were then aligned to the human genome reference (hg19 version) using the BWA Multi-Vision software package (0.7.17) and samtools (1.10). The output files from the alignment were used to conduct sequencing coverage and depth analysis of the target region, single-nucleotide variants (SNVs), and INDEL calling. To detect SNVs and indels, we utilized the GATK software (4.1.9.0), and all SNVs and indels were filtered and evaluated using multiple databases, including NCBI dbSNP, 1000 human genome dataset (phase 3), ClinVar (2020-03-16), ESP6500 (V2), GnomAD (v2.1.1), ExAC (r0.3.1), and SecondaryFinding_Var* (V1.1_2020.3, an in-house database of BGI Genomics). To predict the impact of missense variants, we employed dbNSFP, which consists of seven well-established in silico prediction programs such as SIFT, Polyphen2, LRT, MutationTaster, and PhyloP. To analyze the variants near the splicing sites, we used SpliceAI (1.3) for prediction. Lastly, variant classification was conducted following the American Society for Medical Genetics and Genomics (ACMG) guidelines, and reported as pathogenic, likely pathogenic, variant of unknown significance (VUS), likely benign, or benign.
Sanger verification
Conventional Sanger sequencing methods were used to validate all mutations and potential pathogenic variants. If DNA was available from family members, segregation analysis was performed (24).
Results
Clinical phenotype of the probands and their family members
Pedigree A includes a 48-year-old male proband who showed signs of chronic kidney disease stage 2 (CKD2) and hypertension (HT), as determined by clinical data. Abdominal CT results revealed an increase in the size of both kidneys with low and slightly high-density shadows of varying sizes. Multiple round low-density shadows in the liver suggested the presence of polycystic liver (PL) (Table 2). The proband’s father had also been diagnosed with ADPKD in middle age, and the proband’s son was found to be a carrier of the PKD1 mutation gene but appeared to be asymptomatic, suggesting autosomal dominant inheritance in the family (Table 3).
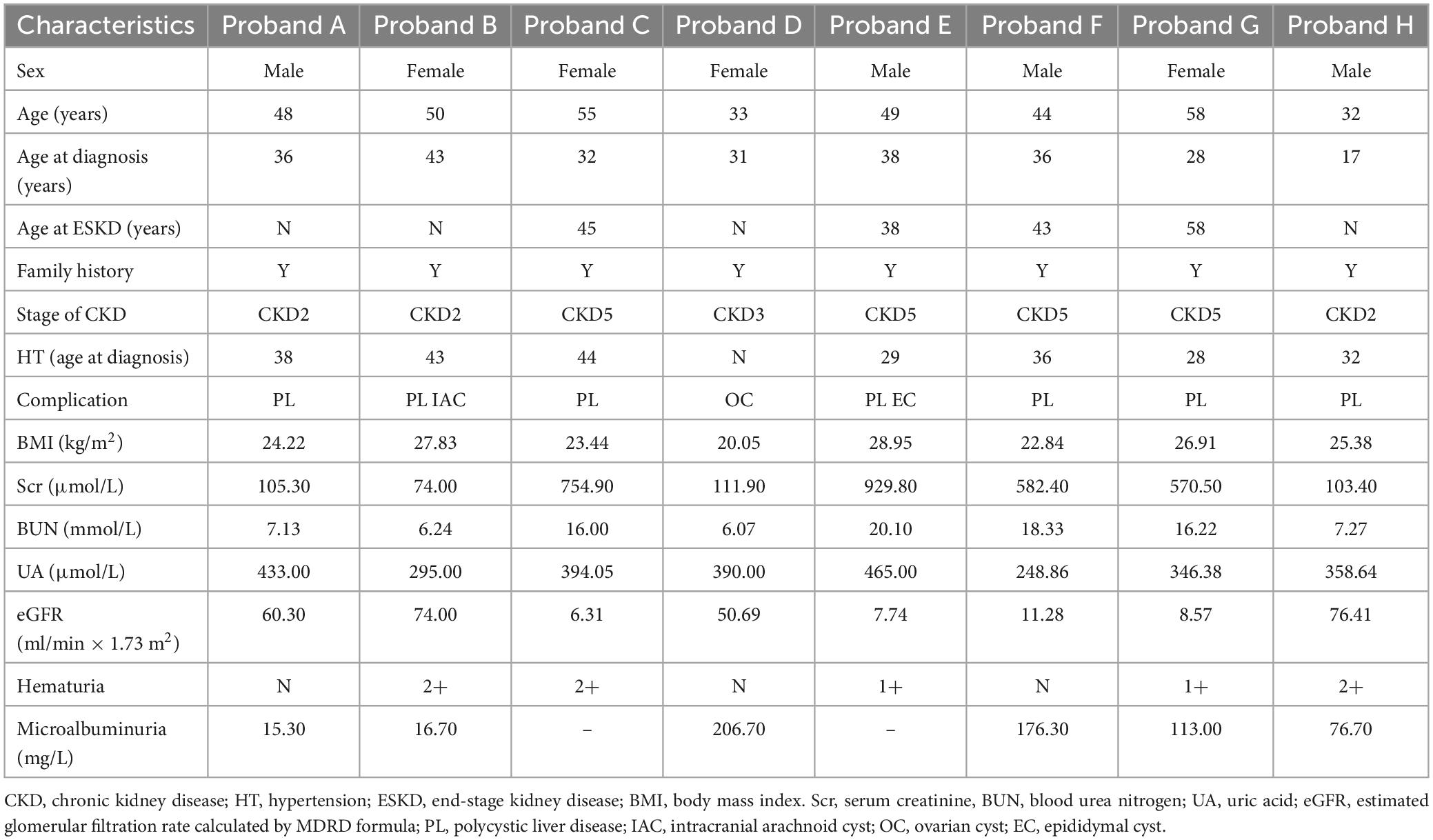
Table 2. Clinical features of probands with autosomal dominant polycystic kidney disease.
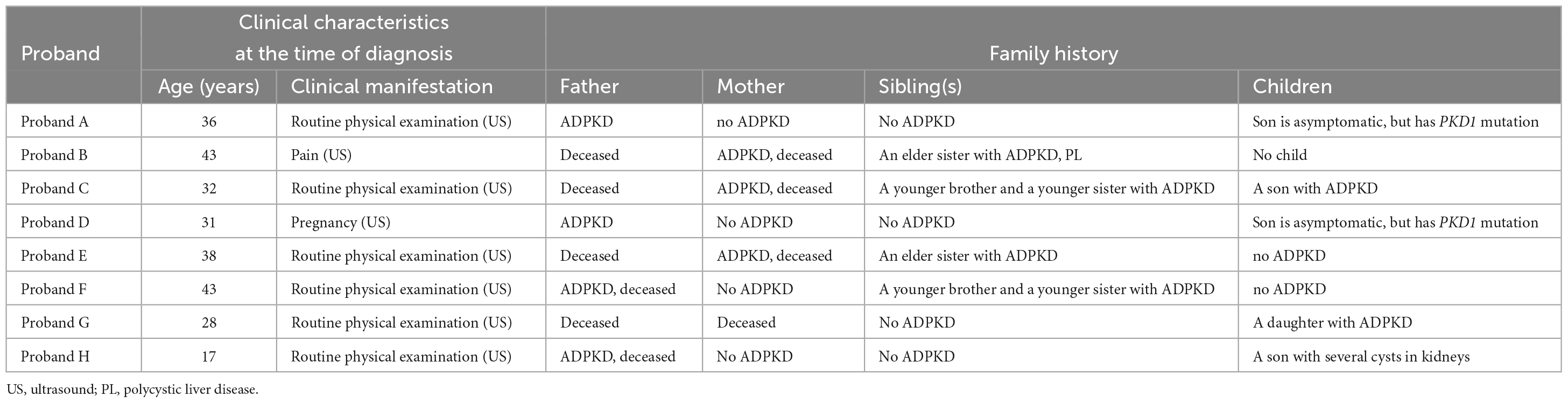
Table 3. Family history in probands with autosomal dominant polycystic kidney disease.
Pedigree B includes a 50-year-old female proband who showed symptoms of CKD2 with hematuria, HT, PL, and intracranial arachnoid cyst (IAC). Imaging results revealed an increase in the size of both kidneys with many anechoic areas of varying sizes, as well as an abnormal liver with anechoic areas of different sizes. Cranial MRI showed arachnoid cysts in the right forehead and left temporal region (Table 2). The proband’s mother and sister were also diagnosed with ADPKD, and the nephew was found to carry the PKD1 mutation gene but was asymptomatic, indicating autosomal dominant inheritance in the family (Table 3).
Pedigree C includes a 55-year-old female proband who showed signs of CKD5 with hematuria, HT, and PL. Abdominal CT scan showed enlarged bilateral kidneys with kidney cysts, it also showed innumerable cysts in the liver (Table 2). In this family, the proband’s mother, younger brother, younger sister, and son were diagnosed with ADPKD, and the proband’s mother had passed away (Table 3). These findings suggest an autosomal dominant inheritance pattern in the family.
