 Audrey Cleuren
Audrey Cleuren Grietje Molema
Grietje Molema
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 09 November 2023
Sec. Intensive Care Medicine and Anesthesiology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1252021
This article is part of the Research TopicEndothelial Activation and Microcirculatory Disorders in Sepsis and Critical Illness - Volume IIView all 9 articles
In the last decades, it has become evident that endothelial cells (ECs) in the microvasculature play an important role in the pathophysiology of sepsis-associated multiple organ dysfunction syndrome (MODS). Studies on how ECs orchestrate leukocyte recruitment, control microvascular integrity and permeability, and regulate the haemostatic balance have provided a wealth of knowledge and potential molecular targets that could be considered for pharmacological intervention in sepsis. Yet, this information has not been translated into effective treatments. As MODS affects specific vascular beds, (organotypic) endothelial heterogeneity may be an important contributing factor to this lack of success. On the other hand, given the involvement of ECs in sepsis, this heterogeneity could also be leveraged for therapeutic gain to target specific sites of the vasculature given its full accessibility to drugs. In this review, we describe current knowledge that defines heterogeneity of organ-specific microvascular ECs at the molecular level and elaborate on studies that have reported EC responses across organ systems in sepsis patients and animal models of sepsis. We discuss hypothesis-driven, single-molecule studies that have formed the basis of our understanding of endothelial cell engagement in sepsis pathophysiology, and include recent studies employing high-throughput technologies. The latter deliver comprehensive data sets to describe molecular signatures for organotypic ECs that could lead to new hypotheses and form the foundation for rational pharmacological intervention and biomarker panel development. Particularly results from single cell RNA sequencing and spatial transcriptomics studies are eagerly awaited as they are expected to unveil the full spatiotemporal signature of EC responses to sepsis. With increasing awareness of the existence of distinct sepsis subphenotypes, and the need to develop new drug regimen and companion diagnostics, a better understanding of the molecular pathways exploited by ECs in sepsis pathophysiology will be a cornerstone to halt the detrimental processes that lead to MODS.
Forming a barrier between the blood and underlying parenchyma, endothelial cells (ECs) cover the inner lining of all blood vessels. Together with circulating immune cells, they actively engage in the host response to bacterial and viral infections. While this is initially a localised process aimed at eliminating the pathogen and restoring tissue homeostasis, an excessive and dysregulated response can eventually result in sepsis (1). Despite improvements in patient care, there is currently still no effective treatment directly improving clinical outcome (2), and sepsis remains a global health problem with incidence estimates nearing 50 million cases per year and 11 million sepsis-related deaths reported in 2017 (3).
Progressive organ dysfunction is common in sepsis pathogenesis, leading to multiple organ dysfunction syndrome (MODS) when two or more organs are affected (Figure 1). In the clinic, this typically presents as diminished creatinine clearance, elevated blood urea nitrogen and oliguria/anuria; acute respiratory distress syndrome (ARDS); elevated bilirubin levels, dysregulated production of plasma coagulation factors and increased production of acute-phase proteins; malabsorption in the gut; lower consciousness; shock, cardiac dysfunction and/or thrombocytopenia and disseminated intravascular coagulation (4–7). Furthermore, patients with sepsis have systemically elevated levels of vascular cell adhesion molecule 1 (VCAM-1), vascular endothelial cadherin (VE-cadherin), EC-derived coagulation factors such as von Willebrand factor (VWF) and soluble thrombomodulin, as well as angiopoietin-1 and -2, thus strongly implying engagement of the endothelium in the body’s response in sepsis (8–12). Indeed, the microvasculature, which represents the largest surface of the vascular tree, is not only a main target but also a contributor to the sepsis-associated pathophysiological processes underlying MODS since it is the predominant site of leukocyte recruitment and blood vessel permeability, and is important for maintaining the haemostatic balance (5, 7, 13).
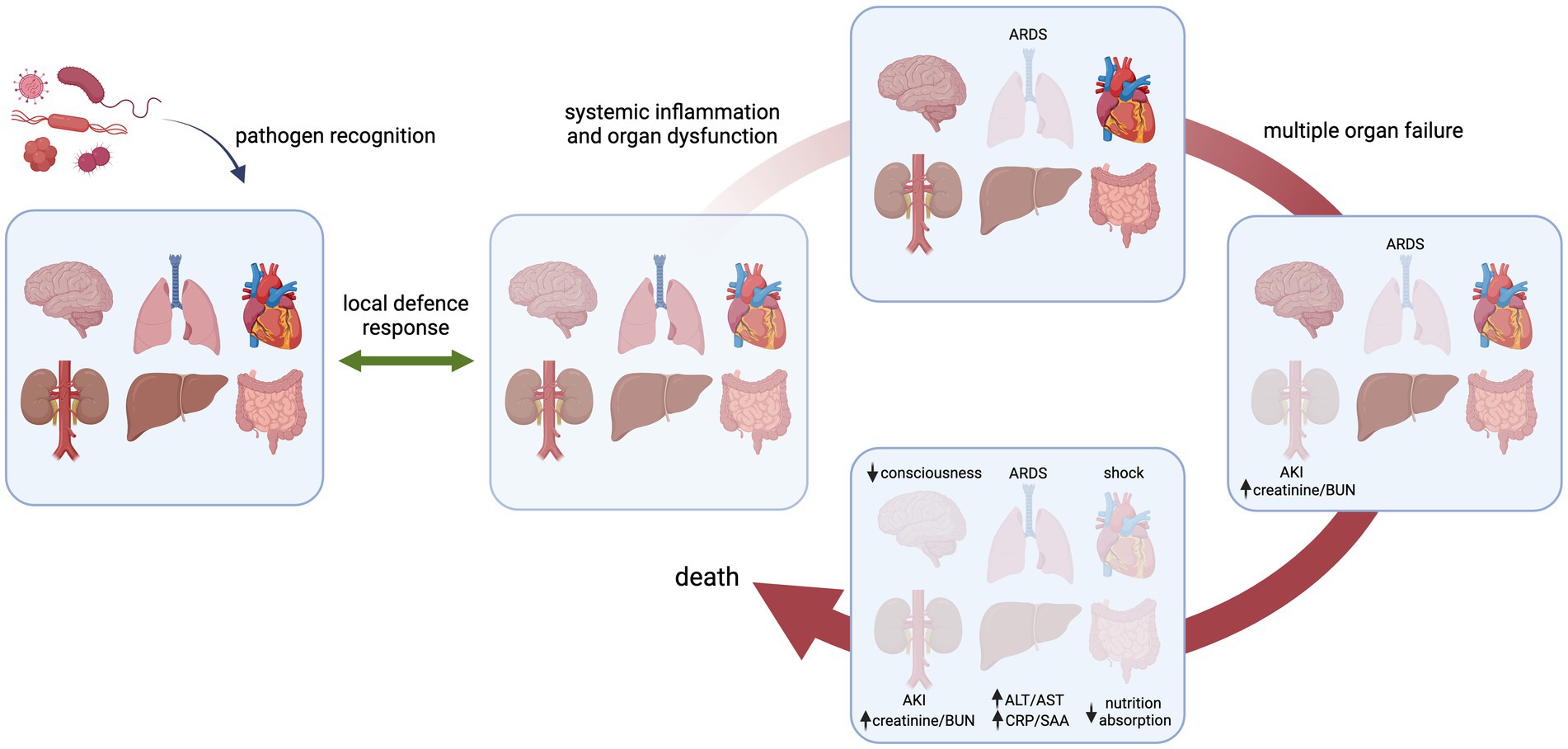
Figure 1. Schematic presentation of progression of organ loss in sepsis in response to an invading pathogen. Initially, the host senses the infectious microorganism that results in a local defence response to eliminate the pathogen and restore tissue homeostasis. (A dysregulated host response can lead to systemic inflammation and organ dysfunction, exemplified here for the lung, eventually leading to failure of multiple organs and death. Examples of specific organ failure as seen in the clinic are listed. The order and kinetics of organ systems affected are arbitrarily chosen and do not occur in a sequential fashion per se. ARDS, acute respiratory distress syndrome; AKI, acute kidney injury; BUN, blood urea nitrogen; ALT/AST, alanine transaminase/aspartate transaminase; CRP/SAA, C-reactive protein/serum amyloid A.
Given the strong dependence of ECs on their microenvironment, studying their role necessitates looking at their behaviour in an in vivo context, since enzymatic dissociation or culturing ECs leads to dramatic shifts in gene expression profiles (14–16). The organ- and microenvironment-specific behaviour also precludes the extrapolation of observations from one vascular bed to another without further validation (17, 18). This does not only hold true for changes observed under pathophysiological conditions, but also for studying pharmacological interventions aimed at the microvasculature (19). Although it is still technically challenging to study ECs in vivo, recent technological advances have created exciting new opportunities in this respect. Based on these developments we have chosen to focus this review on a combination of hypothesis-driven and unbiased -omics studies reporting in vivo endothelial responses in sepsis. While the former typically interrogate a single molecule or pathway, the latter provide a more comprehensive approach that has the potential to unveil previously understudied pathways important in sepsis pathology, and could thus lead to the identification of novel therapeutic targets.
We first provide a brief introduction on blood vessel and endothelial cell heterogeneity, as well as on relevant functions of the microvascular endothelium. We will then describe our current understanding of endothelial responses in sepsis and sepsis-related conditions based on studies performed in tissue samples from patients who died of sepsis, and in animals subjected to experimental sepsis, with a special focus on EC reactivity across organs. Both the differences and similarities in endothelial responses between tissues may provide opportunities for the design of biomarker panels that can be measured in blood or urine to determine the kinetics of microvascular engagement in response to infection. A better understanding of organ-specific molecular reactions of the microvascular endothelium in sepsis is also essential for a rational design of (combination) therapies that interfere with organotypic EC dysfunction. By providing a perspective that combines knowledge from the past with technological innovations of today, we hope the long-standing notion of sepsis being “the graveyard of pharmaceutical industry” (20) will become invalid.
All blood vessels in the body consist of an endothelial cell monolayer that is in direct contact with the blood and supported by mural cells (smooth muscle cells in larger veins and arteries, pericytes in the microvasculature). The structural distinction along the different branches of the vascular tree is predominantly dependent on mechanical forces (shear stress), whereas structural differences within the microvasculature are mostly dictated by the needs of the underlying parenchyma (Figure 2). For example, the liver sinusoidal endothelium is highly permeable as it consists of discontinuous ECs that lack a basement membrane and have open fenestrae to allow free transfer of fluid, nutrients, and both small and large molecules (21, 22). Endo- and exocrine glands, intestinal mucosa, and dedicated microvascular segments in the kidney all contain fenestrated endothelium to accommodate efficient secretion into and filtration of the blood (23–26). Microvessels in the heart, skin and lung are less permeable and contain continuous, non-fenestrated endothelium hallmarked by the presence of caveoli and vesiculo-vacuolar organelles (VVOs). While caveoli assist in the passage of macromolecules from blood into tissue (27), VVOs are involved in macromolecular extravasation and typically located in the ECs of postcapillary venules from organs that are sensitive to permeability-increasing signals induced by for example, vascular endothelial growth factor (VEGF) and histamine (28).
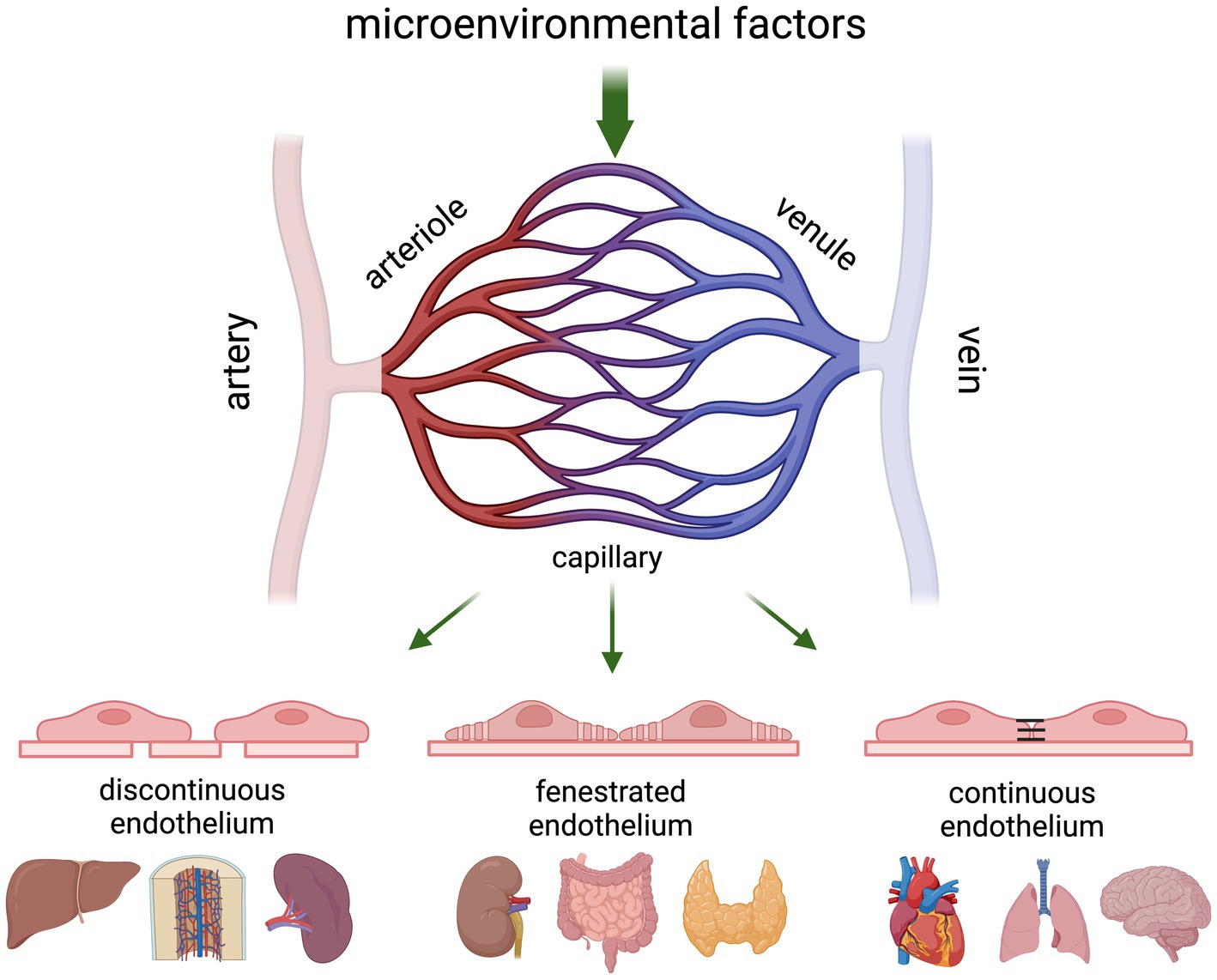
Figure 2. Schematic presentation of the gross architecture of blood vessels in an adult vertebrate. The cellular composition of blood vessels changes along the vascular tree where arterioles transport the blood from arteries into the tissue capillaries, and postcapillary venules transport it back to the bigger veins. Vascular permeability and leukocyte recruitment is predominantly regulated at the level of the capillaries and postcapillary venules, which show organ-specific structural differences based on inter-endothelial connections with continuous capillaries allowing the most controlled passage of blood and soluble components in the blood, and discontinuous capillaries allowing free passage. Although vessels are often categorised based on diameter, scRNA-seq studies have shown that there is a continuum of transcriptional states in ECs across the different branches of the vascular tree.
The correlation between structure and function of the endothelium is also clearly illustrated in the blood-brain barrier, where adherens junctions and tight junctions form strong connections between ECs to avoid noxious stimuli present in the circulation from entering the central nervous system. Particularly the presence of tight junctions, which are composed of occludin and members of the claudin, zona occludens and junction-associated molecule families, limits transcytosis. Therefore, to enable selective transfer of solutes from the blood into the brain parenchyma, these ECs express many highly specialised transporters (29, 30).
As illustrated above, ECs need to tailor their form and function to the microenvironment they reside in. While this has long been recognized, understanding the observed heterogeneity at the molecular level was initially limited to studies evaluating specific genes and proteins in a low-throughput manner via histological approaches or the use of transgenic animals (31). Although crucial in laying the foundation for discerning the role of the endothelium in regulating immune cell recruitment and extravasation, barrier function and vascular permeability, and its contribution to coagulation, it was the introduction of unbiased -omics approaches that has greatly enhanced our knowledge on the molecular mechanisms underlying the structural and functional heterogeneity of ECs. Particularly high-throughput transcriptomics combined with improvements in methods to select ECs (32–34), EC-associated transcripts (16, 35–37) or luminal proteins (38, 39), has made it possible to evaluate the endothelium directly from its native in vivo environment in an unbiased manner (Table 1).
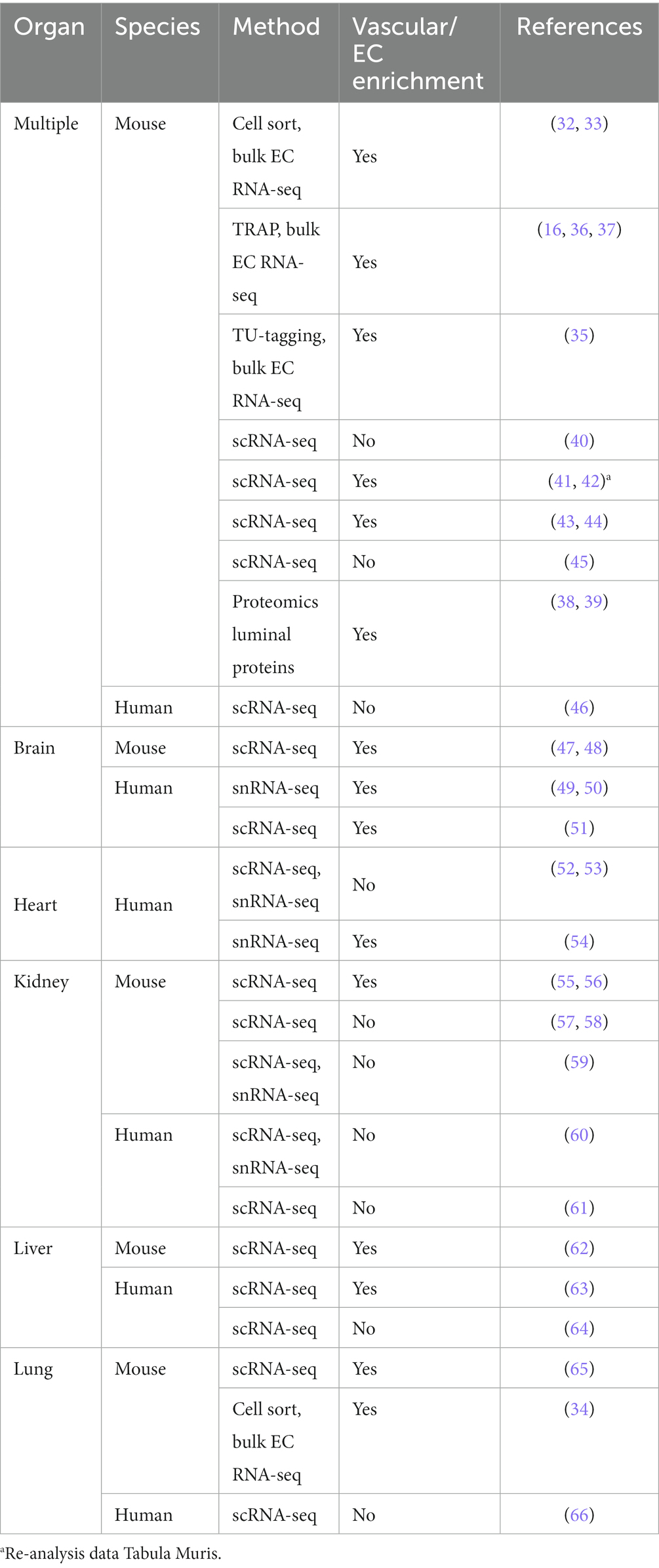
Table 1. Overview of studies evaluating organ-specific EC expression profiles.
Despite being invaluable in establishing organotypic EC heterogeneity, it is important to note that these EC-enrichment methods do not allow the deconvolution of distinct endothelial subpopulations within a tissue. Instead, the data form an averaged endothelial expression profile per organ. This limitation has at least in part been overcome by the introduction of single cell and single nucleus RNA sequencing (RNA-seq), which has further improved our understanding of EC heterogeneity by providing increased cellular resolution. This has enabled identification of the EC compartment from organ-specific single cell RNA-seq (scRNA-seq) data based on the expression of known EC markers (41, 42, 45, 46, 52–54, 57–61, 64, 66). Furthermore, by performing scRNA-seq analysis on preselected ECs or vasculature-enriched samples (43, 44, 47–51, 55, 56, 62, 63, 65), transcriptional differences across EC subpopulations within organs and along the arterio-venous axis have been established as recently reviewed (24, 67–70), with available studies and datasets summarised in Table 1. The revealed molecular heterogeneity of the endothelium between, and even within organs, has further strengthened the conclusions that observations from one tissue or vascular segment cannot simply be extrapolated to the next.
Although our knowledge on EC heterogeneity has vastly improved in recent years, it is still not fully understood how heterogeneous responses in pathological conditions are controlled at the molecular level. Yet, their rapid responses to fluctuations in the local milieu and their unique anatomical position make ECs both an early target and contributor to many diseases, including sepsis (71). As such, the endothelium produces and secretes proteins into the circulation that could be used in the development of biomarkers, while on the other hand it makes them attractive targets for pharmacological interventions (72). In the following paragraphs, we will provide a brief overview of processes essential to the function of ECs in maintaining tissue homeostasis, discuss the impact of EC heterogeneity on these processes, and how they are perturbed under sepsis conditions.
Leukocyte recruitment from the vasculature into the parenchyma in response to inflammatory conditions as present in sepsis involves sequential leukocyte tethering, rolling, adhesion, and transmigration through the endothelial layer. Under normal conditions, ECs are covered by the glycocalyx, a layer consisting of proteoglycans, glycosaminoglycans, and incorporated plasma- and EC-derived proteins that protects the endothelium from directly interacting with blood cells (73, 74). In the presence of cytokines such as TNFα, interleukin (IL)-6 and IL-8 the glycocalyx can be degraded, thereby exposing (upregulated) adhesion molecules including P- and E-selectin as well as VCAM-1 and ICAM-1 (75–78). These adhesion molecules, in addition to inflammation-induced chemokines (e.g., MCP-1), recognize their cognate receptors on circulating immune cells, and guide leukocyte tethering and adhesion under pathological conditions (79, 80). Leukocyte transmigration on the other hand, is largely dependent on dynamics of EC adherens junctions and involve pan-EC molecules like PECAM-1 (also known as CD31) and junctional adhesion molecules, which also play an important role in regulating vascular permeability.
Nowadays it is widely accepted that the recruitment of (subsets of) immune cells into the parenchyma is a complex concerted action that takes place predominantly in the microvasculature, and that this process is dependent on the molecular make-up of the local tissue environment. For example, T lymphocyte recruitment into the brain in response to experimental encephalomyelitis has been shown to be dependent on the local presence of laminin α4 (81), while leukocytes expressing the chemokine receptor CCR10 preferentially home to skin endothelium, and CCR7-positive leukocytes migrate into secondary lymphoid organs (82). Furthermore, early studies using radiolabelled antibodies specific for VCAM-1 and ICAM-1 have shown that constitutive expression of these 2 adhesion molecules is different across organs, with ICAM-1 levels being higher than VCAM-1 in brain and heart (83). These observations have been recently confirmed in bulk RNA-seq data of EC transcripts after translating ribosome affinity purification, which in addition showed the opposite result for kidney and to a lesser extent lung endothelium (16). Subsequent single cell RNA-seq data identified aerocytes (a specific EC subset in the lung) as the main ICAM-1-positive capillary cell (65), while the cerebral cortex contains a specialized postcapillary EC population characterized by constitutive expression of adhesion molecules, including ICAM-1 (47). Not surprisingly, for both organs these EC subsets have been shown to be the preferred site of immune interactions and leukocyte transmigration (47, 65).
As mentioned, microvascular ECs have distinct morphological characteristics reflecting the permeability control required to exert their physiological function (27, 30). In addition to claudins and occludin that encompass tight junctions, VE-cadherin (encoded by CDH5) has been well-established as a gate-keeper of endothelial integrity in which EC–EC interactions are being formed by adherens junctions (84, 85). The dynamic expression of VE-cadherin is dependent on its phosphorylation status, with phosphorylation via VEGFR2 (KDR) or PECAM-1 leading to its endocytosis and degradation, resulting in increased vascular permeability (86).
Stabilization of the microvasculature is further influenced by the receptor tyrosine kinase TIE2 (TEK) and its ligands angiopoietin (ANGPT)-1 and ANGPT-2 (Figure 3). In quiescent endothelium, locally produced ANGPT-1 binds TIE2 and can subsequently activate 2 distinct intracellular signalling cascades that lead to stabilization of EC-EC junctions via VE-cadherin and suppression of inflammation-induced EC activation (91, 92). When ECs become activated they release Weibel–Palade bodies that contain high concentrations of ANGPT-2 (93). This rapidly tips the ANGPT balance in favour of ANGPT2, which via competitive binding to TIE2 leads to destabilization of the EC layer, a process that also involves the orphan receptor TIE1 (94). The ANGPT-induced destabilization furthermore enables a third well-studied molecular system important for vascular permeability: the VEGF pathway, in which VEGF interacts with VEGFR2, thereby leading to increased vascular leakage (95).
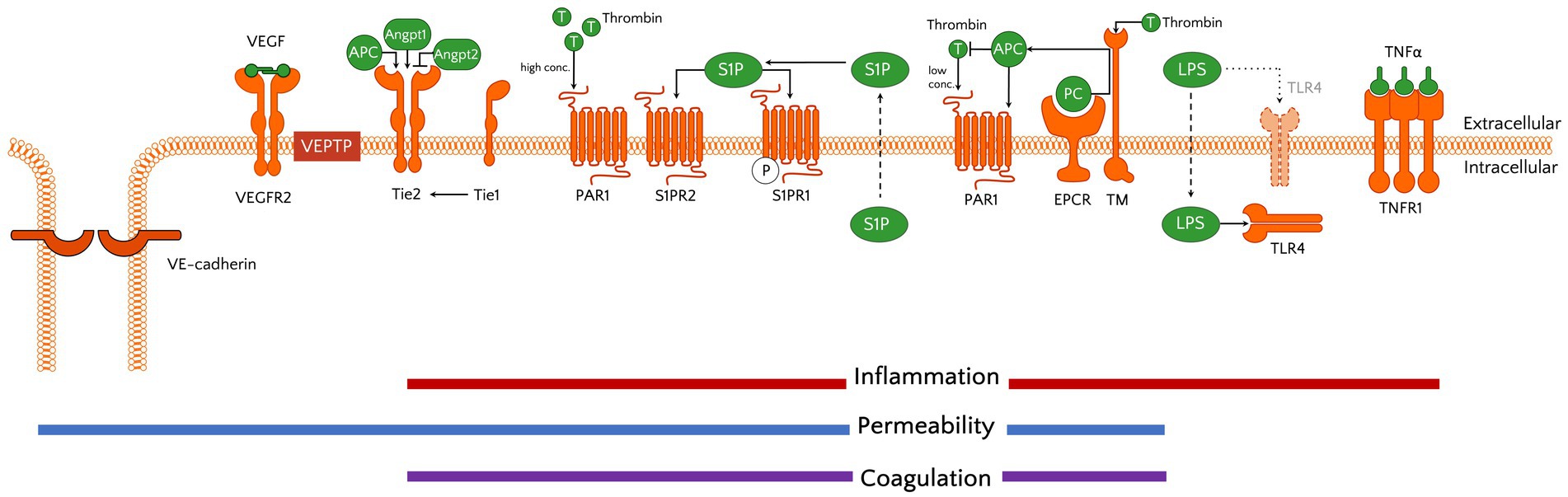
Figure 3. Schematic showing molecular systems important in endothelial (patho)physiology. Activation of receptors provides the microvascular ECs a pro-inflammatory, permeable, and coagulant status. The receptors and their ligands include examples discussed in the text, yet are not all encompassing. Of note, although TLR4 is traditionally known as a transmembrane receptor in myeloid cells, previous studies have demonstrated its intracellular location and function in ECs (87, 88). The reader is referred to Luxen et al. (90) for more detailed information on the signal transduction cascades that are activated by the receptors shown. APC, activated protein C; angpt, angiopoietin; EPCR, endothelial protein C receptor; LPS, lipopolysaccharide; PAR1, protease-activated receptor 1; PC, protein C; S1P(R), spingosine-1-phosphate (receptor); T, thrombin; Tie, tunica intima endothelial kinase; TLR, Toll-like receptor; TM, thrombomodulin; TNF(R), tumour necrosis factor (receptor); VEGF(R2), vascular endothelial growth factor (receptor 2).
In addition to these 3 main pathways regulating vascular permeability, other proteins have been identified to contribute to this process. Most notably is vascular endothelial phosphatase VE-PTP (PTPRB), which controls barrier function through its substrates that include VE-cadherin, TIE2 and VEGFR2 (96–98). Furthermore, sphingosine 1-phosphate (S1P) regulates endothelial integrity via binding to its G-protein coupled receptor S1PR1 that is expressed on ECs, thereby stabilizing adherens junctions (99, 100) (Figure 3). Interestingly, S1P binding to S1P receptor type 2 (S1PR2) was shown to be associated with increased permeability in conditions of acute inflammation (101).
Studies using conditional knock-out mice lacking endothelial VE-cadherin showed distinct patterns of organotypic microvascular leakage, with increased permeability in the heart and lung, while skin and brain vessels were not affected by loss of VE-cadherin (96). This could possibly be explained by differences in basal expression levels, as RT-qPCR and EC-enriched RNA-seq data showed that CDH5 is more abundant in the lung and heart as compared to kidney, brain and liver (16, 102). Besides organotypic differences in expression of genes important for maintaining vascular integrity, heterogeneous phenotypes can also exist within tissues. For example, it has been shown that in quiescent conditions, VE-cadherin contains phosphorylated residues in veins but not arteries, which might prime the protein for rapid internalization under inflammatory conditions to allow the required increase in vascular permeability (86). In addition, the tight junction protein claudin 5 (CLDN5) is highly expressed in the brain as compared to the other organs, while a recent scRNA-seq study showed that the decrease in CLDN5 levels along the arteriovenous axis in the brain inversely correlated with histamine-induced vascular leakage (103). Similarly, ANGPT-2 is barely detectable in murine renal arterioles and postcapillary venules, while it is abundantly expressed in the glomerular microvasculature (19). Interestingly, although this ANGPT-2 expression in glomeruli coincides with high levels of TIE2, relatively low levels of ANGPT-1, and robust expression of VEGF and VEGFR2—a combination that is normally associated with promotion of barrier degradation and neovascularization—this vascular segment is not particularly leaky nor proliferatively active (19).
Under physiological conditions, ECs have an anticoagulant phenotype as they express tissue factor pathway inhibitor (TFPI), thrombomodulin (TM) and the endothelial protein C receptor (EPCR; PROCR) (104, 105). Their anticoagulant property is further supported by the presence of fibrinolytic proteins including tissue-type plasminogen activator (tPA) and the plasminogen activator inhibitor PAI-1 (SERPINE1). Endothelial activation causes a procoagulant shift and leads to the release of VWF, which is required for platelet binding and activation that enhances thrombin formation. Although thrombin is mostly known as a procoagulant factor, it can also serve as an anticoagulant via binding to TM to activate protein C (leading to the formation of activated protein C, or APC), a process that is further facilitated by EPCR (106) (Figure 3). In addition to its anticoagulant function, APC also exerts anti-inflammatory and barrier protection properties (107–109), and has therefore gained a lot of interest in the sepsis field as a target for pharmacological interventions in the past (see below). Besides activating protein C, thrombin can also bind and activate protease-activated receptors (PARs), with binding to PAR1 playing a role in regulating microvascular permeability (110). Additional crosstalk between regulation of haemostasis and vascular integrity also takes place via ANGPT-2, which was recently shown to inhibit TM-mediated formation of APC (111).