In pedigree D, a 33-year-old female proband displayed CKD3 symptoms and ovarian cyst (OC). Abdominal ultrasound results showed normal size and shape of both kidneys, but there were many anechoic areas of different sizes (Table 2). The proband’s father was diagnosed with ADPKD, and the son was a carrier of the PKD1 mutation gene but appeared normal, indicating that the inheritance pattern is autosomal dominant in the family (Table 3).
In pedigree E, a 49-year-old male proband presented with CKD5 with hematuria, HT, PL, and epididymal cyst (EC). Abdominal ultrasound showed that the right kidney was significantly enlarged with multiple anechoic areas of varying sizes. The left kidney had been removed due to a renal cyst rupture and hemorrhage 4°years prior (Table 2). The proband’s mother and elder sister both have ADPKD, and the proband’s mother passed away. However, the proband’s child did not inherit the disease. These findings suggest an autosomal dominant inheritance pattern in the family (Table 3).
In pedigree F, a 44-year-old male proband was diagnosed with CKD5, PL, and HT. Abdominal ultrasound showed significantly enlarged kidneys with multiple anechoic areas of different sizes and anechoic lesions in the right lobe of the liver (Table 2). The proband’s father had ADPKD and passed away, and the proband’s younger brother and younger sister were also diagnosed with ADPKD (Table 3).
In pedigree G, a 58-year-old female proband presented with CKD5 with hematuria, HT, and PL. Abdominal ultrasound showed increased kidney size with multiple anechoic areas, as well as several anechoic lesions in the liver (Table 2). The proband’s daughter was diagnosed with ADPKD, while her siblings did not have the condition (Table 3).
In pedigree H, a 32-year-old male proband presented with CKD2 with hematuria, HT, and PL. Abdominal ultrasound showed enlarged kidneys with multiple anechoic areas and anechoic lesions in the liver (Table 2). The proband’s father had a history of ADPKD and passed away, and his son was a carrier of the PKD1 mutation gene with several kidney cysts despite having a normal appearance, suggesting an autosomal dominant inheritance pattern (Table 3).
Genotype of the probands and their family members
In Pedigree A, WES analysis revealed that the proband had one likely pathogenic mutation and one uncertain mutation in the PKD1 gene (Supplementary Table 1). The analysis showed a heterozygous missense mutation in exon 23 of PKD1: c.8447T > C (p.Leu2816Pro), which has been reported as likely pathogenic (25–27) and is also indicated by the ACMG guidelines. Another heterozygous missense mutation was detected in exon 15 of PKD1: c.4247T > G (p.Phe1416Cys). There is no relevant literature on the pathogenicity of this variation. Sanger sequencing confirmed that proband’s father (I-1) was a PKD patient (79 years old) with PKD1: c.8447T > C, while III-1 (21 years old) carries PKD1: c.4247T > G and is currently an asymptomatic carrier of the mutation (Figure 1).
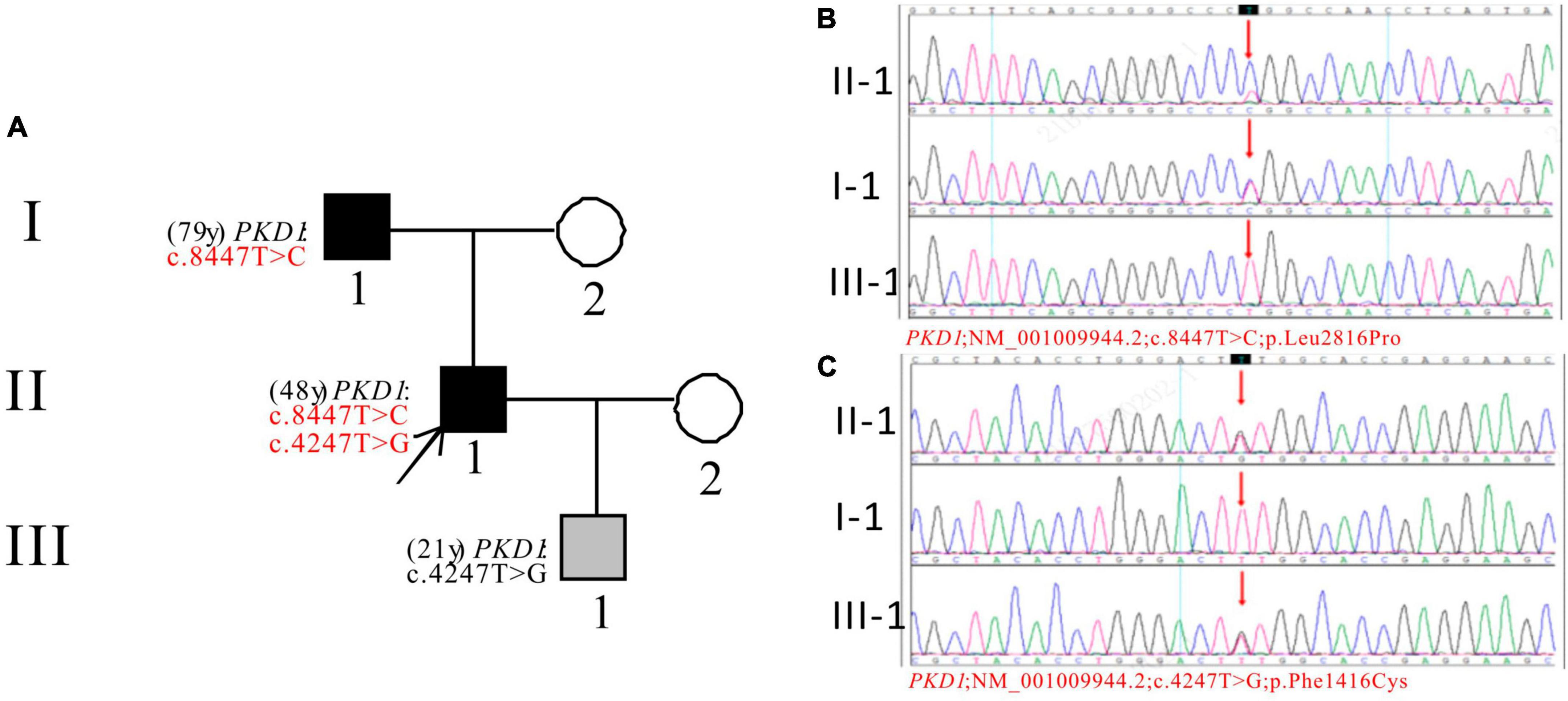
Figure 1. Pedigree diagram and sequencing results of proband A. (A) Pedigree diagram. The WES identified a heterozygous missense mutation in exon 23 (c.8447T > C) (B) and a heterozygous missense mutation in exon 15 (c.4247T > G) (C) of the PKD1 gene in the proband (II-1) and affected family members (I-1 and III-1). The mutation sites were indicated by a red arrows in panels (B,C).
In Pedigree B, WES analysis revealed a likely pathogenic mutation in the PKD1 gene in the proband (Supplementary Table 1). Specifically, a heterozygous deletion mutation in exon 15 of PKD1 was identified: c.3298_3301delGAGT (p.Glu1100Thrfs*3). Sanger sequencing was performed to verify the results and it was found that the proband’s sister (II-2) (53 years old) also carries the same mutation and was diagnosed with ADPKD at the age of 45. Her kidney function began to decline at the age of 52, reaching CKD3. Additionally, the proband’s nephew (III-1) (21 years old) seemingly asymptomatic, but has the same PKD1 mutation (Figure 2).
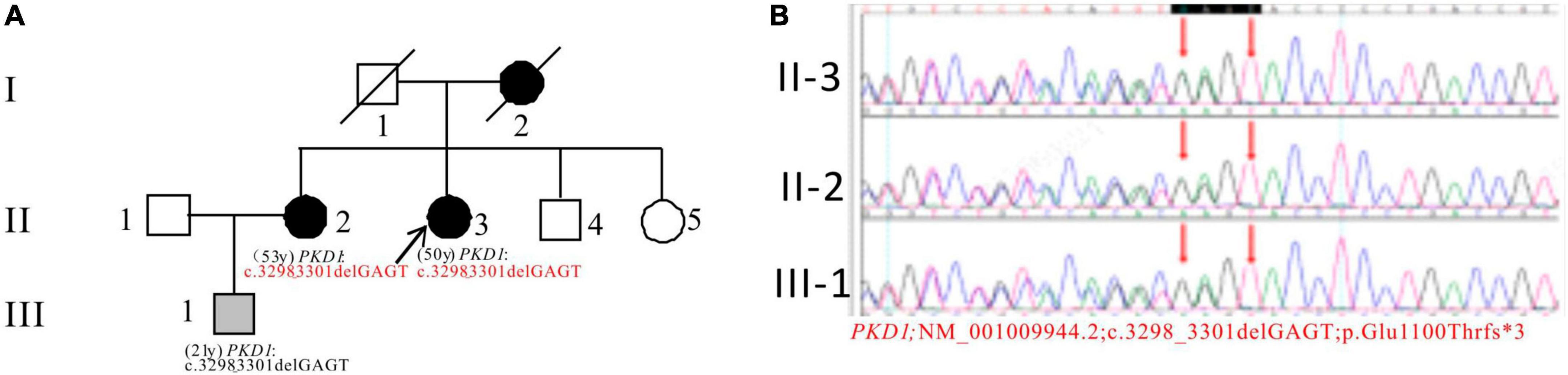
Figure 2. Pedigree diagram and sequencing results of proband B. (A) Pedigree diagram. (B) WES identified a heterozygous deletion mutation in exon 15 (c.3298_3301delGAGT) of the PKD1 gene in the proband (II-3) and affected family members (II-2 and III-1). The mutation sites were indicated by a red arrows panel in panel (B).