Endothelial heterogeneity with respect to coagulation factors has received substantial attention, with studies in the 1990s already showing distinct expression patterns for EPCR, TFPI and tPA across organs as well as along the vascular tree [recently reviewed in (31)]. Similarly, VWF expression has been shown to be particularly high in large blood vessels, but low in capillaries (31, 112, 113). VWF serves as a carrier protein for coagulation factor VIII (F8), and in the past decade it has become evident that FVIII is also synthesized in the endothelium, particularly in the liver and kidney (114, 115). These initial organotypic observations have since been validated in numerous studies, including those using scRNA-seq analyses such as the Tabula Muris Consortium (40). Evaluation of VWF/F8 expression levels across different organs in this dataset showed that lung also contains a specific EC subpopulation that expresses F8, but interestingly these ECs do not express VWF. In line with this, a recent scRNA-seq analysis of alveolar capillary endothelium showed that 2 different EC types (aerocyte vs. “general” capillary ECs) each produce their own unique set of pro- and anticoagulant factors. Whereas expression of TFPI and PAI-1 was restricted to aerocytes, general capillary ECs expressed VWF and tPA (65). These data suggest a division of labour, where different ECs serve different functions in regulating haemostasis, which could potentially also explain the observations regarding F8/VWF expression in the lung (40).
Multiple organ dysfunction syndrome in sepsis patients is a complex disorder resulting from an aberrant host response to pathogen invasion, characterized by a derailed immune response leading to organ injury (116) (Figure 1). The endothelium actively engages in the establishment of MODS, as evidenced by clinical studies reporting increased circulating levels of EC-derived proteins including soluble E-selectin, VCAM-1, VE-cadherin, PECAM-1, VEGFR1, ANGPT-2, IL-8, VWF, TM, tPA, PAI-1, and glycocalyx constituents in sepsis patients (8–11, 117–123). Also in the human endotoxaemia model (124), a consistent rapid increase in many of these plasma markers were established after LPS administration, strongly suggesting a direct link between pathogen exposure and EC response (119, 125, 126).
Although important for diagnostic purposes, changes in EC-derived proteins present in the circulation do not provide direct information on the extent of organ-specific microvascular contributions. Being able to address the latter depends directly on tissue samples, yet studies reporting molecular and functional behaviour throughout different vascular beds have been scarce and are typically limited to those evaluating samples of patients who succumbed to sepsis. Nevertheless, histological analyses have shown the presence of immune cells in the periportal regions of the liver and kidney glomeruli (127, 128), and neutrophil infiltration in the lung has long been considered an important contributor to sepsis-related ARDS (129, 130). In addition to neutrophils, the number of macrophages and B- and T-lymphocyte are also increased in lungs of patients who died of Neisseria meningitidis septic shock (131). A direct comparison between lung and heart samples of these patients revealed that the influx of several leukocyte populations was 30–80% higher in the lung as compared to heart, whereas the brain was devoid of enhanced leukocyte recruitment (132). Despite this latter, haemostatic abnormalities have been observed in brain samples of sepsis patients, with areas displaying hypercoagulability as well as haemorrhaging, indicative of consumptive coagulopathy (128). In addition, enhanced coagulation has also been noted in the kidney, as illustrated by fibrin depositions in glomeruli and peritubular capillaries in a subset of sepsis patients (127, 128).
Besides evaluating cellular contributions, immunohistochemical analyses indicated increased levels of MCP-1 and PAI-1 in the alveolar space in the lung, likely expressed by alveolar capillary ECs, whereas in heart these proteins were upregulated and co-localized in the arteriolar microvascular segment (132). Furthermore, evaluation of kidney biopsies of septic patients for markers known for their role in vascular permeability regulation showed a decrease in ANGPT-1 levels while ANGPT-2 was increased and TIE2 remained unchanged. At the same time, expression of VEGF and VEGFR2 was also reduced (102). These changes are all expected to contribute to kidney failure associated with sepsis.
In a recent study, microarray analysis was performed in tissue samples of meningococcal septic shock patients, which provides an opportunity to evaluate transcriptional changes in an unbiased manner and determine differences and similarities in organotypic responses (132). Although all organs included in the study showed changes under septic conditions as compared to samples from non-inflammatory origin, the number of differentially expressed genes was highest in kidney, followed by lung, heart, liver, and was lowest in the spleen. Not surprisingly, many of these genes were related to the host’s inflammatory response. In addition, a decrease in transcripts associated with metabolism and energy production was also observed across multiple organs (132). Interestingly, short-term changes in metabolism can be beneficial to the host as a temporal increase in energy is required to eliminate the pathogen. However, uncontrolled and prolonged disruption of the metabolic balance is detrimental since at the same time energy should be preserved because of poor nutritional input (133).
While studies based on human tissue biopsies have been informative, interpretation of results is difficult as they are often performed on bulk tissue, which precludes identification of the cell-specific contribution to the observed molecular changes. Even for histopathological and immunohistochemical analysis, correlating spatiotemporal cellular and molecular changes in the microvasculature to clinical symptoms is challenging. This is partly due to the intrinsic heterogeneity in the patient population itself (age, underlying co-morbidities, etcetera), and also because it is often unknown when the patient got infected prior to presentation in the hospital (see Challenges below). It thus remains elusive whether EC responses occur prior to the establishment of MODS, or whether EC activation forms an integral part of its pathogenesis. Therefore, animal models are invaluable to provide a rationale for validation studies in the sparse clinical material available to date as they recapitulate processes underlying sepsis and allow for detailed cellular and molecular analyses in an organ- and cell type-specific manner over time.
Non-human primates are highly similar to humans in their anatomy and physiology, as well as their hemodynamic and cytokine responses to infection. Also the ability to provide supportive care similar to that in septic patients makes them an important model to study sepsis, and as such they have provided crucial insights in disease aetiology (134–136). Other larger animals including pigs and sheep have also been used as they too share many aspects of sepsis that are similar to humans (134). However, from an economical and feasibility perspective, mice are much more attractive due to the availability of genetically engineered animals and low costs. Most importantly, the relative ease of experimental models allows a more reproducible evaluation of sepsis pathology. Although the use of mouse models in sepsis research has been a topic of debate (137, 138) as they do not fully recapitulate the clinical complexity, they have significantly contributed to our molecular understanding of the host response to infection and form the cornerstone of translational research.
The most frequently used models to induce a sepsis-like phenotype are based on the administration of either a toxin (e.g., lipopolysaccharide; LPS) or viable pathogen, or by breaching an endogenous protective barrier such as done in the caecal ligation and puncture (CLP) model (139–141). Whereas LPS is often used because of its technical ease and reproducibility, it has also been criticised for not capturing the intricacies of sepsis. On the other hand, CLP results in a polymicrobial infection, leading to a response that is more representative for sepsis (142). Although considered the “golden standard,” it is more variable and the severity of disease is highly dependent on the experimental parameters. Despite these differences, both models display signs of MODS as indicated by an increase in serum creatinine and blood urea levels reflecting renal dysfunction, AST and ALT levels indicating hepatocellular injury, and lung myeloperoxidase representing increased neutrophil infiltration. In addition, these models lead to a decrease in body temperature and heart rate, and metabolic derailment of organs has also been reported in both the CLP model and LPS-induced endotoxaemia (143–146).
For the remainder of this review, we focus where possible on studies that reported sepsis-induced changes in more than one organ to best reflect human MODS. Moreover, this circumvents limitations in interpretation of combined results from different studies which would be complicated due to methodological variations in execution of experiments, such as the use of different dosages and administration routes of LPS, differences in experimental execution of the CLP model, and/or differences in the time frame in which molecular and functional changes were assessed. We will first discuss results from hypothesis-driven studies that directly evaluated molecules involved in leukocyte recruitment, regulation of vascular permeability, and maintenance of the haemostatic balance. This will be followed by an overview of animal studies that used unbiased transcriptomics approaches in order to get a more comprehensive overview of the vascular bed-specific changes that occur during, and contribute to, sepsis pathology.
EC activation in sepsis can occur via the LPS-induced Toll-like receptor (TLR)-4 signalling cascade, interaction with circulating cytokines, and via changes in blood flow leading to differences in shear stress (147, 148). Histological analyses showed that leukocytes, predominantly neutrophils, infiltrate lung, and to a lesser extent liver and intestine, with the lowest neutrophil content observed in kidney and heart 4 h after LPS exposure (149). In the polymicrobial CLP model of sepsis a similar increase in neutrophil accumulation occurred in major organs including lung, liver and kidney, which lasted until at least 24 h after surgery (150–152). Despite the comparable neutrophil influx across these organs, formation of neutrophil extracellular traps (NETs), which has been associated with loss of organ function (153), was most prominent in lung and least in kidney (154).
In general, endotoxaemia models all result in an upregulation of E-selectin, P-selectin, ICAM-1 and VCAM-1 (18, 155–158). As expected, induction of gene expression occurs early, within 4 h after LPS exposure, and with the exception of E-selectin in the liver, all these changes were still present after 24 h (18, 157, 158). It is important to note that lung showed the least increase in expression as compared to other organs tested, which might relate to the relatively higher abundance of these proteins under basal conditions (157, 158).
In line with these observations, a previous study of N. meningitidis bacteraemia in pigs also showed increased levels of E-selectin, ICAM-1 and VCAM-1 in kidney, liver and lung with results in the latter being the most modest (159). Escherichia coli infection in mice identified the heart as the least responsive organ regarding the up-regulation of VCAM-1 as compared to changes in the lung, kidney and liver (160). Also in the CLP model, increases in E- and P-selectin occurred early, after 6 h (161), and their expression remained high in brain, heart and lung up to at least 24 h after sepsis induction (18, 162). Effects of CLP-induced sepsis on ICAM-1 and VCAM-1 expression were less prominent at these early time points in these organs. Wen et al. (163) showed that 14 days after CLP surgery ICAM-1 levels in kidney were elevated. However, it was not investigated whether this late stage increase in expression occurred in other organs as well.
Besides differences in expression levels across organs, immunohistochemical analysis of the lung and kidney indicated that 16 h post-CLP surgery E-selectin was primarily present in the bigger vessels in the lung (164). In the kidney, VCAM-1 was predominantly induced in arterioles whereas ICAM-1 was most abundant in glomeruli and peritubular capillaries (164).
Together these data indicate that spatiotemporal changes are unique to each organ and disease model, suggesting specialized functions and recruitment of potentially different (subsets of) immune cells across organs as well as different branches of the vascular tree within organs (158, 165, 166). Despite some differences in results between studies that were likely due to differences in pathogenic stimulus, in general selectins appear to respond more strongly than VCAM-1 and ICAM-1.
Endotoxaemic mice display increased permeability in the kidney, lung, heart and spleen at 6 h after LPS administration (101, 149). For kidney and lung, this increase sustained at least until 24 h post-LPS, whereas this was not the case for heart and spleen (158, 167). Collectively, these studies indicated that there was significant leakage in main organs in the first 24 h after LPS exposure, although data on vascular leakage in the liver was inconclusive at the 6 h timepoint. Interestingly, LPS did not affect cerebrovascular permeability. On the other hand, CLP surgery increased vascular permeability in all major organs, including the brain (168, 169). Mice infected with E.coli or Staphylococcus aureus to simulate peritonitis or pneumonia, respectively, also displayed microvascular leakage in the kidney, lung, liver and heart at 6–7 h after infection (160, 170). The brain was not assessed in these latter studies.
The molecular changes underlying the observed increase in sepsis-induced vascular permeability are predominantly regulated via VEGF/VEGFR and angiopoietin/TIE2 receptor interactions (30). Although VEGF is not produced by ECs, it has a major impact on their function via binding to the EC-expressed VEGFR1 (FLT1) and VEGFR2 (KDR). Changes in VEGF expression under septic conditions showed a spatiotemporal expression signature. For example in the kidney, LPS caused an increase in VEGF mRNA levels 4 h post-LPS, while after 8 h a significant reduction was observed and levels were normalized 24 h after LPS administration (102). Protein levels followed this kinetics, though at 24 h after LPS challenge VEGF protein in kidney remained reduced compared to control conditions (158). In lung, mRNA decrease only occurred later in time, at 8 h after LPS exposure, and remained low until at least 24 h post-LPS, coinciding with reduced protein levels at 24 h (102, 158). In heart on the other hand, VEGF protein levels were significantly increased in the early hours of endotoxaemia, and returned to basal levels at 24 h (158). In contrast to VEGF, VEGF receptors are expressed by the endothelium. In endotoxaemia, only in lung the expression of VEGFR1 and VEGFR2 decreased and remained low until 24 h post-LPS. In kidney, expression of neither receptor was significantly affected in time. Also in mouse CLP-sepsis, VEGFR2 expression remained constant in the kidney, while it decreased in a time-dependent manner in heart, liver and lung (171).
Regarding the angiopoietin/TIE2 axis, loss of TIE2 expression occurred in all major organs at 4–8 h after LPS exposure, with the kidney and lung being most affected (157, 172). These changes coincided with a transient decrease in ANGPT-1 levels in kidney at 8 h, while in lung ANGPT-1 was already reduced after 4 h, and remained low until at least 24 h after LPS administration (102). At the same time, ANGPT-2 levels were increased at the 4 h and 8 h timepoint in both organs, but normalized after 24 h. In line with these data from LPS studies, ANGPT-1 levels were also downregulated in CLP-induced sepsis (171). However, in contrast to LPS, CLP resulted in a decrease in ANGPT-2 in heart, kidney, liver, and lung between 12 and 24 h after surgery.
Given its importance in adhesion junctions, VE-cadherin is also often evaluated with regard to changes in vascular permeability, and endotoxaemia has been shown to result in significantly decreased levels in the lung (164). Also in the CLP model, a reduction in VE-cadherin was present in the heart, lung, kidney and liver, which was associated with increased vascular leakage in the latter two (173).
Overall, it can be concluded that there is a general increase in microvascular permeability across different experimental sepsis models, though there is variation in the kinetics and extent of leakage between organs, as well as variation in the responses observed in specific pathways associated with vascular barrier function.
Dysregulation of the haemostatic balance is a common feature of sepsis, which can lead to disseminated intravascular coagulation contributing to the development of organ dysfunction (174). This procoagulant shift is reflected by increased levels of circulating thrombin-antithrombin complexes, tissue factor, PAI-1 and D-dimer levels (175), with the latter indicating fibrin degradation. LPS-induced endotoxaemia results in fibrin depositions in the liver, lung and kidney, where particularly bigger vessels and a subset of capillaries were affected in liver and lung 8 h post-LPS (175, 176). In kidney, fibrin was present in all microvascular segments at 24 h after LPS exposure (177). In a rat LPS model, fibrin deposition was reported to occur in liver capillaries, glomeruli and peritubular capillaries but not in the lung (178). However, they noted that LPS-induced endothelial injury was dependent on the dose and duration of LPS treatment. Finally, fibrin deposition was also observed in the liver after CLP-induced sepsis (176).
The procoagulant shift of the endothelium is greatly influenced by the downregulation of anticoagulant proteins such as TFPI and thrombomodulin (TM). For example, in response to LPS TFPI mRNA levels decreased in heart, kidney, and lung at the 8 h timepoint, with levels returning back to normal within 24 h (179). TM in rat liver and lung capillaries was strongly reduced within 2–4 h after LPS exposure, and a similar trend was observed in kidney peritubular capillaries, although TM expression in glomeruli remained high (178).
PAI-1 inhibits fibrinolysis, and in line with the previously mentioned increase in plasma PAI-1 levels, LPS also upregulated its local expression in heart (mostly in capillaries), kidney, lung and brain (predominantly in venules). Increase in PAI-1 levels was highest in heart, whereas brain and lung showed only a modest increase (158, 179). Although the origin of procoagulant tissue factor has been controversial (175, 180), a (transient) increase in expression was observed across multiple organs in a rabbit LPS model with the highest induction in brain, kidney, and lung at 2–3 h after LPS exposure (180). In mice, on the other hand, LPS caused increased TF expression in kidney and lung, but not in liver, heart or brain (101, 181). Interestingly, the observed increase in kidney appeared to be almost completely regulated via S1PR2 signaling, whereas changes in the lung were only partly regulated by S1PR2 (101), indicating an additional connection between coagulation and vascular barrier protection, besides the protein C pathway (107–109).