In Pedigree C, WES analysis revealed a pathogenic mutation and an uncertain mutation in the PKD1 gene in the proband (Supplementary Table 1). Specifically, a heterozygous nonsense mutation in exon 15 of PKD1 was identified: c.6406C > T (p.Gln2136*), which has been reported as pathogenic (28) and is also indicated by the ACMG guidelines. Another heterozygous missense mutation was also detected in exon 15 of PKD1: c.4798A > G (p.Thr1600Ala), which has an uncertain significance according to the ACMG guidelines and currently lacks reports on its pathogenicity. Sanger sequencing confirmed that the proband’s son (III-1) (30 years old) carried both mutations and was diagnosed with ADPKD at the age of 20. His kidney function began to decline at the age of 29, reaching CKD2. The proband’s mother (I-2), sister (II-3), and brother (II-4) were all diagnosed with ADPKD. Among them, the proband’s sister (II-3) reached ESKD at the age of 45, received a kidney transplant at the age of 52, and is currently in generally good condition (Figure 3).
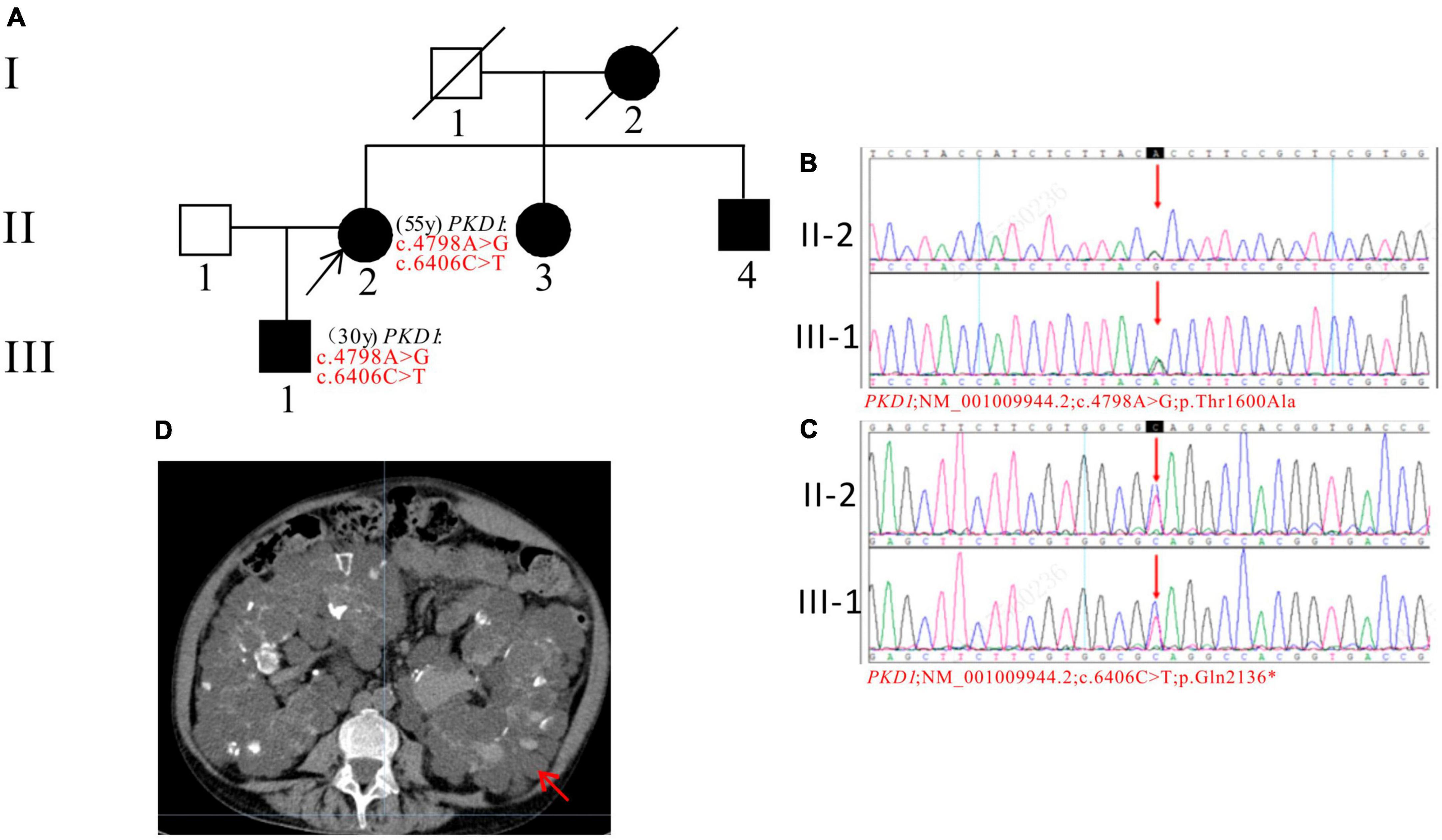
Figure 3. Pedigree diagram, sequencing and abdominal CT scan result of proband C. (A) Pedigree diagram. The WES identified a heterozygous nonsense mutation in exon 15 (c.6406C > T) (B) and a heterozygous missense mutation (c.4798A > G) (C) of the PKD1 gene in the proband (II-2) and affected family member (III-1). The mutation sites were indicated by a red arrows in panels (B,C). (D) Abdominal CT scan performed in proband C at 55y showed enlarged bilateral kidneys with kidney cysts. The kidney cysts is indicated by a red arrow.
In Pedigree D, WES analysis revealed a mutation in the PKD1 gene in the proband (Supplementary Table 1). Specifically, a heterozygous missense mutation in exon 19 of the PKD1 gene was identified: c.7567G > A (p.Glu2523Lys), which is of unknown significance according to the ACMG guidelines and currently lacks reports on its pathogenicity. Sanger sequencing confirmed that the father of the proband (I-1) (72 years old) and the proband’s son (III-1) (4 years old) carried the same mutation. I-1 was diagnosed with PKD at the age of 72 and currently has normal kidney function, while III-1 is seemingly asymptomatic now (Figure 4).
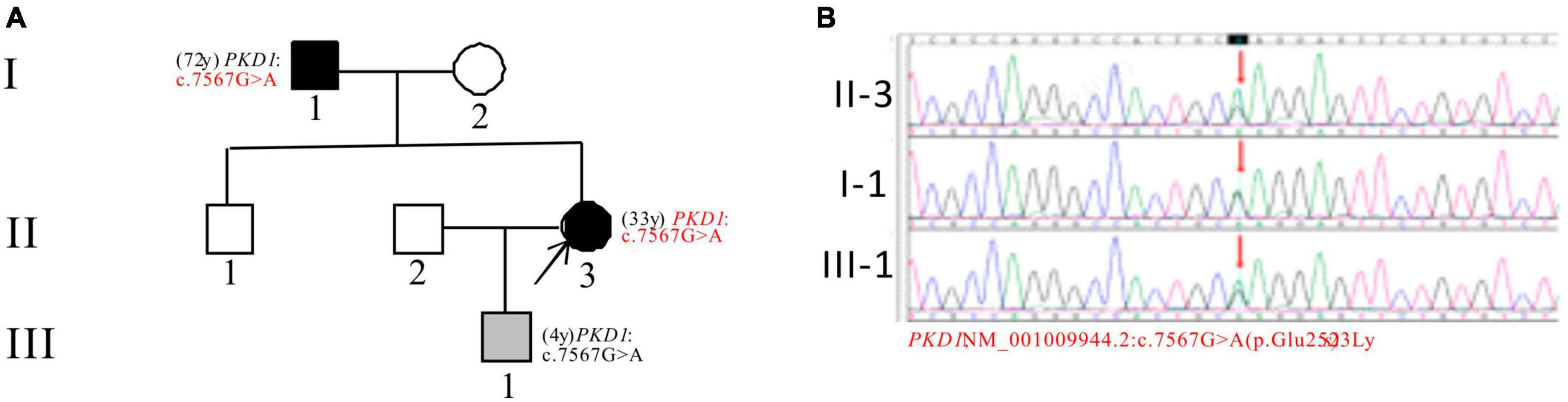
Figure 4. Pedigree diagram and sequencing results of proband D. (A) Pedigree diagram. (B) WES identified a heterozygous missense mutation in exon 19 (c.7567G > A) of the PKD1 gene in the proband (II-3) and affected family members (I-1 and III-1). The mutation sites were indicated by a red arrows in panel (B).
In Pedigree E, the WES analysis of the proband revealed a pathogenic variation and a uncertain variation in the PKD1 gene (Supplementary Table 1). Specifically, a heterozygous nonsense mutation in exon 39 of PKD1 was identified: c.11215C > T (p.Gln3739*), which has been classified as pathogenic according to the ACMG guidelines. Another heterozygous missense mutation was detected in exon 43 of PKD1: c.11717G > C (p.Cys3906Ser), which has an uncertain significance according to the ACMG guidelines and currently lacks reports on its pathogenicity. The proband’s father (I-1) and sister (II-2) were diagnosed with ADPKD. However, the corresponding PKD1 gene mutation was negative for the proband’s son (III-2) (11 years old) and proband’s nephew (III-1) (29 years old), both of whom were apparently normal population (Figure 5).
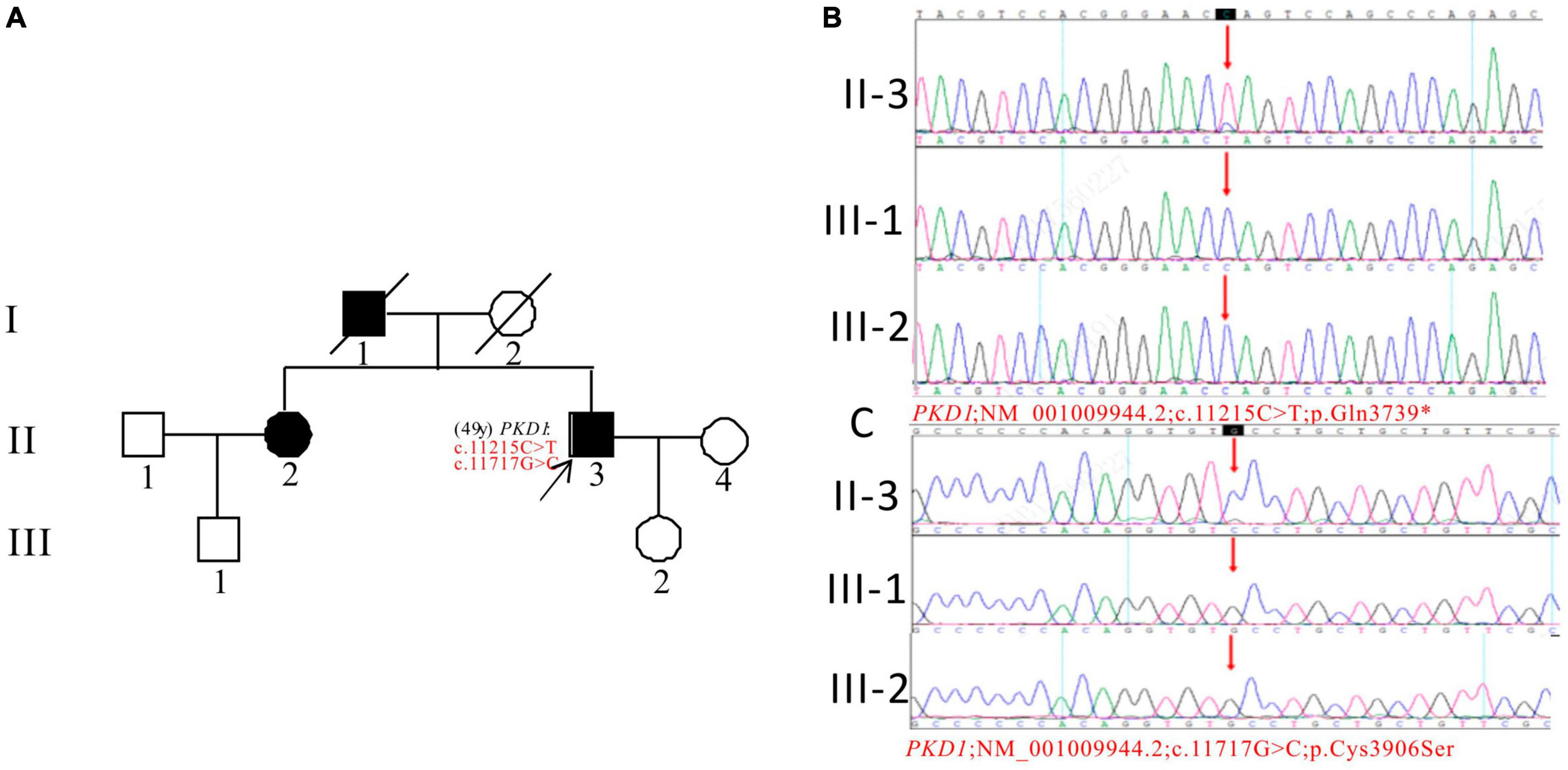
Figure 5. Pedigree diagram and sequencing results of proband E. (A) Pedigree diagram. The WES identified a heterozygous nonsense mutation in exon 39 (c.11215C > T) (B) and a heterozygous missense mutation (c.11717G > C) in exon 43 (C) of the PKD1 gene in the proband, and the results of family members (III-1 and III-2) were displayed. The mutation sites were indicated by a red arrows in panels (B,C).
In Pedigree F, the proband was found to have a heterozygous intronic mutation in the PKD1 gene: c.7703 + 5G > C (Supplementary Table 1). This mutation is located within an intron of the gene and its significance is currently unknown according to the ACMG guidelines. There are no reports available regarding its pathogenicity. The proband’s father (I-1) was a PKD patient who passed away. Sanger sequencing confirmed that one of the proband’s brothers (II-4) (37 years old) and sisters (II-6) (33 years old) carried the same mutation. II-4 was diagnosed with ADPKD at 36 years old, entered ESKD at the age of 37. II-6 was diagnosed with ADPKD at the age of 32 and currently maintains normal kidney function. However, the mutation in the PKD1 gene was not detected in the proband’s daughter (III-1) (16 years old) (Figure 6).
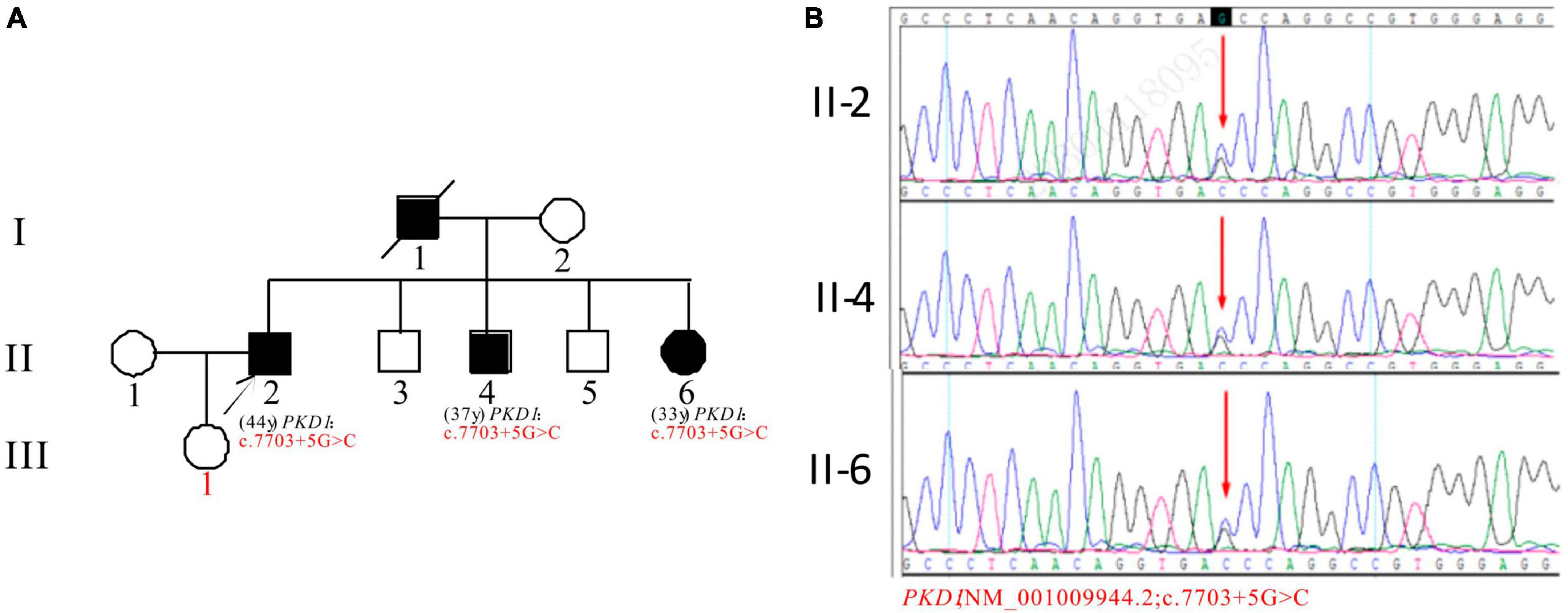
Figure 6. Pedigree diagram and sequencing results of proband F. (A) Pedigree diagram. (B) A heterozygous intron variation (c.7703 + 5G > C) of the PKD1 gene in the proband (II-2) and affected family members (II-4 and II-6) were identified. The mutation sites are indicated by a red arrows in panel (B).