In summary, these results indicate that rapid increases in tissue factor and PAI-1, together with decreased levels of anticoagulant proteins such as TFPI and TM, contribute to a procoagulant shift that can lead to fibrin deposition. Changes occur primarily at the level of the microvasculature, although distinct organotypic differences exist.
Based on the previously discussed hypothesis-driven studies, direct comparison of EC behaviour across organs revealed organ-specific differences in kinetics of molecular changes, yet literature on spatiotemporal responses of ECs is scarce. In addition, a big gap in knowledge exists to date regarding the location of molecular changes in microvascular branches in organs in response to sepsis conditions.
Unbiased approaches have become one of the most powerful tools to evaluate molecular characteristics of cells and organs in a systematic manner, and have provided valuable information on changes occurring in biological processes under pathophysiological conditions. Especially the application of single cell RNA-seq and spatial transcriptomics to help unravel protein–protein interactions important for identifying novel pathways contributing to disease pathogenesis, is eagerly awaited as this could enable the identification of potential diagnostic and prognostic biomarkers, as well as targets that would facilitate the development of therapeutics (182). The following section will discuss studies that used -omics analyses in order to get a better understanding of (organotypic) sepsis pathophysiology, and the contribution of the endothelium in this process. We will particularly focus on studies performing unbiased RNA-seq analysis of mRNA species in intact tissue (bulk RNA-seq), mRNAs bound to actively translating ribosomes isolated from endothelial cells specifically using a translating ribosome affinity purification (TRAP) approach followed by RNA-seq, and studies utilizing single cell RNA-seq that allows the evaluation of gene expression profiles at the individual cell level. We refer the reader to (31, 183, 184) for more information on these different techniques and their application in studying the vasculature.
One of the first studies using an unbiased approach to evaluate sepsis in an organ-specific way employed a rat CLP model, and identified subsets of genes that had both shared and unique expression patterns across different tissues that were temporally regulated (185). As expected, these included genes involved in inflammation and coagulation, but in addition they identified several transcripts that were not previously linked to sepsis-induced responses. For example, downregulation of genes known to be related to maintaining extracellular matrix (ECM) was observed in the lung and spleen, which suggests inadequate tissue repair. Furthermore, changes in transcripts important for antioxidant defence mechanisms were present in multiple tissues, including decreased levels of several members of the glutathione S-transferase gene family, of which glutathione S-transferase pi has been shown to play a role in regulating vascular permeability (186). Finally, they identified prominent alterations in genes associated with lipid metabolism across organs, and it is now well-established that lipid dysregulation in sepsis can affect the immune system and regulate the immune response by clearing bacterial toxins, reduce inflammation and inhibit the expression of adhesion molecules (187).
Many of these initial observations based on microarray-based data have since been confirmed using RNA-seq analysis. A recent study using a murine CLP model also identified significant changes in lipid metabolism and ECM remodelling in at least 2 organs, in addition to the expected (early) effects on the immune system and haemostasis (171). Metabolic dysregulation in the liver was already affected 6 h post-CLP, and changes sustained for at least 24 h. The kidney and lung also showed significant metabolic alterations at 12, respectively, 24 h after surgery, whereas the metabolic processes in the heart did not appear to be affected (171). This study furthermore validated the decrease in ECM genes in the lung, but interestingly found a CLP-induced overrepresentation of these transcripts in the liver. Overall, the number of significant differentially expressed genes for each organ was highest 12 h after CLP, with only the heart being almost completely normalized 24 h post-surgery.
Together, these studies illustrate spatiotemporal changes in gene expression patterns in sepsis in an unbiased way (171, 185). However, although they indicated a potential role for the endothelium in sepsis-related pathology, it must be noted that these studies used bulk tissue RNA for their analyses. The importance of studying transcriptional changes in a specific cell type, particularly those that, like ECs, are present in organs in low numbers, was initially shown using a translating ribosome affinity purification (TRAP) approach to determine EC-specific changes in an endotoxaemia model (16, 37). Similar to the apparent lack of CLP-induced changes in the brain in the rat CLP study (185), short-term LPS exposure in the mouse also resulted in minimal effects in the brain based on bulk tissue RNA-seq (16). However, when specifically evaluating expression profiles of the endothelial cells in the brain, the number of genes significantly affected was much higher and remarkably similar to those observed in other organs. Furthermore, with the exception of the brain, other target organs including the kidney, liver, lung and heart showed fewer EC-enriched genes to be upregulated than downregulated after LPS treatment, with the upregulated genes displaying a high degree of tissue-specificity. On the other hand, EC-enriched genes with reduced expression in LPS-treated mice were more commonly shared between organs (16).
EC-TRAP studies have confirmed many of the previously discussed organotypic alterations in expression levels of endothelial genes involved in leukocyte recruitment, cell junctions, and haemostasis (16, 37). In addition, gene ontology analyses based on transcripts significantly affected by LPS have indicated a consistent overrepresentation of genes involved in the regulation of metabolism across tissues. This has not only been observed after short-term LPS exposure, but a timecourse evaluating early and late inflammatory responses as well as changes occurring during the resolution phase also showed unique spatiotemporal expression patterns of metabolic genes (37). For example, brain EC-specific glycolysis genes were upregulated by LPS with expression levels remaining high throughout the course of inflammation progression and resolution, while glycolytic genes in ECs of the heart were not affected. In lung endothelium, the key glycolytic enzyme Pfkfb3 showed a more dynamic pattern over time, with only a temporary rise in transcript levels. Targeting dysregulated EC metabolism has recently (re)gained increased interest as a therapeutic strategy for sepsis (188, 189), and it is worth noting that several studies have shown beneficial effects of PFKFB3 inhibition in acute sepsis-induced lung injury models (190, 191).
Besides evaluating transcript levels, recent studies have used in vivo biotinylation combined with proteomics to characterize the luminal surface of vascular cells (38, 39). These studies have confirmed organotypic differences in surface proteins under basal conditions as well as those induced by S. aureus infection. Not surprisingly, proteins that changed across multiple organs due to infection showed (organ-specific) enrichment in acute phase reactants. More specifically, these included haptoglobin in brain, and serum amyloid A proteins in kidney, liver and heart, which are both known to have predictive value for the diagnosis of systemic inflammatory processes (192). Methicillin-resistant S. aureus infection affected predominantly the liver vasculature, with a strong and tissue-specific increase in PRG4 levels (39). Although a role in sepsis has not been previously demonstrated, PRG4 has been established as an anti-inflammatory competitor for hyaluronic acid binding and it could therefore influence how immune cells interact with activated ECs (193). Besides, PRG4 has recently been suggested to serve as a potential therapeutic and biomarker in sepsis (194).
A follow-up study using vascular proteomics in the same model also showed a decrease in proteins involved in lipid metabolism in the liver, as well as a time-dependent procoagulant and antifibrinolytic shift of the endothelium. Interestingly, the changes in haemostatic proteins occurred well before signs of organ dysfunction and thrombosis were observed (38).
Organotypic changes in animal models of sepsis are now well-established, but the previous studies evaluated the EC population within an organ as a whole, which prohibits determining whether changes follow a distinct pattern along different branches of the vascular tree. Using scRNA-seq analysis, a recent study determined cell-specific changes in acute kidney injury during a 48 h course after LPS injection (195). Although this study did not specifically focus on the endothelium, they did notice that changes in ECs already started to appear 1 h post-LPS, whereas changes in expression profiles of proximal tubule epithelial cells took 4 h to occur. Moreover, based on gene regulatory networks and receptor-ligand crosstalk analyses, it was shown that there is general cell–cell communication failure around 16 h after LPS exposure, which plays not only a role in the progression to organ dysfunction, but also in activating recovery pathways (195).
A scRNA-seq study looking at CLP-induced changes in non-parenchymal cells of the liver was able to identify 4 distinct EC subpopulations, which were all uniquely affected by CLP (196). Out of these 4 subsets, 2 were nearly exclusively present under sepsis conditions. Although they both displayed a proinflammatory phenotype, they presented at different stages during infection, with expression data suggesting that one subset likely plays a role in EC activation while the other contributes to the adaptive immune response to infection as antigen presenting cells (196). Similarly, scRNA-seq of non-neuronal cells of the mouse cerebral cortex identified 6 main EC subsets under basal conditions, with an additional minor subpopulation of venous ECs characterized by the presence of Icam-1 and Vcam-1 (47). The significance of the latter was evaluated 2 h after LPS exposure, where based on immunohistochemical analyses it was suggested that this EC subpopulation predominantly exists in the postcapillary venules and may serve as an initial site for leukocyte recruitment. Finally, scRNA-seq of lung endothelium established 2 major EC populations under naïve conditions, with one subpopulation expressing immune response genes while the other expressed genes associated with development and regeneration (197). Evaluation of these subsets after LPS exposure showed distinct transcriptional changes in the early and late inflammatory response. Interestingly, during the recovery phase (3 days post-LPS) a third population of proliferative ECs emerged from the developmental subset, which is presumably important for vascular regeneration.
Taken together, these scRNA-seq studies have established several endothelial subsets per organ, which show unique properties depending on their location along the vascular tree as well as their function. Sepsis-induced transcriptional changes imply that these subpopulations participate in distinct but complementary tasks.
Because of the direct contact between the endothelium and blood, EC-derived proteins easily emerge in plasma, and can therefore be used as potential biomarkers. Over the years, hundreds of biomarkers in relation to sepsis for diagnostic and/or prognostic applications have been proposed, typically focusing on inflammation, coagulation, endothelial damage or vital organ function, although only a small fraction of these have been evaluated in larger patient cohorts or validated across multiple studies (198). Several of these are indeed EC-derived, and not surprisingly, they are often related to leukocyte adhesion, vascular permeability, or coagulation processes (Table 2). It is important to note, however, that despite the observed differences in plasma levels and their reported associations with disease severity, none of them (either alone or in combination) are truly specific or sensitive enough to accurately diagnose sepsis or predict sepsis outcome (198). Furthermore, many of these markers are expressed by ECs in multiple organs, and changes in blood levels may thus represent either general EC dysfunction or organ-specific EC damage. Especially the latter would be valuable for diagnostic and prognostic purposes since several studies have indicated that EC dysfunction precedes organ failure (195, 196). Therefore, being able to identify organotypic EC-derived proteins in the circulation could be used to predict organ dysfunction so preventive measures can be taken. Unbiased -omics studies using transcriptomics and proteomics analyses as described here could guide the identification of such biomarkers.
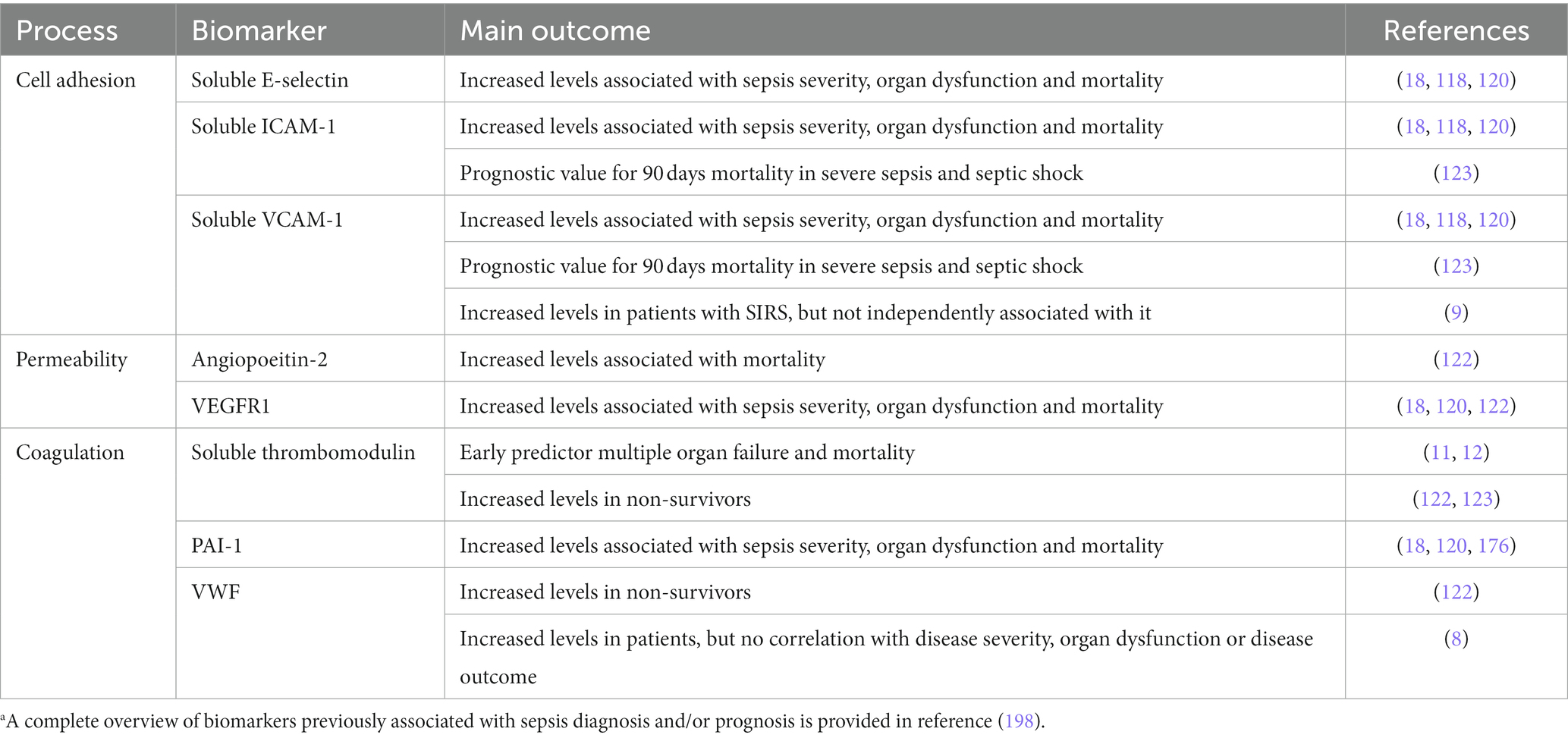
Table 2. Overview of EC-derived biomarkers and their main diagnostic/prognostic value based on selected references.a
Besides identifying biomarkers, -omics studies are also likely to generate information from which new candidate molecules for drug interventions can be derived, as the easy access and early responses of the endothelium provides a unique potential for therapeutic intervention. Historically, proposed treatments for sepsis have focused on inhibiting inflammation (e.g., anti-TNFα or IL-1Ra therapy), promoting vascular integrity (via TIE2 stabilization), or counteracting the procoagulant shift of the endothelium (e.g., by using activated protein C, or recombinant soluble thrombomodulin) (199–205). However, none of these have resulted in approved drugs currently used in the clinic. There are several plausible explanations for this (see also next section), including the now widely recognized organotypic EC heterogeneity which could have contributed to their lack of success, as many of these drugs might have been developed under the assumption that circulating biomarkers would represent a general dysfunctional EC phenotype. Based on current data, this is likely incorrect, and these early developed strategies may therefore cause more harm than good under certain conditions, as EC activation during inflammation is first and foremost a beneficial response aimed at maintaining tissue homeostasis by eliminating the pathogen and limiting damage (206).
It is important to obtain a comprehensive understanding of the differences in kinetics of EC activation and dysfunction across vascular beds, as well as the presence of unique molecules that can distinguish between affected EC subsets since this would support the development of targeted delivery approaches to limit unwanted side-effects. Trying to leverage EC heterogeneity for therapeutic gain, drug delivery approaches have employed antibodies coupled with therapeutic agents or targeted nanobodies against EC surface markers such as PECAM, E-selectin, and VCAM-1 (207–211). Yet, while intended to target dysfunctional ECs, in reality many non-dysfunctional ECs also express these markers and thus molecular information at a high cellular resolution will be required to establish specific cargo transfer of these drug delivery systems. Besides the decision on which receptor to target (Figure 3), the choice of drug treatment needs to be carefully considered to avoid that risks associated with treatment outweigh the benefits. Therefore, we need to get a better understanding of the kinetics of (downstream) intracellular pathways affected in sepsis, and their status in different microvascular beds. This pharmacological Gordian knot can in theory, and possibly also experimentally in the near future, be disentangled by combining transcriptomics and proteomics with, for example, protein kinase activity platform techniques (kinomics) to study activation status of multiple signal transduction cascades (212).