In Pedigree G, WES analysis revealed a likely pathogenic mutation and an uncertain mutation in the PKD1 gene (Supplementary Table 1). Specifically, a heterozygous missense mutation in exon 23 of the PKD1 gene was identified: c.8312A > T (p.Glu2771Val), which has been indicated as likely pathogenic by the ACMG guidelines, and its pathogenicity had been reported (29). Another heterozygous missense mutation was detected in exon 15 of PKD1: c.3296G > A (p.Gly1099Asp), which is of unknown significance according to the ACMG guidelines and currently lacks reports on its pathogenicity. Sanger sequencing confirmed that the proband’s daughter (III-2) (31 years old) was a ADPKD patient with the same mutations and is currently with normal kidney function. The corresponding PKD gene mutation in the son of the proband (III-1) (36 years old) was negative (Figure 7).
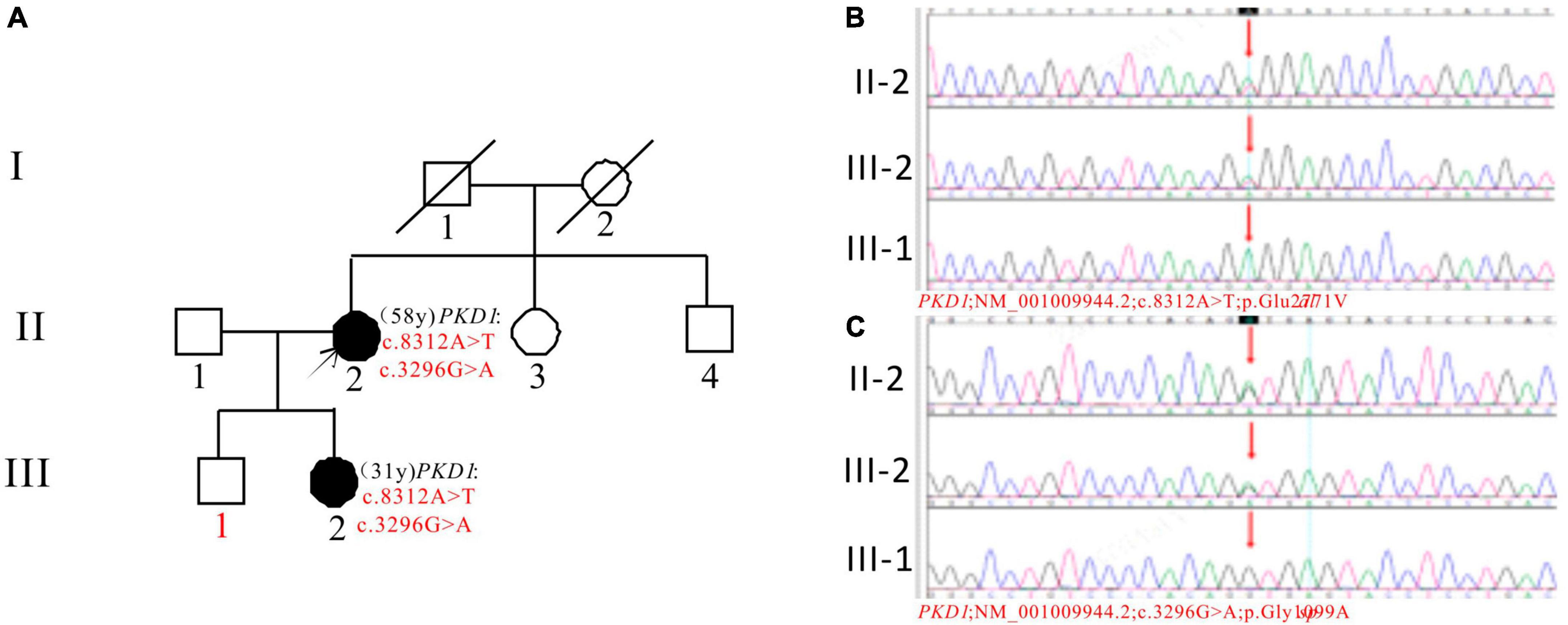
Figure 7. Pedigree diagram and sequencing results of proband G. (A) Pedigree diagram. The WES identified a heterozygous missense mutation in exon 23 (c.8312A > T) (B) and a heterozygous missense mutation in exon 15 (c.3296G > A) (C) of the PKD1 gene in the proband (II-2), and the results in family members (III-1 and III-2) were displayed. The mutation sites are indicated by a red arrows in panels (B,C).
In Pedigree H, WES analysis revealed a pathogenic and an uncertain mutation in the PKD1 gene (Supplementary Table 1). Specifically, a heterozygous frameshift mutation in exon 23 of the PKD1 gene was identified: c.8515_8516insG (p.Ile2839Serfs*98), which has been indicated as pathogenic by the ACMG guidelines. Another heterozygous missense mutation was detected in exon 15 of PKD1: c.5524C > A (p.Pro1842Thr), which has been indicated as having uncertain significance according to the ACMG guidelines. At present, there are no reports on their pathogenicities. The proband’s father was an ADPKD patient who passed away at the age of 50. Sanger sequencing confirmed that the son of the proband (III-1) (3 years old) carries the mutations and is an apparently normal mutation carrier (Figure 8).
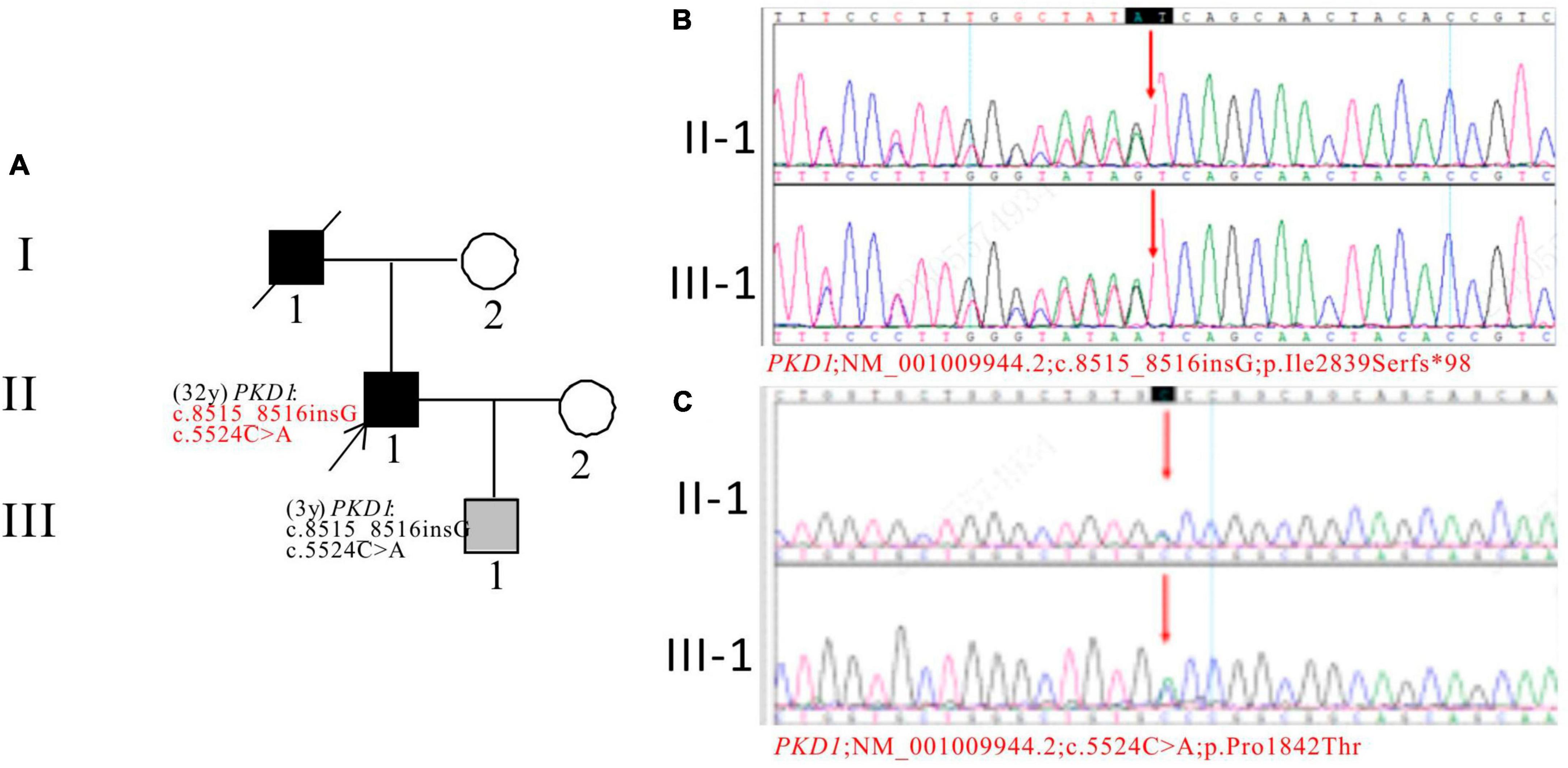
Figure 8. Pedigree diagram and sequencing results of proband H. (A) Pedigree diagram. The WES identified a heterozygous frameshift mutation in exon 23 (c.8515_8516insG) (B) and a heterozygous missense mutation in exon 15 (c.5524C > A) (C) of the PKD1 gene in the proband (II-1) and affected family member (III-1). The mutation sites were indicated by a red arrows in panels (B,C).
Discussion
In this study, we aimed to analyze the gene mutations in 8 Chinese families with ADPKD. This investigation is crucial to expand the genetic diversity data of different ethnic groups, improve the ADPKD mutation database, and explore its underlying pathogenesis. Due to the differences in genetic backgrounds and environmental factors, there is considerable phenotypic variability among ADPKD patients. Key factors that determine the progression of ADPKD include age, PKD genotype (including mutation gene, mutation type, gene mutation location, and the presence of multiple gene mutations), urine protein, estimated glomerular filtration rate (eGFR), and highly adjusted total kidney volume (HtTKV) (30).
Two variants of the PKD1 gene were found in Pedigree A: c.8447T > C (p.Leu2816Pro) and c.4247T > G (p.Phe1416Cys). The first variant is known to cause ADPKD in several patients and families (26, 27). The pathogenicity of the second variant is unknown, and no information on its frequency in the population was found in the GnomAD databases. The proband and their father had ADPKD, with the father having the pathogenic mutation. The proband’s son is a carrier of the second variant with a normal phenotype. Further study is needed to determine the role of the second variant in ADPKD.
Pedigree B’s proband was diagnosed with ADPKD and has a frameshift mutation c.3298_3301delGAGT (p.Glu1100Thrfs*3) in PKD1 gene. The mutation is not reported in relevant databases, and leads to a truncated PC1 protein affecting its function (31). No information on its frequency in the population was found in the GnomAD databases. The mutation is suspected to be pathogenic based on ACMG guidelines and is inherited from the proband’s mother who died of ADPKD. The proband’s sister also has ADPKD and PL. The proband’s nephew carries the mutation but is currently asymptomatic. Further follow-up is ongoing.
In pedigree C, two variants were found in the PKD1 gene. One of the variants, c.6406C > T, has been reported as pathogenic (28). The other variant, c.4798A > G, is new and its pathogenicity is uncertain. The population frequency in the GnomAD is 0.000016. The variants may work together to cause the disease in the family. Family members with the variants show co-segregation of genotype and phenotype, with early onset of ADPKD and kidney failure. The role of the new variant in ADPKD needs further confirmation.
A new PKD1 gene mutation c.7567G > A (p.Glu2523Lys) was detected in pedigree D, with the proband, father, and son carrying it. The proband has ADPKD and CKD3, while the father was diagnosed with ADPKD in middle age and has normal kidney function. The mutation is predicted to have harmful effects on the gene and is considered pathogenic in this family, although its significance is uncertain according to ACMG guidelines. The population frequency of this mutation in the GnomAD is 0.000051. The proband’s son is a carrier and is being closely monitored. Further research is needed to determine the mutation’s role in ADPKD.
The proband in pedigree E have PKD1 gene mutations, including a harmful c.11215C > T(p.Gln3739*) nonsense mutation in exon 39 and a newly discovered c.11717G > C(p.Cys3906Ser) missense mutation in exon 43, which is of uncertain significance. The mutation frequency of c.11717G > C in the GnomAD databases is 0.000011. The proband’s sister is an ADPKD patient. Their father had ADPKD, suggesting inheritance. The proband’s daughter and nephew did not inherit the mutations. Both proband and his sister developed ESKD at an early age, and the two mutations are considered pathogenic when present together.
In pedigree F, the PKD1 gene of the proband and his younger brother with the disease was found to have a novel intron variation c.7703 + 5G > C. However, the proband’s daughter did not inherit these mutations. No reports on the mutation frequency of the population at this locus in the GnomAD databases. The mutation frequency of the population at this locus is unknown. Predictive analysis indicates harmful effects on the gene and mRNA splicing. The mutation is predicted to be pathogenic based on genetic characteristics and clinical manifestations. However, it is still uncertain based on ACMG guidelines. The proband and his siblings may have inherited the mutation from their father who had PKD. Further observation and follow-up are needed.
Pedigree G has a proband who developed ADPKD in their youth and entered ESKD 30 years later. The proband’s daughter has ADPKD but normal kidney function. One of the mutations, c.8312A > T (p.Glu2771Val), has been reported previously (29), while the other mutation, c.3296G > A, is likely a new mutation, the mutation frequency in the GnomAD databases is 0.000061. Both mutations are predicted to be harmful and affect PKD1 mRNA splicing. According to ACMG guidelines, the mutations are classified as of uncertain significance. The proband’s son is negative for the mutations and apparently normal. The mutations are considered pathogenic and require further follow-up observation.
In pedigree H, the proband and his son have two mutations in PKD1: a frameshift mutation and a missense mutation, which are likely new mutations. The frameshift mutation is predicted to be harmful and classified as pathogenic, no reports on the mutation frequency of the population at this locus in the GnomAD databases. The missense mutation is predicted to be harmful by one software tool (SpliceAI) and polymorphic/neutral by others, with uncertain significance, the mutation frequency in the GnomAD databases is 0.000006 in the GnomAD databases. The proband’s father had ADPKD, suggesting inheritance of the mutations. The proband has early onset ADPKD and his son has a single renal cyst with normal liver and kidney functions, currently under monitoring.
In this study, 13 mutation sites were identified in the PKD1 gene, including 9 new mutations not previously reported, from 8 ADPKD probands (Figure 9). Among the 8 probands, 4 had progressed to ESKD, with ages ranging from 38 to 58 years old, and all had chosen hemodialysis as their kidney replacement therapy. Hypertension is a common occurrence in ADPKD patients, with about 60% diagnosed before kidney function decline (32, 33). The average age of hypertension diagnosis in PKD1 mutation patients is around 39 years old (26). Seven of our 8 probands had hypertension, except for probands E and D, who were diagnosed at the same time or after the ADPKD diagnosis. It remains uncertain whether hypertension in ADPKD is caused by the primary angiopathy of polycystic protein gene mutation or the activation of a renal renin-angiotensin-aldosterone system (RAAS) due to cyst expansion, and further research is needed. Therefore, gene detection can offer valuable insights for the diagnosis and treatment of ADPKD with hypertension. ADPKD typically occurs after the age of 40 and 100% by age 80, making it a late-onset disease (34). In this study, PKD1-related mutations were also found in family members who appeared clinically normal and were younger than 21 years old. The absence of related clinical manifestations in these family members can be explained by the age-dependent delayed dominance of ADPKD, which requires further follow-up and observation.