Despite all efforts to develop therapies to improve sepsis outcome, at present only supportive organ care can be offered to sepsis patients. This lack of pharmacologically effective treatment strategies to counteract the pathophysiological processes in sepsis is in stark contrast to the successes observed in preclinical studies that interfere with dedicated molecular pathways. The following part will discuss some of the challenges sepsis research has faced, and is still facing, that could explain this discrepancy, together with future directions to help move the sepsis field forward.
The complexity of sepsis requires the use of experimental animal models to study (cell-specific) mechanisms underlying pathophysiology. However, even in an intact animal such as the mouse, it is difficult to accurately recapitulate sepsis pathobiology. This is partly due to physiological differences between mice and humans, as well as their respective response to a septic insult, as mice are more resistant and show greater resilience, and are typically relatively young and healthy. Especially this latter is opposite from the “average” septic patient, who is generally older with more comorbidities (2). Furthermore, particularly in small animal models, it is not feasible to incorporate organ support that is typically given to sepsis patients.
A previous study compared 3 different murine sepsis models and showed clear differences in kinetics and magnitude of the inflammatory response (146). These data indicate that the model strongly influences the host response and findings from one model do not necessarily reflect general sepsis patho(physio)logy. Although sepsis models thus need to be carefully chosen based on the goal of the study, these differences can also be used to our advantage: in order to standardize preclinical studies, a systematic multi-model approach should be developed in which drug candidates are tested throughout models with greater levels of complexity (i.e., LPS followed by CLP in mice, followed by studies in bigger animal models in which care can be given as applied in the ICU) as research progresses. In addition, therapeutic interventions should also be tested at different time points after insult to fully address their pharmacological effectiveness when drugs are administered at different stages of sepsis progression, as each stage is likely associated with distinct changes in the host response. Furthermore, attention should be paid to pharmacokinetics/pharmacodynamics in these pre-clinical studies, to prohibit misinterpretation of treatment outcome due to differences in drug half-life and/or metabolism, for example. Addressing cell type-specific pharmacodynamics furthermore aids in understanding which effects of drug treatment occur in cells considered to be the target of therapeutic intervention versus cells that engage in the pathophysiology but are not target cells per se. scRNA-seq techniques as well as isolation of cellular subsets by laser microdissection prior to RNA-seq (213) or spatial transcriptomics, which allows evaluation of cellular interactions in situ and thus provides information on how the alterations in ECs affect underlying parenchyma, will be critical technological advancements for this purpose, and eventually create a full “pharmacolomics” view in which drug effects are related to pathophysiological changes. This approach is of crucial importance to make a rational decision to enter a clinical testing phase, and to provide substantial information to make a well-informed choice of biomarkers to measure in the restricted clinical samples available.
A retrospective analysis of >20,000 sepsis patients identified 4 distinct sepsis phenotypes that correlated with host response patterns and clinical outcome (214). Interestingly, this study suggested that including patients in trials based on the specific sepsis phenotype could lead to drastic differences in outcome depending on the clinical intervention. For example, they showed that treatment with activated protein C (Xigris) which, despite its dual action as an anticoagulant and involvement in protecting the vascular barrier, was discontinued as a therapy for sepsis due to an increased bleeding risk (203, 215), would improve outcome in patients with sepsis-induced disseminated intravascular coagulopathy whereas for other phenotypes it was indicated to be harmful (214). A similar result was described in a reanalysis of sepsis patients treated with recombinant thrombomodulin, which was shown to only benefit patients with a severe coagulopathy phenotype (216). These data illustrate that unbiased patient inclusion can cause conflicting results as interventions can be beneficial or detrimental to certain patient populations. Therefore, it has been suggested to revisit previous sepsis treatments that failed in clinical trials, and re-evaluate them in better defined patient groups.
Another clinical challenge pertains to the use of biomarkers to guide clinical decisions. Although a consistent difference in biomarker patterns was observed between sepsis phenotypes (214), it required data obtained from several biomarkers as not one by itself is specific or sensitive enough to diagnose sepsis reliably. Besides their lack of specificity regarding cellular and organ origin, biomarker levels display large inter-patient variability and they are time-dependent as they fluctuate based on disease progression. Thus, the absence of a significantly elevated protein in serum does not automatically preclude sepsis diagnosis, and it is therefore important to continuously monitor patients to accurately assess the pathophysiological stage of sepsis, starting as soon as the patient presents with clinical symptoms through at least until they get discharged from the ICU. This is not only to initially determine the course of sepsis, but also to monitor effects of organ support, and possibly drug treatment regimens.
Based on their early response, ECs have been consistently suggested as a target for therapeutics. Even though better biomarkers and patient stratification will be helpful in determining a treatment plan, it is unlikely that targeting only one molecule or pathway will be sufficient to ensure vascular protection and facilitate restoration of EC function since the molecular mechanisms underlying sepsis are so complex. This complexity has become even more fully appreciated with the introduction of unbiased -omics approaches.
In light of the latter, it is interesting to take a closer look at the efforts taken to study SARS-CoV-2 infection leading to COVID-19. Once the true impact of this disease became apparent, the scientific community quickly rallied to employ methods such as (single cell) RNA-seq and spatial transcriptomics to generate testable hypotheses leading to a better understanding of the molecular mechanisms driving the host response and general patho(physio)logy of this devastating disease (90, 217–222). Although several parallels can be drawn between COVID-19 and sepsis, including the presence of endotheliopathy (219, 221), it is interesting to note that studies using these relatively new technologies have only started to emerge recently in the sepsis field (195, 196). Nonetheless, these are expected to make a great stride forward in elucidating the complexity of sepsis.
Traditional transcriptomics approaches are invaluable for understanding the dynamics of gene expression, but they do not tell the full story. miRNAs, including EC-derived miRNAs, are increasingly recognized for their role in sepsis and potential as biomarker (223–226). Although studying miRNAs in an unbiased manner has been historically more difficult due to technical limitations (227), our recent studies combining RNA-seq and miRNA-seq data obtained from distinct microvascular segments in the kidney have demonstrated its potential to predict RNA-miRNA relations in the complexity of the vasculature in an in vivo setting (213). Thus, these data could serve as a starting point for the identification of targets for therapeutic interventions. Furthermore, epigenetic regulation of gene expression and the resulting proteome also provide important information, particularly the proteome of the vessel wall (38, 39) and the plasma proteome as these could lead to the identification of markers used for therapeutics or diagnostic/prognostic purposes. While a few translational research teams have actively sought to create a biobank of biological materials including plasma, serum, urine, and tissue samples obtained from sepsis patients, a broad concerted strategy that can be incorporated into the daily workflow of clinical departments is at present lacking. In addition, miniaturization of -omics analyses is key to make the desired progress in unravelling the molecular basis of sepsis and sepsis-related multiple organ dysfunction and to make -omics not compete with standard assessment of clinically relevant parameters but become an add-on assessment instead (112). Even though it is currently not feasible (yet) to base a treatment plan on -omics data generated from patient samples given its relatively slow turn-around time whereas sepsis rapidly progresses, eventually the integration of -omics data will be required to map the spatiotemporal changes occurring during sepsis progression. This will create opportunities for a systems biology approach to get a holistic picture of sepsis pathophysiology required for developing rational therapeutic intervention strategies.
Endothelial cells are often referred to as gatekeepers of tissue homeostasis given their unique position between the blood and parenchyma, and their interspersed distribution throughout the body. This makes ECs a particularly relevant cell type for therapeutic strategies since they are not only early responders and active contributors to disease pathophysiology, including sepsis, but are also easily accessible. However, their heterogeneity across vascular beds under both physiological and pathological conditions makes this notion more complicated and may explain, at least partly, the lack of success in our quest of developing effective treatment options for sepsis aimed at the endothelium. The introduction of -omics approaches has enabled the field to get a better understanding of organ- and (endothelial) cell-specific changes that occur during sepsis progression. This has not only contributed to our general knowledge of pathogen-host interactions, but it is also expected that it will open the doors for identifying new therapeutic targets, molecules for targeted drug delivery, and biomarkers with diagnostic, prognostic and/or predictive value. Together with patient stratification, -omics data from pathophysiological and pharmacological studies is expected to unveil the molecular treasure trove to advance disentangling, or maybe even cutting, the Gordian knot of EC heterogeneity in the contribution to sepsis pathology.
AC and GM developed the concept of the article and performed literature searches. Both authors contributed to the article and approved the submitted version.
The authors would like to thank Dr. Luxen for sharing the basis for Figure 3, which was adapted from (89) with permission. All other figures were generated using Biorender. The authors apologize to the scientists whose contributions to the field could not be included in this review due to space limitations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Singer, M, Deutschman, CS, Seymour, CW, Shankar-Hari, M, Annane, D, Bauer, M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
2. Cavaillon, JM, Singer, M, and Skirecki, T. Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO Mol Med. (2020) 12:e10128. doi: 10.15252/emmm.201810128
3. Rudd, KE, Johnson, SC, Agesa, KM, Shackelford, KA, Tsoi, D, Kievlan, DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the global burden of disease study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
4. Asim, M, Amin, F, and El-Menyar, A. Multiple organ dysfunction syndrome: contemporary insights on the clinicopathological spectrum. Qatar Med J. (2020) 2020:22. doi: 10.5339/qmj.2020.22
5. Gourd, NM, and Nikitas, N. Multiple organ dysfunction syndrome. J Intensive Care Med. (2020) 35:1564–75. doi: 10.1177/0885066619871452
6. Jacobi, J. The pathophysiology of sepsis—2021 update: part 2, organ dysfunction and assessment. Am J Health Syst Pharm. (2022) 79:424–36. doi: 10.1093/ajhp/zxab393
7. Lelubre, C, and Vincent, JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. (2018) 14:417–27. doi: 10.1038/s41581-018-0005-7
8. Kremer Hovinga, JA, Zeerleder, S, Kessler, P, Romani de Wit, T, van Mourik, JA, Hack, CE, et al. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost. (2007) 5:2284–90. doi: 10.1111/j.1538-7836.2007.02743.x
9. Mikacenic, C, Hahn, WO, Price, BL, Harju-Baker, S, Katz, R, Kain, KC, et al. Biomarkers of endothelial activation are associated with poor outcome in critical illness. PLoS One. (2015) 10:e0141251. doi: 10.1371/journal.pone.0141251
10. Uhel, F, Peters-Sengers, H, Falahi, F, Scicluna, BP, van Vught, LA, Bonten, MJ, et al. Mortality and host response aberrations associated with transient and persistent acute kidney injury in critically ill patients with sepsis: a prospective cohort study. Intensive Care Med. (2020) 46:1576–89. doi: 10.1007/s00134-020-06119-x
11. Zhou, G, Liu, J, Zhang, H, Wang, X, and Liu, D. Elevated endothelial dysfunction-related biomarker levels indicate the severity and predict sepsis incidence. Sci Rep. (2022) 12:21935. doi: 10.1038/s41598-022-26623-y
12. Johansen, ME, Johansson, PI, Ostrowski, SR, Bestle, MH, Hein, L, Jensen, AL, et al. Profound endothelial damage predicts impending organ failure and death in sepsis. Semin Thromb Hemost. (2015) 41:16–25. doi: 10.1055/s-0034-1398377
13. De Backer, D, Ricottilli, F, and Ospina-Tascon, GA. Septic shock: a microcirculation disease. Curr Opin Anaesthesiol. (2021) 34:85–91. doi: 10.1097/ACO.0000000000000957
14. Afshar, Y, Ma, F, Quach, A, Jeong, A, Sunshine, HL, Freitas, V, et al. Transcriptional drifts associated with environmental changes in endothelial cells. eLife. (2023) 12:12. doi: 10.7554/eLife.81370
15. Amatschek, S, Kriehuber, E, Bauer, W, Reininger, B, Meraner, P, Wolpl, A, et al. Blood and lymphatic endothelial cell-specific differentiation programs are stringently controlled by the tissue environment. Blood. (2007) 109:4777–85. doi: 10.1182/blood-2006-10-053280
16. Cleuren, ACA, van der Ent, MA, Jiang, H, Hunker, KL, Yee, A, Siemieniak, DR, et al. The in vivo endothelial cell translatome is highly heterogeneous across vascular beds. Proc Natl Acad Sci U S A. (2019) 116:23618–24. doi: 10.1073/pnas.1912409116
17. Leone, M, Boutiere, B, Camoin-Jau, L, Albanese, J, Horschowsky, N, Mege, JL, et al. Systemic endothelial activation is greater in septic than in traumatic-hemorrhagic shock but does not correlate with endothelial activation in skin biopsies. Crit Care Med. (2002) 30:808–14. doi: 10.1097/00003246-200204000-00015
18. Shapiro, NI, Yano, K, Sorasaki, M, Fischer, C, Shih, SC, and Aird, WC. Skin biopsies demonstrate site-specific endothelial activation in mouse models of sepsis. J Vasc Res. (2009) 46:495–502. doi: 10.1159/000210662
19. Yan, R, van Meurs, M, Popa, ER, Li, R, Zwiers, PJ, Zijlstra, JG, et al. Early heterogenic response of renal microvasculature to hemorrhagic shock/resuscitation and the influence of NF-kappaB pathway blockade. Shock. (2019) 51:200–12. doi: 10.1097/SHK.0000000000001126
20. Riedemann, NC, Guo, RF, and Ward, PA. Novel strategies for the treatment of sepsis. Nat Med. (2003) 9:517–24. doi: 10.1038/nm0503-517
21. Gracia-Sancho, J, Caparros, E, Fernandez-Iglesias, A, and Frances, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol. (2021) 18:411–31. doi: 10.1038/s41575-020-00411-3
22. Koch, PS, Lee, KH, Goerdt, S, and Augustin, HG. Angiodiversity and organotypic functions of sinusoidal endothelial cells. Angiogenesis. (2021) 24:289–310. doi: 10.1007/s10456-021-09780-y
23. Aird, WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. (2007) 100:174–90. doi: 10.1161/01.RES.0000255690.03436.ae