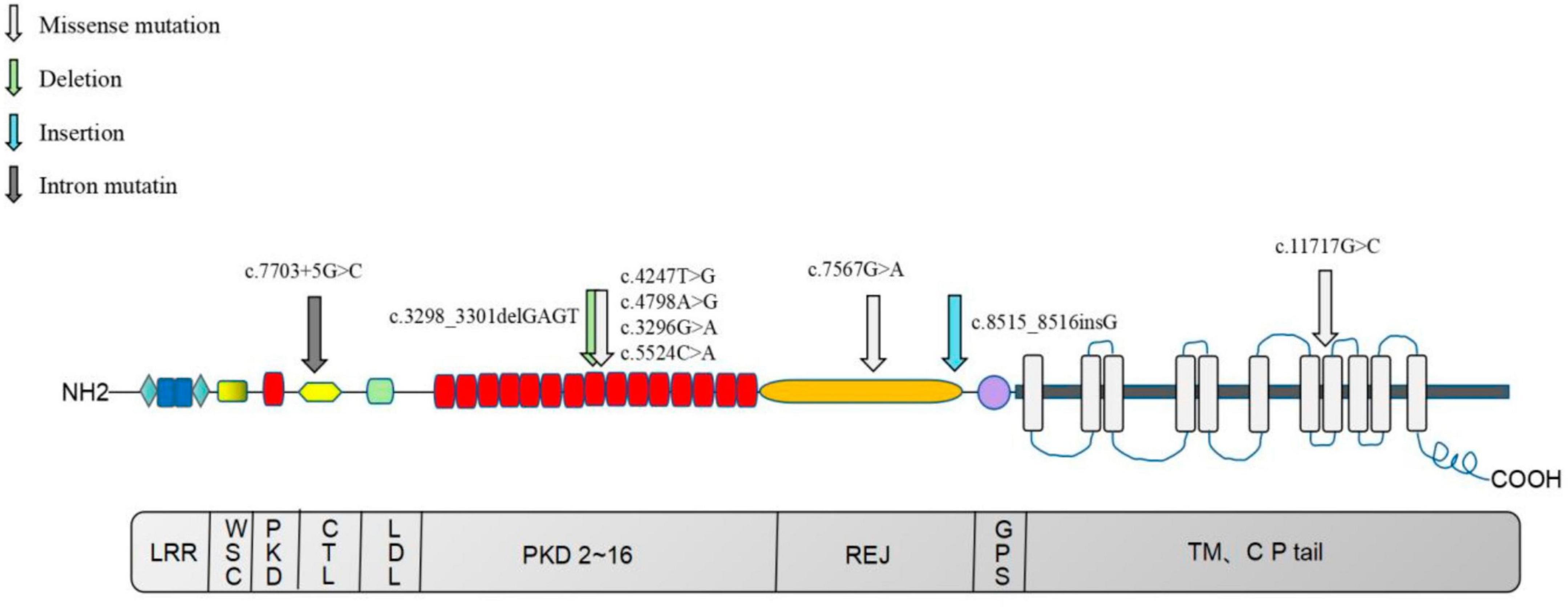
Figure 9. Distribution map of nine newly identified PKD1 mutations in eight ADPKD patients. The upper section of the image illustrates the PC-1 domain, which includes 11 transmembrane domains, a large extracellular N-terminal domain, and a short intracellular C-terminal tail region. The lower section of the image depicts the coding region of the PKD1 gene. LRR, Leucine-rich repeats; PKD, PKD functional area; CTL, C-type lectin motif, C-type phytohemagglutinin region; LDL, LDL-A like domain, rich in cysteine low-density lipoprotein receptor region; REJ, receptor egg jelly, the homologous region of sea urchin egg glue receptor; GPS, G-proteolytic site; TM, transmembrane, transmembrane domain; CP tail, cytoplasmic tail.
The human PKD1 gene is made up of 46 exons with complex structures. However, the presence of six pseudogenes, which are highly homologous to exons 1 to 33 of PKD1, poses a challenge to the molecular diagnosis of ADPKD (35). Second-generation sequencing technology has revolutionized genetic analysis, enabling disease group screening, WES, and even whole genome analysis, replacing single gene analysis based on Sanger sequencing. To date, the ADPKD mutation database has recorded 2,322 pathogenic PKD1 mutations. However, most of these mutations occur only once in a single family, with few duplications. Therefore, it is necessary to investigate more families to confirm known mutations and discover new ones. Gene decoding-based basic and clinical studies of related drugs have shown that cystic disease progression can be delayed to varying degrees. Identifying specific pathogenic genes and alleles can help determine the disease prognosis, influencing disease management and the formulation of treatment plans (9, 36). If gene testing can help predict the benefits that drug therapy, represented by tolvaptan, can bring to ADPKD, it can create greater value for patients (37). However, slowing down cyst progression is not a long-term solution. The fundamental strategy is to reverse the functional re-expression of the pathogenic PKD gene at the physiological level in vivo. Stefan Somlo’s research team at Yale University has established a mouse model of PKD inactivation and reactivation to examine the impact of PKD inactivation on renal structure and function. They found that the re-expression of the PKD gene in the cystic kidney can rapidly reverse ADPKD, reducing cyst cell proliferation, activating autophagy, and restoring the cystic tubules to a normal lumen lined by cubic cells. Since the ADPKD phenotype is reversible, at least partially controlled by the function of the ADPKD gene (38), gene screening plays a crucial role in early diagnosis and intervention. Early diagnosis can benefit patients through lifestyle changes, drug intervention, and understanding of kidney transplantation information.
Kidney transplantation is considered the most effective surgical treatment for end-stage ADPKD (39, 40). Studies have shown that the recurrence rate of ADPKD after transplantation is similar to that of other non-diabetic kidney transplant recipients (41). Moreover, the volume of an autologous polycystic kidney tends to decrease after kidney transplantation (42). Therefore, in cases where there is enough space for kidney transplantation and no risk of bleeding or infection, it may not be necessary to remove the other kidney, which can improve the quality of life of patients. In ADPKD families, gene testing can help select suitable unaffected relatives as potential kidney transplant donors, promoting more transplantation evaluations and preemptive transplantation opportunities for ADPKD patients (43). This approach can improve patient outcomes, as preemptive transplantation before the onset of end-stage renal disease can prevent the need for dialysis, reduce post-operative complications, and improve long-term patient survival rates.
In this study, DNA was extracted from the subject’s blood, but the possibility of interpretation bias caused by chimerism cannot be ruled out. Moreover, the sample size was too small to eliminate all possible biases. In addition, this study did not provide a detailed molecular-level understanding of how new genes affect the functional changes of PC1. Therefore, larger clinical and basic research studies are needed to support more comprehensive genetic testing of ADPKD patients. Follow-up studies of patients and their family members have been extended to observe the relationship between genotype and phenotype more closely over time.
In summary, the findings from this study contribute to the growing body of knowledge on ADPKD genetics and highlight the importance of genetic testing in the diagnosis and management of this disease. By identifying new mutation sites and analyzing their effects on genotype and phenotype, this research provides a better understanding of ADPKD pathogenesis and opens up new avenues for further exploration. Second-generation sequencing technology has proven to be a valuable tool for detecting gene mutations in ADPKD patients, and its use should be encouraged in clinical practice and research. Ultimately, genetic testing can lead to more accurate diagnoses, better disease management, and improved genetic counseling for affected families. Further studies are needed to confirm and expand upon these findings and to elucidate the underlying mechanisms of ADPKD pathogenesis.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: The National Omics Data Encyclopedia (NODE), accession number: OEP004096.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Xinjiang Uygur Autonomous Region People’s Hospital confirms the approval of the project titled “Genotype-Phenotype Characteristics of Nine Novel PKD1 Mutations in Eight Chinese Patients with Autosomal Dominant Polycystic Kidney Disease” with the approval number KY2023032201. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
HJ: Conceptualization, Investigation, Methodology, Validation, Supervision, Data curation, Resources, Writing – review and editing, Funding acquisition. XT: Conceptualization, Methodology, Validation, Supervision, Writing – review and editing. JiZ: Investigation, Data curation, Writing – original draft, Methodology, Formal analysis, Software, Resources, Validation. AA: Investigation, Data curation, Writing – original draft, Methodology, Formal analysis, Software, Resources, Validation. DY: Investigation, Methodology, Data curation, Formal analysis, Writing – review and editing. JuZ: Investigation, Methodology, Data curation, Formal analysis, Writing – review and editing. XW: Investigation, Methodology, Data curation, Formal analysis, Writing – review and editing. YR: Investigation, Visualization, Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Xinjiang Uygur Autonomous Region Project “Mechanistic study of RNA binding protein HSPB1 regulation of MMP14 transcript stability in ECM deposition in lupus nephritis” (2022D01C631).