24. Parab, S, Setten, E, Astanina, E, Bussolino, F, and Doronzo, G. The tissue-specific transcriptional landscape underlines the involvement of endothelial cells in health and disease. Pharmacol Ther. (2023) 246:108418. doi: 10.1016/j.pharmthera.2023.108418
25. Perez-Gutierrez, L, Li, P, and Ferrara, N. Endothelial cell diversity: the many facets of the crystal. FEBS J. (2022). doi: 10.1111/febs.16660
26. Potente, M, and Makinen, T. Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol. (2017) 18:477–94. doi: 10.1038/nrm.2017.36
27. Aird, WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. (2007) 100:158–73. doi: 10.1161/01.RES.0000255691.76142.4a
28. Dvorak, AM, and Feng, D. The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem. (2001) 49:419–32. doi: 10.1177/002215540104900401
29. Armulik, A, Genove, G, Mae, M, Nisancioglu, MH, Wallgard, E, Niaudet, C, et al. Pericytes regulate the blood-brain barrier. Nature. (2010) 468:557–61. doi: 10.1038/nature09522
30. Claesson-Welsh, L, Dejana, E, and McDonald, DM. Permeability of the endothelial barrier: identifying and reconciling controversies. Trends Mol Med. (2021) 27:314–31. doi: 10.1016/j.molmed.2020.11.006
31. Van der Ent, MA, Svilar, D, and Cleuren, ACA. Molecular analysis of vascular gene expression. Res Pract Thromb Haemost. (2022) 6:e12718. doi: 10.1002/rth2.12718
32. Nolan, DJ, Ginsberg, M, Israely, E, Palikuqi, B, Poulos, MG, James, D, et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. (2013) 26:204–19. doi: 10.1016/j.devcel.2013.06.017
33. Sabbagh, MF, Heng, JS, Luo, C, Castanon, RG, Nery, JR, Rattner, A, et al. Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. eLife. (2018) 7:7. doi: 10.7554/eLife.36187
34. Schlereth, K, Weichenhan, D, Bauer, T, Heumann, T, Giannakouri, E, Lipka, D, et al. The transcriptomic and epigenetic map of vascular quiescence in the continuous lung endothelium. eLife. (2018) 7:7. doi: 10.7554/eLife.34423
35. Gay, L, Miller, MR, Ventura, PB, Devasthali, V, Vue, Z, Thompson, HL, et al. Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes Dev. (2013) 27:98–115. doi: 10.1101/gad.205278.112
36. Hupe, M, Li, MX, Gertow Gillner, K, Adams, RH, and Stenman, JM. Evaluation of TRAP-sequencing technology with a versatile conditional mouse model. Nucleic Acids Res. (2014) 42:e14. doi: 10.1093/nar/gkt995
37. Jambusaria, A, Hong, Z, Zhang, L, Srivastava, S, Jana, A, Toth, PT, et al. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife. (2020) 9:9. doi: 10.7554/eLife.51413
38. Sorrentino, JT, Golden, GJ, Morris, C, Painter, CD, Nizet, V, Campos, AR, et al. Vascular proteome responses precede organ dysfunction in a murine model of Staphylococcus aureus bacteremia. mSystems. (2022) 7:e0039522. doi: 10.1128/msystems.00395-22
39. Toledo, AG, Golden, G, Campos, AR, Cuello, H, Sorrentino, J, Lewis, N, et al. Proteomic atlas of organ vasculopathies triggered by Staphylococcus aureus sepsis. Nat Commun. (2019) 10:4656. doi: 10.1038/s41467-019-12672-x
40. The Tabula Muris Consortium, Overall Coordination, Logistical Coordination, Organ Collection and Processing, Library Preparation and Sequencing, Computational Data Analysis, Cell Type Annotation, Writing Group, Supplemental Text Writing Group & Principal Investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. (2018) 562:367–72. doi: 10.1038/s41586-018-0590-4
41. Feng, W, Chen, L, Nguyen, PK, Wu, SM, and Li, G. Single cell analysis of endothelial cells identified organ-specific molecular signatures and heart-specific cell populations and molecular features. Front Cardiovasc Med. (2019) 6:165. doi: 10.3389/fcvm.2019.00165
42. Paik, DT, Tian, L, Williams, IM, Rhee, S, Zhang, H, Liu, C, et al. Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. (2020) 142:1848–62. doi: 10.1161/CIRCULATIONAHA.119.041433
43. He, L, Vanlandewijck, M, Mae, MA, Andrae, J, Ando, K, Del Gaudio, F, et al. Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci Data. (2018) 5:180160. doi: 10.1038/sdata.2018.160
44. Kalucka, J, de Rooij, L, Goveia, J, Rohlenova, K, Dumas, SJ, Meta, E, et al. Single-cell transcriptome atlas of murine endothelial cells. Cells. (2020) 180:e720:764–779.e20. doi: 10.1016/j.cell.2020.01.015
45. Han, X, Wang, R, Zhou, Y, Fei, L, Sun, H, Lai, S, et al. Mapping the mouse cell atlas by microwell-Seq. Cells. (2018) 172:e1017:1091–1107.e17. doi: 10.1016/j.cell.2018.02.001
46. Tabula Sapiens, C, Jones, RC, Karkanias, J, Krasnow, MA, Pisco, AO, Quake, SR, et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science. (2022) 376:eabl4896. doi: 10.1126/science.abl4896
47. Jeong, HW, Dieguez-Hurtado, R, Arf, H, Song, J, Park, H, Kruse, K, et al. Single-cell transcriptomics reveals functionally specialized vascular endothelium in brain. eLife. (2022) 11:11. doi: 10.7554/eLife.57520
48. Vanlandewijck, M, He, L, Mae, MA, Andrae, J, Ando, K, Del Gaudio, F, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. (2018) 554:475–80. doi: 10.1038/nature25739
49. Garcia, FJ, Sun, N, Lee, H, Godlewski, B, Mathys, H, Galani, K, et al. Single-cell dissection of the human brain vasculature. Nature. (2022) 603:893–9. doi: 10.1038/s41586-022-04521-7
50. Yang, AC, Vest, RT, Kern, F, Lee, DP, Agam, M, Maat, CA, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. (2022) 603:885–92. doi: 10.1038/s41586-021-04369-3
51. Winkler, EA, Kim, CN, Ross, JM, Garcia, JH, Gil, E, Oh, I, et al. A single-cell atlas of the normal and malformed human brain vasculature. Science. (2022) 375:eabi7377. doi: 10.1126/science.abi7377
52. Koenig, AL, Shchukina, I, Amrute, J, Andhey, PS, Zaitsev, K, Lai, L, et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat Cardiovasc Res. (2022) 1:263–80. doi: 10.1038/s44161-022-00028-6
53. Litvinukova, M, Talavera-Lopez, C, Maatz, H, Reichart, D, Worth, CL, Lindberg, EL, et al. Cells of the adult human heart. Nature. (2020) 588:466–72. doi: 10.1038/s41586-020-2797-4
54. Tucker, NR, Chaffin, M, Fleming, SJ, Hall, AW, Parsons, VA, Bedi, KC Jr, et al. Transcriptional and cellular diversity of the human heart. Circulation. (2020) 142:466–82. doi: 10.1161/CIRCULATIONAHA.119.045401
55. Barry, DM, McMillan, EA, Kunar, B, Lis, R, Zhang, T, Lu, T, et al. Molecular determinants of nephron vascular specialization in the kidney. Nat Commun. (2019) 10:5705. doi: 10.1038/s41467-019-12872-5
56. Dumas, SJ, Meta, E, Borri, M, Goveia, J, Rohlenova, K, Conchinha, NV, et al. Single-cell RNA sequencing reveals renal endothelium heterogeneity and metabolic adaptation to water deprivation. J Am Soc Nephrol. (2020) 31:118–38. doi: 10.1681/ASN.2019080832
57. Karaiskos, N, Rahmatollahi, M, Boltengagen, A, Liu, H, Hoehne, M, Rinschen, M, et al. A single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol. (2018) 29:2060–8. doi: 10.1681/ASN.2018030238
58. Park, J, Shrestha, R, Qiu, C, Kondo, A, Huang, S, Werth, M, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. (2018) 360:758–63. doi: 10.1126/science.aar2131
59. Miao, Z, Balzer, MS, Ma, Z, Liu, H, Wu, J, Shrestha, R, et al. Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat Commun. (2021) 12:2277. doi: 10.1038/s41467-021-22266-1
60. Muto, Y, Wilson, PC, Ledru, N, Wu, H, Dimke, H, Waikar, SS, et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat Commun. (2021) 12:2190. doi: 10.1038/s41467-021-22368-w
61. Young, MD, Mitchell, TJ, Vieira Braga, FA, Tran, MGB, Stewart, BJ, Ferdinand, JR, et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science. (2018) 361:594–9. doi: 10.1126/science.aat1699
62. Su, T, Yang, Y, Lai, S, Jeong, J, Jung, Y, McConnell, M, et al. Single-cell transcriptomics reveals zone-specific alterations of liver sinusoidal endothelial cells in cirrhosis. Cell Mol Gastroenterol Hepatol. (2021) 11:1139–61. doi: 10.1016/j.jcmgh.2020.12.007
63. Aizarani, N, Saviano, A, Sagar, ML, Durand, S, Herman, JS, Pessaux, P, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. (2019) 572:199–204. doi: 10.1038/s41586-019-1373-2
64. MacParland, SA, Liu, JC, Ma, XZ, Innes, BT, Bartczak, AM, Gage, BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
65. Gillich, A, Zhang, F, Farmer, CG, Travaglini, KJ, Tan, SY, Gu, M, et al. Capillary cell-type specialization in the alveolus. Nature. (2020) 586:785–9. doi: 10.1038/s41586-020-2822-7
66. Travaglini, KJ, Nabhan, AN, Penland, L, Sinha, R, Gillich, A, Sit, RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. (2020) 587:619–25. doi: 10.1038/s41586-020-2922-4
67. Becker, LM, Chen, SH, Rodor, J, de Rooij, L, Baker, AH, and Carmeliet, P. Deciphering endothelial heterogeneity in health and disease at single-cell resolution: progress and perspectives. Cardiovasc Res. (2023) 119:6–27. doi: 10.1093/cvr/cvac018
68. Chavkin, NW, and Hirschi, KK. Single cell analysis in vascular biology. Front Cardiovasc Med. (2020) 7:42. doi: 10.3389/fcvm.2020.00042
69. Trimm, E, and Red-Horse, K. Vascular endothelial cell development and diversity. Nat Rev Cardiol. (2023) 20:197–210. doi: 10.1038/s41569-022-00770-1
70. Wakabayashi, T, and Naito, H. Cellular heterogeneity and stem cells of vascular endothelial cells in blood vessel formation and homeostasis: insights from single-cell RNA sequencing. Front Cell Dev Biol. (2023) 11:1146399. doi: 10.3389/fcell.2023.1146399
71. Aird, WC. Endothelium as a therapeutic target in sepsis. Curr Drug Targets. (2007) 8:501–7. doi: 10.2174/138945007780362782
72. Molema, G. Heterogeneity in endothelial responsiveness to cytokines, molecular causes, and pharmacological consequences. Semin Thromb Hemost. (2010) 36:246–64. doi: 10.1055/s-0030-1253448
73. Lupu, F, Kinasewitz, G, and Dormer, K. The role of endothelial shear stress on haemodynamics, inflammation, coagulation and glycocalyx during sepsis. J Cell Mol Med. (2020) 24:12258–71. doi: 10.1111/jcmm.15895
74. Reitsma, S, Slaaf, DW, Vink, H, van Zandvoort, MA, and oude Egbrink, MGA. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. (2007) 454:345–59. doi: 10.1007/s00424-007-0212-8
75. Iba, T, and Levy, JH. Derangement of the endothelial glycocalyx in sepsis. J Thromb Haemost. (2019) 17:283–94. doi: 10.1111/jth.14371
76. Kang, S, and Kishimoto, T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. (2021) 53:1116–23. doi: 10.1038/s12276-021-00649-0
77. Matsushima, K, Yang, D, and Oppenheim, JJ. Interleukin-8: an evolving chemokine. Cytokine. (2022) 153:155828. doi: 10.1016/j.cyto.2022.155828
78. Springer, TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cells. (1994) 76:301–14. doi: 10.1016/0092-8674(94)90337-9
79. Muller, WA. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol. (2014) 184:886–96. doi: 10.1016/j.ajpath.2013.12.033
80. Speyer, CL, and Ward, PA. Role of endothelial chemokines and their receptors during inflammation. J Investig Surg. (2011) 24:18–27. doi: 10.3109/08941939.2010.521232
81. Wu, C, Ivars, F, Anderson, P, Hallmann, R, Vestweber, D, Nilsson, P, et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. (2009) 15:519–27. doi: 10.1038/nm.1957
82. Graham, GJ, Handel, TM, and Proudfoot, AEI. Leukocyte adhesion: reconceptualizing chemokine presentation by glycosaminoglycans. Trends Immunol. (2019) 40:472–81. doi: 10.1016/j.it.2019.03.009
83. Henninger, DD, Panes, J, Eppihimer, M, Russell, J, Gerritsen, M, Anderson, DC, et al. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. (1997) 158:1825–32.
84. Broermann, A, Winderlich, M, Block, H, Frye, M, Rossaint, J, Zarbock, A, et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med. (2011) 208:2393–401. doi: 10.1084/jem.20110525
85. Giannotta, M, Trani, M, and Dejana, E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. (2013) 26:441–54. doi: 10.1016/j.devcel.2013.08.020
86. Orsenigo, F, Giampietro, C, Ferrari, A, Corada, M, Galaup, A, Sigismund, S, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. (2012) 3:1208. doi: 10.1038/ncomms2199
87. Dunzendorfer, S, Lee, HK, Soldau, K, and Tobias, PS. TLR4 is the signaling but not the lipopolysaccharide uptake receptor. J Immunol. (2004) 173:1166–70. doi: 10.4049/jimmunol.173.2.1166
88. Moser, J, Heeringa, P, Jongman, RM, Zwiers, PJ, Niemarkt, AE, Yan, R, et al. Intracellular RIG-I signaling regulates TLR4-independent endothelial inflammatory responses to endotoxin. J Immunol. (2016) 196:4681–91. doi: 10.4049/jimmunol.1501819
89. Luxen, M, van Meurs, M, and Molema, G. Unlocking the untapped potential of endothelial kinase and phosphatase involvement in sepsis for drug treatment design. Front Immunol. (2022) 13:867625. doi: 10.3389/fimmu.2022.867625
90. Bass, A, Liu, Y, and Dakshanamurthy, S. Single-cell and bulk RNASeq profiling of COVID-19 patients reveal immune and inflammatory mechanisms of infection-induced organ damage. Viruses. (2021) 13:13. doi: 10.3390/v13122418
91. Hellenthal, KEM, Brabenec, L, and Wagner, NM. Regulation and dysregulation of endothelial permeability during systemic inflammation. Cells. (2022) 11:11. doi: 10.3390/cells11121935
92. Hughes, DP, Marron, MB, and Brindle, NP. The antiinflammatory endothelial tyrosine kinase Tie2 interacts with a novel nuclear factor-kappaB inhibitor ABIN-2. Circ Res. (2003) 92:630–6. doi: 10.1161/01.RES.0000063422.38690.DC
93. Fiedler, U, Scharpfenecker, M, Koidl, S, Hegen, A, Grunow, V, Schmidt, JM, et al. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel–Palade bodies. Blood. (2004) 103:4150–6. doi: 10.1182/blood-2003-10-3685
94. Korhonen, EA, Lampinen, A, Giri, H, Anisimov, A, Kim, M, Allen, B, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest. (2016) 126:3495–510. doi: 10.1172/JCI84923
95. Bates, DO. Vascular endothelial growth factors and vascular permeability. Cardiovasc Res. (2010) 87:262–71. doi: 10.1093/cvr/cvq105
96. Frye, M, Dierkes, M, Kuppers, V, Vockel, M, Tomm, J, Zeuschner, D, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med. (2015) 212:2267–87. doi: 10.1084/jem.20150718
97. Nawroth, R, Poell, G, Ranft, A, Kloep, S, Samulowitz, U, Fachinger, G, et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. (2002) 21:4885–95. doi: 10.1093/emboj/cdf497
98. Shirakura, K, Baluk, P, Nottebaum, AF, Ipe, U, Peters, KG, McDonald, DM, et al. Shear stress control of vascular leaks and atheromas through Tie2 activation by VE-PTP sequestration. EMBO Mol Med. (2023) 15:e16128. doi: 10.15252/emmm.202216128
99. Gaengel, K, Niaudet, C, Hagikura, K, Lavina, B, Muhl, L, Hofmann, JJ, et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev Cell. (2012) 23:587–99. doi: 10.1016/j.devcel.2012.08.005
100. Wang, L, and Dudek, SM. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res. (2009) 77:39–45. doi: 10.1016/j.mvr.2008.09.005
101. Zhang, G, Yang, L, Kim, GS, Ryan, K, Lu, S, O’Donnell, RK, et al. Critical role of sphingosine-1-phosphate receptor 2 (S1PR2) in acute vascular inflammation. Blood. (2013) 122:443–55. doi: 10.1182/blood-2012-11-467191
102. Aslan, A, van Meurs, M, Moser, J, Popa, ER, Jongman, RM, Zwiers, PJ, et al. Organ-specific differences in endothelial permeability-regulating molecular responses in mouse and human sepsis. Shock. (2017) 48:69–77. doi: 10.1097/SHK.0000000000000841
103. Richards, M, Nwadozi, E, Pal, S, Martinsson, P, Kaakinen, M, Gloger, M, et al. Claudin5 protects the peripheral endothelial barrier in an organ and vessel-type-specific manner. eLife. (2022) 11:11. doi: 10.7554/eLife.78517
104. Neubauer, K, and Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. (2022) 387:391–8. doi: 10.1007/s00441-021-03471-2
105. Pober, JS, and Sessa, WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
106. Van de Wouwer, M, Collen, D, and Conway, EM. Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol. (2004) 24:1374–83. doi: 10.1161/01.ATV.0000134298.25489.92
107. Esmon, CT. Protein C anticoagulant system—anti-inflammatory effects. Semin Immunopathol. (2012) 34:127–32. doi: 10.1007/s00281-011-0284-6
108. Lay, AJ, Donahue, D, Tsai, MJ, and Castellino, FJ. Acute inflammation is exacerbated in mice genetically predisposed to a severe protein C deficiency. Blood. (2007) 109:1984–91. doi: 10.1182/blood-2006-07-037945
109. Shahzad, K, Kohli, S, Al-Dabet, MM, and Isermann, B. Cell biology of activated protein C. Curr Opin Hematol. (2019) 26:41–50. doi: 10.1097/MOH.0000000000000473
110. Li, Z, Yin, M, Zhang, H, Ni, W, Pierce, RW, Zhou, HJ, et al. BMX represses thrombin-PAR1-mediated endothelial permeability and vascular leakage during early sepsis. Circ Res. (2020) 126:471–85. doi: 10.1161/CIRCRESAHA.119.315769
111. Hultstrom, M, Fromell, K, Larsson, A, Persson, B, Nilsson, B, Quaggin, SE, et al. Angiopoietin-2 inhibition of thrombomodulin-mediated anticoagulation-a novel mechanism that may contribute to hypercoagulation in critically ill COVID-19 patients. Biomedicine. (2022) 10:10. doi: 10.3390/biomedicines10061333
112. Langenkamp, E, Kamps, JA, Mrug, M, Verpoorte, E, Niyaz, Y, Horvatovich, P, et al. Innovations in studying in vivo cell behavior and pharmacology in complex tissues—microvascular endothelial cells in the spotlight. Cell Tissue Res. (2013) 354:647–69. doi: 10.1007/s00441-013-1714-7
113. Molema, G, Zijlstra, JG, van Meurs, M, and Kamps, J. Renal microvascular endothelial cell responses in sepsis-induced acute kidney injury. Nat Rev Nephrol. (2022) 18:95–112. doi: 10.1038/s41581-021-00489-1
114. Everett, LA, Cleuren, AC, Khoriaty, RN, and Ginsburg, D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. (2014) 123:3697–705. doi: 10.1182/blood-2014-02-554501
115. Fahs, SA, Hille, MT, Shi, Q, Weiler, H, and Montgomery, RR. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. (2014) 123:3706–13. doi: 10.1182/blood-2014-02-555151
116. Delano, MJ, and Ward, PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. (2016) 126:23–31. doi: 10.1172/JCI82224
117. Hippensteel, JA, Anderson, BJ, Orfila, JE, McMurtry, SA, Dietz, RM, Su, G, et al. Circulating heparan sulfate fragments mediate septic cognitive dysfunction. J Clin Invest. (2019) 129:1779–84. doi: 10.1172/JCI124485
118. Kjaergaard, AG, Dige, A, Nielsen, JS, Tonnesen, E, and Krog, J. The use of the soluble adhesion molecules sE-selectin, sICAM-1, sVCAM-1, sPECAM-1 and their ligands CD11a and CD49d as diagnostic and prognostic biomarkers in septic and critically ill non-septic ICU patients. APMIS. (2016) 124:846–55. doi: 10.1111/apm.12585
119. Kumpers, P, van Meurs, M, David, S, Molema, G, Bijzet, J, Lukasz, A, et al. Time course of angiopoietin-2 release during experimental human endotoxemia and sepsis. Crit Care. (2009) 13:R64. doi: 10.1186/cc7866
120. Skibsted, S, Jones, AE, Puskarich, MA, Arnold, R, Sherwin, R, Trzeciak, S, et al. Biomarkers of endothelial cell activation in early sepsis. Shock. (2013) 39:427–32. doi: 10.1097/SHK.0b013e3182903f0d
121. Ware, LB, Eisner, MD, Thompson, BT, Parsons, PE, and Matthay, MA. Significance of von Willebrand factor in septic and nonseptic patients with acute lung injury. Am J Respir Crit Care Med. (2004) 170:766–72. doi: 10.1164/rccm.200310-1434OC
122. Hou, PC, Filbin, MR, Wang, H, Ngo, L, Huang, DT, Aird, WC, et al. Endothelial permeability and hemostasis in septic shock: results from the ProCESS trial. Chest. (2017) 152:22–31. doi: 10.1016/j.chest.2017.01.010
123. Fang, Y, Li, C, Shao, R, Yu, H, and Zhang, Q. The role of biomarkers of endothelial activation in predicting morbidity and mortality in patients with severe sepsis and septic shock in intensive care: a prospective observational study. Thromb Res. (2018) 171:149–54. doi: 10.1016/j.thromres.2018.09.059
124. Dickson, K, and Lehmann, C. Inflammatory response to different toxins in experimental sepsis models. Int J Mol Sci. (2019) 20:20. doi: 10.3390/ijms20184341
125. Tang, AL, Peng, Y, Shen, MJ, Liu, XY, Li, S, Xiong, MC, et al. Prognostic role of elevated VEGF in sepsis: a systematic review and meta-analysis. Front Physiol. (2022) 13:941257. doi: 10.3389/fphys.2022.941257
126. van Poelgeest, EP, Dillingh, MR, de Kam, M, Malone, KE, Kemper, M, Stroes, ESG, et al. Characterization of immune cell, endothelial, and renal responses upon experimental human endotoxemia. J Pharmacol Toxicol Methods. (2018) 89:39–46. doi: 10.1016/j.vascn.2017.10.004
127. Aslan, A, van den Heuvel, MC, Stegeman, CA, Popa, ER, Leliveld, AM, Molema, G, et al. Kidney histopathology in lethal human sepsis. Crit Care. (2018) 22:359. doi: 10.1186/s13054-018-2287-3
128. Garofalo, AM, Lorente-Ros, M, Goncalvez, G, Carriedo, D, Ballen-Barragan, A, Villar-Fernandez, A, et al. Histopathological changes of organ dysfunction in sepsis. Intensive Care Med Exp. (2019) 7:45. doi: 10.1186/s40635-019-0236-3
129. Kinoshita, M, Mochizuki, H, and Ono, S. Pulmonary neutrophil accumulation following human endotoxemia. Chest. (1999) 116:1709–15. doi: 10.1378/chest.116.6.1709
130. Kumar, V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. (2020) 11:1722. doi: 10.3389/fimmu.2020.01722
131. Brusletto, BS, Hellerud, BC, Loberg, EM, Goverud, IL, Vege, A, Berg, JP, et al. Traceability and distribution of Neisseria meningitidis DNA in archived post mortem tissue samples from patients with systemic meningococcal disease. BMC Clin Pathol. (2017) 17:10. doi: 10.1186/s12907-017-0049-9
132. Brusletto, BS, Loberg, EM, Hellerud, BC, Goverud, IL, Berg, JP, Olstad, OK, et al. Extensive changes in transcriptomic “fingerprints” and immunological cells in the large organs of patients dying of acute septic shock and multiple organ failure caused by Neisseria meningitidis. Front Cell Infect Microbiol. (2020) 10:42. doi: 10.3389/fcimb.2020.00042
133. Van Wyngene, L, Vandewalle, J, and Libert, C. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med. (2018) 10:e8712. doi: 10.15252/emmm.201708712
134. Chen, L, Welty-Wolf, KE, and Kraft, BD. Nonhuman primate species as models of human bacterial sepsis. Lab Anim. (2019) 48:57–65. doi: 10.1038/s41684-018-0217-2
135. Lupu, F, Keshari, RS, Lambris, JD, and Coggeshall, KM. Crosstalk between the coagulation and complement systems in sepsis. Thromb Res. (2014) 133:S28–31. doi: 10.1016/j.thromres.2014.03.014
136. Taylor, FB Jr, Kinasewitz, GT, and Lupu, F. Pathophysiology, staging and therapy of severe sepsis in baboon models. J Cell Mol Med. (2012) 16:672–82. doi: 10.1111/j.1582-4934.2011.01454.x
137. Seok, J, Warren, HS, Cuenca, AG, Mindrinos, MN, Baker, HV, Xu, W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. (2013) 110:3507–12. doi: 10.1073/pnas.1222878110
138. Takao, K, and Miyakawa, T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. (2015) 112:1167–72. doi: 10.1073/pnas.1401965111
139. Buras, JA, Holzmann, B, and Sitkovsky, M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. (2005) 4:854–65. doi: 10.1038/nrd1854
140. Dyson, A, and Singer, M. Animal models of sepsis: why does preclinical efficacy fail to translate to the clinical setting? Crit Care Med. (2009) 37:S30–7. doi: 10.1097/CCM.0b013e3181922bd3
141. Lewis, AJ, Seymour, CW, and Rosengart, MR. Current murine models of sepsis. Surg Infect. (2016) 17:385–93. doi: 10.1089/sur.2016.021
142. Dejager, L, Pinheiro, I, Dejonckheere, E, and Libert, C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. (2011) 19:198–208. doi: 10.1016/j.tim.2011.01.001
143. Coldewey, SM, Rogazzo, M, Collino, M, Patel, NS, and Thiemermann, C. Inhibition of IkappaB kinase reduces the multiple organ dysfunction caused by sepsis in the mouse. Dis Model Mech. (2013) 6:1031–42. doi: 10.1242/dmm.012435
144. Coletta, C, Modis, K, Olah, G, Brunyanszki, A, Herzig, DS, Sherwood, ER, et al. Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: preclinical studies in a murine model of cecal ligation and puncture. Crit Care. (2014) 18:511. doi: 10.1186/s13054-014-0511-3
145. Miao, H, Chen, S, and Ding, R. Evaluation of the molecular mechanisms of sepsis using proteomics. Front Immunol. (2021) 12:733537. doi: 10.3389/fimmu.2021.733537
146. Seemann, S, Zohles, F, and Lupp, A. Comprehensive comparison of three different animal models for systemic inflammation. J Biomed Sci. (2017) 24:60. doi: 10.1186/s12929-017-0370-8
147. Garcia-Cardena, G, Comander, J, Anderson, KR, Blackman, BR, and Gimbrone, MA Jr. Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc Natl Acad Sci U S A. (2001) 98:4478–85. doi: 10.1073/pnas.071052598
148. Shao, R, Yang, Y, Zhang, Y, Zhao, S, Zheng, Z, and Chen, G. The expression of thioredoxin-1 and inflammatory cytokines in patients with sepsis. Immunopharmacol Immunotoxicol. (2020) 42:280–5. doi: 10.1080/08923973.2020.1755309
149. Ye, X, Ding, J, Zhou, X, Chen, G, and Liu, SF. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med. (2008) 205:1303–15. doi: 10.1084/jem.20071393
150. Margotti, W, Goldim, MPS, Machado, RS, Bagio, E, Dacoregio, C, Bernades, G, et al. Oxidative stress in multiple organs after sepsis in elderly rats. Exp Gerontol. (2022) 160:111705. doi: 10.1016/j.exger.2022.111705
151. Theobaldo, MC, Llimona, F, Petroni, RC, Rios, EC, Velasco, IT, and Soriano, FG. Hypertonic saline solution drives neutrophil from bystander organ to infectious site in polymicrobial sepsis: a cecal ligation and puncture model. PLoS One. (2013) 8:e74369. doi: 10.1371/journal.pone.0074369
152. Zhang, H, Zhi, L, Moochhala, SM, Moore, PK, and Bhatia, M. Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. J Leukoc Biol. (2007) 82:894–905. doi: 10.1189/jlb.0407237
153. Silva, CMS, Wanderley, CWS, Veras, FP, Sonego, F, Nascimento, DC, Goncalves, AV, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. (2021) 138:2702–13. doi: 10.1182/blood.2021011525
154. Rossaint, J, Berger, C, Kraft, F, Van Aken, H, Giesbrecht, N, and Zarbock, A. Hydroxyethyl starch 130/0.4 decreases inflammation, neutrophil recruitment, and neutrophil extracellular trap formation. Br J Anaesth. (2015) 114:509–19. doi: 10.1093/bja/aeu340
155. Hickey, MJ, Granger, DN, and Kubes, P. Inducible nitric oxide synthase (iNOS) and regulation of leucocyte/endothelial cell interactions: studies in iNOS-deficient mice. Acta Physiol Scand. (2001) 173:119–26. doi: 10.1046/j.1365-201X.2001.00892.x
156. Jongman, RM, Zwiers, PJ, van de Sluis, B, van der Laan, M, Moser, J, Zijlstra, JG, et al. Partial deletion of Tie2 affects microvascular endothelial responses to critical illness in a vascular bed and organ-specific way. Shock. (2019) 51:757–69. doi: 10.1097/SHK.0000000000001226
157. Kurniati, NF, van Meurs, M, Vom Hagen, F, Jongman, RM, Moser, J, Zwiers, PJ, et al. Pleiotropic effects of angiopoietin-2 deficiency do not protect mice against endotoxin-induced acute kidney injury. Nephrol Dial Transplant. (2013) 28:567–75. doi: 10.1093/ndt/gfs336
158. Yano, K, Liaw, PC, Mullington, JM, Shih, SC, Okada, H, Bodyak, N, et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med. (2006) 203:1447–58. doi: 10.1084/jem.20060375
159. Hellerud, BC, Olstad, OK, Nielsen, EW, Troseid, AM, Skadberg, O, Thorgersen, EB, et al. Massive organ inflammation in experimental and in clinical meningococcal septic shock. Shock. (2015) 44:458–69. doi: 10.1097/SHK.0000000000000441
160. Xu, H, Ye, X, Steinberg, H, and Liu, SF. Selective blockade of endothelial NF-kappaB pathway differentially affects systemic inflammation and multiple organ dysfunction and injury in septic mice. J Pathol. (2010) 220:490–8. doi: 10.1002/path.2666
161. Lush, CW, Cepinskas, G, Sibbald, WJ, and Kvietys, PR. Endothelial E- and P-selectin expression in iNOS- deficient mice exposed to polymicrobial sepsis. Am J Physiol Gastrointest Liver Physiol. (2001) 280:G291–7. doi: 10.1152/ajpgi.2001.280.2.G291
162. van Meurs, M, Castro, P, Shapiro, NI, Lu, S, Yano, M, Maeda, N, et al. Adiponectin diminishes organ-specific microvascular endothelial cell activation associated with sepsis. Shock. (2012) 37:392–8. doi: 10.1097/SHK.0b013e318248225e
163. Wen, X, Li, S, Frank, A, Chen, X, Emlet, D, Hukriede, NA, et al. Time-dependent effects of histone deacetylase inhibition in sepsis-associated acute kidney injury. Intensive Care Med Exp. (2020) 8:9. doi: 10.1186/s40635-020-0297-3
164. David, S, Park, JK, Meurs, M, Zijlstra, JG, Koenecke, C, Schrimpf, C, et al. Acute administration of recombinant angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine. (2011) 55:251–9. doi: 10.1016/j.cyto.2011.04.005
165. Maas, SL, Soehnlein, O, and Viola, JR. Organ-specific mechanisms of transendothelial neutrophil migration in the lung, liver, kidney, and aorta. Front Immunol. (2018) 9:2739. doi: 10.3389/fimmu.2018.02739
166. Rossaint, J, and Zarbock, A. Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J Innate Immun. (2013) 5:348–57. doi: 10.1159/000345943
167. Carrithers, M, Tandon, S, Canosa, S, Michaud, M, Graesser, D, and Madri, JA. Enhanced susceptibility to endotoxic shock and impaired STAT3 signaling in CD31-deficient mice. Am J Pathol. (2005) 166:185–96. doi: 10.1016/S0002-9440(10)62243-2
168. Margraf, A, Herter, JM, Kuhne, K, Stadtmann, A, Ermert, T, Wenk, M, et al. 6% hydroxyethyl starch (HES 130/0.4) diminishes glycocalyx degradation and decreases vascular permeability during systemic and pulmonary inflammation in mice. Crit Care. (2018) 22:111. doi: 10.1186/s13054-017-1846-3
169. Vandewalle, J, Steeland, S, Van Ryckeghem, S, Eggermont, M, Van Wonterghem, E, Vandenbroucke, RE, et al. A study of cecal ligation and puncture-induced sepsis in tissue-specific tumor necrosis factor receptor 1-deficient mice. Front Immunol. (2019) 10:2574. doi: 10.3389/fimmu.2019.02574
170. Bomsztyk, K, Mar, D, An, D, Sharifian, R, Mikula, M, Gharib, SA, et al. Experimental acute lung injury induces multi-organ epigenetic modifications in key angiogenic genes implicated in sepsis-associated endothelial dysfunction. Crit Care. (2015) 19:225. doi: 10.1186/s13054-015-0943-4
171. Rumienczyk, I, Kulecka, M, Ostrowski, J, Mar, D, Bomsztyk, K, Standage, SW, et al. Multi-organ transcriptome dynamics in a mouse model of cecal ligation and puncture-induced polymicrobial sepsis. J Inflamm Res. (2021) 14:2377–88. doi: 10.2147/JIR.S307305
172. van Meurs, M, Kurniati, NF, Wulfert, FM, Asgeirsdottir, SA, de Graaf, IA, Satchell, SC, et al. Shock-induced stress induces loss of microvascular endothelial Tie2 in the kidney which is not associated with reduced glomerular barrier function. Am J Physiol Renal Physiol. (2009) 297:F272–81. doi: 10.1152/ajprenal.00137.2009
173. Yang, K, Fan, M, Wang, X, Xu, J, Wang, Y, Gill, PS, et al. Lactate induces vascular permeability via disruption of VE-cadherin in endothelial cells during sepsis. Sci Adv. (2022) 8:eabm8965. doi: 10.1126/sciadv.abm8965
174. Levi, M, and van der Poll, T. Coagulation and sepsis. Thromb Res. (2017) 149:38–44. doi: 10.1016/j.thromres.2016.11.007
175. Shi, J, Tang, Y, Liang, F, Liu, L, Liang, N, Yang, X, et al. NLRP3 inflammasome contributes to endotoxin-induced coagulation. Thromb Res. (2022) 214:8–15. doi: 10.1016/j.thromres.2022.04.001
176. Vago, JP, Zaidan, I, Perucci, LO, Brito, LF, Teixeira, LC, Silva, CMS, et al. Plasmin and plasminogen prevent sepsis severity by reducing neutrophil extracellular traps and systemic inflammation. JCI Insight. (2023) 8:8. doi: 10.1172/jci.insight.166044
177. Valladolid, C, Martinez-Vargas, M, Sekhar, N, Lam, F, Brown, C, Palzkill, T, et al. Modulating the rate of fibrin formation and clot structure attenuates microvascular thrombosis in systemic inflammation. Blood Adv. (2020) 4:1340–9. doi: 10.1182/bloodadvances.2020001500
178. Terada, Y, Eguchi, Y, Nosaka, S, Toba, T, Nakamura, T, and Shimizu, Y. Capillary endothelial thrombomodulin expression and fibrin deposition in rats with continuous and bolus lipopolysaccharide administration. Lab Investig. (2003) 83:1165–73. doi: 10.1097/01.lab.0000080606.96797.a5
179. Jesmin, S, Gando, S, Zaedi, S, Prodhan, SH, Sawamura, A, Miyauchi, T, et al. Protease-activated receptor 2 blocking peptide counteracts endotoxin-induced inflammation and coagulation and ameliorates renal fibrin deposition in a rat model of acute renal failure. Shock. (2009) 32:626–32. doi: 10.1097/SHK.0b013e3181a5359c
180. Erlich, J, Fearns, C, Mathison, J, Ulevitch, RJ, and Mackman, N. Lipopolysaccharide induction of tissue factor expression in rabbits. Infect Immun. (1999) 67:2540–6. doi: 10.1128/IAI.67.5.2540-2546.1999
181. Mackman, N, Sawdey, MS, Keeton, MR, and Loskutoff, DJ. Murine tissue factor gene expression in vivo. Tissue and cell specificity and regulation by lipopolysaccharide. Am J Pathol. (1993) 143:76–84.