Acknowledgments
We thank the families and coordinators for involvement in the study. We thank the technique support from Research Center of Clinical Medicine of Xinjiang Uygur Autonomous Region. The study was supported by Xinjiang Uygur Autonomous Region Project Application of bone disease information processing system in end-stage kidney disease (2020E02118) to HJ. We also thank the Genetic analysis performed by the BGI Genomics, China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2023.1268307/full#supplementary-material
References
1. Lanktree MB, Haghighi A, Guiard E, Iliuta IA, Song X, Harris PC, et al. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol. (2018) 29:2593–600. doi: 10.1681/ASN.2018050493
2. Willey CJ, Blais JD, Hall AK, Krasa HB, Makin AJ, Czerwiec FS. Prevalence of autosomal dominant polycystic kidney disease in the European Union. Nephrol Dial Transplant. (2017) 32:1356–63. doi: 10.1093/ndt/gfw240
3. Lanktree MB, Haghighi A, di Bari I, Song X, Pei Y. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. (2021) 16:790–9. doi: 10.2215/CJN.02320220
4. Ong AC, Devuyst O, Knebelmann B, Walz G, ERA-EDTA Working Group for Inherited Kidney Diseases. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. (2015) 385:1993–2002. doi: 10.1016/S0140-6736(15)60907-2
5. Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. (2019) 393:919–35. doi: 10.1016/S0140-6736(18)32782-X
6. Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, et al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet. (2018) 102:832–44. doi: 10.1016/j.ajhg.2018.03.013
7. Delbarba E, Econimo L, Dordoni C, Martin E, Mazza C, Savoldi G, et al. Expanding the variability of the ADPKD-GANAB clinical phenotype in a family of Italian ancestry. J Nephrol. (2022) 35:645–52. doi: 10.1007/s40620-021-01131-w
8. Senum SR, Li YSM, Benson KA, Joli G, Olinger E, Lavu S, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet. (2022) 109:136–56. doi: 10.1016/j.ajhg.2021.11.016
9. Cornec-Le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol. (2018) 29:13–23. doi: 10.1681/ASN.2017050483
10. Spithoven EM, Kramer A, Meijer E, Orskov B, Wanner C, Caskey F, et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. (2014) 86:1244–52. doi: 10.1038/ki.2014.120
11. Suwabe T, Shukoor S, Chamberlain AM, Killian JM, King BF, Edwards M, et al. Epidemiology of autosomal dominant polycystic kidney disease in olmsted county. Clin J Am Soc Nephrol. (2020) 15:69–79. doi: 10.2215/CJN.05900519
12. Mei CL, Xue C, Yu SQ, Dai B, Chen JH, Li Y, et al. Executive summary: clinical practice guideline for autosomal dominant polycystic kidney disease in China. Kidney Dis. (2020) 6:144–9. doi: 10.1159/000506288
13. Kim DY, Park JH. Genetic mechanisms of ADPKD. Adv Exp Med Biol. (2016) 933:13–22. doi: 10.1007/978-981-10-2041-4_2
14. Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. (2018) 4:50. doi: 10.1038/s41572-018-0047-y
15. Groopman EE, Rasouly HM, Gharavi AG. Genomic medicine for kidney disease. Nat Rev Nephrol. (2018) 14:83–104. doi: 10.1038/nrneph.2017.167
16. Brook-Carter PT, Peral B, Ward CJ, Thompson P, Hughes J, Maheshwar MM, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease–a contiguous gene syndrome. Nat Genet. (1994) 8:328–32. doi: 10.1038/ng1294-328
17. Hwang YH, Conklin J, Chan W, Roslin NM, Liu J, He N, et al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. (2016) 27:1861–8. doi: 10.1681/ASN.2015060648
18. Rossetti S, Harris PC. Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol. (2007) 18:1374–80. doi: 10.1681/ASN.2007010125
19. Rossetti S, Hopp K, Sikkink RA, Sundsbak JL, Lee YK, Kubly V, et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol. (2012) 23:915–33. doi: 10.1681/ASN.2011101032
20. Cornec-Le Gall E, Audrézet MP, Rousseau A, Hourmant M, Renaudineau E, Charasse C, et al. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. (2016) 27:942–51. doi: 10.1681/ASN.2015010016
21. Gimpel C, Bergmann C, Bockenhauer D, Breysem L, Cadnapaphornchai MA, Cetiner M, et al. International consensus statement on the diagnosis and management of autosomal dominant polycystic kidney disease in children and young people. Nat Rev Nephrol. (2019) 15:713–26. doi: 10.1038/s41581-019-0155-2
22. Reiterová J, Štekrová J, Merta M, Kotlas J, Elišáková V, Lněnička P, et al. Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol. (2013) 14:59. doi: 10.1186/1471-2369-14-59
23. Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. (2009) 20:205–12. doi: 10.1681/ASN.2008050507
24. Zhuang J, Zhao Z, Zhang C, Song X, Lu C, Tian X, et al. Case report: successful outcome of treatment using rituximab in an adult patient with refractory minimal change disease and β-thalassemia complicating autoimmune hemolytic anemia. Front Med. (2022) 9:1059740. doi: 10.3389/fmed.2022.1059740
25. Rossetti S, Strmecki L, Gamble V, Burton S, Sneddon V, Peral B, et al. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet. (2001) 68:46–63. doi: 10.1086/316939
26. Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. (2013) 24:1006–13. doi: 10.1681/ASN.2012070650
27. Cai Y, Fedeles SV, Dong K, Anyatonwu G, Onoe T, Mitobe M, et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J Clin Invest. (2014) 124:5129–44. doi: 10.1172/JCI67273
28. Ranjzad F, Aghdami N, Tara A, Mohseni M, Moghadasali R, Basiri A. Identification of three novel frameshift mutations in the PKD1 gene in iranian families with autosomal dominant polycystic kidney disease using efficient targeted next-generation sequencing. Kidney Blood Press Res. (2018) 43:471–8. doi: 10.1159/000488471
29. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
30. Benz EG, Hartung EA. Predictors of progression in autosomal dominant and autosomal recessive polycystic kidney disease. Pediatr Nephrol. (2021) 36:2639–58. doi: 10.1007/s00467-020-04869-w
31. Fang ZW, Xu SZ, Wang YD, Sun LE, Feng YE, Guo YI, et al. Pathogenicity analysis of novel variations in Chinese Han patients with polycystic kidney disease. Gene. (2017) 626:433–41. doi: 10.1016/j.gene.2017.05.046
32. Schrier R, Abebe K, Perrone R, Torres V, Braun W, Steinman T, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. (2014) 371:2255–66. doi: 10.1056/NEJMoa1402685
33. Rahbari-Oskoui F, Williams O, Chapman A. Mechanisms and management of hypertension in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. (2014) 29:2194–201. doi: 10.1093/ndt/gft513
34. Murphy EL, Droher ML, DiMaio MS, Dahl NK. Preimplantation genetic diagnosis counseling in autosomal dominant polycystic kidney disease. Am J Kidney Dis. (2018) 72:866–72. doi: 10.1053/j.ajkd.2018.01.048
35. Bergmann C. Recent advances in the molecular diagnosis of polycystic kidney disease. Expert Rev Mol Diagn. (2017) 17:1037–54. doi: 10.1007/s00467-013-2706-2
36. Radhakrishnan Y, Duriseti P, Chebib FT. Management of autosomal dominant polycystic kidney disease in the era of disease-modifying treatment options. Kidney Res Clin Pract. (2022) 41:422–31. doi: 10.23876/j.krcp.21.309
37. Chang MY, Ong AC. Mechanism-based therapeutics for autosomal dominant polycystic kidney disease: recent progress and future prospects. Nephron Clin Pract. (2012) 120:c25–34. doi: 10.1159/000334166
38. Dong K, Zhang C, Tian X, Coman D, Hyder F, Ma M, et al. Renal plasticity revealed through reversal of polycystic kidney disease in mice. Nat Genet. (2021) 53:1649–63. doi: 10.1038/s41588-021-00946-4
39. Bhutani G, Astor BC, Mandelbrot DA, Mankowski-Gettle L, Ziemlewicz T, Wells SA, et al. Long-term outcomes and prognostic factors in kidney transplant recipients with polycystic kidney disease. Kidney360. (2020) 2:312–24. doi: 10.34067/KID.0001182019
40. Spithoven EM, Kramer A, Meijer E, Orskov B, Wanner C, Abad JM, et al. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival–an analysis of data from the ERA-EDTA Registry. Nephrol Dial Transplant. (2014) 29 Suppl 4(Suppl. 4):iv15–25. doi: 10.1093/ndt/gfu017
41. Jung Y, Irazabal MV, Chebib FT, Harris PC, Dean PG, Prieto M, et al. Volume regression of native polycystic kidneys after renal transplantation. Nephrol Dial Transplant. (2016) 31:73–9. doi: 10.1093/ndt/gfv227
42. Yamamoto T, Watarai Y, Kobayashi T, Matsuda Y, Tsujita M, Hiramitsu T, et al. Kidney volume changes in patients with autosomal dominant polycystic kidney disease after renal transplantation. Transplantation. (2012) 93:794–8. doi: 10.1097/TP.0b013e318246f910
Keywords: second-generation sequencing, autosomal dominant polycystic kidney disease, PKD1 gene mutation, genotype-phenotype characteristics, genetic kidney disease
Citation: Zhuang J, Aierken A, Yalikun D, Zhang J, Wang X, Ren Y, Tian X and Jiang H (2023) Case report: Genotype-phenotype characteristics of nine novel PKD1 mutations in eight Chinese patients with autosomal dominant polycystic kidney disease. Front. Med. 10:1268307. doi: 10.3389/fmed.2023.1268307
Received: 27 July 2023; Accepted: 18 September 2023;
Published: 11 October 2023.
Edited by:
Jing Miao, Mayo Clinic, United StatesReviewed by:
Pamela Tran, University of Kansas Medical Center, United StatesDavid M. Feliciano, Clemson University, United States
Copyright © 2023 Zhuang, Aierken, Yalikun, Zhang, Wang, Ren, Tian and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Jiang, amFuZ2gteXRAMTYzLmNvbQ==; Xuefei Tian, eHVlZmVpLnRpYW5AeWFsZS5lZHU=