182. Zeng, Q, Mousa, M, Nadukkandy, AS, Franssens, L, Alnaqbi, H, Alshamsi, FY, et al. Understanding tumour endothelial cell heterogeneity and function from single-cell omics. Nat Rev Cancer. (2023) 23:544–64. doi: 10.1038/s41568-023-00591-5
183. Hu, Y, Zhang, Y, Liu, Y, Gao, Y, San, T, Li, X, et al. Advances in application of single-cell RNA sequencing in cardiovascular research. Front Cardiovasc Med. (2022) 9:905151. doi: 10.3389/fcvm.2022.905151
184. Paik, DT, Cho, S, Tian, L, Chang, HY, and Wu, JC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. (2020) 17:457–73. doi: 10.1038/s41569-020-0359-y
185. Chinnaiyan, AM, Huber-Lang, M, Kumar-Sinha, C, Barrette, TR, Shankar-Sinha, S, Sarma, VJ, et al. Molecular signatures of sepsis: multiorgan gene expression profiles of systemic inflammation. Am J Pathol. (2001) 159:1199–209. doi: 10.1016/S0002-9440(10)62505-9
186. Yang, Y, Yin, F, Hang, Q, Dong, X, Chen, J, Li, L, et al. Regulation of endothelial permeability by glutathione S-transferase pi against actin polymerization. Cell Physiol Biochem. (2018) 45:406–18. doi: 10.1159/000486918
187. Barker, G, Leeuwenburgh, C, Brusko, T, Moldawer, L, Reddy, ST, and Guirgis, FW. Lipid and lipoprotein dysregulation in sepsis: clinical and mechanistic insights into chronic critical illness. J Clin Med. (2021) 10:10. doi: 10.3390/jcm10081693
188. Van den Berghe, G. Should glucocorticoid-induced hyperglycemia be treated in patients with septic shock? JAMA. (2010) 303:365–6. doi: 10.1001/jama.2010.41
189. Wei, JX, Jiang, HL, and Chen, XH. Endothelial cell metabolism in sepsis. World J Emerg Med. (2023) 14:10–6. doi: 10.5847/wjem.j.1920-8642.2023.019
190. Gong, Y, Lan, H, Yu, Z, Wang, M, Wang, S, Chen, Y, et al. Blockage of glycolysis by targeting PFKFB3 alleviates sepsis-related acute lung injury via suppressing inflammation and apoptosis of alveolar epithelial cells. Biochem Biophys Res Commun. (2017) 491:522–9. doi: 10.1016/j.bbrc.2017.05.173
191. Wang, L, Cao, Y, Gorshkov, B, Zhou, Y, Yang, Q, Xu, J, et al. Ablation of endothelial Pfkfb3 protects mice from acute lung injury in LPS-induced endotoxemia. Pharmacol Res. (2019) 146:104292. doi: 10.1016/j.phrs.2019.104292
192. Mithal, LB, Palac, HL, Yogev, R, Ernst, LM, and Mestan, KK. Cord blood acute phase reactants predict early onset neonatal sepsis in preterm infants. PLoS One. (2017) 12:e0168677. doi: 10.1371/journal.pone.0168677
193. Das, N, Schmidt, TA, Krawetz, RJ, and Dufour, A. Proteoglycan 4: from mere lubricant to regulator of tissue homeostasis and inflammation: does proteoglycan 4 have the ability to buffer the inflammatory response? BioEssays. (2019) 41:e1800166. doi: 10.1002/bies.201800166
194. Richendrfer, H, and Jay, GD. Lubricin as a therapeutic and potential biomarker in sepsis. Crit Care Clin. (2020) 36:55–67. doi: 10.1016/j.ccc.2019.08.005
195. Janosevic, D, Myslinski, J, McCarthy, TW, Zollman, A, Syed, F, Xuei, X, et al. The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. eLife. (2021) 10:10. doi: 10.7554/eLife.62270
196. Chen, G, Ren, C, Xiao, Y, Wang, Y, Yao, R, Wang, Q, et al. Time-resolved single-cell transcriptomics reveals the landscape and dynamics of hepatic cells in sepsis-induced acute liver dysfunction. JHEP Rep. (2023) 5:100718. doi: 10.1016/j.jhepr.2023.100718
197. Zhang, L, Gao, S, White, Z, Dai, Y, Malik, AB, and Rehman, J. Single-cell transcriptomic profiling of lung endothelial cells identifies dynamic inflammatory and regenerative subpopulations. JCI Insight. (2022) 7:7. doi: 10.1172/jci.insight.158079
198. Pierrakos, C, Velissaris, D, Bisdorff, M, Marshall, JC, and Vincent, JL. Biomarkers of sepsis: time for a reappraisal. Crit Care. (2020) 24:287. doi: 10.1186/s13054-020-02993-5
199. Bernard, GR, Francois, B, Mira, JP, Vincent, JL, Dellinger, RP, Russell, JA, et al. Evaluating the efficacy and safety of two doses of the polyclonal anti-tumor necrosis factor-alpha fragment antibody AZD9773 in adult patients with severe sepsis and/or septic shock: randomized, double-blind, placebo-controlled phase IIb study*. Crit Care Med. (2014) 42:504–11. doi: 10.1097/CCM.0000000000000043
200. Besnier, E, Brakenhielm, E, Richard, V, and Tamion, F. Does anti-VEGF bevacizumab improve survival in experimental sepsis? Crit Care. (2017) 21:163. doi: 10.1186/s13054-017-1734-x
201. Jeong, SJ, Han, SH, Kim, CO, Choi, JY, and Kim, JM. Anti-vascular endothelial growth factor antibody attenuates inflammation and decreases mortality in an experimental model of severe sepsis. Crit Care. (2013) 17:R97. doi: 10.1186/cc12742
202. Opal, SM, Laterre, PF, Francois, B, LaRosa, SP, Angus, DC, Mira, JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. (2013) 309:1154–62. doi: 10.1001/jama.2013.2194
203. Ranieri, VM, Thompson, BT, Barie, PS, Dhainaut, JF, Douglas, IS, Finfer, S, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. (2012) 366:2055–64. doi: 10.1056/NEJMoa1202290
204. Rice, TW, Wheeler, AP, Bernard, GR, Vincent, JL, Angus, DC, Aikawa, N, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. (2010) 38:1685–94. doi: 10.1097/CCM.0b013e3181e7c5c9
205. Vincent, JL, Francois, B, Zabolotskikh, I, Daga, MK, Lascarrou, JB, Kirov, MY, et al. Effect of a recombinant human soluble thrombomodulin on mortality in patients with sepsis-associated coagulopathy: the SCARLET randomized clinical trial. JAMA. (2019) 321:1993–2002. doi: 10.1001/jama.2019.5358
206. Meizlish, ML, Franklin, RA, Zhou, X, and Medzhitov, R. Tissue homeostasis and inflammation. Annu Rev Immunol. (2021) 39:557–81. doi: 10.1146/annurev-immunol-061020-053734
207. Asgeirsdottir, SA, Kamps, JA, Bakker, HI, Zwiers, PJ, Heeringa, P, van der Weide, K, et al. Site-specific inhibition of glomerulonephritis progression by targeted delivery of dexamethasone to glomerular endothelium. Mol Pharmacol. (2007) 72:121–31. doi: 10.1124/mol.107.034140
208. Ding, BS, Hong, N, Christofidou-Solomidou, M, Gottstein, C, Albelda, SM, Cines, DB, et al. Anchoring fusion thrombomodulin to the endothelial lumen protects against injury-induced lung thrombosis and inflammation. Am J Respir Crit Care Med. (2009) 180:247–56. doi: 10.1164/rccm.200809-1433OC
209. Hennigs, JK, Matuszcak, C, Trepel, M, and Korbelin, J. Vascular endothelial cells: heterogeneity and targeting approaches. Cells. (2021) 10:10. doi: 10.3390/cells10102712
210. Kowalski, PS, Zwiers, PJ, Morselt, HW, Kuldo, JM, Leus, NG, Ruiters, MH, et al. Anti-VCAM-1 SAINT-O-somes enable endothelial-specific delivery of siRNA and downregulation of inflammatory genes in activated endothelium in vivo. J Control Release. (2014) 176:64–75. doi: 10.1016/j.jconrel.2013.12.029
211. Li, R, Kowalski, PS, Morselt, HWM, Schepel, I, Jongman, RM, Aslan, A, et al. Endothelium-targeted delivery of dexamethasone by anti-VCAM-1 SAINT-O-somes in mouse endotoxemia. PLoS One. (2018) 13:e0196976. doi: 10.1371/journal.pone.0196976
212. Dussaq, AM, Kennell, T Jr, Eustace, NJ, Anderson, JC, Almeida, JS, and Willey, CD. Kinomics toolbox-a web platform for analysis and viewing of kinomic peptide array data. PLoS One. (2018) 13:e0202139. doi: 10.1371/journal.pone.0202139
213. Luxen, M, Zwiers, PJ, Meester, F, Jongman, RM, Kuiper, T, Moser, J, et al. Unique miRNome and transcriptome profiles underlie microvascular heterogeneity in mouse kidney. Am J Physiol Renal Physiol. (2023) 325:F299–316. doi: 10.1152/ajprenal.00005.2023
214. Seymour, CW, Kennedy, JN, Wang, S, Chang, CH, Elliott, CF, Xu, Z, et al. Derivation, validation, and potential treatment implications of novel clinical phenotypes for sepsis. JAMA. (2019) 321:2003–17. doi: 10.1001/jama.2019.5791
215. Marti-Carvajal, AJ, Sola, I, Lathyris, D, and Cardona, AF. Human recombinant activated protein C for severe sepsis. Cochrane Database Syst Rev. (2012):CD004388. doi: 10.1002/14651858.CD004388.pub5
216. Kudo, D, Goto, T, Uchimido, R, Hayakawa, M, Yamakawa, K, Abe, T, et al. Coagulation phenotypes in sepsis and effects of recombinant human thrombomodulin: an analysis of three multicentre observational studies. Crit Care. (2021) 25:114. doi: 10.1186/s13054-021-03541-5
217. Alexander, MP, Mangalaparthi, KK, Madugundu, AK, Moyer, AM, Adam, BA, Mengel, M, et al. Acute kidney injury in severe COVID-19 has similarities to sepsis-associated kidney injury: a multi-omics study. Mayo Clin Proc. (2021) 96:2561–75. doi: 10.1016/j.mayocp.2021.07.001
218. Delorey, TM, Ziegler, CGK, Heimberg, G, Normand, R, Yang, Y, Segerstolpe, A, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. (2021) 595:107–13. doi: 10.1038/s41586-021-03570-8
219. Fernandez, S, Moreno-Castano, AB, Palomo, M, Martinez-Sanchez, J, Torramade-Moix, S, Tellez, A, et al. Distinctive biomarker features in the Endotheliopathy of COVID-19 and septic syndromes. Shock. (2022) 57:95–105. doi: 10.1097/SHK.0000000000001823
220. Filbin, MR, Mehta, A, Schneider, AM, Kays, KR, Guess, JR, Gentili, M, et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell Rep Med. (2021) 2:100287. doi: 10.1016/j.xcrm.2021.100287
221. Ito, T, Kakuuchi, M, and Maruyama, I. Endotheliopathy in septic conditions: mechanistic insight into intravascular coagulation. Crit Care. (2021) 25:95. doi: 10.1186/s13054-021-03524-6
222. Muus, C, Luecken, MD, Eraslan, G, Sikkema, L, Waghray, A, Heimberg, G, et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med. (2021) 27:546–59. doi: 10.1038/s41591-020-01227-z
223. Antonakos, N, Gilbert, C, Theroude, C, Schrijver, IT, and Roger, T. Modes of action and diagnostic value of miRNAs in sepsis. Front Immunol. (2022) 13:951798. doi: 10.3389/fimmu.2022.951798
224. Benz, F, Roy, S, Trautwein, C, Roderburg, C, and Luedde, T. Circulating microRNAs as biomarkers for sepsis. Int J Mol Sci. (2016) 17:17. doi: 10.3390/ijms17010078
225. Formosa, A, Turgeon, P, and Dos Santos, CC. Role of miRNA dysregulation in sepsis. Mol Med. (2022) 28:99. doi: 10.1186/s10020-022-00527-z
226. Szilagyi, B, Fejes, Z, Pocsi, M, Kappelmayer, J, and Nagy, B Jr. Role of sepsis modulated circulating microRNAs. EJIFCC. (2019) 30:128–45.
Keywords: sepsis, multiple organ dysfunction syndrome, endothelial cell heterogeneity, human studies, animal models, -omics, pharmacology, biomarkers
Citation: Cleuren A and Molema G (2023) Organotypic heterogeneity in microvascular endothelial cell responses in sepsis—a molecular treasure trove and pharmacological Gordian knot. Front. Med. 10:1252021. doi: 10.3389/fmed.2023.1252021
Edited by:
W. Conrad Liles, University of Washington, United StatesReviewed by:
Eiji Kawamoto, Mie University Hospital, JapanCopyright © 2023 Cleuren and Molema. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Audrey Cleuren, QXVkcmV5LUNsZXVyZW5Ab21yZi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.